

Abstract
Background
Doxorubicin (DOX) is one of the most effective anti-neoplastic drugs although its clinical use is limited by the severe cardiotoxicity. Apoptosis and defective autophagy are believed to contribute to DOX-induced cardiotoxicity. Here we explored the effect of curcumin (Cur) on DOX-induced cardiac injury and the mechanism involved with a focus on oxidative stress, autophagy and pyroptosis.
Methods
Kunming mice were challenged with DOX (3 mg·kg−1, i.p. every other day) with cohorts of mice receiving Cur at 50, 100, 200 and 400 mg·kg−1 via gavage daily. Serum levels of cardiac enzymes, such as aspartate amino transferase (AST), lactate dehydrogenase (LDH), creatine kinase (CK), and heart homogenate oxidative stress markers, such as superoxide dismutase (SOD) and malondialdehyde (MDA) were determined. Echocardiographic and cardiac contraction were examined. Apoptosis, pyroptosis, autophagy and Akt/mTOR-signalling proteins were detected using western blot or electron microscopy. Cardiac contractile properties were assessed including peak shortening, maximal velocity of shortening/relengthening (± dL/dt), time-to-PS, and time-to-90% relengthening (TR90). Superoxide levels were evaluated using DHE staining. GFP-LC3 was conducted to measure autophagosomes.
Results
Our study showed that Cur protected against cardiotoxicity manifested by a significant decrease in serum myocardial enzymes and improvement of anti-oxidative capacity. Cur inhibited autophagy and offered overt benefit for cardiomyocyte survive against DOX-induced toxicity. Cur attenuated DOX-induced cardiomyocyte pyroptosis as evidenced by NLR family pyrin domain containing 3 (NLRP3), Caspase-1, and interleukin-18 levels. DOX impaired cardiac function (reduced fractional shortening, ejection fraction, increased plasma cTnI level and TR90, decreased PS and ± dL/dt), the effects of which were overtly reconciled by 100 mg·kg−1 but not 50 mg·kg−1 Cur. H9c2 cells exposure to DOX displayed increased intracellular reactive oxygen species (ROS) and autophagy, the effects of which were nullified by Cur. Autophagy activator rapamycin cancelled off Cur-induced protective effects.
Conclusions
Our finding suggested that Cur rescued against DOX-induced cardiac injury probably through regulation of autophagy and pyroptosis in a mTOR-dependent manner.
Keywords: Doxorubicin (DOX), curcumin, autophagy, oxidative stress, apoptosis
Introduction
Doxorubicin (DOX), commonly known as an anthracycline antibiotic, is a potent anti-neoplastic agent employed in a variety of carcinomas including breast cancer, multiple myeloma, neuroblastoma and leukemia (1,2). However, its clinical application has been greatly hindered by its detrimental cardiotoxicity. The risk prevalence is estimated to be approximately 5% with a cumulative dose of DOX 400 mg·m−2, 16% at a dose of 500 mg·m−2, 26% with 550 mg·m−2 and 48% with 700 mg·m−2, resulting in severe left ventricular dysfunction, dilated cardiomyopathy and even congestive heart failure in humans (3,4). Given the essential role for DOX in chemotherapy, intense efforts have been engaged to combat the cardiotoxicity of DOX without compromising its anticancer efficacy. However, clinical usage of DOX remains dismal with regards to the proper management of DOX cardiotoxicity.
Up-to-date, a number of mechanisms have been postulated and confirmed for DOX-induced cardiac damage. In particular, DOX is capable of promoting oxidative stress and apoptosis through production of ROS and lipid peroxidation while decreasing levels of antioxidant and sulfhydryl groups. Production of ROS directly damages cell membrane, leading to subsequent release of apoptotic initiating proteins, en route to the DOX-induced cardiotoxicity (5). To overcome this obstacle, various agents including medicinal compounds are employed to inhibit DOX-induced cardiotoxicity. Recent studies also indicated that autophagic dysregulation plays an important role in DOX-induced cardiotoxicity (6). In eukaryotic cells, autophagy is responsible for the degradation or recycling of misfolded or damaged proteins, in an effort to maintain cellular homeostasis and survival (7). In the heart, low levels of autophagy are beneficial for the maintenance of cardiac function and geometry. However, excessive induction of autophagy, such as in the case of DOX challenge, leads to cardiac cell death (8,9). Curcumin (Cur) is a vibrant yellow spice derived from rhizome of Curcuma longa and has been used for a long time as a coloring and flavoring additive in foods in India. A numerous studies have demonstrated that Cur offers potent beneficial effects against cancer, Alzheimer’s disease, and cystic fibrosis (10). Earlier evidence from our own group indicated that Cur alleviates hyperglycemia, inhibits oxidative stress and rescues cardiomyopathy in streptozotocin-induced diabetes and high glucose-induced cardiomyocyte apoptosis (11,12). To better understand the effect of Cur on DOX-induced cardiac damage, this study was designed to evaluate the cardioprotective role, if any, of Cur against cardiac damage triggered by DOX in murine hearts and cardiomyocytes and the underlying molecular mechanism involved with a focus on mTOR-dependent autophagy.
We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/cdt-19-707).
Methods
Animal model
All protocols were approved by the Committee of Experimental Animals of the Hubei University of Science and Technology (HBUST-IACUC-2016-002). All experimental subjects were treated according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996). In brief, SPF, healthy male kunming mice (n=120, weight 22±2 g, 4–5 weeks) were purchased from Laboratory Animal Center of preventive medicine academy of the Hubei province. One week after acclimation in house with at a temperature of 22–26 °C and a humidity of 40–60% under a 12 h light/dark cycle, mice were randomly divided into six groups (n=20 per group). Mice in DOX-treated group were intraperitoneally injected with the DOX at 3 mg·kg−1 every other day with a cumulative dosage of 24 mg·kg−1 (13). Control group (Con) received intraperitoneal injection with an equivalent volume of saline. Mice in Cur treatment groups were intragastric administered different dosage of Cur (100, 200, and 400 mg·kg−1, mixed with 0.5% sodium carboxymethyl cellulose) daily with simultaneous injection of DOX every other day. Mice in Cur group (Cur) were administered with Cur (400 mg·kg−1) daily. In addition, a cohort of mice were randomly divided into six groups, including control, DOX, Con-Cur (50 mg·kg−1), DOX-Cur (50 mg·kg−1), Con-Cur (100 mg·kg−1) and DOX-Cur (100 mg·kg−1) groups. On the 17th day, echocardiographic or cardiomyocyte functional assessment were performed prior to collection of blood samples under anesthesia for analysis of cardiac enzymes. Hearts were then used for electron microscopic detection. The remaining hearts were frozen at −80 °C for western blot analysis.
Cellular model
H9c2 (American Type Culture Collection, ATCC) were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere (5% CO2 and 95% air). Cells were divided into 4 groups: Control, DOX (2 µmol·L−1 DOX), Con-Cur and DOX-Cur. DOX-damaged H9c2 model was cultured with DOX for 24 h. To assess whether Cur affects autophagy, H9c2 cells were exposed to DOX in the absence or presence of Cur (10 µmol·L−1) or the autophagy inducer rapamycin (100 nmol·L−1) (14).
Cell shortening/relengthening
Contractile properties of cardiomyocytes were detected using a SoftEdge MyoCam® system (IonOptix Corporation, Milton, MA) as described (15). In brief, cells were placed in a Warner chamber mounted on an inverted microscope (Olympus, IX-70) and superfused (~1 mL·min−1 at 25 °C) with a buffer containing (in mmol·L−1): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 Glucose, 10 HEPES, at pH 7.4. Myocytes were stimulated at 0.5 Hz with cell shortening and relengthening evaluated using the following indices: peak shortening (PS), time to peak shortening (TPS), time to 90% relengthening (TR90), and maximal velocities of shortening/relengthening (± dL/dt). To evaluate the role of Cur on DOX-induced changes in contractile function, cells were cultured for 4 h in a KHB solution containing DOX (10 µmol·L−1) with or without Cur (10 µmol·L−1) prior to the assessment of cardiomyocyte mechanical properties.
Measurement of biochemical index
Myocardial injury marker enzymes such as serum aspartate transaminase (AST), lactate dehydrogenase (LDH), creatine kinase (CK) and heart homogenate oxidative stress markers such as superoxide dismutase (SOD) and malondialdehyde (MDA) were measured using kits from Nanjing Jiancheng Institute of Biological Engineering. The detailed assay procedures were performed according to the manufacturer’s instructions.
Electron microscopy detection
Approximate 1 mm3 left ventricular tissues were fixed in 2.5% glutaraldehyde fixative for more than 2 h and post-fixed with 1% osmium tetraoxide, then tissues were rinsed with phosphoric acid rinse solution and dehydrated in ascending alcohol series. Subsequently tissues were embedded in Epon. Finally, samples were cut on ultramicrotome and were double-stained with uranyl acetate and lead citrate. Images were captured using transmission electron microscopy (FEI Tecnai G212).
Plasma cTnI level detection
Plasma troponin I (cTnI) level was evaluated using the chemiluminescent immunoassay (Beckman, A98264, USA). In brief, paramagnetic microparticles coated with an alkaline phosphatase-conjugated monoclonal cTnI antibody were mixed with plasma samples and a chemiluminescent substrate was added to the reaction tube. Levels of light generated in the reaction were measured using a luminescence detector. The level of cTnI in plasma samples was determined based on the stored multi-point calibration curve, with a window of 0.02–100 ng·mL−1.
Echocardiographic assessment
Mice were anaesthetized with ~1% isoflurane inhalation and guided by M-mode echocardiography equipped with a 15–6 MHz linear transducer (Phillips Sonos 5500). LV anterior and posterior wall dimensions during diastole and systole were recorded from three consecutive cycles in M-mode. Heart rates were averaged over 10 cardiac cycles. Fractional shortening was calculated from LV end diastolic (LVEDD) and end-systolic (LVESD) diameters using the equation (LVEDD − LVESD)/LVEDD ×100%. Ejection fraction was calculated using the equation (LVEDD3 − LVESD3)/LVEDD3 ×100% (16).
Reactive oxygen species (ROS) measurement
Intracellular superoxide anions were measured using the dihydroethidium (DHE) fluorescence probe. H9c2 cells were incubated in a light-impermeable chamber at 37 °C for 30min after application of 10 µmol·L−1 DHE (Life Technology, USA) and were then cultured with 5 µg·mL−1 4',6-diamidino-2-phenylindole (DAPI, Sigma, USA) for 5 min. The images of H9c2 cardiomyocytes were captured and analyzed immediately under an inverted fluorescence microscopy (Olympus BX51, Japan). The intracellular ROS level was quantified using a redox-sensitive dye carboxy 2'7'-dichlorodihydrofluorescein diacetate (H2DCF-DA) obtained from Molecular Probes (Life Technology, USA). H2DCF-DA is non-fluorescent, after meeting oxidation by ROS it converts to fluorescent marker 2'7'-dichlorofluorescein (DCF) which emits green fluorescence. In brief, H9c2 cells were treated with 1 µmol·L−1 H2DCF-DA for 30 min in a conventional incubator (37 °C, 5% CO2). Cardiomyocytes were rinsed with phosphate buffer saline (PBS) and DCF fluorescence intensity was measured using a fluorescence plate reader (Molecular Devices, Sunnyvale, CA) with 480 nm excitation and 530 nm emission filters (17).
LC3-GFP-adenovirus production and infection in H9c2
Autophagy was assessed in H9c2 cells using GFP fluorescence. Adenovirus containing GFP-LC3 construct was kindly provided by Dr. Cindy Miranti from the University of Arizona and propagated using HEK293 cell line. Cells were transfected with GFP-LC3 adenovirus for 8 h and were then refreshed with normal medium. Cells were visualized for autophagy using fluorescence microscopy and were treated with DOX in the absence or presence of Cur and rapamycin. For autophagy evaluation, cells were captured under a fluorescence microscope (Olympus BX51) and percentage of GFP-LC3 positive cells (with >10 GFP-LC3 puncta dots per cell) were achieved as described (18). About 400–500 cells in each group were calculated.
Western blot analysis
Myocardial tissues were homogenized in RIPA buffer and were centrifuged at 12,000× rpm for 15 min at 4 °C. Equal concentration of protein was separated by different concentration SDS-PAGE gels and then transferred to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk in TBST and then incubated with primary antibodies against Bax, Bcl-2, Beclin1, LC3, p-AKT (ser473), AKT, p-mTOR (s2808), mTOR, GAPDH (Cell Signaling Technology, CA, USA) and NLR family pyrin domain containing 3 (NLRP3), Caspase-1, interleukin-1β (IL-1β), IL-18 (Bioss, China) at 4 °C overnight. After washing with TBST for three times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody at a 1:5,000 dilution at room temperature for 1 h. Blots were visualized with ECL kits (Pierce Biosciences, USA) and intensity of protein bands was assessed and quantified using Quantity One software from Bio-Rad (16).
HepG2 cell survival assay
HepG2 cells at a density of 5,000 cells per well were plated in a 96-well plate and incubated with DOX or Cur for 24 h. Cell viability was measured by an MTT assay according to the manufacturer’s instructions.
Data analysis
Results are presented as mean ± SD. Significant differences (P<0.05) were compared using a one-way analysis of variance (ANOVA) followed by a Tukey post hoc analysis.
Results
Cur alleviated DOX-induced myocardial injury and changes in body and heart weights
As shown in Figure 1A,B,C, DOX significantly reduced body and heart weight as well as the heart-to-body weight (HW-to-BW) ratio, the effects of which were spared by Cur. Cur itself did not produce any effects on body or heart weight (or the heart-to-body weight ratio). In addition, DOX significantly increased serum levels of cardiac enzymes including CK, AST and LDH, all of which were significantly attenuated or mitigated by 100, 200 and 400 mg·kg−1 doses of Cur, with little effect from Cur itself (Figure 1D,E,F).
Figure 1.

Effect of Cur and DOX on biochemical and biometric parameters. (A) Body weights (BW); (B) heart weights (HW); (C) HW-to-BW ratio; (D) serum CK release; (E) serum AST release; (F) serum LDH content; (G) SOD activity; and (H) MDA content. Mean ± SD, n=14 (panels A, B, C) or 9–10 (panels D, E, F, G, H) per group. *, P<0.05 between the indicated groups. Con, Control group; DOX, DOX group; DOX-Cur [100], DOX with Cur treatment at 100 mg·kg−1; DOX-Cur [200], DOX with Cur treatment at 200 mg·kg−1; DOX-Cur [400], DOX with Cur treatment at 400 mg·kg−1; Con-Cur [400], 400 mg·kg−1 Cur treatment in control group; BW, body weight; HW, heart weight; CK, creatine kinase; AST, aspartate amino transferase; LDH, lactate dehydrogenase; SOD, superoxide dismutase; MDA, malondialdehyde.
Cur attenuated DOX-induced oxidative stress
SOD activity and MDA content are essential oxidative markers. As shown in Figure 1G,H, DOX dramatically increased MDA content, the effect of which was significantly ameliorated by 200 and 400 mg·kg−1 doses of Cur, with little effect from Cur itself. Neither DOX nor DOX in combination with Cur overtly affected SOD activity. High dose of Cur (400 mg·kg−1) increased SOD activity by itself.
Cur reduced DOX-induced cardiomyocyte apoptosis and pyroptosis in mice
In order to determine the effect of Cur on apoptosis, apoptosis-related proteins were monitored using western blot analysis. Data shown in Figure 2A,B,C,D suggested that DOX challenge remarkably increased Bax while slightly decreased Bcl-2 level and enhanced the Bax-to-Bcl-2 ratio, which were notably reversed by Cur treatment (100, 200 or 400 mg·kg−1), with the exception of Bcl-2 level at 400 mg·kg−1 Cur. Cur itself yielded little effect on these apoptotic markers.
Figure 2.

Effect of Cur on apoptosis and pyroptosis protein markers in DOX-treated mice. (A) Representative gel blots of Bcl-2 and Bax proteins using specific antibodies (GAPDH as loading control); (B) Bax; (C) Bcl-2; (D) Bax-to-Bcl-2 ratio; (E) representative gel blots of NLRP3, Caspase-1, IL-18 and IL-1β proteins using specific antibodies expression (GAPDH as loading control); (F) NLRP3; (G) Caspase-1; (H) IL-18; and (I) IL-1β. Mean ± SD, n=3–4 per group, *, P<0.05 between the indicated groups. Bcl-2, B-cell lymphoma-2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Bax, Bcl-2 associated X protein; NLRP3, NLR family pyrin domain containing 3; IL-18, interleukin-18; IL-1β, interleukin-1β.
To understand if Cur inhibits NLRP3-mediated pyroptosis in DOX-treated mice, if any, levels of pyroptosis-related proteins were examined using western blot. As shown in Figure 2E,F,G,H,I, DOX treatment triggered elevated pyroptosis, as manifested by upregulation of pyroptosis protein markers including NLRP3, Caspase-1 and IL-18 without affecting IL-1β. Cur partially reversed DOX-evoked changes in NLRP3, Caspase-1 and IL-18 without affecting that of IL-1β. Cur yielded little effect on pyroptosis markers by itself.
Cur inhibited DOX-induced cardiomyocyte autophagy in mice
Autophagy plays an important role in cardiomyocyte survival (16). Autophagosomes, sarcomere and mitochondria ultrastructure were evaluated using transmission electronic microscopy. As shown in Figure 3A, DOX challenge provoked overt cytoarchitectural aberrations such as mitochondrial swelling, dissolving myofibers and accumulation of double-membrane vacuoles, the response of which was greatly lessened by 100 or 200 mg·kg−1 Cur, while 400 mg·kg−1 Cur treatment seemed to exhibit an worsened ultrastructural change compared with 100 or 200 mg·kg−1 Cur treatment. Moreover, autophagy protein markers including Beclin1 and LC3 were determined using western blot. As revealed in Figure 3B,C,D,E,F, level of Beclin1 and ratio of LC3II-to-LC3I were markedly elevated following DOX challenge, the effects of which were obliterated by 100 or 200 mg·kg−1 Cur.
Figure 3.

Effect of Cur on autophagy in DOX-treated mice. (A) Mitochondria and sarcomere ultrastructure by transmission electron microscopy; (B) representative gel blots of Beclin1 and LC3 proteins using specific antibodies (GAPDH as loading control); (C) Beclin1; (D) LC3I; (E) LC3II; and (F) LC3II-to-LC3I ratio. Mean ± SD, n=4–5 per group, *, P<0.05 between the indicated groups.
Cur activated the Akt/mTOR pathway in DOX-treated mice
To explore the potential mechanism of action behind apoptotic and autophagy responses, activation of the Akt/mTOR signaling cascade was detected using western blot. As shown in Figure 4A,B,C,D,E,F,G, DOX downregulated levels of phosphorylation of Akt and mTOR, the effects of which were notably reversed by 100 and 200 mg·kg−1 doses of Cur. Cur itself failed to exhibit any notable effect in phosphorylation of Akt and mTOR expression.
Figure 4.

Effect of Cur on the Akt/mTOR pathway in DOX-treated mice. (A) Representative gel blots of p-Akt, Akt, p-mTOR and mTOR proteins using specific antibodies (GAPDH as loading control); (B) p-Akt; (C) Akt; (D) p-Akt-to-Akt ratio; (E) p-mTOR; (F) mTOR; and (G) p-mTOR-to-mTOR ratio. Mean ± SD, n=4 per group, *, P<0.05 between the indicated groups.
Cur ameliorated DOX-induced changes in plasma cTnI level, echocardiographic and cardiomyocyte contractile properties
In order to further discern whether lower dose Cur possesses any protective effect against DOX-induced cardiotoxicity, we examined the possible effect of low doses of Cur treatment (50 or 100 mg·kg−1) on DOX-induced cardiac toxicity using plasma cTnI, echocardiographic and cardiomyocyte contractile properties. Echocardiographic evaluation revealed that DOX significantly increased LVESD and decreased left ventricular fractional shortening as well as ejection fraction, all of which were partly attenuated by 100 but not 50 mg·kg−1 Cur treatment (Figure 5A,B,C,D,E,F). Moreover, plasma cTnI level was overtly elevated in DOX mice, the effect of which was significantly ablated by 100 mg·kg−1 Cur treatment, while 50 mg·kg−1 dose of Cur failed to offer any effect on these changes (Figure 5G).
Figure 5.

Effect of Cur on myocardial echocardiographic properties and plasma cTnI level in DOX-treated mice. (A) Heart rate; (B) LV posterior wall thickness in diastole; (C) LV end diastolic diameter (LVEDD); (D) LV end systolic diameter (LVESD); (E) fractional shortening; (F) ejection fraction; and (G) cardiac troponin I (cTnI). Mean ± SD; n=6–8 mice per group, *, P<0.05 between the indicated groups. Con, Control group; DOX, DOX group; Con-Cur [50], 50 mg·kg−1 Cur treatment in control group; DOX-Cur [50], DOX with Cur treatment at 50 mg·kg−1; Con-Cur [100], 100 mg·kg−1 Cur treatment in control group; DOX-Cur [100], DOX with Cur treatment at 100 mg·kg−1.
Consistent with echocardiographic findings, Cardiomyocytes from DOX mice showed markedly dampened contractile function, as manifested by decreased peak shortening (PS) and maximal velocity of shortening/relengthening (± dL/dt), as well as prolonged TR90 without any change in resting cell length and TPS. 100 mg·kg−1 Cur treatment ameliorated DOX-induced cardiomyocyte contractile anomalies without any effect itself, while the effect of which were unaffected by 50 mg·kg−1 dose of Cur (Figure 6A,B,C,D,E,F).
Figure 6.

Contractile properties of cardiomyocytes from mice treated with or without Cur. (A) Resting cell length; (B) peak shortening (PS); (C) maximal velocity of shortening (+dL/dt); (D) maximal velocity of relengthening (−dL/dt); (E) time-to-PS (TPS); and (F) time-to-90% relengthening (TR90). Mean ± SD, n=45 cells per group, *, P<0.05 between the indicated groups.
Cur alleviated DOX-induced cardiomyocyte dysfunction
To determine the possible mechanism of action behind Cur-induced myocardial response in the face of DOX, the effects of Cur on contractile function were measured in murine cardiomyocytes in the absence of presence of certain pharmacological regulators. As shown in Figure 7A,B,C,D,E,F, acute exposure (4 hours) of DOX led to compromised cardiomyocyte contractile capacity (suppressed ± dL/dt, PS, and prolonged TR90, with unchanged resting cell length and TPS), Cur effectively restored DOX-induced cardiomyocyte dysfunction with little response itself. Interestingly, the class III PtdIns3K inhibitor wortmannin and the mTOR inhibitor rapamycin effectively abolished Cur-induced protection against DOX-induced cardiomyocyte mechanical dysfunction.
Figure 7.

Effect of Cur on cardiomyocyte shortening in ventricular myocytes isolated from adult mouse hearts. (A) Resting cell length; (B) peak shortening (PS); (C) maximal velocity of shortening (+ dL/dt); (D) maximal velocity of relengthening (-dL/dt); (E) time-to-PS (TPS); and (F) time-to-90% relengthening (TR90). Mean ± SD, n=40–60 cells per group, *, P<0.05 between the indicated groups. Con, Control group; DOX, DOX group; Con-Cur, 10 µmol·L−1 Cur treatment in control group; DOX-Cur, DOX with Cur treatment at 10 µmol·L−1; Con-Rap, Rapamycin treatment in control group; DOX-Cur-Rap, DOX with Cur and rapamycin treatment; Con-Wor, Wortmannin treatment in control group; DOX-Cur-Wor, DOX with Cur and wortmannin treatment.
Cur attenuated DOX-induced ROS/O2− production
As shown in Figure 8A, O2− production was assessed using DHE fluorescence. Our data revealed that DOX significantly elevated O2− levels in H9c2 cells, although Cur did not affect O2− levels in control groups, it nullified DOX-induced elevation in O2− level. Furthermore, DOX also enhanced DCF fluorescent intensity of DCF in H9c2 cells, the effect of which was marginally reduced by 10 µmol·L−1 Cur treatment, with little effect from Cur itself (Figure 8B).
Figure 8.

Effect of Cur on accumulation of ROS in H9c2 cells. (A) Representative images of DHE staining; (B) pooled data of DCF quantification. Mean ± SD, n=14 cultures per group, *, P<0.05 between the indicated groups. DHE, dihydroethidium; DCF, 2'7'-dichlorodihydrofluorescein diacetate.
Cur attenuated DOX-induced autophagy in H9c2
Increased initial autophagosome formation and impaired autophagosome-lysosome clearance may both lead to LC3II accumulation. As shown in Figure 9A,B, H9c2 cells exhibited an increase in the number of punctate GFP-LC3 structures upon exposure to DOX. However, decreased number of punctate GFP-LC3 was detected after Cur treatment. Furthermore, rapamycin inhibited the effect of LC3II induced by Cur. We then determined the autophagy protein marker LC3 expression and found that DOX significantly increased the LC3II- to-LC3I ratio, the effect of which was negated by Cur (Figure 9C,D,E,F).
Figure 9.

Effect of Cur on autophagy in H9c2 cells. (A) Representative pictures of H9c2 cells transfected with GFP-LC3 expression vector; (B) pooled data of GFP-LC3 punctate; (C) representative gel blot of LC3 protein using specific antibodies (GAPDH as loading control); (D) LC3I; (E) LC3II; and (F) LC3II-to-LC3I ratio. Mean ± SD, n=9 fields of independent cultures per group (panels A, B) or 6 (panels C, D, E, F) per group, *, P<0.05 between the indicated groups.
Cur decreased the DOX-induced cell viability in HepG2
In order to determine if Cur affects the anti-cancer effects of DOX, HepG2 cell viability was monitored using MTT method. As shown in Figure 10, DOX partially or overtly decreased cell viability in HepG2 cells. Interesting, the combination of DOX and Cur (10 µmol·L−1) was superior to DOX treatment alone.
Figure 10.
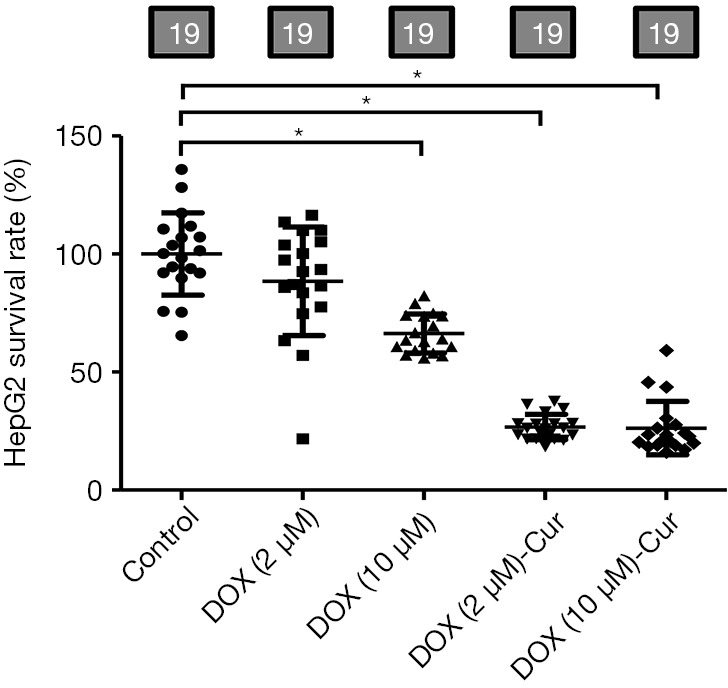
Cur decreased the DOX-induced cell viability in HepG2. Pooled data of MTT quantification. Mean ± SD, n=19 cultures per group, *, P<0.05 between the indicated groups. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide.
Discussion
The salient findings from our present study suggested that Cur exerts a protective effect on DOX-induced cardiac function, ROS/O2− production and pyroptosis. At the same time, Cur plays a synergistic response in DOX-mediated antitumor effect through decreased tumor cell viability, which is consistent with a recent report (19). DOX toxicity-induced change in oxidative stress and autophagy is believed to be responsible for ventricular dysfunction under DOX toxicity. Thus, once developed, DOX cardiotoxicity carries a poor prognosis and may be frequently fatal. While treatment of DOX cardiomyopathy remains challenging, our work suggested that Cur may serve as an alternative avenue for cardiovascular complications in DOX-induced cardiomyopathy and a likely role for autophagy and pyroptosis in Cur offered beneficial effects.
Our findings suggested that DOX challenge compromised cardiac contractile function (echocardiographic and cardiomyocyte contraction), promoted accumulation of ROS and O2− as well as apoptosis, in a manner somewhat similar to earlier reports (20,21). In our hands, pronounced cardiac injury was noted following acute DOX challenge, the effect of which was partly or overtly reversed by different doses of Cur. Oxidative stress is considered the main contributing factors in the pathogenesis of DOX cardiotoxicity (22,23). Thus, anti-oxidative therapy with antioxidants and over-expression of anti-oxidative enzymes is deemed effective against DOX-induced cardiotoxicity. Up-to-date, dexrazoxane is the only drug approved to protect against DOX-induced cardiotoxicity through inhibition of iron-dependent DOX-based oxidative stress. However, dexrazoxane was shown to preferentially mitigate adverse changes in female children and has raised clinical safety concerns, at least in children (24,25). Our observation of the oxidative stress biomarkers, DHE and DCF staining supported the earlier notion of oxidative stress upon DOX challenge, the effects of which were attenuated by Cur. Oxidative stress evoked by DOX triggers cardiomyocyte apoptosis, which can be primarily mediated via mitochondrial apoptosis cascade (21). Bcl-2 is an anti-apoptotic protein to suppress mitochondrial apoptosis via antagonism of Bax oligomerization and later cytochrome c release (26). DOX incubation promoted level of Bax and partially inhibited level of Bcl-2 in mouse hearts, while after Cur administered, the ratio of Bax/Bcl-2 was remarkably decreased. The current study proposed that DOX induces a new form of inflammatory-mediated cell death called pyroptosis in the heart (27). Caspase-1 was known as the major pyroptotic marker, which initiates pyroptosis in response to an activated inflammasome and converts pro-IL-1β and pro-IL-18 into their biologically active forms (28). Finding from our study indicated that Cur effectively reversed pro-pyroptosis alteration of NLRP3, Caspase-1 and IL-18 in DOX-induced cardiac damage. However, 400 mg·kg−1 dose of Cur in combination with DOX treatment showed worse outcomes (as shown by elevation of apoptosis and pyroptosis) compared with 100 and 200 mg·kg−1 doses of Cur treatment. It is thus plausible to speculate that sustained pyroptosis and apoptosis in the face of Cur treatment contributes to the inhibition the ROS generation.
Autophagy, an evolutionarily conserved process, is essential to cellular homeostasis and involves in cell survival under physiological and pathological conditions through restoring energy status and clearing damaged or unnecessary proteins and organelles (29). Proper autophagy helps to maintain cardiac homeostasis (30,31), however, excessive or unchecked autophagy is deemed another form of cell death to participate in myocardial remodeling and heart failure. DOX impaired autophagic processes, prompting cardiotoxicity. It was reported that up-regulated autophagy preceded DOX-induced cardiotoxicity and inhibition of autophagy improved the survival rate of cardiomyocytes treated with DOX (32). To this end, DOX-induced cardiomyocyte autophagy might be protective or detrimental depending on the stress or dosage levels (33). In our study, elevated autophagy was noted in DOX-challenged cardiomyocytes, these effects of which were restored by Cur. This is supported several experimental data. (I) In vitro study, electron microscopic examination indicated that DOX treatment dramatically promotes accumulation of double-membrane vacuoles, when mice were exposed simultaneously to DOX and 100 mg·kg−1 dose of Cur, the autophagosome production was significant reduced, while DOX-challenged mice treated with a high dosage of Cur (400 mg·kg−1) demonstrated toxic effect on autophagosome formation. (II) In our hands, autophagosome formation (GFP-LC3 puncta) was remarkably increased by DOX incubation in H9c2 cells, the effect of which was strikingly decreased with Cur, in consistence with the in vivo data. Interestingly, Cur inactivation-elicited autophagosome formation against DOX was cancelled off by the autophagy inducer rapamycin. On the other hand, Beclin1 is a core part of the class III PI3K protein and serves as an initiator for multiple autophagy-related protein recruitment process (34). Emerging evidence also suggested that cross-talk exists between autophagic and apoptotic pathways. For example, the antiapoptotic protein Bcl-2 inhibits autophagy through binding and sequestering Beclin1 away from the class III PI3K complex, eventually suppressing Beclin1-mediated autophagy (35). LC3 or MAP1LC3 is an indicator for autophagosome formation through conversion of cytosolic LC3I to autophagosome-bound LC3II (36). Our western blot analysis revealed increased Beclin1 and LC3II-to-LC3I ratio in DOX challenged mice, the effects of which were reversed by 100 and 200 mg·kg−1 Cur. Our further examination noted overtly elevated LC3II-to-LC3I ratio when H9c2 cells were exposure to DOX, in accordance with the experimental results in vivo. Thus, it is plausible to speculate that Cur might recover the constitutive high levels of autophagy under DOX challenge.
The PI3K-Akt-mTOR pathway is required for the pro-survival signalling cascade under a variety of circumstances. In previous studies, it was revealed that DOX induced myocardial apoptosis by down-regulating the PI3K/Akt/mTOR survival pathway (37). In addition, the PI3K/Akt/mTOR pathway is confirmed to be a negative regulator of autophagy via downstream targets (38). The class III PI3K inhibitor 3-MA and other approaches such as ghrelin, a metabolic regulatory peptide, were found to suppress DOX-induced autophagy and attenuate cardiomyocyte apoptosis (8). Our earlier study revealed that Cur protects against high glucose-induced cardiomyopathy through stimulating Akt signal cascade (11). As a result, it is intriguing to unveil whether manipulation of Akt/mTOR signalling pathway by Cur contributes to its cardioprotective effects against DOX. Mechanistically, Cur increased phosphorylation of Akt, a downstream mediator of mTOR signaling pathway in the heart. Therefore, the Cur-mediated reduction of DOX-induced pyroptosis and autophagy might be attributed to the activation of Akt/mTOR pathway.
In summary, findings from our study provided evidence that Cur rescues against DOX-induced cardiac function, oxidative stress and pyroptosis possibly through regulation of autophagy. Cur may offer beneficial effect through up-regulation of phosphorylation of Akt and subsequently mTOR. These findings should shed some lights towards a better understanding of the utility of Cur in DOX-induced cardiotoxicity. It is noteworthy that our study suffers from a few experimental limitations such as lack of data from clinical research and animal tumorigenesis examination. In this context, the ultimate clinical utilization of Cur merits further scrutiny to better define its effect in a wide range of patients.
Acknowledgments
Funding: This work was supported by grants from the Hubei Province Natural Science Fund Project (No. 2019CFB609). JR is a recipient of the Hubei Province Chutian Scholar Program.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All protocols were approved by the Committee of Experimental Animals of the Hubei University of Science and Technology (HBUST-IACUC-2016-002). All experimental subjects were treated according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996).
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Reporting Checklist: The authors present the study in accordance with the ARRIVE reporting checklist. Available at http://dx.doi.org/10.21037/cdt-19-707
Data Sharing Statement: Available at http://dx.doi.org/10.21037/cdt-19-707
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/cdt-19-707). The authors have no conflicts of interest to declare.
References
- 1.Argenziano M, Gigliotti CL, Clemente N, et al. Improvement in the Anti-Tumor Efficacy of Doxorubicin Nanosponges in In Vitro and in Mice Bearing Breast Tumor Models. Cancers (Basel) 2020;12:162. 10.3390/cancers12010162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan S, Zhao Y, Lu J, et al. Second-generation proteasome inhibitor carfilzomib sensitizes neuroblastoma cells to doxorubicin-induced apoptosis. Oncotarget 2016;7:75914-25. 10.18632/oncotarget.12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 2003;97:2869-79. 10.1002/cncr.11407 [DOI] [PubMed] [Google Scholar]
- 4.Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol 2020;29:101394. 10.1016/j.redox.2019.101394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan BYH, Roczkowsky A, Cho WJ, et al. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodeling. Cardiovasc Res 2020. [Epub ahead of print]. 10.1093/cvr/cvaa017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol 2017;104:1-8. 10.1016/j.yjmcc.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 7.Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069-75. 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Wang XL, Chen HL, et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem Pharmacol 2014;88:334-50. 10.1016/j.bcp.2014.01.040 [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Yang L, Ma J, et al. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim Biophys Acta Mol Basis Dis 2017;1863:1904-11. [DOI] [PubMed]
- 10.Ren J, Sowers JR. Application of a novel curcumin analog in the management of diabetic cardiomyopathy. Diabetes 2014;63:3166-8. 10.2337/db14-0863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W, Zha W, Ke Z, et al. Curcumin Protects Neonatal Rat Cardiomyocytes against High Glucose-Induced Apoptosis via PI3K/Akt Signalling Pathway. J Diabetes Res 2016;2016:4158591. [DOI] [PMC free article] [PubMed]
- 12.Yu W, Wu J, Cai F, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS One 2012;7:e52013. 10.1371/journal.pone.0052013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vijay V, Moland CL, Han T, et al. Early transcriptional changes in cardiac mitochondria during chronic doxorubicin exposure and mitigation by dexrazoxane in mice. Toxicol Appl Pharmacol 2016;295:68-84. 10.1016/j.taap.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 14.Lin C, Zhang M, Zhang Y, et al. Helix B surface peptide attenuates diabetic cardiomyopathy via AMPK-dependent autophagy. Biochem Biophys Res Commun 2017;482:665-71. 10.1016/j.bbrc.2016.11.091 [DOI] [PubMed] [Google Scholar]
- 15.Guo R, Hu N, Kandadi MR, et al. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy 2012;8:593-608. 10.4161/auto.18997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Ge W, Harns C, et al. Ablation of toll-like receptor 4 attenuates aging-induced myocardial remodeling and contractile dysfunction through NCoRI-HDAC1-mediated regulation of autophagy. J Mol Cell Cardiol 2018;119:40-50. 10.1016/j.yjmcc.2018.04.009 [DOI] [PubMed] [Google Scholar]
- 17.Turdi S, Fan X, Li J, et al. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010;9:592-606. 10.1111/j.1474-9726.2010.00586.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu W, Zha W, Ren J. Exendin-4 and Liraglutide Attenuate Glucose Toxicity-Induced Cardiac Injury through mTOR/ULK1-Dependent Autophagy. Oxid Med Cell Longev 2018;2018:5396806. [DOI] [PMC free article] [PubMed]
- 19.Zhao G, Sun Y, Dong X. Zwitterionic Polymer Micelles with Dual Conjugation of Doxorubicin and Curcumin: Synergistically Enhanced Efficacy against Multidrug-Resistant Tumor Cells. Langmuir 2020;36:2383-95. 10.1021/acs.langmuir.9b03722 [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Wang M, Liu J, et al. Inhibition of TRPA1 Attenuates Doxorubicin-Induced Acute Cardiotoxicity by Suppressing Oxidative Stress, the Inflammatory Response, and Endoplasmic Reticulum Stress. Oxid Med Cell Longev 2018;2018:5179468. [DOI] [PMC free article] [PubMed]
- 21.Huang PC, Kuo WW, Shen CY, et al. Anthocyanin Attenuates Doxorubicin-Induced Cardiomyotoxicity via Estrogen Receptor-alpha/beta and Stabilizes HSF1 to Inhibit the IGF-IIR Apoptotic Pathway. Int J Mol Sci 2016;17:1588. 10.3390/ijms17091588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalilzadeh M, Abdollahi A, Abdolahi F, et al. Protective effects of magnesium sulfate against doxorubicin induced cardiotoxicity in rats. Life Sci 2018;207:436-41. 10.1016/j.lfs.2018.06.022 [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Lei T, Yuan J, et al. GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol 2018;17:25-34. 10.1016/j.redox.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timm KN, Tyler DJ. The Role of AMPK Activation for Cardioprotection in Doxorubicin-Induced Cardiotoxicity. Cardiovasc Drugs Ther 2020;34:255-69. 10.1007/s10557-020-06941-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narayan HK, Putt ME, Kosaraju N, et al. Dexrazoxane preferentially mitigates doxorubicin cardiotoxicity in female children with sarcoma. Open Heart 2019;6:e001025. 10.1136/openhrt-2019-001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan W, Jiang X, Li X, et al. Silencing of angiotensin-converting enzyme by RNA interference prevents H9c2 cardiomyocytes from apoptosis induced by anoxia/reoxygenation through regulation of the intracellular renin-angiotensin system. Int J Mol Med 2013;32:1380-6. 10.3892/ijmm.2013.1525 [DOI] [PubMed] [Google Scholar]
- 27.Tavakoli Dargani Z, Singla DK. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am J Physiol Heart Circ Physiol 2019;317:H460-H471. 10.1152/ajpheart.00056.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Man SM, Karki R, Briard B, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep 2017;7:45126. 10.1038/srep45126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011;13:495-504. 10.1016/j.cmet.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu S, Chen L, Wu Y, et al. Gastrodin pretreatment alleviates myocardial ischemia/reperfusion injury through promoting autophagic flux. Biochem Biophys Res Commun 2018;503:2421-8. 10.1016/j.bbrc.2018.06.171 [DOI] [PubMed] [Google Scholar]
- 31.Xiao J, Ke ZP, Shi Y, et al. The cardioprotective effect of thymoquinone on ischemia-reperfusion injury in isolated rat heart via regulation of apoptosis and autophagy. J Cell Biochem 2018;119:7212-7. 10.1002/jcb.26878 [DOI] [PubMed] [Google Scholar]
- 32.Luo P, Zhu Y, Chen M, et al. HMGB1 contributes to adriamycin-induced cardiotoxicity via up-regulating autophagy. Toxicol Lett 2018;292:115-22. 10.1016/j.toxlet.2018.04.034 [DOI] [PubMed] [Google Scholar]
- 33.Carresi C, Musolino V, Gliozzi M, et al. Anti-oxidant effect of bergamot polyphenolic fraction counteracts doxorubicin-induced cardiomyopathy: Role of autophagy and c-kit(pos)CD45(neg)CD31(neg) cardiac stem cell activation. J Mol Cell Cardiol 2018;119:10-8. 10.1016/j.yjmcc.2018.04.007 [DOI] [PubMed] [Google Scholar]
- 34.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009;186:773-82. 10.1083/jcb.200907014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 2010;17:268-77. 10.1038/cdd.2009.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirkin V. History of the Selective Autophagy Research: How Did It Begin and Where Does It Stand Today? J Mol Biol 2020;432:3-27. 10.1016/j.jmb.2019.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee BS, Oh J, Kang SK, et al. Insulin Protects Cardiac Myocytes from Doxorubicin Toxicity by Sp1-Mediated Transactivation of Survivin. PLoS One 2015;10:e0135438. 10.1371/journal.pone.0135438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang ZG, Wang Y, Huang Y, et al. bFGF regulates autophagy and ubiquitinated protein accumulation induced by myocardial ischemia/reperfusion via the activation of the PI3K/Akt/mTOR pathway. Sci Rep 2015;5:9287. 10.1038/srep09287 [DOI] [PMC free article] [PubMed] [Google Scholar]