

Abstract
Background/Aims
N-acetylcysteine (NAC) affects signaling pathways involved in apoptosis, angiogenesis, cell growth and arrest, redox-regulated gene expression, and the inflammatory response. However, it is not known how the signal mechanism for tight junctional protein claudin (CLDN) 18 is regulated in asthma patients.
Methods
To investigate the effects of NAC on CLDN18 expression in a mouse model of asthma, and to assess plasma levels of CLDN18 in asthma patients. A murine model of asthma induced by ovalbumin (OVA) was established using wild-type BALB/c female mice, and the levels of CLDNs, phosphorylated-pyruvate dehydrogenase kinase 1 (p-PDK1), and protein kinase B (Akt) pathway proteins following NAC treatment were examined by Western blotting and immunohistochemistry. In addition, the plasma levels of CLDN18 were evaluated in asthmatic patients and control subjects.
Results
NAC diminished OVA-induced airway hyper-responsiveness and inflammation. Levels of CLDN18 protein were higher in lung tissue from OVA mice than tissue from control mice, and were increased by treatment with NAC or dexamethasone. Treatment with NAC or dexamethasone decreased the OVA-induced increase in interleukin-1α protein levels. Although treatment with NAC increased OVA-induced p-PDK1 protein levels, it decreased phosphorylated Akt (pAkt)/Akt levels. Soluble CLDN18 levels were lower in patients with asthma than in controls and were correlated with the percentage of neutrophils, forced expiratory volume in 1 second (FEV1)/forced vital capacity % (FVC%) and FEV1%.
Conclusions
CLDN18 plays a role in the pathogenesis of asthma and NAC diminishes airway inflammation and responsiveness by modulating CLDN18 expression.
Keywords: Asthma, Claudin 18, N-acetylcysteine, Plasma, Tight junction
INTRODUCTION
N-acetylcysteine (NAC), the precursor of L-cysteine and reduced glutathione, functions as an antioxidant in vivo [1]. It acts as a direct scavenger of reactive oxygen species to regulate redox status and affects signaling pathways involved in apoptosis, angiogenesis, cell growth and arrest, redox-regulated gene expression, and the inflammatory response [2,3]. It has an indirect antioxidant effect by synthesizing glutathione, which serves as primary intracellular defense against toxic agents [4]. NAC may not only protect against the direct injurious effects of oxidants but may also alter inflammatory events in a beneficial manner [5].
Tight junctions (TJs) act as a semipermeable barrier (or gate) to the paracellular transport of ions, solutes, water, and cells, and function as a fence dividing the apical and basolateral domains of the plasma membrane [6-8]. TJ cleavage and repair occur following exposure of epithelial cells to proteinase and peptidase of dermatophagoides pteronyssinus 1 (Der p1), which suggests that disruption of this barrier may be an important event in allergic sensitization and asthma [9-11].
Claudins (CLDNs), which are expressed in various types of airway epithelial cells, are structural molecules of TJs responsible for changes in the electrolyte and solute permeability of cell layers [12]. However, it is unclear how the signal mechanism for CLDN18 affecting the permeability barrier in the airways is regulated, and the impact of NAC on CLDN18 in asthma patients is unknown. In this study, we evaluated the contribution of CLDN18 to asthma, and whether CLDN18 expression is modulated by NAC in bronchial asthma in a mouse model. In addition, the plasma CLDN18 levels of patients with asthma were analyzed.
METHODS
Experimental design
Eight BALB/c mice were exposed to saline (control) or ovalbumin (OVA; OVA/OVA) and treated with NAC or dexamethasone (Fig. 1A). We evaluated the impact of NAC and CLDN18 on the levels of cytokines, CLDNs, and signal-pathway proteins. The patient study was approved by the Soonchunhyang University Institutional Review Board (SCHBC-2014-10-011). Written informed consent was obtained from all patients. The animal study was approved by the Soonchunhyang University Institutional Animal Care and Use Committee (SCHBC-animal-2014-13).
Figure 1.

(A) Effect of N-acetylcysteine (NAC) or dexamethasone on airway hyperresponsiveness (AHR) and inflammation in a mouse model of asthma. (B) AHR measured after inhalation of increasing doses of methacholine. (C) Numbers of total cells, macrophages, eosinophils, and neutrophils per milliliter bronchoalveolar lavage fluid (BALF). Data are means ± SEMs (n = 6 to 7 per group). OVA, ovalbumin; I.P, intraperitoneal; DEX, dexamethasone. ap < 0.05 compared to sham. bp < 0.05 compared to OVA/OVA.
Patients and control subjects
Biospecimens and clinical data were provided by the biobank of Soonchunhyang University, Bucheon Hospital, a member of the Korea Biobank Network. All subjects had a clinical diagnosis of asthma according to Global Initiative for Asthma (GINA) guidelines [13] with modification. All subjects underwent standardized assessments that included complete blood cell and differential counts, immunoglobulin E (IgE) quantification, chest posteroanterior radiography, skin prick allergy testing, and spirometry (Table 1). All data were collected at the time of diagnosis, before administration of asthma medication. Normal control subjects were recruited from among spouses of the subjects or members of the general population.
Table 1.
Clinical characteristics in control subjects and patients with asthma
| Characteristic | Control subjects | Asthma patients |
|
|---|---|---|---|
| Stable | Exacerbated | ||
| No. of subjects | 15 | 20 | 17 |
| Sex, male/female | 2/13 | 8/12 | 7/10 |
| Age, yr | 69 (56–78) | 49 (23–74) | 63 (42–83) |
| Onset of age, yr | - | 41 (23–74) | 49.80 (20–74) |
| Asthma period, yr | - | 6.55 ± 10.40a | 9.82 ± 16.64a |
| Smoke, NS/ES/SM | 15/0/0 | 14-4-2 | 9/5/3 |
| Smoke amount, pack-yr | 0 | 6.8 ± 13.57a | 14.35 ± 19.47a |
| Initial lung function | |||
| FVC% | 92.93 ± 15.41 | 87.5 ± 19.90 | 81.06 ± 16.15 |
| FEV1% | 109.80 ± 16.48 | 85.15 ± 24.22a | 81.41 ± 22.78a |
| FEV1/FVC | 83.07 ± 6.82 | 73.5 ± 12.40a | 71.88 ± 9.00a |
| Stable and exacerbate lung function | |||
| FVC% | - | 86.91 ± 16.52 | 66.00 ± 15.92b |
| FEV1% | - | 86.82 ± 19.25 | 64.13 ± 16.09b |
| FEV1/FVC | - | 74.15 ± 9.73 | 69.69 ± 9.26 |
| BMI | 24.56 ± 2.04 | 25.54 ± 2.63 | 26.29 ± 3.32 |
| Initial PC20 | - | 9.29 ± 11.11 | 9.72 ± 10.29 |
| Total IgE | 14.55 ± 28.02 | 561.12 ± 903.50a | 495.00 ± 729.07a |
| Skin test positive | 1 (6.6) | 11 (55) | 7 (41.2) |
| Blood WBC, µL | 5,798.67 ± 1,400.65 | 6,388 ± 2,000.41 | 7,474.71 ± 2,400.96a |
| Blood eosinophil, % | 2.99 ± 2.72 | 4.35 ± 4.72 | 4.31 ± 4.75 |
Values are presented as median (range), mean ± SD, or number (%). Inhaled corticosteroid potency (fluticasone propionate: budesonide = 1.5:1) PC20 methacholine; the concentration of methacholine required to decrease the FEV1 by 20%.
NS, non-smoker; ES, ex-smoker; SM, smoker; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; BMI, body mass index; PC20, provocative concentration 20; IgE, immunoglobulin E; WBC, white blood cell.
p < 0.01 compared with control subjects.
p < 0.05 compared with stable as.
All subjects had a clinical diagnosis of asthma that was supported by one or more of the following criteria: variability in the maximum diurnal peak expiratory flow greater than 20% over the course of 14 days; an increase in the forced expiratory volume in 1 second (FEV1) greater than 15% after inhalation of 200 to 400 μg albuterol; or a positive provocative concentration 20 (PC20) response, defined as a 20% reduction in FEV1 in response to an inhalation of < 10 mg/mL methacholine. Asthma exacerbation was defined, based on the GINA guidelines [13], as episodes of progressive increases in shortness of breath, cough, wheezing, or chest tightness; some combination of these symptoms, accompanied by decreases in expiratory airflow and use of systemic corticosteroids (tablets, suspension, or injection); or an increase from a stable maintenance dose for at least 3 days and a hospitalization or emergency department visit because of asthma, requiring systemic corticosteroids.
All data were collected at the time of diagnosis, before the administration of asthma medication, and during the period of exacerbation. Asthmatics from the hospital cohort were matched to healthy subjects according to age, sex, and body mass index. These healthy control subjects were included based on the following criteria: negative responses on a screening questionnaire for respiratory symptoms and other allergic diseases, FEV1 values greater than 80% of predicted values, PC20 methacholine values > 10 mg/mL, and normal findings in simple chest radiographs. The exclusion criteria included respiratory infection during sputum induction, chronic obstructive pulmonary disease, vocal cord dysfunction, obstructive sleep apnea, Churg-Strauss syndrome, cardiac dysfunction, allergic bronchopulmonary aspergillosis, or poor compliance with treatment.
Animals
All experimental animal methods followed a protocol approved by the Institutional Animal Care and Use Committee of the Soonchunhyang University Bucheon Hospital. Female BALB/c mice (6 weeks old) were sensitized and challenged with OVA. Airway responsiveness was evaluated, bronchoalveolar lavage fluid (BALF) was collected, and lung tissue was harvested for protein extraction, hematoxylin and eosin (H&E) staining and immunohistochemistry.
NAC and dexamethasone treatments
NAC (100 mg/kg) (Boryung, Seoul, Korea) inhalation treatment was performed 1 hour after each challenge with OVA for 6 consecutive days. Additional groups of mice were treated with intraperitoneally dexamethasone (3 mg/kg) (Sigma-Aldrich, St. Louis, MO, USA) 1 hour before each OVA challenge daily for 3 days.
Western blotting
For Western blot analysis, Protein concentration in the homogenates of lung tissue was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Proteins were separated and transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% bovine serum albumin in 0.1% Tween 20 in Tris-buffered saline, and incubated with anti-CLDN7 and CLDN18 antibodies (1:200, Abcam, Cambridge, MA, USA) (4°C, overnight) followed by the horseradish peroxidase-conjugated secondary antibody. Bands were visualized using the WESTZOL plus Western blotting Detection System (iNtRon, Seongnam, Korea). The relative abundance of proteins was determined by quantitative densitometry with normalization to β-actin (Sigma-Aldrich).
Histological assessment of lung tissue
Lung sections were deparaffinized, rehydrated in an ethanol series, and stained with H&E. For detection of CLDNs, Lung sections were incubated with anti-CLDN7 and CLDN18 antibodies (1:200, Abcam) at 4°C overnight. Next, the sections were incubated with an avidin and biotinylated horseradish peroxidase macromolecular complex (Vector Laboratories, Burlingame, CA, USA). Color was developed by staining with liquid DAB + substrate kit (Golden Bridge International Inc., Mukilteo, WA, USA). After immunohistochemical staining, the slides were counterstained with Harris’s hematoxylin for 1 min. Images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay
The levels of IL-1α, IL-10, and tumor necrosis factor-α (TNF-α) in BALF from mice were determined using the Multi Analyte ELISArray Kit (Qiagen, Valencia, CA, USA) and CLDN18 levels in human plasma were measured using an enzyme-linked immunosorbent assay (ELISA) (USCN, Wuhan, China). The lower detection limit was set at 32.5 pg/mL (IL-1α), 59.4 pg/mL (IL-10), 30.5 pg/mL (TNF-α), or 0.055 ng/mL (CLDN18), respectively, based on the manufacturer’s recommendations.
Statistical analyses
Data are expressed as means ± standard errors of the mean (SEMs), and the Mann-Whitney U test was used to evaluate the significance of differences between groups. A p < 0.05 was deemed to indicate statistical significance.
RESULTS
Effects of NAC on AHR and inflammation in a mouse model of asthma
OVA-sensitized/challenged mice had a significantly greater airway hyperresponsiveness (AHR) compared to sham mice (Fig. 1B), which was significantly reduced by treatment with NAC or dexamethasone. The numbers of total inflammatory cells, macrophages, eosinophils, and neutrophils in BALF were significantly increased in OVA/OVA mice compared to sham mice (p < 0.05) (Fig. 1C) and were significantly reduced by NAC or dexamethasone.
NAC and dexamethasone decrease IL-1α and TNF-α levels in BALF
Next, we evaluated the levels of mononuclear phagocyte-produced cytokines in BALF. IL-1α levels tended to be higher in OVA/OVA mice compared to sham mice and were reduced by NAC or dexamethasone (Fig. 2). IL-10 and TNF-α levels tended to be higher in OVA/OVA compared to sham mice and were reduced by dexamethasone (Fig. 2).
Figure 2.

N-acetylcysteine (NAC) induces the production of inflammatory cytokines. Cytokine levels in bronchoalveolar lavage fluid according to the ELISArray kit. (A) Interleukin α (IL-1a), (B) IL-10, (C) tumor necrosis factor-α (TNF-α). OVA, ovalbumin; DEX, dexamethasone. ap < 0.05 compared to OVA/OVA.
Effects of NAC on CLDN expression in lung tissue
CLDN18 levels were significantly higher in the lungs of OVA/OVA mice than in sham mice and were increased further by NAC or dexamethasone (Fig. 3A). Histopathologic examination revealed epithelium loss and increased infiltration of inflammatory cells in the lungs of OVA/OVA mice, which were decreased by NAC or dexamethasone. CLDN18 levels were increased in the lung tissue of OVA/OVA mice compared to control mice and were increased further by NAC (Fig. 3B).
Figure 3.

Effects of N-acetylcysteine (NAC) and dexamethasone on claudins (CLDNs). (A) Levels of CLDN18 in lung homogenates. Densitometry of three immunoblots with normalization to β-actin. (B) Immunohistochemistry staining (×200) of lung sections of the sham and ovalbumin (OVA; OVA/OVA) groups. Tight junctions were disrupted in OVA sensitized/challenged mice but recovered after treatment with NAC or dexamethasone. Data are means ± SEMs (n = 6 to 7 per group). DEX, dexamethasone. ap < 0.05 compared to sham. bp < 0.05 compared to OVA/OVA.
Effects of NAC on signaling pathways in lung tissue
Activation of Akt and phosphorylated-pyruvate dehydrogenase kinase 1 (p-PDK1) was assessed by Western blotting (Fig. 4). Treatment with NAC increased and decreased the p-PDK1 and p-Akt/Akt protein levels, respectively. NAC and dexamethasone suppressed the phosphorylation of Akt in lungs.
Figure 4.

(A) Claudin 18 (CLDN18) is involved the phosphorylation of pyruvate dehydrogenase kinase 1 (PDK1) and protein kinase B (Akt) after N-acetylcysteine (NAC) or dexamethasone treatment. (B-E) Densitometry of with normalization to β-actin. Data are means ± SEMs (n = 6 to 7 per group). p-PDK1, phosphorylated-PDK1; DEX, dexamethasone. ap < 0.05 compared to sham. bp < 0.05 compared to ovalbumin (OVA)/OVA.
CLDN18 expression in patients with asthma
In total, 20 patients with stable asthma (median age [range], 49 years [23 to 74]), 17 patients with exacerbated asthma (63 years [42 to 83]), and 15 control subjects (69 years [56 to 78]) were recruited (Table 1). Initial FEV1% and FEV1/forced vital capacity (FVC) were significantly lower in patients with asthma than in controls. Total IgE levels and amount of smoking (pack-years) were significantly higher in patients with asthma than in controls. The body mass index was similar between patients and controls. FEV1% and FVC% were significantly lower in patients with exacerbated asthma than in those with stable asthma.
Plasma levels of CLDN18 were lower in asthma patients than in healthy controls (p < 0.05). The levels tended to be lower in patients with exacerbated asthma than in those with stable asthma (p > 0.05) (Fig. 5), and were correlated with the percentage of neutrophils, FEV1/FVC%, and FEV1% (neutrophils, ρ = 0.286, p = 0.040; FEV1/FVC%, ρ = –0.463, p = 0.040; FEV1%, ρ = –0.576, p = 0.019) (Fig. 6).
Figure 5.
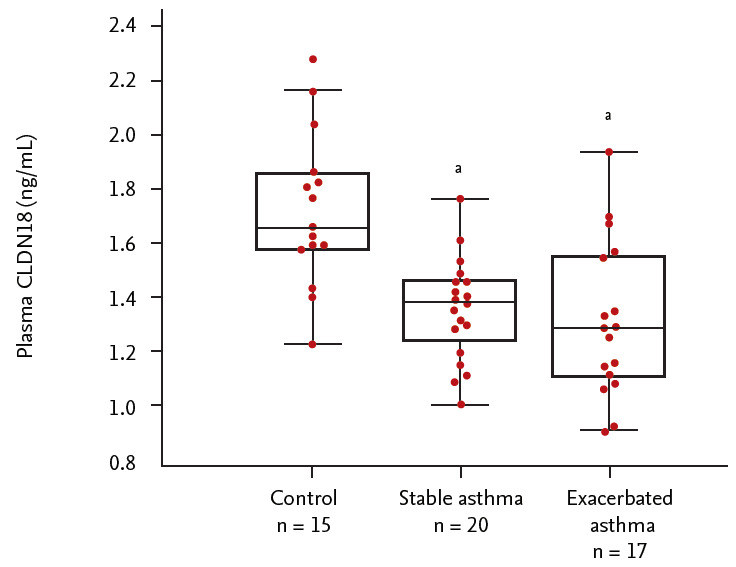
Plasma levels of claudin 18 (CLDN18) in control subjects and asthma patients. ap < 0.05, control vs. asthma patients.
Figure 6.

Correlation between plasma claudin 18 (CLDN18) levels and the percentage of (A) neutrophils, (B) forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC), and (C) FEV1% in asthmatic patients.
DISCUSSION
We evaluated whether NAC enhances CLDN18 expression in a mouse model of asthma. OVA/OVA mice had a significantly greater AHR and larger numbers of total inflammatory cells, macrophages, eosinophils, and neutrophils in BALF compared to sham mice. NAC treatment reduced AHR, the numbers of inflammatory cells, and the levels of cytokines compared to OVA/OVA mice. It also increased CLDN18 expression by altering phospho-PDK1 and Akt signaling in the lung, similar to the results of dexamethasone treatment. Acetylcysteine is the NAC derivative of the naturally occurring amino acid cysteine. The compound is a white crystalline powder with a molecular formula of C5H9NO3S and a molecular weight of 163.2 Da.
Apical junctional complexes (AJCs) in polarized epithelia make up adherens junctions (AJs) and TJs [14,15], which mediate intercellular adhesion and paracellular permeability to ions and solutes, respectively. TJs, the most apical component of AJCs, are composed of integral membrane proteins [16,17] linked to the actin cytoskeleton via accessory proteins [18]. CLDNs are a family of integral membrane proteins essential for TJ formation and integrity [16,19-21]. The permeability properties of various epithelia tissues are determined by cell-specific patterns of CLDN expression. CLDN18 is one of the most highly expressed CLDN family members in lung alveolar epithelia [22,23]. It is expressed at low levels in airway epithelia and is not expressed in lung endothelia [24].
CLDNs, TJ proteins that regulate paracellular permeability and cell polarity, are frequently dysregulated in cancer [23,24]; however, their role in asthma is unclear. In this study, soluble CLDN18 expression was altered in the plasma of asthma patients and lung CLDN18 was significantly upregulated in mice with asthma compared to sham mice; it was also correlated with AHR and inflammation.
The contribution of CLDN18 deficiency to aeroantigen sensitization and airway hyper-responsiveness was examined in CLDN18 null mice using the intratracheal aspergillus sensitization model. The results support the hypothesis that CLDN18 is an essential contributor to the airway epithelial barrier to aeroantigens. Furthermore, TH2 inflammation suppresses CLDN18 expression, potentially promoting sensitization and AHR [25]. CLDN18 also regulates lung epithelial cell proliferation and motility by inhibiting the phosphorylation of PDK1 and Akt [26].
In the lung tissue of mice with asthma, increase in CLDN18 was more increased following NAC. Therefore, CLDN18 react to NAC and dexamethasone; further studies of this phenomenon are warranted. NAC and dexamethasone impacted p-Akt and p-PDK1 protein levels, suggesting that those signal pathways are involved in CLDN18 regulation in mouse lungs.
In conclusion, lung CLDN18 activates p-Akt and p-PDK1 signaling and affects AHR and inflammation, which suggests involvement in the pathogenesis of asthma. NAC treatment may modulate airway inflammation by impacting CLDN18.
KEY MESSAGE
1. Plasma levels of soluble claudin 18 (CLDN18) decrease in asthma patients, while lung levels are highly expressed in an ovalbumin (OVA) sensitization/challenge mouse model of asthma.
2. CLDN18 activates phosphorylated-protein kinase B (p-Akt) and phosphorylated-pyruvate dehydrogenase kinase 1 (p-PDK1) signaling and affects airway hyperresponsiveness and inflammation, suggesting that it is involved in the pathogenesis of asthma.
3. N-acetylcysteine (NAC) decreases OVA-induced airway hyper-responsiveness and inf lammation by modulating CLDN18.
Acknowledgments
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health and Welfare, Republic of Korea (HI15C2032) and Soonchunhyang University Hospital.
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Harkness LM, Weckmann M, Kopp M, Becker T, Ashton AW, Burgess JK. Tumstatin regulates the angiogenic and inflammatory potential of airway smooth muscle extracellular matrix. J Cell Mol Med. 2017;21:3288–3297. doi: 10.1111/jcmm.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ebina M, Yaegashi H, Chiba R, Takahashi T, Motomiya M, Tanemura M. Hyperreactive site in the airway tree of asthmatic patients revealed by thickening of bronchial muscles: a morphometric study. Am Rev Respir Dis. 1990;141:1327–1332. doi: 10.1164/ajrccm/141.5_Pt_1.1327. [DOI] [PubMed] [Google Scholar]
- 3.Wilson J. The bronchial microcirculation in asthma. Clin Exp Allergy. 2000;30 Suppl 1:51–53. doi: 10.1046/j.1365-2222.2000.00098.x. [DOI] [PubMed] [Google Scholar]
- 4.Davies DE. Epithelial barrier function and immunity in asthma. Ann Am Thorac Soc. 2014;11 Suppl 5:S244–S251. doi: 10.1513/AnnalsATS.201407-304AW. [DOI] [PubMed] [Google Scholar]
- 5.Park CS, Kim YY, Kang SY. Collection between RAST and skin test for inhalant offending allergens. Allergy. 1983;3:1–9. [Google Scholar]
- 6.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134:509–520. doi: 10.1016/j.jaci.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin HJ, Park HS. Claudin may be a potential biomarker for epithelial barrier dysfunction in asthma. Allergy Asthma Immunol Res. 2018;10:4–5. doi: 10.4168/aair.2018.10.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–1284. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 11.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Meer G, Gumbiner B, Simons K. The tight junction does not allow lipid molecules to diffuse from one epithelial cell to the next. Nature. 1986;322:639–641. doi: 10.1038/322639a0. [DOI] [PubMed] [Google Scholar]
- 13.Global Initiative for Asthma . Global strategy for asthma management and prevention 2014 [Internet] Fontana (WI): Global Initiative for Asthma; 2014. [cited 2019 Nov 28]. Available from: http://www.ginasthma.org/ [Google Scholar]
- 14.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cereijido M, Contreras RG, Shoshani L, Flores-Benitez D, Larre I. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim Biophys Acta. 2008;1778:770–793. doi: 10.1016/j.bbamem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cording J, Berg J, Kading N, et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci. 2013;126:554–564. doi: 10.1242/jcs.114306. [DOI] [PubMed] [Google Scholar]
- 19.Van Itallie CM, Anderson JM. Claudin interactions in and out of the tight junction. Tissue Barriers. 2013;1:e25247. doi: 10.4161/tisb.25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75:551–567. doi: 10.1146/annurev-physiol-030212-183809. [DOI] [PubMed] [Google Scholar]
- 21.Schlingmann B, Molina SA, Koval M. Claudins: gatekeepers of lung epithelial function. Semin Cell Dev Biol. 2015;42:47–57. doi: 10.1016/j.semcdb.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L193–L205. doi: 10.1152/ajplung.00349.2010. [DOI] [PubMed] [Google Scholar]
- 23.Ouban A, Ahmed AA. Claudins in human cancer: a review. Histol Histopathol. 2010;25:83–90. doi: 10.14670/HH-25.83. [DOI] [PubMed] [Google Scholar]
- 24.Soini Y. Expression of claudins 1, 2, 3, 4, 5 and 7 in various types of tumours. Histopathology. 2005;46:551–560. doi: 10.1111/j.1365-2559.2005.02127.x. [DOI] [PubMed] [Google Scholar]
- 25.Sweerus K, Lachowicz-Scroggins M, Gordon E, et al. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J Allergy Clin Immunol. 2017;139:72–81. doi: 10.1016/j.jaci.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimobaba S, Taga S, Akizuki R, et al. Claudin-18 inhibits cell proliferation and motility mediated by inhibition of phosphorylation of PDK1 and Akt in human lung adenocarcinoma A549 cells. Biochim Biophys Acta. 2016;1863:1170–1178. doi: 10.1016/j.bbamcr.2016.02.015. [DOI] [PubMed] [Google Scholar]