

Abstract
Ceratonova shasta is an important myxozoan pathogen affecting the health of salmonid fishes in the Pacific Northwest of North America. Ceratonova shasta exists as a complex of host-specific genotypes, some with low to moderate virulence, and one that causes a profound, lethal infection in susceptible hosts. High throughput sequencing methods are powerful tools for discovering the genetic basis of these host/virulence differences, but deep sequencing of myxozoans has been challenging due to extremely fast molecular evolution of this group, yielding strongly divergent sequences that are difficult to identify, and unavoidable host contamination. We designed and optimized different bioinformatic pipelines to address these challenges. We obtained a unique set of comprehensive, host-free myxozoan RNA-seq data from C. shasta genotypes of varying virulence from different salmonid hosts. Analyses of transcriptome-wide genetic distances and maximum likelihood multigene phylogenies elucidated the evolutionary relationship between lineages and demonstrated the limited resolution of the established Internal Transcribed Spacer marker for C. shasta genotype identification, as this marker fails to differentiate between biologically distinct genotype II lineages from coho salmon and rainbow trout. We further analyzed the data sets based on polymorphisms in two gene groups related to virulence: cell migration and proteolytic enzymes including their inhibitors. The developed single-nucleotide polymorphism-calling pipeline identified polymorphisms between genotypes and demonstrated that variations in both motility and protease genes were associated with different levels of virulence of C. shasta in its salmonid hosts. The prospective use of proteolytic enzymes as promising candidates for targeted interventions against myxozoans in aquaculture is discussed. We developed host-free transcriptomes of a myxozoan model organism from strains that exhibited different degrees of virulence, as a unique source of data that will foster functional gene analyses and serve as a base for the development of potential therapeutics for efficient control of these parasites.
Keywords: Myxozoa, aquaculture, cell migration/motility, proteases, SNPs
Introduction
Myxozoans are a group of cnidarians that emerged as parasites of aquatic invertebrate hosts (annelids and byrozoans) ∼650 Ma (Holzer et al. 2018). Following the emergence of vertebrates, myxozoans acquired these as secondary hosts, predominantly fish, which fostered massive host-associated diversification and led to the distinct success of the Myxozoa. Their presently known diversity accounts for approximately a fifth of all cnidarian species (Atkinson, Bartholomew, et al. 2018) but may be greatly underestimated (Hartikainen et al. 2016). Myxozoans are extremely reduced in size and body plan, produce spores as transmission stages, but retain the nematocysts (polar capsules) present in all Cnidaria, both free-living and parasitic (Holland et al. 2011; Shpirer et al. 2018). Myxozoans are especially known for the diseases they can cause in wild and cultured fish.
Salmonid fishes are of significant economic and cultural value in the Pacific Northwest of North America. In many rivers, these fish are exposed to the myxozoan parasite, Ceratonova shasta (Noble, 1950) (syn. Ceratomyxa shasta), which affects wild and artificially reared salmon and trout. This microscopic cnidarian has a life cycle that alternates between actinospores, which develop in the annelid host, and myxospores, which develop in the vertebrate host—various species of Pacific salmon and trout (Bartholomew et al. 1997). The parasite penetrates through the gills of the fish, enters the bloodstream, then invades all layers of the gut, where it proliferates, and can cause severe enteronecrosis with gross lesions of swollen, necrotic, and hemorrhagic intestine (Bjork and Bartholomew 2009a, 2010). Ceratonova shasta is one of the most virulent myxozoans known, with mortalities approaching 100% in susceptible species/stocks (Hallett and Bartholomew 2012), with an infectious dose of a single parasite actinospore capable of causing a lethal infection (Bjork and Bartholomew 2009a).
Several abiotic and biotic factors affect the severity of disease caused by C. shasta: host stock origin (sympatric or allopatric with the parasite), water temperature, water flow, actinospore density (Bjork and Bartholomew 2009a, 2009b; Hallett et al. 2012; Ray et al. 2012), and parasite genotype (Hurst and Bartholomew 2012). The parasite occurs across the Pacific Northwest in at least three host-specific genotypes (0, I, and II; Atkinson, Hallett, et al. 2018; Stinson et al. 2018), which have different levels of virulence (i.e., mortality, proliferation, and pathogenicity) in different salmonid species and strains. These genotypes are presently identified based on single-nucleotide polymorphisms (SNPs) in the Internal Transcribed Spacer region 1 (ITS-1) of the ribosomal DNA array (Atkinson and Bartholomew 2010a, 2010b). Although dependent on parasite dose and fish strain, the most virulent genotypes are type I (specific for Chinook salmon, Oncorhynchus tschawytscha) and type II, a generalist that can infect up to six different species but is dominant in coho salmon (Oncorhynchus kisutch) (Stinson et al. 2018). Two biotypes of genotype II, IIC, and IIR are differentiated by host specificity and virulence: Both cause disease in susceptible allopatric rainbow trout (Oncorhynchus mykiss), but IIC causes a dose-dependent mortality in sympatric coho salmon, whereas IIR can only infect coho salmon to a limited extent (Hurst and Bartholomew 2012); there are no ITS-1 molecular differences to distinguish these biotypes. The most genetically distinct strain, genotype 0, is less virulent and produces chronic infections in O. mykiss stocks, both sympatric and allopatric with the parasite, with low proliferation rates and few clinical signs (Atkinson and Bartholomew 2010b; Stinson et al. 2018). The mechanisms accounting for these virulence differences in C. shasta genotypes are unknown.
Virulence factors are molecules responsible for the pathogenicity of an organism—its ability to cause disease and mortality in the host (e.g., Casadevall and Pirofski 1999). These factors are important in different aspects of the life history of a parasite, for example, adhesion, invasion, migration, or host immune evasion (e.g., McKerrow et al. 2006; Bouzid et al. 2013). Motility genes and proteases are generally considered candidate virulence factors (e.g., Barragan and Sibley 2002; McKerrow et al. 2006; Bouzid et al. 2013). Virulence factors vary both among and within species of parasites: Different factors may be expressed by different genotypes or strains, with parasite populations often able to be ranked from highly virulent to avirulent (e.g., Ali et al. 2007; Dardé 2008). Transcriptomic and genomic technologies are enabling novel approaches to compare parasite strains and identify potential virulence factors (e.g., Eichenberger et al. 2017), which can then become targets for drug and chemotherapeutic strategies for disease control (Seib et al. 2009; Mennerat et al. 2010).
Only recently have genomic and transcriptomic data become available for myxozoans (e.g., Yang et al. 2014; Chang et al. 2015; Foox et al. 2015). Myxozoans are unculturable, obligate endoparasites, thus unambiguous sequencing and data analysis have proven extremely difficult, given the presence of contaminating host tissue. We solved several fundamental challenges of host contamination in our ‘omics studies of different genotypes of C. shasta by taking advantage of a novel aspect of its biology: that less-contaminated, metabolically active parasite material is present in ascitic fluid, produced in systemic infections of type IIR in susceptible rainbow trout. Herein, we report a novel method for removal of host contamination in a two-step bioinformatic process, at both read and transcript levels. We then used this workflow to characterize genetic variation among RNA-seq data from genotypes I, IIC, and IIR, with the aim to identify 1) if there are genetic differences that support the separation of biotype II into IIR and IIC and 2) which category of transcript (proteases or motility factors) is most variable between genotypes, and thereby identify which category of genes is under highest selection pressure. For broader context, we reconstructed the evolutionary history of C. shasta genotypes based on phylogenomic analyses.
Materials and Methods
Sampling and ITS-1 Ribosomal DNA Genotyping
We collected different C. shasta genotypes from infected fish: allopatric rainbow trout O. mykiss (Roaring River Hatchery stock, ascitic fluid, genotype II, biotype IIR); sympatric Chinook salmon Oncorhynchus tshawytscha (Iron Gate Hatchery stock, intestine, genotype I); and sympatric coho salmon Oncorhynchus kisutch (Iron Gate Hatchery stock, intestine, genotype II, biotype IIC) (table 1). All fish were euthanized with an overdose of buffered MS-222. Ascitic fluid was collected by syringe from the abdominal cavity (fig. 1) and intestinal tissue was dissected out. Wet mounts of tissues were examined by microscope to confirm presence of parasite developmental stages, that is, presporogonic, sporogonic, and/or mature spores of C. shasta. Ascitic fluid was centrifuged at 8,000 rpm for 2 min. The supernatant was removed, and the pellet was suspended in RNA later (Qiagen, Valencia, CA) and stored at −80 °C until RNA extraction. Similarly, ∼10-mm sections of intestines were stored in RNA later. Replicates of all samples were taken for DNA analyses and stored at −20 °C until extraction, using the DNeasy Blood and Tissue kit (Qiagen). The parasite ITS-1 genotype composition in each sample was confirmed by polymerase chain reaction amplification and Sanger sequencing (GenBank accession numbers: MN173024 Cs-genotype-I and MN173025 Cs-genotype II; Atkinson and Bartholomew 2010a). This study was carried out in accordance to the recommendations of Oregon State University Institutional Animal Care and Use Committee under approval ACUP #4666.
Table 1.
Sequencing, Filtering, and Read Mapping Results from Transcriptomes Obtained During This Study.
| Ceratonova shasta Genotype | IIR Reference (RBT6) | IIR (RBTC16) | IIR (RBTJ7) | IIC | I |
|---|---|---|---|---|---|
| SRA Run Acc. Num. BioProject: PRJNA241036 | SRR6782113 | SRR1575205 | SRR1205836 | SRR1573049 | SRR1461768 |
| Tissue | Ascitic fluid | Intestine | |||
| Fish host | Allopatric rainbow trout (Oncorhynchus mykiss) | Sympatric coho salmon (Oncorhynchus kisutch) | Sympatric Chinook salmon (Oncorhynchus tshawytscha) | ||
| Fish origin (stock) | Roaring River Hatchery, OR | Iron Gate Fish Hatchery, CA | |||
| Location of exposure | Willamette River (WR), OR | Keno Eddy, Upper Klamath River (UKR), CA | Lower Klamath River (LKR), CA | Lower Klamath River (LKR), CA | Lower Klamath River (LKR), CA |
| Year | 2015 | 2014 | 2014 | 2014 | 2014 |
| Illumina sequencer | HiSeq 3000 | HiSeq 2000 | |||
| Total number of reads (paired) | 788,875,442 | 15,639,718 | 16,728,880 | 41,920,212 | 44,310,970 |
| Reads after quality filtering | 759,877,916 | 3,027,123 | 6,039,071 | 12,139,060 | 17,729,817 |
| Reads C. shasta origin | 452,498,284 | 1,829,473 | 1,968,164 | 1,552,718 | 787,739 |
| Reads fish host origin | 118,745,658 | 287,874 | 1,949,421 | 6,890,494 | 12,448,482 |
| Reads NHP (neither host nor parasite) | 182,816,267 | 900,957 | 2,112,865 | 3,681,236 | 4,487,947 |
| Reads match both C. shasta and host equally well | 5,817,707 | 8,819 | 8,621 | 14,612 | 5,649 |
Fig. 1.
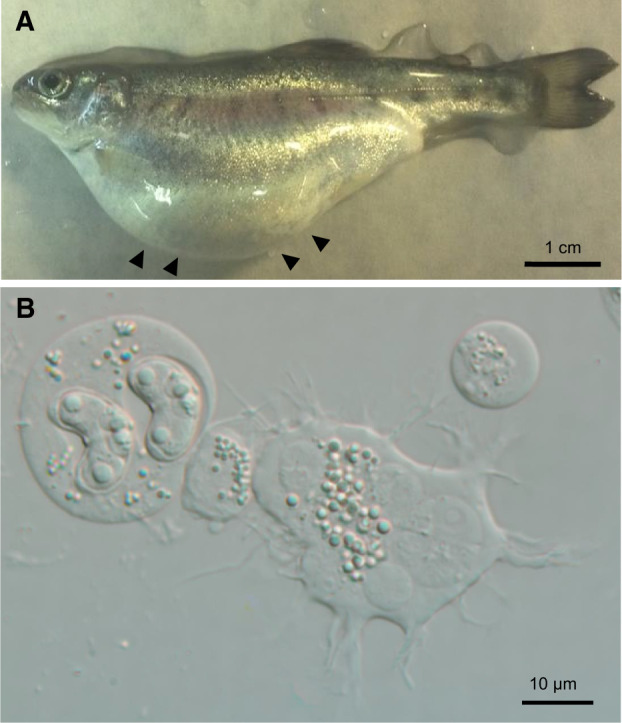
(A) Rainbow trout infected with Ceratonova shasta genotype IIR, showing swollen abdomen due to accumulation of ascitic fluid in the visceral cavity. (B) Ascitic fluid rich in C. shasta presporogonic, sporogonic, and spore stages.
Transcriptome Library Prep and Next-Generation Sequencing
RNA from each of the five fish samples was extracted using a High Pure RNA Tissue Kit (Roche, Switzerland) and treated on-column with DNAse I to remove genomic DNA. RNA was quantified using a spectrophotometer (NanoDrop Technologies, Wilmington, DE) and 1 µg of total RNA was used downstream. Samples (table 1) were analyzed in two sets: a deeply sequenced C. shasta genotype IIR reference transcriptome from one rainbow trout (RBT6; Willamette River) and low coverage C. shasta transcriptomes from genotypes I (Chinook salmon, Lower Klamath River [LKR]), IIC (Coho salmon, LKR), 2× IIR (RBTC16—Upper Klamath River [UKR]; RBTJ7—LKR, both from rainbow trout).
Reference Transcriptome of C. shasta Genotype IIR
RNA (RBT6) from rainbow trout was submitted to Oregon State University’s Center for Genome Research and Biocomputing (OSU CGRB) for directional library preparation and sequencing. A cDNA library was made using the PrepXTM mRNA Strand Specific Library Prep Kit with poly-A selection in an Apollo 324 Next-Generation Sequencing Library Prep System (Wafergen, Fremont, CA). The library was quantified and size checked using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA) and Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). The reference library was pair-end sequenced on one lane of HiSeq 3000 (Illumina, San Diego, CA).
Lower-Coverage Transcriptomes of Genotypes I, IIC, and 2 × IIR
Four normalized custom libraries were prepared following Meyer et al. (2009). In summary, first strand cDNA was synthesized using SuperScript II Reverse Transcriptase (Life Technologies, Carlsbad, CA), then normalized using DSN (double-strand-specific nuclease, Evrogen, Moscow, Russia) to decrease the prevalence of the most abundant, repeated transcripts and increase discovery of rare transcripts; we considered that this was important due to contamination by host RNA. Following normalization, the cDNA was amplified and fragmented using NEBNext dsDNA Fragmentase (New England Biolabs, Ipswich, MA). Oligonucleotide adaptors were ligated to the fragmented cDNA. Constructs were amplified and barcoded with HiSeq oligos and selected by size (350–550 bp) on a 2% Tris-acetate-EDTA agarose gel. The four libraries were pair-end sequenced by Illumina HiSeq 2000 at CGRB, using one-sixth of a lane for each.
Custom scripts used in this study were written by E.M. (available at https://github.com/Eli-Meyer, last accessed October 1, 2015 or in supplementary material 1, Supplementary Material online). Analyses were performed on the OSU CGRB computing cluster. We used bit score thresholds instead of e-values for our BLAST searches to avoid potential problems with different database sizes (Pearson 2013). Our bioinformatic pipeline is summarized in figure 2.
Fig. 2.

The workflow developed during this study for host contaminant filtration, assembly, and annotation. We filtered out host contamination at both the read level before assembly, using read mappings to reference genomes of the parasite Ceratonova shasta and host Oncorhynchus mykiss, and at the contig level after assembly, using BlastN against the same reference genomes. We produced two versions of the reference transcriptome: 1) C. shasta only, which is the more conservative assembly from only those reads that mapped to the parasite genome and 2) C. shasta + NHP, which was assembled from both the reads that mapped to the C. shasta genome and those that mapped to NHP (neither host nor parasite).
Removal of Host Contamination at Read Level: Filtering and Mapping of Reads
Raw Illumina pair-end reads from the five libraries were filtered for adaptor sequences (score 15–20), low-quality base calls (>20–60 bases with quality score below 20), and homopolymer poly-A (>50 homopolymer repeats), using custom Perl scripts. We then filtered out host reads prior to assembly or SNPs calling, by mapping against two references: 1) combined O. mykiss genome (Berthelot et al. 2014; http://www.genoscope.cns.fr/trout/data/, last accessed April 21, 2016; BioProject accession number PRJEB4421 version GCA_900005704.1) and the mitochondrial genome (Zardoya et al. 1995; BioProject accession number PRJNA11824 version NC_001717.1); 2) our reference C. shasta genome, which was obtained from purified myxospores and prefiltered using BlastN (score = 150) against the same host reference (Version Velvet2015-93, 14,586 sequences, 185–452,519 bp length, N50 = 36,283, total size 69.8 Mb; Atkinson S., Alama-Bermejo G., Bartholomew J.L., in preparation). We mapped the high-quality, filtered transcriptome reads against the references using gmapper (Shrimp v 2.0; David et al. 2011), reporting one hit per read and using the local alignment option. We then filtered the resulting .sam file to exclude ambiguous, short, and weak matches: minimum match length 45 bp in any 50 bp alignment. We compared the filtered .sam files to determine if reads best matched host, parasite, or neither host nor parasite (NHP) and binned reads into respective .fastq files. Ceratonova shasta and NHP reads from each of the five data sets were used for subsequent analyses; host reads were excluded.
Reference Transcriptome De Novo Assembly and Contaminant Removal at the Contig Level
We generated two reference assemblies of the deeply sequenced IIR data set (RBT6): a more conservative assembly of only reads that best matched the C. shasta genome assembly and a less conservative version with reads that matched the C. shasta genome, plus the NHP reads (fig. 2). The less conservative assembly was made to recover parasite transcripts missed due to lack of completeness/coverage of the C. shasta genome. Reads were de novo assembled into contigs using the Trinity v2.2.0 (2016) pipeline (Haas et al. 2013).
We used a second level of contaminant filtering, by BlastN comparison of our assembled transcriptomes against the same reference genome databases used for read filtering (rainbow trout and C. shasta). We used a custom script that identified the most likely contig origin (bit score >100). The script assigned contigs to one of three bins based on the best match: 1) target (=C. shasta), 2) contaminant (=rainbow trout), and 3) NHP (=bit score below threshold for both databases). Additionally, we used taxon ID annotations to remove contigs of prokaryotes, viruses, fungi, algae, and protists. For this purpose, we downloaded the nonredundant (nr) database (March 3, 2014) and removed all taxa belonging to the before-mentioned groups by filtering on phylum level (level 4 in National Center for Biotechnology Information [NCBI]), using a >50-bit score.
Fragments per kilobase of transcript per million mapped reads and transcripts per million were calculated using an alignment-based quantification method, RNA-Seq by Expectation Maximization (Li and Dewey 2011).
Reference Transcriptome Annotation and Completeness
We used three methods to annotate contigs in the two assemblies: gene names (putative functional annotations), Gene Ontology (GO) terms, and taxon identification. Gene names were assigned by comparison with the UniProt database (Release 2015), using BlastX (NCBI; cutoff e-value 10−6). We used UniProt over larger databases such as nr because it has more accurate and fewer redundant gene function annotations. We assigned gene names to contigs based on the best BLAST match after excluding database hits whose annotations included uninformative terms, for example, “unknown,” “uncharacterized,” and “predicted protein” (ambiguous terms filtering step; full list specified in supplementary material 2, Supplementary Material online). We assigned GO terms (http://geneontology.org/, last accessed June 23, 2015) using the same best BLAST hits from UniProt. UniProt is equipped with GO terms and annotations; hence, it was efficient to use one database for both gene and GO annotations. We annotated contigs with available taxon identification to phylum level (level 4 in NCBI) using the best hits from a BlastX search of the NCBI nr database (bit score > 50).
We ran the Core Eukaryotic Genes Mapping Approach (CEGMA) analysis (Parra et al. 2007) using the 248 core eukaryotic proteins to test for completeness of the reference transcriptome, with default settings, except for specifying max_intron size of 2,630 to account for known intron size information of the myxozoan Myxobolus cerebralis (see Chang et al. 2015).
Phylogenomic Analyses
The raw reads were checked for quality using FastQC (http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/, accessed August 14, 2019) and adapters and low-quality sites were removed from the reads using Trimmomatic v0.38. The transcriptomic data sets for Kudoa iwatai (SRR1300899), Thelohanellus kitauei (SRR1103279), and Polypodium hydriforme (SRR1336770) were downloaded from the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). All the reads were assembled using rnaSPAdes v3.13.1 using default settings. Protein coding sequences were predicted using default settings in program “LongestOrfs” implemented in TransDecoder v5.5.0.
The phylogenomic tree was built using a data set of 78 ribosomal protein coding genes comprising diverse eukaryote taxa (Yahalomi et al. 2020, https://datadryad.org/stash/dataset/doi:10.5061/dryad.v15dv41sm, last accessed January 29, 2020). Fifty-one genes of these 78 candidate sequences from our transcriptomes were identified by a TBlastN search, e-value threshold 1e−10. These selected nucleotide sequences were then added to the respective gene alignments and aligned individually using the translation align function in Geneious Prime 2019.2.1 using MAFFT algorithms and the translation frame 1 (Katoh and Standley 2013). Poorly aligned regions were then removed using trimAl v1.2 (with 0.01 gap rate cutoff), and preliminary trees were constructed using RAxML v8.2.12 with the GTRGAMMA model and 100 rapid bootstrap replicates. Single-gene trees were manually inspected to select orthologous sequences, and paralogous sequences (see supplementary material 3, Supplementary Material online, for more detail) and host contamination were removed from the data sets. The remaining orthologs were realigned following the previously described method and concatenated into a supermatrix using catfasta2phyml (https://github.com/nylander/catfasta2phyml/, accessed September 12, 2019). The resulting alignment was subjected to maximum likelihood (ML) phylogenetic analysis in RAxML v8.2.12 using the GTRGAMMA model and 100 rapid bootstrap replicates.
Candidate Virulence Gene Searches
We chose cell migration genes and proteases/inhibitors as candidate virulence factors, due to their relevance in host–pathogen interactions (e.g., Barragan and Sibley 2002; McKerrow et al. 2006; Bouzid et al. 2013). Given the low amount of functional (GO) annotation in our largest assembly, C. shasta + NHP (see Results), we used BlastX to search for homologs of genes of interest in two custom concatenated databases that comprised the Cell Migration Knowledge Database (CMKB) which includes proteins, families, and complexes involved in cell migration (http://www.cellmigration.org/index.shtml, last accessed February 5, 2015; ∼7,600 protein sequences), and the MEROPS database, which consists of a nr library of full-length sequences of peptidases and peptidase inhibitors (http://merops.sanger.ac.uk/, last accessed December 2, 2014; ∼450,000 sequences; Rawlings et al. 2016). We searched using the longest representatives for each gene in the C. shasta + NHP assembly (23,418 contigs; IIR-RBT6) then parsed matches (bit score > 100) and posteriorly classified homologs to proteases or motility genes matching the specific databases. We then selected only genes with the same annotation in UniProt (determined using the same standards: no ambiguous terms filtering, bit score > 100) to confirm gene identity. Due to disagreements between annotations (CMKB vs. UniProt, and MEROPS vs. UniProt), we curated gene lists manually, removing genes that matched one or more of the following criteria: 1) no genetic distances available (only available for reference); 2) disagreements between annotations from the different databases, after checking for synonyms or function similarity; 3) annotations that contained terms from UniProt “Uncharacterized protein” and “Predicted protein” (ambiguous terms), and whose identity could not be confirmed; and 4) annotations that contained nonspecific terms, such as “heat shock protein” or “ribosomal protein.” For genetic distance analyses and inference of SNPs-based phylogenetic trees, we created lists of homologs that met different sets of the above criteria (strict: 1–4 criteria; permissive-filtered: 1, 2, and 4; and permissive: 1 and 4 criteria).
Analysis of Genetic Variation between C. shasta Genotypes Using SNPs from RNA-seq Data
The pipeline for finding (“calling”) SNPs from RNA-seq data employs samtools v1.9 (Li et al. 2009) and bcftools v1.9 (Li 2011) and is described in E.M.’s wiki page (http://sites.science.oregonstate.edu/∼meyere/wiki/index.php/RNASeqSNPs, last accessed February 19, 2017). To quantify genetic variation between different genotypes of C. shasta, parasite and NHP reads of the different transcriptomes (genotypes I, IIC, and the three IIR) were mapped against the nr assembly (23,418 sequences/genes) of the reference IIR-RBT6 transcriptome (C. shasta + NHP, filtered for host and other contaminants). We mapped reads using gmapper as detailed before but reported only the three top hits per read, followed by more stringent filtering of the .sam file with a minimum 99 bp matches over a 101-bp-length sequence alignments (allowing for only two mismatches), to recover only true SNPs. Highly expressed genes were resampled to avoid genotyping errors, using a maximum coverage of 100×. We used samtools and bcftools (see above) to convert .sam files to .bam files, then sorted, indexed, and called SNPs from the alignments generating .vcf (variant call format) files. Genotypes were called from nucleotide frequencies with a minimum coverage of 5× and heterozygosity threshold of 0.25. We combined results into tables, which consisted of a list of loci (labeled homozygous or heterozygous according to the parameters selected) for each C. shasta transcriptome. These tables were combined as ten pairwise comparisons. Overall genetic transcriptome-wide distances were calculated as 1 – (proportion of shared alleles) (Bowcock et al. 1994). We estimated these distances for all genes (22,755), for genes available across the different transcriptomes (593) and for cell migration (141) and proteases and inhibitors (41) strictly curated data sets. Genetic distances of selected genes from the cell migration and proteases and inhibitors data sets were parsed, and hierarchical cluster analyses were performed using hclust function in Rstudio (v3.6.0, Inc, Boston, MA) (Supplementary MM, Supplementary Material online, for the R code). Intratranscriptomic variation was calculated by calling genotypes from the same alignments with at least 20 reads, in order to confirm genotype results and to resolve any genotypic variation in the C. shasta population infecting the sampled fish. The heterozygosity threshold was set at 0.1 (at least 2 reads out of 20 to call a locus heterozygous). A ratio was calculated for each transcriptome: heterozygous loci divided by total sites (homozygous + heterozygous). Due to the limited number of replicates for each genotype, no statistical comparison was performed between transcriptomes.
Using the strictly curated lists of homologs (of motility and proteases genes), we parsed the genetic distances, then calculated the relative frequency (%) of genes for two categories: perfectly conserved genes and genes with genetic divergence. We calculated which subset of genes of interest was less conserved by subtracting the overall relative frequency (%) of perfectly conserved genes for each transcriptome’s pairwise comparison, from the relative frequency (%) of perfectly conserved cell migration or proteases/inhibitors genes.
SNPs-Based Phylogenetic Analyses
Using the genetic variation data, we explored whether SNPs-based phylogenetic analyses would resolve phylogenetic relationships between C. shasta genotypes. SNP genotypes matrices of all transcriptomes were converted to a FASTA-format alignment using the “gt2fas.pl” (https://github.com/em-bellis/2brad_utilities, last accessed October 23, 2019). The mask alignment option in Geneious Prime was used to remove identical bases from previously obtained alignments and a ML trees were constructed using RAxML v8.2.12 (ASC_GTRGAMA model) with 1,000 bootstrap replicates. Up to eight different trees were generated from the data sets with different levels of completeness or curation mentioned above.
Results
Sequencing and Reads Composition: Removal of Host Reads
Read data were archived in the NCBI Sequence Read Archive (table 1; accession number PRJNA241036). Reference transcriptomes were assembled from a single deeply sequenced sample (RBT6): comprising 788 million raw, paired 101-bp reads, with only 3.8% of reads filtered out as low-quality, homopolymer repeats or adaptor sequences. The remaining 759 million high-quality reads were mapped separately against both host and parasite to assess contamination: 59.6% (452 million) mapped best to C. shasta, 24.1% (182 million) matched neither of the two databases, 0.8% (5 million) were ties, and 15.6% (118 million) had host origin and were discarded (table 1 and fig. 3).
Fig. 3.

Results of first stage filtering: the percentages of reads that best matched our draft Ceratonova shasta genome, rainbow trout genome, or NHP (neither host nor parasite) for each of the five C. shasta genotype transcriptome libraries.
The other four isolates (table 1) were sequenced at lower coverage and yielded on average 29.6 (15.6–44.3) million paired 101-bp reads each. Only 19.3–40.0% of reads from these libraries were kept after quality and adaptor filtering steps. The two ascitic fluid-derived data sets (IIR) had the highest proportions of parasites reads (32.6–60.4%), whereas intestine-derived transcriptomes had the lowest: I (4.5%) and IIC (12.8%). The proportion of reads in the NHP bins was similar across transcriptomes (24.9–35.1%). Host contamination was 9.5–70.2%, with genotypes I and IIC having the highest amount (56.8–70.2%) (fig. 3).
Reference Transcriptome Assembly and Functional Annotations
We produced two reference transcriptomes from IIR-RBT6, using either C. shasta reads or C. shasta reads + NHP reads (fig. 2). The 452 million reads that mapped best to the C. shasta genome were assembled into 44,986 transcripts (“C. shasta only” assembly), and the 635 million reads that mapped best to both the C. shasta genome and NHP were assembled into 75,087 transcripts (“C. shasta + NHP” assembly) (table 2).
Table 2.
Assembly and Annotation Statistics for De Novo Assemblies of Reference Transcriptome of IIR (RBT6) Ceratonova shasta from Ascitic Fluid: C. shasta Only (More Conservative) and C. shasta + NHP (neither host nor parasite; Less Conservative)
| Ceratonova shasta Only | Ceratonova shasta + NHP | |
|---|---|---|
| Host filtered reads into assembly | 452,498,284 | 452,498,284 + 182,816,267 = 635,314,551 |
| Assembled transcripts | 44,986 | 75,087 |
| Length min–max | 201–10,626 | 201–18,696 |
| Average length | 589 | 550 |
| N50 | 871 | 810 |
| Size of assembly (Mb) | 26.5 | 41.3 |
| Contigs after redundancy | 18,253 | 28,503 |
| Gene annotation (UniProt) | 8,460 (18.8%) | 15,906 (21.2%) |
| GO annotations (% total UniProt BLAST matches; % total assembly) | 5,229 (61.8%; 11.6%) | 11,219 (70.5%; 14.9%) |
| Taxon ID annotation (nr) | 15,831 (35.2%) | 29,560 (39.4%) |
| Taxon ID Cnidaria (% total Taxon ID annotations; % total assembly) | 1,530 (9.7%; 3.4%) | 2,126 (7.2%; 2.8%) |
| Filtration postassembly: transcripts | ||
| Best match to C. shasta | 43,395 (96.4%) | 48,824 (65%) |
| Neither | 1,476 (3.3%) | 14,635 (19.5%) |
| Best match to rainbow trout | 115 (0.3%) | 11,628 (15.5%) |
| Other taxa contamination (microorganisms) | 5,465 (12.1%) | 6,669 (8.9%) |
| Final version assemblies (read, contig, and other taxa filtration) | 39,407 | 56,876 |
| Length min–max | 201–10,626 | 201–18,696 |
| Average length | 569 | 579 |
| N50 | 825 | 870 |
| Size of assembly (Mb) | 22.4 | 32.9 |
| Gene annotation (UniProt) | 7,074 (18%) | 10,679 (18.8%) |
| GO annotations (% total UniProt BLAST matches; % total assembly) | 4,525 (64%; 11.5%) | 7,280 (68.2%; 12.8%) |
| Taxon ID annotation (nr) | 10,291 (26.1%) | 15,503 (27.3%) |
| Taxon ID Cnidaria (% total Taxon ID annotations; % total assembly) | 1,530 (14.9%; 3.9%) | 2,116 (13.6%; 3.7%) |
BLAST homology searches against UniProt annotated 18.8/21.2% of transcripts (C. shasta only/C. shasta + NHP; table 2 and supplementary file 1, Supplementary Material online) with annotations for 7.6/13.1% of transcripts <400 bp, 20.9/21.1% of transcripts 400–1,000 bp, and 59.8/61.2% of transcripts >1,000 bp. We assigned GO terms to 61.8/70.5% of transcripts that had matches with UniProt, which represented only 11.6/14.9% of the total assembly (table 2 and supplementary file 1, Supplementary Material online). Taxonomic origin obtained by BLAST search to NCBI nr showed that approximately one-third (35.2/39.4%; 15,831/29,560) of transcripts matched taxa in the nr database: 9.7% (1,530) were annotated as Cnidaria in the C. shasta only assembly, whereas 7.2% (2,126 sequences) were annotated as Cnidaria in the C. shasta + NHP assembly (table 2 and supplementary file 1, Supplementary Material online).
Host and Other Contamination Removal at Contig Level
Using Taxon ID annotation, we removed nonfish contamination (prokaryotes, viruses, fungi, protists, and algae) from the final data sets: In C. shasta, only we removed 12.1% (5,465 transcripts) and in C. shasta + NHP, we removed 8.9% (6,669 transcripts).
Secondary host filtering at the transcript level, by comparing transcripts from both reference assemblies against C. shasta and O. mykiss genomes, showed that the C. shasta only assembly had 96.4% (43,395) of transcripts matching the parasite genome (fig. 4A), with only 0.3% (115) transcripts matching fish; 3.3% (1,476) of transcripts had no match. Ceratonova shasta transcripts were the longest (av. length 600 bp, range 201–10,626 bp), whereas the fish transcripts still present after assembly were short (av. length 234 bp, range 201–481 bp), and no match transcripts were also short (av. length 278 bp, range 201–1,136 bp).
Fig. 4.

Results of second stage filtering: the percentages and size distributions of assembled contigs that best matched to the Ceratonova shasta genome, rainbow trout genome, or NHP (neither host nor parasite) for the two versions of the reference transcriptome: (A) C. shasta only and (B) C. shasta + NHP.
In the C. shasta + NHP assembly, 65% (48,824) of transcripts matched the parasite genome and 15.5% (11,628) matched rainbow trout (fig. 4B). No match transcripts represented 19.5% (14,635). Size distribution showed that both C. shasta and no match transcripts had similar length distributions: av. length 609 bp (range 201–18,696 bp) and 582 bp (201–12,037 bp), respectively, whereas fish transcripts were shorter: 262 bp (201–2,278 bp).
After these filtering steps, the final reference transcriptomes had the following compositions: C. shasta only: 39,407 transcripts (av. length 569 bp, range 201–10,626 bp, N50 825); C. shasta + NHP: 56,876 transcripts (av. length 579 bp, range 201–18,696 bp, N50 870). UniProt and GO terms annotation rates remained similar to nonfish + other contaminants filtered assemblies. By size, long transcripts were more highly represented in the annotated genes proportion (UniProt), due to the removal of short O. mykiss transcripts (6.5/7.4% of transcripts <400 bp in length, 19.6/20.1% of transcripts 400/1000 bp in length, and 65.5/66.4% of transcripts >1000 bp) (fig. 4). Taxon ID annotations rate was lower (>10%), due to removal of nonfish contamination (microorganisms).
CEGMA analysis of the C. shasta only assembly identified 46% complete core eukaryotic proteins present (50% if partial matches were included). For the cleaned C. shasta + NHP assembly, CEGMA identified 71% complete core eukaryotic proteins (76% if partial matches were included).
Phylogenomic Analyses of C. shasta Genotypes
ML tree reconstruction based on 51 genes from eight taxa and 29,730 sites (fig. 5A) showed that C. shasta genotypes cluster in two well-supported clades (96–100 bootstrap support), independent from geographic origin: one containing genotypes I and IIC from coho and Chinook salmon and a monophyletic group accommodating all transcriptomes of genotype IIR from rainbow trout. Within the IIR clade, IIR genotypes from the Klamath River (LKR and UKR) appear to be more closely related to each other than to IIR genotype from Willamette River. SNP-based ML analyses showed similar clustering when using the full SNP data set (22,755 genes and 918 SNPs; fig. 5B) or a subset of genes present in all ten pairwise comparisons (593 genes and 235 SNPs; fig. 5C).
Fig. 5.

Maximum likelihood phylogenetic trees. (A) Phylogenomic tree showing Ceratonova shasta genotypes relationships. Alignment based on 51/78 genes from Yahalomi et al. 2020. Polypodium hydriforme was set as the outgroup. (B, C) SNPs-based trees of transcriptomes of C. shasta genotypes using different subset of genes: (B) all genes (N = 22,755) and (C) only genes present across all ten transcriptomes pairwise comparisons (N = 593). Values at nodes indicate bootstrap support. LKR: Lower Klamath River; UKR: Upper Klamath River; WR: Willamette River.
Overall Transcriptome-Wide Genetic Distances between Genotypes
The number of genes used to calculate genetic distances between transcriptomes varied depending on library completeness (657–3,857; supplementary table 1 and file 2, Supplementary Material online). We found the highest genetic transcriptome-wide distances between genotypes I and II (2.1 × 10−3–2.4 × 10−3). IIC was closest to IIR (1.7 × 10−3–1.8 × 10−3), but the distances were the same magnitude between IIC and I (2.1 × 10−3). IIR intragenotype genetic distances were an order of magnitude lower (1.1 × 10−4–1.5 × 10−4) compared with the distances observed between genotypes I and II (22,755 genes, see supplementary table 2, Supplementary Material online, for genetic distances of the other genes data sets).
Within Transcriptome/Genotype Diversity
Heterozygosity levels varied 2–10-fold between transcriptomes. The transcriptome with the highest variability was genotype I with 1 variant in every 208 loci (63 heterozygous loci/13,116 total sites) followed by the IIR (RBT6) with 1/432 variant loci (3,348 heterozygous loci/1,445,297 total sites). IIC showed 1/614 variant loci (113 heterozygous loci/69,226 total sites). The least variable libraries were the two IIR transcriptomes from the Klamath River, IIR (RBTC16) with 1/1,869 variant loci (48 heterozygous loci/9,670 total sites) and IIR (RBTJ7), with the lowest heterozygous loci ratio, with 1/2,159 variant loci (25 heterozygous loci/53,948 total sites).
Genetic Distances and SNP-Based Phylogenetic Analyses of Putative Virulence Genes in Different C. shasta Genotypes
From the longest representatives for each gene in the C. shasta + NHP assembly (23,418 contigs), 431 matched cell migration genes in CMKB and 507 had homologs to proteases and inhibitors in the MEROPS database. We identified many discrepancies between annotations from MEROPS and UniProt (without ambiguous terms filtering). Manual curation produced a subset of 41 homologous genes to MEROPS and 14 SNPs (strictly curated) for proteases and inhibitors, 56 genes, 15 SNPs (permissive_filtered data set), and 110 genes, 26 SNPs (permissive data set), listed in supplementary files 3 and 4, Supplementary Material online. Fewer discrepancies were observed between CMKB and UniProt annotations, but we removed genes with annotations seemingly unrelated to cell migration (e.g., heat shock proteins and ribosomal proteins). After curation, 164 gene homologs to CMKB, 40 SNPs remained in the strictly annotated data set, 271 genes, 71 SNPs in the permissive_filtered one, and 325 genes, 83 SNPs in the permissive one (supplementary files 3 and 4, Supplementary Material online). We compared subsets of these among genotypes, depending on the completeness of the transcriptomes (supplementary table 1, Supplementary Material online).
Relative frequencies of perfectly conserved transcripts (i.e., with no SNPs) are given in supplementary table 3, Supplementary Material online. Relative frequency values of conserved genes of interest (cell migration or proteases/inhibitors) against overall conserved genes in each transcriptome are shown in figure 6A and supplementary table 4, Supplementary Material online. Cell migration genes were less conserved between genotypes I and II (a negative relative value in I/IIC and I/IIR comparisons) but more conserved in IIC/IIR comparisons, and in intragenotype IIR comparisons (fig. 6A; positive relative values). Conversely, proteases and inhibitors were more conserved for I versus IIC, but less conserved for I versus IIR (fig. 6A), but these results varied depending on which IIR data set was used (supplementary fig. 1, Supplementary Material online). I/IIR-RBT6 and I/IIR-RBTC16 showed negative relative values, whereas I/IIR-RBTJ7 showed slightly positive value. Protease and inhibitor genes were in general less conserved for IIC/IIR (negative value) except for IIC/IIR-RBTC16, which showed a slight positive relative frequency. We observed lower relative frequencies between IIR transcriptomes, except for IIR-RBTJ7/IIR-RBTC16 (supplementary fig. 1, Supplementary Material online).
Fig. 6.

Relative conservation of motility and protease/inhibitor gene sets based on pairwise genetic difference comparisons and on SNPs-based phylogenetic analyses of Ceratonova shasta genotypes. (A) Chart shows relative frequency of conserved genes in pairwise comparisons between data sets, subtracting relative frequencies of all conserved genes. Negative values indicate that the subset of genes is less conserved than average. Positive values indicate that the genes are more conserved than average; (B, C) SNP-based ML trees of transcriptomes of C. shasta genotypes using strictly curated data sets of (B) cell migration genes (N = 164) and (C) proteases and inhibitor genes (N = 41). Values at nodes indicate bootstrap support. LKR: Lower Klamath River; UKR: Upper Klamath River; WR: Willamette River.
Cell migration SNP-based phylogenetic analyses (strictly curated to permissive data sets, fig. 6B and supplementary fig. 2, Supplementary Material online) showed the same tree topology as the larger transcriptomic SNP data set of all genes (22,755 and 593 genes, respectively, fig. 5B and C). Topology had high nodal support and indicated a close relationship of genotypes I and IIC, with all IIR transcriptomes forming a monophyletic sister clade to these two genotypes. The subset of proteases and inhibitors genes revealed an unstable tree topologies with both the strictly curated (fig. 6C) and the permissive_filtered (supplementary fig. 2, Supplementary Material online) data sets, having low nodal support likely due to the low number of genes and fewer informative sites within them. ML analyses of the permissive cell proteases/inhibitors data set resulted in the same clustering as in phylogenomic analyses and motility-based SNP analyses, though with low nodal support.
SNP Analyses Highlight Specific Virulence Gene Candidates
When comparing cell migration subsets in I versus II, we observed nonzero genetic distances (based on SNPs) in actin cytoskeleton proteins (ARP2/3, several actin isoforms, and F-actin capping protein), tubulin alpha chain and microtubule force-producing proteins (dynamin), actin binding (coronin), actin and tubulin folding molecules (T-complex protein 1), and Rho family (small GTPases involved in migration regulation) genes (supplementary file 3 and fig. 3, Supplementary Material online).
For the protease subset, up to ten α and β subunits of the proteasome complex had genetic distances between all genotypes except for comparisons between the different IIR isolates. A cathepsin Z-like cysteine protease, homologous to cathepsin X of M. cerebralis, showed genetic differences between all I and II comparisons (except for any comparison to transcriptome IIR-RBTJ7), with the highest genetic distance for I/IIR-RBT6. A methionyl aminopeptidase (homolog to methionyl aminopeptidase 2 of Hydra vulgaris syn. Hydra magnipapillata) had genetic differences between I and two of the IIR data sets (IIR-RBTJ7 and IIR-RBT6). Other methionyl aminopeptidase showed differences between IIC/IIR-RBTC16 and IIC/IIR-RBTJ7. Genetic differences were observed for a serpin or serine protease inhibitor between I and II (except IIR-RBTJ7), IIR/IIC, and IIR-RBTJ7/IIR-RBT6 (supplementary file 3 and fig. 4, Supplementary Material online).
Discussion
The Challenge of Myxozoan ‘Omics: Parasite Identification and Host Filtration
Myxozoans are unculturable, and throughout most of their life cycle are composed of only a few cells. Only some species have spore-forming stages with macroscopic plasmodia, which represent clonal parasite material that can be physically separated from host tissue; many other species are more broadly distributed within hosts and intimately associated with host cells. These properties make myxozoan ‘omics research fundamentally challenging. Thus, most genome and transcriptome data from myxozoans are from spores or spore-forming stages (Jiménez-Guri et al. 2007; Holland et al. 2011; Nesnidal et al. 2013; Yang et al. 2014; Chang et al. 2015; Foox et al. 2015). In a parallel study, we have sequenced the C. shasta genome from purified myxospores, which had been separated physically from contaminating host cells, to obtain a high proportion of parasite DNA (Atkinson S., Alama-Bermejo G., Bartholomew J.L., in preparation). In the present study, we investigated transcriptome-wide genetic differences among closely related C. shasta genotypes from different host fish. Given that C. shasta is an obligate parasite that does not form cysts, its metabolically active developmental stages typically occur in intimate contact with host intestinal tissues, which presents an inherent challenge to obtaining samples with high proportions of parasite transcripts. We succeeded in obtained high-quality RNA from C. shasta stages, utilizing a unique feature of the parasite’s biology: Virulent genotype IIR can cause systemic infection in allopatric stocks of rainbow trout and induce the production of ascitic fluid in the body cavity (fig. 1). This ascitic fluid is rich in manifold C. shasta developmental stages (visibly motile presporogonic and sporogonic stages, and mature myxospores) and thus provided excellent material for transcriptomics. The data that we obtained from a single ascitic fluid sample (RBT6) had sufficient coverage and sequence depth to build a reference transcriptome, against which we then compared the lower-coverage transcriptome data from the other genotypes, sampled from intestinal tissues, and which presented the expected challenges of host contamination and low read depth.
Given the mixed host–parasite read data, we developed bioinformatic pipelines to separate transcripts by species origin. Host genomes and transcriptomes, especially for fish species important to aquaculture, are available (e.g., Berthelot et al. 2014; Tine et al. 2014; Xu et al. 2014; Lien et al. 2016). Two myxozoan studies have included host filtering steps in assembly pipelines (Chang et al. 2015; Foox et al. 2015). Foox et al. (2015) filtered the transcriptome of Myxobolus pendula using an in silico hybridization pipeline of iterative BLAST searches against a filtered myxozoan/cnidarian query set and a close relative fish host. Chang et al. (2015) filtered genomic and transcriptomic data using BLAST searches of the specific fish host genomes. We attempted postassembly filtration using predicted proteins of these available myxozoans/cnidarians versus fish hosts and found this gave only a poor recovery rate of C. shasta sequences (data not shown). However, as we had genomes of both host (O. mykiss; public data) and parasite (C. shasta; our draft data, unpublished), we used these in a novel approach of combined preassembly read filtering, and postassembly contig filtering. Removal of host reads prior to assembly greatly reduces assembly of host contigs and minimizes creation of chimeric transcripts (Daly et al. 2015). Myxozoans have some of the most derived genomes of all metazoans (Chang 2013; Holzer et al. 2018), and host–parasite sequence chimeras can be a frequent and insidious component of contaminated assemblies; hence, a host removal step is essential for comparative approaches and may be further improved once long-read sequences become available. The requirement for host and parasite reference genomes may be considered a limitation in our pipeline but we found that this approach gave improved yields of transcriptome libraries with maximum expressed gene recovery and minor host contamination. During postassembly filtering of contigs using BLAST, we created distinct bins of host or parasite transcripts, and a third bin of NHP, which allowed us to recover additional contigs that could not be identified by mapping against the references (i.e., either true parasite genes missing from the draft C. shasta genome, or nonparasite, nonhost contaminants). Interestingly, the similar size distribution of transcripts in both C. shasta and NHP bins suggested parasite origin for the majority of NHP transcripts. CEGMA analyses showed that the combined C. shasta + NHP assembly had more matches (71%) than C. shasta alone (46%), further supporting the validity of combining both contigs that matched to the parasite genome and nonfish matches. The difference in completeness of the C. shasta only and C. shasta + NHP assemblies revealed a limitation of our pipeline and demonstrated the need to include NHP reads in our analyses to avoid losing parasite genes, which could not be matched to our presumably incomplete reference genome. The CEGMA completeness of C. shasta + NHP (71–76%) of identifiable core eukaryotic genes was similar to results from other cnidarian transcriptomes: 77–84% for Kudoa iwatai, 76–90% for five corallimorpharians, 87% for Hydra vulgaris syn. Hydra magnipapillata, 91% for Calliactis polypus, and 90–92% for three scleractinian corals (Chang et al. 2015; Rodrigues et al. 2016; Kenkel and Bay 2017; Lin et al. 2017; Stewart et al. 2017).
We then used taxon ID assignments from NCBI database searches to further remove contaminating contigs attributable to microorganisms. This is the first time that multiple bioinformatics filters have been used to remove significant, unavoidable contaminants from myxozoan high throughput sequence data sets. Overall, we consider that our bioinformatic pipeline is optimal for generating transcriptomes of nonmodel organisms within host tissues, as it prevented both the assembly of chimeric sequences and the loss of unknown and highly derived parasite genes.
Functional Myxozoan Annotations
Myxozoan genomes are highly divergent from both their free-living cnidarian relatives and all other metazoans, even considering conserved genes (Hartigan et al. 2016). Thus, myxozoan ‘omics data sets can be functionally annotated only poorly at present: for example, 5.6% (Foox et al. 2015), 19% (Chang 2013), 21% (Yang et al. 2014; specified as 3,500/16,638 proteins), and 11–13% (this study). Even annotation of genes by comparison with other myxozoan species is difficult, given that they are as strongly divergent from each other as from other cnidarians, and other metazoans. Development of a comprehensively annotated myxozoan reference will still require considerable manual effort to identify more unambiguous myxozoan genes and provide meaningful functional annotations.
The lack of functional annotations for C. shasta made enrichment analysis or parsing of the data using GO terms unfeasible, as several functional categories of interest did not have any hits in the transcriptome. We then switched to publicly available databases to parse genes of interest; however, we observed discrepancies between UniProt and MEROPS annotations for our protease and inhibitor gene subsets of interest. This was in concordance with previous reports of missannotations from public databases, for example, Schnoes et al. (2009) who show that >80% of contigs in 10 out of 37 enzyme families were misannotated. The main sources of these errors have been linked to overprediction and error propagation (Jones et al. 2007; Schnoes et al. 2009), particularly in nonmodel species (Clark and Greenwood 2016). As C. shasta, and myxozoans in general, are only distantly related to any model organism (even the free-living cnidarian Nematostella vectensis), we were not surprised by both the low rate and error-prone nature of the annotation using general organismal databases. For the cell migration database, CMBK, we obtained annotations that agreed substantially with those from UniProt (which was expected given CMBK is a subset of the NCBI database). However, we found that CMBK contained genes with a questionable link to cell motility (e.g., ribosomal genes and heat shock proteins), which suggested that present curation of this database is too permissive. Thus, we regarded manually curated subsets of genes as the best option for exploring our data. Future work should concentrate on better functional annotations of myxozoan genes of interest (virulence and structure) and cnidarian genes in general.
Ceratonova shasta Genotypes: Transcriptomic Data Elucidate Intergenomic Relationships and Pave the Way to New Markers
Ceratonova shasta represents an excellent myxozoan model parasite for several reasons: It affects both wild and cultured salmonid fishes of economic and cultural value; its life cycle is known and can be maintained in the laboratory (Bartholomew et al. 1997); and it is the only myxozoan species for which host-specific genotypes have been characterized and associated with different virulence patterns (Atkinson and Bartholomew 2010a, 2010b; Hurst and Bartholomew 2012; Stinson et al. 2018). Although C. shasta genotypes are characterized by ITS sequence differences, this marker has several drawbacks: It does not resolve observed virulence differences between coho salmon and rainbow trout (“biotypes” IIC and IIR; Hurst and Bartholomew 2012); it can have intraorganism variation (Atkinson, Hallett, et al. 2018); and it cannot differentiate between geographic isolates (Stinson et al. 2018). Additional markers for resolution of different C. shasta strains are needed to better characterize spatial and temporal variation of the parasite in environmental samples, particularly when assessing risk for endangered coho salmon populations in the Pacific Northwest (Williams et al. 2016).
Our phylogenomic analyses of C. shasta provided unexpected results regarding the relationships between the different genotypes. We provide, for the first time, significant support for the separation of IIC and IIR into two independent genotypes, which are not resolved by ITS data alone. Phylogenomics showed that genotypes I and IIC share a closer relationship with each other than with IIR, which correlates with the evolutionary distance between their fish hosts, with coho (IIC) and Chinook (I) salmon being more closely related to each other than to rainbow trout (IIR; Domanico et al. 1997; Crête-Lafrenière et al. 2012). We hypothesize that the high virulence of IIR in a naïve host (allopatric rainbow trout) is a consequence of a relatively recent host switch from a common ancestor of genotypes I and IIC.
Though based on a limited number of isolates, we also observed some phylogeographic signal, in addition to host association, in clustering of the genotypes. The two sympatric IIR isolates from the Klamath River clustered together, whereas the IIR isolate from the Willamette River (some 500 km distant) was distinct (fig. 5). This signal of sympatry is consistent with data from studies showing that salmonids show strong site fidelity (e.g., Minakawa and Kraft 2005; Dittman et al. 2010), which can lead to establishment of local pathogen strains, for example, in IHN virus (Kurath et al. 2003) and in another myxozoan, Parvicapsula minibicornis (Atkinson et al. 2011).
Characterizing Virulence in C. shasta Genotypes
Heterozygosity
We examined patterns of C. shasta genotypic diversity and virulence by comparing heterozygosity levels between isolates from different geographic localities and hosts. We found no obvious relationship between parasite heterozygosity and a particular geographic site, though our analyses were limited by geographic sampling from only three sites (LKR, UKR, and Willamette River). We observed isolates of low (IIR-RBTJ7), medium (IIC), and high (I) heterozygosity within the Klamath River. High virulence of pathogen strains is related to higher levels of heterozygosity (Cogliati et al. 2012). This may be the case for C. shasta genotype I, which is highly diverse and relatively pathogenic in a native host (Chinook salmon); however, genotype IIR, which is highly virulent in allopatric rainbow trout stocks, had low (IIR-RBTJ7 and IIR-RBTC16) or moderately high diversity (IIR-RBT6). Hence, we found no evidence to suggest that greater heterozygosity correlates with the higher virulence of C. shasta genotype IIR.
Cell Migration
Cell motility in parasites is important for host invasion, adhesion to host cells, and migration through tissues (Barragan and Sibley 2002; Lentini et al. 2015) and hence can affect virulence. For example, the capacity of Toxoplasma gondii for migration, both across tissues and over long distances, characterizes its virulence (Barragan and Sibley 2002). Developmental stages of C. shasta are motile and capable of producing different cell protrusions (filopodia, lamellipodia, and blebs), with specific functions such as anchoring/adhesion, crawling, and blebbing (Alama-Bermejo et al. 2019). For C. shasta, we hypothesized that differences in parasite migration and proliferation strategies underpin differences in virulence (proliferation rate, spore production, and cumulative mortality rate), as observed previously between genotypes I/IIC and IIR (Hallett et al. 2012: Stinson and Bartholomew 2012). In vivo observations reveal that IIR infection is characterized by rapid proliferation, fast amoeboid bleb-based motility, and high adhesion, with significant differential expression (by quantitative PCR) of key motility and adhesive factors between type 0 (low virulent) and IIR (highly virulent) genotypes in rainbow trout (Alama-Bermejo et al. 2019). In the current study, we curated a “motility” data set based on genes with functions related to migration, proliferation, cytoskeleton, and cell division. Although our genetic distance analyses showed there was a higher frequency of perfectly conserved cell migration genes between IIC and IIR (fig. 6A), our SNPs-based phylogenetic analyses (fig. 6B) indicated a closer evolutionary relationship between I and IIC than either of them with IIR. This was in concordance with the observed general phylogenomic clustering of the genotypes and suggested that these genes are likely under positive selection pressure and linked to virulence. The genetic variation and phylogenetic relationships of SNPs in cell migration genes that we observed provides a valuable genetic framework to understand the observed differences in C. shasta virulence. Future work could explore silencing of selected motility factors in C. shasta to determine their importance and roles in parasite virulence.
Proteolytic Enzymes and Inhibitors
Proteases and their inhibitors comprise 1–5% of infectious organism genomes (Tyndall et al. 2005), and this appears similar in myxozoans: ∼2.5% (422 proteins) in the Thelohanellus kitauei proteome (Yang et al. 2014) and 2.6% of expressed genes in Sphaerospora molnari (Hartigan et al. 2020). Our relative frequency analyses showed that these genes are generally less conserved/more variable in C. shasta than cell migration genes. We observed that they had more variation between genotypes I and IIR, and IIC and IIR but were more conserved between I and IIC, again showing that IIR was distinct. We consider this evidence that proteolytic enzymes are important contributors of virulence in C. shasta. This pattern was not well supported by the SNP data as they did not produce a well-supported SNP tree, but this is likely an artifact of the limited genes and positions available for analyses in the curated and permissive_filtered data set (only 41 and 56 genes, respectively), whereas the largest data set (permissive, 110 genes) supported the same phylogenetic clustering of genotypes as in the phylogenomic study and the cell migration data set (≥164 genes). Whether the observed SNP-based distances are relevant at the functional level is unknown and requires functional analyses of specific genes. Our study is the first to examine genetic distances between biologically different genotypes of a myxozoan and, given associations between relative SNP frequencies and virulence, it is likely that at least some of these genetic differences are translated into differences in protein regulation and activities that affect virulence in different fish hosts.
Biological Relevance of Candidate Virulence Genes in C. shasta: Pinpointing Molecules for Future Interference Studies
Pathogens evolve with their hosts in a stepwise “arms race” of virulence and resistance, mediated by genetic changes (Coletta-Filho et al. 2015). We identified molecular differences between genes related to parasite motility and proteolysis in C. shasta host-associated genotypes. Specifically, we observed that genes encoding several molecules important to the formation and functioning of the cytoskeleton (ARP2/3 and F-actin capping protein) and dynamin had smaller genetic distances between them in the two II genotypes than between either genotypes II and I. These proteins are involved in growth, morphogenesis, migration, cell-to-cell spread and virulence of bacteria (Choe and Welch 2016), fungal plant pathogens (González-Rodríguez et al. 2016), and parasites (Bookwalter et al. 2017). Only a few motility genes in our C. shasta transcriptomes showed smaller genetic distances between genotypes I and IIC, than to IIR, for example coronin, which is an actin filament-binding protein. This protein is essential for the transmission of Plasmodium, as individuals without coronin are unable to invade the salivary glands of mosquitos (Bane et al. 2016). During the invasion of tissues in the systemic phase of C. shasta infections, small changes in the coronin structure could be responsible for a more efficient/faster colonization of tissues by genotype IIR, making it more virulent than IIC and I.
We identified several proteases and protease inhibitors with smaller genetic distances between I and IIR, than between IIC and IIR, particularly proteasome subunits. The proteasome is a protease complex that plays a central role in regulating cellular processes, including cell cycle, apoptosis and differentiation, modulation of immune response, and regulation of gene expression in eukaryotic cells (Konstantinova et al. 2008; Tanaka 2009). In trypanosomes, protein degradation during parasite cell differentiation is primarily proteasome dependent, and proteasome inhibition impedes differentiation and transformation from noninfectious epimastigotes to infective tryptomastigotes (Cardoso et al. 2011; Gupta et al. 2018). Proteasome inhibitors are used routinely in cancer therapy (e.g., multiple myeloma) (Manasanch and Orlowski 2017) and are considered both disease markers and therapeutic targets (Muñoz et al. 2015; Morais et al. 2017; Varga et al. 2017). We observed marked sequence variation in C. shasta proteasome subunit mRNA sequences between different genotypes, which suggested that they are important to C. shasta pathogenesis and they should similarly be considered as both potential molecular markers for C. shasta genotypes, and drug targets for myxozoans. Other proteolytic enzymes and inhibitors with potentially important differences between genotypes were 1) cathepsin Z, whose homolog in M. cerebralis is thought to be involved in tissue invasion and lysis or the initiation of sporogenesis (Kelley et al. 2003); 2) methionyl aminopeptidases, which are enzymes whose inhibition results in antiparasitic activity (Chen et al. 2006; Zheng et al. 2015), for example, by growth inhibition; and 3) protease inhibitors (serpins) which are common in blood-feeding parasites such as ticks, for example, the cattle tick Rhipicephalus microplus encodes at least 24 serpins that inhibit proinflammatory and procoagulatory proteases of the host (Tirloni et al. 2014), processes that are likely to be important in blood and tissue dwelling organisms such as myxozoans. Future molecular and functional studies should test the suitability of these genes as virulence markers and determine their specific role in parasite migration and proteolysis.
Conclusions
The advent of genomics has facilitated the in silico identification of parasite virulence factors, a key objective for therapeutant design. Yet for obligate parasites, creation of high-quality reference genomic and transcriptomic assemblies is made challenging by host contamination, and uncertainties of parasite gene identification and annotation. For myxozoan ‘omics studies, the production of well-curated genomic and transcriptomic databases has been a major hurdle to progress in targeted vaccine approaches for this parasite group. Hence, we designed an optimized bioinformatics pipeline for processing high throughput sequencing data of myxozoans, which recovers a maximum number of putative parasite genes while filtering out sequences of the host and other contaminant organisms. We recommend this repeat-filtration pipeline as a method for cleaning up sequence data of derived nonmodel organisms such as myxozoans. This workflow allowed us to produce transcriptomic data sets of host-associated genotypes of C. shasta having different virulence in their respective hosts. We analyzed genetic distances and SNPs between the transcriptomes, with a focus on candidate virulence factors, motility genes, and proteolytic enzymes including their inhibitors. Phylogenomic analyses supported the observed host-associated clustering of genotypes and suggested an evolutionary history based on host switches, rather than geographic variations. Altogether these results support characterization of genotype II into subtypes IIC (coho salmon) and the derived type IIR (rainbow trout). In the Klamath River, we speculate that IIR evolved as a result of host switching to introduced rainbow trout when its natural coho salmon host was excluded with the construction of barrier dams (Hurst and Bartholomew 2012; Hurst et al. 2012). In this naïve host, genotype IIR is highly virulent and we hypothesize that this indicates a relatively recent host-switch event with insufficient time elapsed for purifying selection among virulence factors, that is, selective removal of gene variants that are deleterious in this genotype, which would occur over time as a result of mutual host–parasite adaptation. We identified variation in genes that are essential for the biology of the parasite and play a role in shaping C. shasta virulence, for example, the observed SNPs in motility and protease genes clearly correlated with the virulent genotype. These findings represent a crucial step toward characterizing the connections between genotype and pathology. By cataloging the pan-genomic SNP diversity of C. shasta, we have created a valuable resource for the development of diagnostic tools and as future targets for therapeutic intervention and vaccine design in salmonid enteronecrosis for this genetically complex parasite.
Data Availability
The following intermediate and final files of all analyses performed in this study are deposited in DRYAD (https://doi.org/10.5061/dryad.tx95x69tt): 1) host filtered parasite and neither reads lists of I, IIC, IIR_RBT7, IIR_RBTC16, and IIR_RBT6 (20 .list files); 2) genotype IIR_RBT6 reference assembled transcriptomes (nonfiltered) (two .fasta files); 3) host and other contaminants (other microorganisms) filtered IIR_RBT6 assemblies (two .fasta files); 4) longest representatives reference IIR_RBT6 cs + neither assembly used for SNPs analyses (one .fasta file); 5) SNPs tables (genotypes called from nucleotide frequencies with a) minimum coverage of 5 and 0.25 heterozygosity threshold [five .tab files] and b) minimum coverage of 20 reads and 0.1 heterozygosity threshold [five .tab files]); and 6) phylogenomic and SNPs-based phylogenetic alignments (nine .nex files and 51 individual genes alignments .fasta used for phylogenomics).
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Ruth Milston-Clements, Ryan Craig, and Richard Holt for fish husbandry at the John L. Fryer Aquatic Animal Health Laboratory; to Charlene Hurst (OSU) for her help with RNA extractions; and to Mark Dasenko at Oregon State University’s Center for Genome Research and Biocomputing (OSU CGRB) for library preparation and next-generation sequencing. We would like to thank to Tamar Lotan (University of Haifa) for critical feedback on this manuscript. We acknowledge the following funding agencies: Czech Science Foundation (Postdoctoral Project 14-28784P to G.A.-B.; EXPRO Grant 19-28399X to A.S.H.), Consellería de Educación, Investigación, Cultura y Deporte, Valencia, Spain (#APOSTD/2013/087 to G.A.-B.), Czech Academy of Sciences-Fellowship Purkyne (to M.K.), and European Regional Development Fund (CZ.02.1.01/0.0/0.0/16_019/0000759 to M.K.). Funds for this project were provided in part by the Bureau of Reclamation, US Department of Interior through Inter-agency Agreement #R15PG00065, as part of its mission to manage, develop, and protect water and related resources in an environmentally and economically sound manner in the interest of the American public. The views in this report are the authors’ and do not necessarily represent the views of Bureau of Reclamation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Mention of trade names does not imply U.S. Government endorsement.
Author Contributions
G.A.-B., S.D.A., A.S.H., and J.L.B. conceived and designed the experiments; G.A.-B. and S.D.A. collected and processed the samples; E.M. wrote the scripts and designed filtering pipeline; G.A.-B., E.M., and S.D.A. analyzed the data; M.M.W. and M.K. performed phylogenetic analyses; and G.A.-B. and A.S.H. drafted the manuscript. All authors edited and approved the final manuscript.
Data deposition: This project has been deposited at the SRA-NCBI repository under the SRA accession SRP040506 and BioProject PRJNA241036. Biosamples represent reads from three genotypes with single (genotypes I and IIC) or triplicate (genotype IIR) isolates: SAMN12275264 (IIR_RBT6; reference transcriptome), SAMN12275265 (IIR_RBTC16), SAMN12275266 (IIR_RBTJ7), SAMN12275267 (IIC), and SAMN12275268 (I).
Literature Cited
- Alama-Bermejo G, Holzer AS, Bartholomew JL.. 2019. Myxozoan adhesion and virulence: Ceratonova shasta on the move. Microorganisms 7(10):397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali IKM, et al. 2007. Evidence for a link between parasite genotype and outcome of infection with Entamoeba histolytica. J Clin Microbiol. 45(2):285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson SD, Bartholomew JL.. 2010. a. Disparate infection patterns of Ceratomyxa shasta (Myxozoa) in rainbow trout Oncorhynchus mykiss and Chinook salmon Oncorhynchus tshawytscha correlate with Internal Transcribed Spacer-1 sequence variation in the parasite. Int J Parasitol. 40(5):599–604. [DOI] [PubMed] [Google Scholar]
- Atkinson SD, Bartholomew JL.. 2010. b. Spatial, temporal and host factors structure the Ceratomyxa shasta (Myxozoa) population in the Klamath River basin. Infect Genet Evol. 10(7):1019–1026. [DOI] [PubMed] [Google Scholar]
- Atkinson SD, Bartholomew JL, Lotan T.. 2018. Myxozoans: ancient metazoan parasites find a home in phylum Cnidaria. Zoology 129:66–68. [DOI] [PubMed] [Google Scholar]
- Atkinson SD, Hallett SL, Bartholomew JL.. 2018. Genotyping of individual Ceratonova shasta (Cnidaria: Myxosporea) myxospores reveals intra-spore ITS-1 variation and invalidates the distinction of genotypes II and III. Parasitology 145(12):1588–1586. [DOI] [PubMed] [Google Scholar]
- Atkinson SD, Jones SR, Adlard RD, Bartholomew JL.. 2011. Geographical and host distribution patterns of Parvicapsula minibicornis (Myxozoa) small subunit ribosomal RNA genetic types. Parasitology 138(8):969–977. [DOI] [PubMed] [Google Scholar]
- Bane KS, et al. 2016. The actin filament-binding protein coronin regulates motility in Plasmodium sporozoites. PLoS Pathog. 12(7):e1005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragan A, Sibley LD.. 2002. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med. 195(12):1625–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew JL, Whipple MJ, Stevens DG, Fryer JL.. 1997. The life cycle of Ceratomyxa shasta, a myxosporean parasite of salmonids, requires a freshwater polychaete as an alternate host. J Parasitol. 83(5):859–868. [PubMed] [Google Scholar]
- Berthelot C, et al. 2014. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun. 5:3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork SJ, Bartholomew JL.. 2009. a. Effects of Ceratomyxa shasta dose on a susceptible strain of rainbow trout and comparatively resistant Chinook and coho salmon. Dis Aquat Org. 86:29–37. [DOI] [PubMed] [Google Scholar]
- Bjork SJ, Bartholomew JL.. 2009. b. The effects of water velocity on the Ceratomyxa shasta infectious cycle. J Fish Dis. 32(2):131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork SJ, Bartholomew JL.. 2010. Invasion of Ceratomyxa shasta (Myxozoa) and comparison of migration to the intestine between susceptible and resistant fish hosts. Int J Parasitol. 40(9):1087–1095. [DOI] [PubMed] [Google Scholar]
- Bookwalter CS, et al. 2017. Reconstitution of the core of the malaria parasite glideosome with recombinant Plasmodium class XIV myosin A and Plasmodium actin. J Biol Chem. 292(47):19290–19303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzid M, Hunter PR, Chalmers RM, Tyler KM.. 2013. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev. 26(1):115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcock AM, et al. 1994. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368(6470):455–457. [DOI] [PubMed] [Google Scholar]
- Cardoso J, et al. 2011. Analysis of proteasomal proteolysis during the in vitro metacyclogenesis of Trypanosoma cruzi. PLoS One 6(6):e21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, Pirofski L.. 1999. Host–pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun. 67(8):3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E. 2013. Transcriptomic evidence that enigmatic parasites Polypodium hydriforme and Myxozoa are cnidarians [dissertation/PhD’s thesis]. [Kansas (KS)]: University of Kansas.
- Chang ES, et al. 2015. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc Natl Acad Sci U S A. 112(48):14912–14917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. 2006. Inhibitors of Plasmodium falciparum methionine aminopeptidase 1b possess antimalarial activity. Proc Natl Acad Sci U S A. 103(39):14548–14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe JE, Welch MD.. 2016. Actin-based motility of bacterial pathogens: mechanistic diversity and its impact on virulence. Pathog Dis. 74(8):ftw099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KF, Greenwood SJ.. 2016. Next-generation sequencing and the crustacean immune system: the need for alternatives in immune gene annotation. Integr Comp Biol. 56(6):1113–1130. [DOI] [PubMed] [Google Scholar]
- Cogliati M, Barchiesi F, Spreghini E, Tortorano AM.. 2012. Heterozygosis and pathogenicity of Cryptococcus neoformans AD-hybrid isolates. Mycopathologia 173(5–6):347–357. [DOI] [PubMed] [Google Scholar]
- Coletta-Filho HD, Bittleston LS, Lopes JRS, Daugherty MP, Almeida RPP.. 2015. Genetic distance may underlie virulence differences among isolates of a bacterial plant pathogen. J Plant Pathol. 97:465–469. [Google Scholar]
- Crête-Lafrenière A, Weir LK, Bernatchez L.. 2012. Framing the Salmonidae family phylogenetic portrait: a more complete picture from increased taxon sampling. PLoS One 7(10):e46662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly GM, et al. 2015. Host subtraction, filtering and assembly validations for novel viral discovery using next generation sequencing data. PLoS One 10(6):e0129059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardé ML. 2008. Toxoplasma gondii, “new” genotypes and virulence. Parasite 15(3):366–371. [DOI] [PubMed] [Google Scholar]
- David M, Dzamba M, Lister D, Ilie L, Brudno M.. 2011. SHRiMP2: sensitive yet practical short read mapping. Bioinformatics 27(7):1011–1012. [DOI] [PubMed] [Google Scholar]
- Dittman AH, et al. 2010. Homing and spawning site selection by supplemented hatchery- and natural-origin Yakima River spring Chinook salmon. Trans Am Fish Soc. 139(4):1014–1028. [Google Scholar]
- Domanico MJ, Phillips RB, Oakley TH.. 1997. Phylogenetic analysis of Pacific salmon (genus Oncorhynchus) using nuclear and mitochondrial DNA sequences. Can J Fish Aquat Sci. 54(8):1865–1872. [Google Scholar]
- Eichenberger RM, Ramakrishnan C, Russo G, Deplazes P, Hehl AB.. 2017. Genome-wide analysis of gene expression and protein secretion of Babesia canis during virulent infection identifies potential pathogenicity factors. Sci Rep. 7(1):3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foox J, Ringuette M, Desser SS, Siddall ME.. 2015. In silico hybridization enables transcriptomic illumination of the nature and evolution of Myxozoa. BMC Genomics. 16(1):840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rodríguez VE, Garrido C, Cantoral JM, Schumacher J.. 2016. The F-actin capping protein is required for hyphal growth and full virulence but is dispensable for septum formation in Botrytis cinerea. Fungal Biol. 120(10):1225–1235. [DOI] [PubMed] [Google Scholar]
- Gupta I, Aggarwal S, Singh K, Yadav A, Khan S.. 2018. Ubiquitin Proteasome pathway proteins as potential drug targets in parasite. Sci Rep. 8(1):8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8(8):1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett SL, Bartholomew JL.. 2012. Myxobolus cerebralis and Ceratomyxa shasta In: Woo PTK, Buchmann K, editors. Fish parasites: pathobiology and protection. Oxfordshire, UK: CABI. p. 141–172. [Google Scholar]
- Hallett SL, et al. 2012. Density of the waterborne parasite Ceratomyxa shasta and its biological effects on salmon. Appl Environ Microbiol. 78(10):3724–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan A, Kosakyan A, Pecková H, Eszterbauer E, Holzer A. 2020. Transcriptome of Sphaerospora molnari (Cnidaria, Myxosporea) blood stages provides proteolytic arsenal as potential therapeutic targets against sphaerosporosis in common carp. BMC Genomics 21(1):404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan A, et al. 2016. New cell motility model observed in parasitic cnidarian Sphaerospora molnari (Myxozoa: Myxosporea) blood stages in fish. Sci Rep. 6:39093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartikainen H, et al. 2016. Assessing myxozoan presence and diversity using environmental DNA. Int J Parasitol. 46(12):781–792. [DOI] [PubMed] [Google Scholar]
- Holland JW, Okamura B, Hartikainen H, Secombes CJ.. 2011. A novel minicollagen gene links cnidarians and myxozoans. Proc R Soc B Biol Sci. 278(1705):546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer AS, et al. 2018. The joint evolution of the Myxozoa and their alternate hosts: a cnidarian recipe for success and vast biodiversity. Mol Ecol. 27(7):1651–1666. [DOI] [PubMed] [Google Scholar]
- Hurst CN, Bartholomew JL.. 2012. Ceratomyxa shasta genotypes cause differential mortality in their salmonid hosts. J Fish Dis. 35(10):725–732. [DOI] [PubMed] [Google Scholar]
- Hurst CN, Holt RA, Bartholomew JL.. 2012. Dam removal and implications for fish health: Ceratomyxa shasta in the Williamson River, Oregon, USA. N Am J Fish Manage. 32(1):14–23. [Google Scholar]
- Jiménez-Guri E, Philippe H, Okamura B, Holland PWH.. 2007. Buddenbrockia is a cnidarian worm. Science 317(5834):116–118. [DOI] [PubMed] [Google Scholar]
- Jones CE, Brown AL, Baumann U.. 2007. Estimating the annotation error rate of curated go database sequence annotations. BMC Bioinf. 8:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GO, Adkison MA, Leutenegger CM, Hedrick RP.. 2003. Myxobolus cerebralis: identification of a cathepsin Z-like protease gene (MyxCP-1) expressed during parasite development in rainbow trout, Oncorhynchus mykiss. Exp Parasitol. 105(3–4):201–210. [DOI] [PubMed] [Google Scholar]
- Kenkel CD, Bay LK.. 2017. Novel transcriptome resources for three scleractinian coral species from the Indo-Pacific. GigaScience 6(9):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinova IM, Tsimokha AS, Mittenberg AG.. 2008. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol. 267:59–124. [DOI] [PubMed] [Google Scholar]
- Kurath G, et al. 2003. Phylogeography of infectious haematopoietic necrosis virus in North America. J Gen Virol. 84(4):803–814. [DOI] [PubMed] [Google Scholar]
- Lentini G, et al. 2015. Identification and characterization of Toxoplasma SIP, a conserved apicomplexan cytoskeleton protein involved in maintaining the shape, motility and virulence of the parasite. Cell Microbiol. 17(1):62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN.. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien S, et al. 2016. The Atlantic salmon genome provides insights into rediploidization. Nature 533(7602):200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, et al. 2017. Analyses of corallimorpharian transcriptomes provide new perspectives on the evolution of calcification in the Scleractinia (Corals). Genome Biol Evol. 9(1):150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasanch EE, Orlowski RZ.. 2017. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 14(7):417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrow J, Caffrey C, Kelly B, Loke P, Sajid M.. 2006. Proteases in parasitic diseases. Annu Rev Pathol Mech Dis. 1(1):497–536. [DOI] [PubMed] [Google Scholar]
- Mennerat A, Nilsen F, Ebert D, Skorping A.. 2010. Intensive farming: evolutionary implications for parasites and pathogens. Evol Biol. 37(2–3):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, et al. 2009. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics. 10(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakawa N, Kraft G.. 2005. Homing behaviour of juvenile coho salmon (Oncorhynchus kisutch) within an off-channel habitat. Ecol Freshw Fish. 14(2):197–201. [Google Scholar]
- Morais ER, et al. 2017. Effects of proteasome inhibitor MG-132 on the parasite Schistosoma mansoni. PLoS One 12(9):e0184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz C, Francisco JS, Gutiérrez B, González J.. 2015. Role of the ubiquitin-proteasome systems in the biology and virulence of protozoan parasites. BioMed Res Int. 2015:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesnidal MP, Helmkampf M, Bruchhaus I, El-Matbouli M, Hausdorf B.. 2013. Agent of whirling disease meets orphan worm: phylogenomic analyses firmly place Myxozoa in Cnidaria. PLoS One 8(1):e54576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I.. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23(9):1061–1067. [DOI] [PubMed] [Google Scholar]
- Pearson WR. 2013. An introduction to sequence similarity (‘homology’) searching. Curr Protoc Bioniformatics Chapter 3:Unit 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Finn RD.. 2016. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44(D1):D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RA, Holt RA, Bartholomew JL.. 2012. Relationship between temperature and Ceratomyxa shasta-induced mortality in Klamath River salmonids. J Parasitol. 98(3):520–526. [DOI] [PubMed] [Google Scholar]
- Rodrigues M, et al. 2016. Profiling of adhesive-related genes in the freshwater cnidarian Hydra magnipapillata by transcriptomics and proteomics. Biofouling 32(9):1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC.. 2009. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 5(12):e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, Dougan G, Rappuoli R.. 2009. The key role of genomics in modern vaccine and drug design for emerging infectious diseases. PLoS Genet. 5(10):e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpirer E, Diamant A, Cartwright P, Huchon D.. 2018. A genome wide survey reveals multiple nematocyst-specific genes in Myxozoa. BMC Evol Biol. 18(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart ZK, Pavasovic A, Hock DH, Prentis PJ.. 2017. Transcriptomic investigation of wound healing and regeneration in the cnidarian Calliactis polypus. Sci Rep. 7:41458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinson MET, Atkinson SD, Bartholomew JL.. 2018. Widespread distribution of Ceratonova shasta (Cnidaria: Myxosporea) genotypes indicates both evolutionary adaptation to its salmonid fish hosts. J Parasitol. 104(6):645–650. [DOI] [PubMed] [Google Scholar]
- Stinson ME, Bartholomew JL.. 2012. Predicted redistribution of Ceratomyxa shasta genotypes with salmonid passage in the Deschutes River, Oregon. J Aquat Anim Health 24(4):274–280. [DOI] [PubMed] [Google Scholar]
- Tanaka K. 2009. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B 85(1):12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tine M, et al. 2014. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat Commun. 5:5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirloni L, et al. 2014. Proteomic analysis of cattle tick Rhipicephalus (Boophilus) microplus saliva: a comparison between partially and fully engorged females. PLoS One 9(4):e94831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyndall JDA, Nall T, Fairlie DP.. 2005. Proteases universally recognize beta strands in their active sites. Chem Rev. 105(3):973–1000. [DOI] [PubMed] [Google Scholar]
- Varga G, et al. 2017. Proteasome subunit beta type 1 P11A polymorphism is a new prognostic marker in multiple myeloma. Clin Lymphoma Myeloma Leuk. 17(11):734–742. [DOI] [PubMed] [Google Scholar]
- Williams TH, et al. 2016. Viability assessment for Pacific salmon and steelhead listed under the Endangered Species Act: Southwest. California: U.S. Department of Commerce, NOAA Technical Memorandum. NMFS-SWFSC-564.
- Xu P, et al. 2014. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet. 46(11):1212–1219. [DOI] [PubMed] [Google Scholar]
- Yahalomi D, et al. . 2020. A cnidarian parasite of salmon (Myxozoa: Henneguya ) lacks a mitochondrial genome. Proc Natl Acad Sci USA. 117(10):5358–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. 2014. The genome of the myxosporean Thelohanellus kitauei shows adaptations to nutrient acquisition within its fish host. Genome Biol Evol. 6(12):3182–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zardoya R, Garrido-Pertierra A, Bautista JM.. 1995. The complete nucleotide sequence of the mitochondrial DNA genome of the rainbow trout, Oncorhynchus mykiss. J Mol Evol. 41(6):942–951. [DOI] [PubMed] [Google Scholar]
- Zheng J, Jia H, Zheng Y.. 2015. Knockout of leucine aminopeptidase in Toxoplasma gondii using CRISPR/Cas9. Int J Parasitol. 45(2–3):141–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following intermediate and final files of all analyses performed in this study are deposited in DRYAD (https://doi.org/10.5061/dryad.tx95x69tt): 1) host filtered parasite and neither reads lists of I, IIC, IIR_RBT7, IIR_RBTC16, and IIR_RBT6 (20 .list files); 2) genotype IIR_RBT6 reference assembled transcriptomes (nonfiltered) (two .fasta files); 3) host and other contaminants (other microorganisms) filtered IIR_RBT6 assemblies (two .fasta files); 4) longest representatives reference IIR_RBT6 cs + neither assembly used for SNPs analyses (one .fasta file); 5) SNPs tables (genotypes called from nucleotide frequencies with a) minimum coverage of 5 and 0.25 heterozygosity threshold [five .tab files] and b) minimum coverage of 20 reads and 0.1 heterozygosity threshold [five .tab files]); and 6) phylogenomic and SNPs-based phylogenetic alignments (nine .nex files and 51 individual genes alignments .fasta used for phylogenomics).