

Abstract
Background
Disruption of β-amyloid (Aβ) homeostasis is the initial culprit in Alzheimer’s disease (AD) pathogenesis. Astrocytes respond to emerging Aβ plaques by altering their phenotype and function, yet molecular mechanisms governing astrocytic response and their precise role in countering Aβ deposition remain ill-defined. Peroxiredoxin (PRDX) 6 is an enzymatic protein with independent glutathione peroxidase (Gpx) and phospholipase A2 (PLA2) activities involved in repair of oxidatively damaged cell membrane lipids and cellular signaling. In the CNS, PRDX6 is uniquely expressed by astrocytes and its exact function remains unexplored.
Methods
APPswe/PS1dE9 AD transgenic mice were once crossed to mice overexpressing wild-type Prdx6 allele or to Prdx6 knock out mice. Aβ pathology and associated neuritic degeneration were assessed in mice aged 10 months. Laser scanning confocal microscopy was used to characterize Aβ plaque morphology and activation of plaque-associated astrocytes and microglia. Effect of Prdx6 gene dose on plaque seeding was assessed in mice aged six months.
Results
We show that hemizygous knock in of the overexpressing Prdx6 transgene in APPswe/PS1dE9 AD transgenic mice promotes selective enticement of astrocytes to Aβ plaques and penetration of plaques by astrocytic processes along with increased number and phagocytic activation of periplaque microglia. This effects suppression of nascent plaque seeding and remodeling of mature plaques consequently curtailing brain Aβ load and Aβ-associated neuritic degeneration. Conversely, Prdx6 haplodeficiency attenuates astro- and microglia activation around Aβ plaques promoting Aβ deposition and neuritic degeneration.
Conclusions
We identify here PRDX6 as an important factor regulating response of astrocytes toward Aβ plaques. Demonstration that phagocytic activation of periplaque microglia vary directly with astrocytic PRDX6 expression level implies previously unappreciated astrocyte-guided microglia effect in Aβ proteostasis. Our showing that upregulation of PRDX6 attenuates Aβ pathology may be of therapeutic relevance for AD.
Keywords: Astrocytes, Alzheimer’s disease, β-Amyloid plaques, Microglia, Neurodegeneration, Peroxiredoxin 6, Plaque seeding, Proteostasis
Background
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, where initially occurring β-amyloid (Aβ) deposition drives neurofibrillary pathology leading to synaptic and neuronal degeneration and gradual cognitive decline [1]. Containment of Aβ deposition or Aβ proteostasis is the initial defense mechanism preventing Aβ accumulation. Aβ plaques attract activated microglia, which are well recognized for their role in Aβ plaque transformation and containment. Microglia phagocytose diffuse Aβ peptide forming the plaque brim and compact Aβ within the plaque amyloid core [2] limiting extent of Aβ-associated neuritic degeneration [3]. AD-associated variants in Triggering Receptor Expressed in Myeloid cells 2 (TREM2) rendering microglia ineffective in Aβ plaque processing have been identified as second to the APOE ε4 allele genetic risk factor for sporadic AD, highlighting the importance of periplaque glia function in Aβ proteostasis and in arresting downstream cascade of AD neurodegeneration [4]. Like microglia, activated astrocytes surround Aβ plaques and penetrate the plaques with their processes. However, in contrast to microglia the role of astrocytes in Aβ proteostasis and plaque formation remains ill-defined, mainly due to a paucity of known factors modulating astrocytic function in AD, and especially those, whose variable expression level would create a tractable experimental model. In this study, we decided to explore function of astrocytes in Aβ proteostasis and plaque formation through modulating expression level of an astrocytic native protein peroxiredoxin (PRDX) 6. PRDX6 is a dual function enzyme with independent glutathione peroxidase (Gpx) and phospholipase A2 (PLA2) activities, which is highly expressed by several cell lineages outside the CNS including alveolar epithelium, endothelium, and macrophages [5]. The PLA2 activity distinguishes PRDX6 from other peroxiredoxins and enables replacement of peroxidatively damaged cell membrane lipids, and cellular signaling [6, 7]. In the CNS, PRDX6 is expressed by astrocytes but no other type of glial cells [8, 9] and its exact function remains largely unexplored. In normal brain, expression of PRDX6 is dormant, while in AD it becomes selectively upregulated in astrocytes, which are associated with Aβ plaques and neurofibrillary tangles [9]. PRDX6 does not accumulate within the plaques, thus it is a reactive but not amyloid associated protein. To explore the function of PRDX6 in Aβ proteostasis we made APPswe/PS1dE9 transgenic (Tg) mice with hemizygous knock in of the overexpressing Prdx6 transgene and those with Prdx6 haplodeficiency. This experimental design showed that PRDX6 governs a protective function of astrocytes in countering Aβ deposition. We found an inverse dependence between Prdx6 gene dose and Aβ plaque load and plaque-associated neuritic degeneration. Prdx6 expression level also showed direct dependence with enticement of astrocytes and microglia to Aβ plaques, penetration of plaques by activated astrocytes, and phagocytic activation of periplaque microglia. Since PRDX6 is an astrocytic protein, the latter observation implies that astrocytes target microglia to Aβ plaques and circuitously regulate microglia dependent Aβ plaque processing.
Methods
Materials and reagents
Primers for polymerase chain reaction protocols were synthesized to order by Sigma-Aldrich (St. Louis, MO). Synthetic Aβ1–40 and Aβ1–42 peptides were produced by the ERI Amyloid Laboratory LLC (Oxford, CT) and handled as previously described [10, 11]. Antibody sources are individually listed below. All chemicals and reagents, were purchased from Sigma-Aldrich unless stated otherwise.
Animals
All mouse care and experimental procedures were approved by Institutional Animal Care and Use Committees of the New York University Grossman School of Medicine. All transgenic mouse lines and C57BL/6 J wild type mice were obtained from The Jackson Laboratories (Bar Harbor, ME). APPswe/PS1dE9 AD model mice (B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax; stock # 34832-JAX) express under the control of the mouse prion protein promoter the mouse amyloid precursor protein (APP) harboring the human Aβ sequence with the double Swedish familial AD mutation K594M/N595L and human presenilin 1 (PS1) with exon 9 deletion (dE9). In this model, the entire APPswe/PS1dE9 transgene is transmitted as a single Mendelian locus [12, 13]. Prdx6 transgenic mice (C57BL/6 J-Tg(Prdx6)153Pgn/Pgn; stock #005902) overexpress the Prdx6129/SvJ-Tg transgene containing the wild type Prdx6 129X1/SvJ allele, typical of the atherosclerosis-resistant 129/SvJ mouse strain, on the endogenous C57BL/6 J Prdx6 expression background [14–16]. Prdx6 knock out mice (B6.129-Prdx6tm1Pgn/Pgn; stock #005974) feature disruption of exons 1 and 2 of the Prdx6 gene using a targeting vector containing the neomycin resistance and lacZ genes [17, 18]. All three transgenic mouse lines have been maintained on C57BL/6 J strain background. Hemizygous APPswe/PS1dE9 mice (APP/PS11/0;Prdx6+/+) were mated with mice homozygous for Prdx6129/SvJ-Tg transgene (Prdx6+/+/Tg1/1), wild type C57BL/6 J mice (Prdx6+/+), or with Prdx6 knock out mice (Prdx6−/−). This cross-breeding strategy depicted in Fig. 1a, produced the following genotypes, which were analyzed in this study: APP/PS11/0;Prdx6+/+/Tg1/0 (thereafter referred as APP/Prdx6Tg), APP/PS11/0;Prdx6+/+ (thereafter referred as APP/Prdx6+/+), and APP/PS11/0;Prdx6+/− (thereafter referred as APP/Prdx6+/−). Littermates, which did not positively genotype for the APPswe/PS1dE9 transgene, were excluded from the study. Mice separated by sex were aged to 6 or 10 months in germ free vivarium with 12-h light/dark cycle and free access to food and water. Mouse health was assessed bi-weekly following standards of good husbandry practice [19]. Each examination started with observation of animals’ behavior in their home cages with attention paid to spontaneous activity, nest building, interaction with cage mates, and general appearance. This was followed by hands-on assessment of hydration status, postural reflexes, mobility, coat integrity, and search for subcutaneous anomalies including masses.
Fig. 1.

Development and characterization of APP/Prdx6Tg and APP/Prdx6+/− lines. a Breeding scheme: hemizygous APPSWE/PS1dE9 AD Tg mice (APP/PS11/0;Prdx6+/+) were mated to mice, which on the background of endogenous Prdx6 transgenically over-express wild type Prdx6129X1/SvJ allele (Prdx6+/+/Tg1/1), wild type C57BL/6 J mice (Prdx6+/+) or Prdx6 knock out mice (Prdx6−/−). Female F1 progeny of the following genotypes APP/PS11/0;Prdx6+/+Tg1/0 (APP/Prdx6Tg), APP/PS11/0;Prdx6+/+ (APP/Prdx6+/+), and APP/PS11/0;Prdx6+/− (APP/Prdx6+/−) were analyzed in this study. b, c Shown is Prdx6 genotyping in parental and F1 generations, respectively. Wild type Prdx6 PCR yields a single 199 bp band, whose variable intensity can side-to-side distinguish endogenous allele from transgenic overexpression, while 315 bp PCR product indicates transgenically disrupted Prdx6 allele. Altered Prdx6 expression does not affect mRNA and protein level for APP and GFAP as evidenced by qRT-PCR analysis in d and quantitative Western blotting in e and f in mice aged 4–6 weeks, respectively. Values in d and e express a fold change relative to APP/Prdx6+/+ line and represent mean (+SEM) from n = 6–8 mice per genotype in d and n = 5–7 mice per genotype in f. Altered Prdx6 expression does not affect load of astrocytes before the onset of Aβ deposition. g Representative microphotographs of coronal cross-sections through the dorsal hippocampus from 3-month-old female mice of indicated genotypes immunolabeled against GFAP. h Quantification of GFAP+ astrocyte load in the dentate hilus. Values represent mean (+SEM) from n = 5 mice per genotype. i ELISA analysis of soluble (DEA-extractable) Aβx-40 and Aβx-42 levels in the brain cortex of mice aged 2 months. Values represent mean (+SEM) from n = 7–10 animals per genotype. p = 0.0008 for Prdx6 mRNA in d and p < 0.0001 for PRDX6 protein in f (ANOVA); *p < 0.05, ***p < 0.001, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). ANOVA for APP and Gfap mRNA in d, APP and GFAP protein in f, GFAP+ astrocyte load in h, and Aβx-40 and Aβx-42 levels in i is not significant. Abbreviations: gl - granular layer, h - hilus. Scale bar: 150 μm in g
Isolation and genotyping of genomic DNA
Genomic DNA was obtained by tail biopsy and genotyped for APPswe/PS1dE9 and Prdx6. Tail tissue was digested overnight at 55° C in the lysis buffer composed of 50 mM Tris-HCl (pH 8.0), 50 mM ethylenediaminetetraacetic acid (EDTA), 50 mM NaCl, 0.5% of N-Lauroyl sarcosine, 0.06% Tween 20, and 0.5 mg/mL of Proteinase K (Roche Life Science, Indianapolis, IN). Digestion product was centrifuged at 9000 x g for 10 min. DNA was precipitated from resulting supernatant with 5 volumes of 100% ethanol, trice washed with 70% ethanol, and dried up in a 55° C heat block. DNA pellets were resuspended in 80–120 μl of PCR grade dH2O and DNA concentration was photometrically determined using Nanodrop 2000 (Thermo Fisher Scientific, Waltham, MA). APPswe/PS1dE9 transgene was identified using real time polymerase chain reaction (RT-PCR) as previously described [10] on a CFX96™ Real-Time System (Bio-Rad Laboratories, Hercules, CA). Prdx6 DNA was amplified using specific primers forward 1: 5′-CAGGATGGAGCCTCTATGCC-3′, forward 2: 5′-TGGCTTCTGAGACGGAAAGAA-3′, and reverse: 5′-CTTTGAACAGAACCAGGCAGG-3′ [18]. The starting amount of genomic DNA was always 500 ng. The PCR protocol was initiated by a 2-min denaturation at 94° C, followed by 40 amplification cycles consisting of 30 s denaturation at 92° C, 30 s annealing at 55° C, and 30 s extension at 70° C, and it was concluded by the final extension step at 72° C for 3 min. Amplified DNA was electrophoresis-separated on a 2% agarose gel for 60 min, visualized with 0.5 μg/ml ethidium bromide and imaged using Gel Doc XR+ (Bio-Rad Laboratories) (Fig. 1 b, c).
Euthanasia and tissue handling
Animals were euthanized with an intraperitoneal injection of sodium pentobarbital (150 mg/kg) and transcardially perfused with 10 mM phosphate-buffered saline (PBS) (pH 7.4) with addition of heparin (1000 units/L). Immediately after perfusion brains were removed from the skulls and divided along the longitudinal fissure into left and right hemispheres. The left hemisphere was dissected under operating microscope on ice to isolate the brain cortex and the hippocampus, which were weighted, flash frozen in a dry ice/methanol cooling bath, and stored at -80° C until further analyses. The right brain hemisphere was fixed by immersion in 4% paraformaldehyde diluted in 100 mM phosphate buffer (PB) for 72 h at 4° C and stored in a mixture of 20% dimethyl sulfoxide and 20% glycerol in 0.01 PBS (pH 7.4) at 4° C until sectioning and immunohistochemistry.
RNA isolation, reverse transcription, and quantitative real time PCR (qRT-PCR) analysis
RNA was extracted from the brain cortex of 4 to 6 weeks old mice immediately after their perfusion using RNeasy Mini Kit (Qiagen Sciences Inc., Germantown, MD). The RNA extract was treated with 2 U of DNAse I (Qiagen Sciences Inc.) and its purity was assessed by electrophoresing 1 μg of RNA on a 1.2% agarose gel for 60 min, staining with 0.5 μg/ml of ethidium bromide and imaging using Gel Doc XR+. Five-hundred nanograms of extracted RNA were reverse transcribed into cDNA using the iScript™ Advanced cDNA Synthesis Kit (Bio-Rad Laboratories). qRT-PCR was performed using SYBR® Green JumpStart™ Taq ReadyMix™ and the following primer sequences: 5′-TTG ATG ATA AGG GCA GGG AC-3′ forward and 5′-CTA CCA TCA CGC TCT CTC CC-3′ reverse for Prdx6 [18], 5′-CGA ACC CTA CGA AGA AGC CAC-3′ forward and 5′-GCT TTC TGG AAA TGG GCA TGT TC-3′ reverse for APP [20], 5′-GCT TTC TGG AAA TGG GCA TGT TC-3′ forward and 5′-TCTGCCTCCAGCCTCAGGTT-3′ reverse for Gfap [21], and 5′-CCG GGG CTG GCA TTG CTC TC-3′ forward and 5′-TGT TGG GGG CCG AGT TGG GA-3′ reverse for murine glyceraldehyde-3-phosphate dehydrogenase (Gapdh) [20]. Amplification efficiency for each primer pair was verified from the regression slope between the cDNA dilution log values and the averaged cycle threshold (CT) value obtained for each cDNA dilution. Expression of target genes: Prdx6, APP, and Gfap was analyzed using 2−ΔΔCt method [22]. Firstly, CT values for each target gene were normalized to the CT values for Gapdh as the reference gene for each animal individually. Resulting ΔCT values were then normalized to averaged ΔCT values for a given target gene in APP/Prdx6+/+ line. Target gene expression in APP/Prdx6Tg and APP/Prdx6+/− lines was calculated as a fold difference relative to these in APP/Prdx6+/+ line. qRT-PCR was performed on a CFX96™ Real-Time System.
Western blot analysis
Samples of the brain cortex stored at -80° C were thawed, weighted, and homogenized (1:10 wt/vol at 4° C) in a buffer consisting of 20 mM Tris-HCL (pH 7.4), 250 mM sucrose, 1 mM egtazic acid, 1 mM EDTA and 10 μg/mL of a Complete Protease Inhibitor Cocktail (Roche Life Science) supplemented with 1 mM phenylmethylsulfonyl fluoride, and leupeptin, antipain, and pepstatin (5 μg/mL each). The three-step homogenization procedure included manual trituration in a pestle grinder, passing the tissue through a 28-gauge needle, and final sonication. Cellular debris were cleared by 3 min centrifugation at 10,000×g and 4° C and protein concentration in the resulting supernatant was measured by bicinchoninic acid (BCA) method using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific), according to the manufacturer provided manual. Aliquots of brain homogenates containing 20 μg of the total protein for PRDX6 and APP and 5 μg for GFAP and β-actin were resolved on 10% SDS-PAGE and electroblotted onto nitrocellulose membranes, which were then blocked with 5% nonfat milk and incubated with the following primary antibodies anti-PRDX6 (rabbit polyclonal 1:10,000; Abcam Inc., Cambridge, MA), anti-GFAP (rabbit polyclonal 1:10,000; DAKO, Denmark), anti-APP (mouse monoclonal, clone C1/6.1 1:3000; BioLegend, San Diego, CA), and anti-β-actin (mouse monoclonal 1:10,000, Sigma-Aldrich). The antigen–antibody complexes were detected using horseradish peroxidase conjugated sheep anti-mouse or anti-rabbit secondary antibodies (1:30,000; GE Healthcare Bio-Sciences Corp. Pittsburgh, PA) and visualized using SuperSignal West Pico PLUS Chemiluminescent Substrates (Thermo Fisher Scientific). Resulting autoradiograph were digitized into a 600 dpi TIFF files and analyzed using NIH ImageJ v1.52a software (Bethesda, MD).
Biochemical analyses of Aβx − 40 and Aβx − 42 levels
Samples of brain cortex homogenate were prepared analogously to those for the Western blot analysis and subjected to diethylamine (DEA) or formic acid (FA) extractions [23, 24], which release soluble or total Aβ peptides, respectively. Concentration of Aβx − 40 and Aβx − 42 in either fraction was determined on ELISA plates coated with HJ2 (anti-Aβ35–40) or HJ7.4 (anti-Aβ37–42) mouse monoclonal antibodies (0.5 μg/well; gift of Dr. DM Holtzman), respectively [20, 25]. Biotinylated 4G8 mouse monoclonal antibody (anti-Aβ17–24,1:5000; BioLegend) [26] was used as the detection antibody. Optic densities of ELISA readouts were converted to the actual concentrations of Aβ peptides based on the standard curves prepared from FA-treated synthetic Aβ1–40 and Aβ1–42 peptides. Final Aβx-40 and Aβx-42 concentrations in the brain tissue were reported in reference to the protein concentration considering all dilutions made during DEA and FA extractions [25].
Histology and immunohistochemistry
The right brain hemisphere was serially sectioned into coronal 40-μm-thick sections using a freezing microtome (Leica Microsystems, Wetzlar, Germany). The sections were alternately collected into 10 series, then randomly selected for the following stainings: 1) Thioflavin-S (ThS), 2) anti-Aβ (mouse monoclonal HJ3.4 directed against the N-terminus of Aβ, 1:100; gift of Dr. DM Holtzman) [20, 25, 27]; 3) anti-GFAP (mouse monoclonal 1:2000; Sigma-Aldrich); 4) anti-Iba1 (rabbit polyclonal 1:1000; Wako Chemicals Inc., Richmond VA); 5) anti-CD68 (rabbit polyclonal 1:200, Abcam Inc., Cambridge, MA), and 6) Gallyas silver staining. For the anti-Aβ immunostaining the sections were pretreated with 44% FA for 10 min to enhance antigen availability [25] and the immunohistochemistry protocol was conducted using peroxidase M.O.M. and 3,3′-diaminobenzidine kits from Vector Laboratories (Burlingame, CA) as per manufacturer manual. For anti-GFAP immunostaining biotinylated horse-anti mouse IgG secondary antibody (1:500; Vector Laboratories) was used, while for anti-CD68 and anti-Iba1 immunostainings the secondary antibody was biotinylated goat anti-rabbit IgG (1:500; Vector Laboratories). The immunostaining protocol was concluded with streptavidin conjugated Cy3 fluorochrome (1:1000; Vector Laboratories) and Th-S counterstaining to visualize relationship between glial cells and fibrillar plaques as previously described [27]. Gallyas silver staining was carried out at 10° C and it was modified from its original protocol [26, 28] by extending the time of alkaline silver iodide enucleation to 40 min.
Specifically, for this project we developed several multi-fluorochrome labeling protocols to interrogate the relationship between Aβ plaques and plaque-associated glia cells. The fibrillar plaque core was labeled first with X-34 (10 μM in 40% ethanol [pH 10]) and then various glial antigens were immunostained with one of the following antibodies: anti-GFAP (rabbit polyclonal 1:2000; Dako/Agilent Technologies; Santa Clara, CA), anti-Iba1 (rabbit polyclonal 1:800; Wako Chemicals USA Inc.) or anti-CD68 (rat monoclonal 1:200; Abcam Inc.). Prior to applying the primary antibody, non-specific staining was blocked with a mixture of 10% normal goat serum, 1% bovine serum albumin (BSA), and 0.3% Triton X-100 in 10 mM PBS (pH 7.4) and in the case of anti-CD68 immunostaining additionally Avidin/Biotin Blocking Kit (Vector Laboratories) was used as per manufacturer manual. The secondary antibodies were Texas Red conjugated goat anti-rabbit IgG (1:500; Vector Laboratories) or goat biotinylated anti-rat IgG (1:500; Abcam Inc.) followed by Cy3 conjugated streptavidin (1:500). Subsequently, we immunolabelled the diffuse (non-fibrillar) Aβ within the plaque brim with HJ3.4 mouse monoclonal antibody (1:100; gift of Dr. D.M. Holtzman) followed by Dylight 488 conjugated horse anti-mouse IgG secondary antibody (1:500; Vector Laboratories). Cell nuclei were optionally stained with 1 μM of DRAQ5 (Thermo Fisher Scientific) as the fourth and the final step of the protocol.
For TREM2 visualization the sections were first pretreated with 10 mM sodium citrate and 0.5% Tween 20 (pH 6.0) for 20 min at 85° C to help the antigen retrieval [29]. Following labeling of the fibrillar plaque core with X-34, non-specific immunostaining was blocked with 5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) and 0.3% Triton X-100. The primary antibodies were anti-TREM2 (sheep polyclonal 1:250; R&D Systems, Minneapolis MN) and anti-Iba1 (rabbit polyclonal 1:1000; Wako Chemicals Inc.), while the secondary antibodies were donkey anti-sheep Alexa Fluor 594 conjugated (1:500; Thermo Scientific Scientific) and donkey anti-rabbit Alexa Fluor 488 conjugated (1:500; Jackson ImmunoResearch) both diluted in 4% normal donkey serum and 0.3% Triton, respectively. For analysis of cellular localization of PRDX6, the sodium citrate antigen retrieval protocol was used too. The primary antibodies were either anti-GFAP (mouse monoclonal 1:1,500; Sigma-Aldrich) or anti-Iba1 (mouse monoclonal 1:100; Sigma-Aldrich) and anti-PRDX6 (rabbit polyclonal 1:250; Abcam Inc.), while the secondary antibodies were goat anti-mouse Alexa Fluor 488 conjugated (1:500; Jackson ImmunoResearch) and goat anti-rabbit Alexa Fluor 594 conjugated (1:500; Jackson ImmunoResearch), respectively. For analysis of Aβ plaque-associated axonal dystrophy antigen retrieval with sodium citrate was used as well. The primary antibodies were anti-Aβ1–16 (rabbit polyclonal 1:500; BioLegend) and anti-Neurofilament (mouse monoclonal [clone SIM 312] 1:300; BioLegend), while the secondary antibodies were goat anti-rabbit Alexa Fluor 488 conjugated (1:500; Jackson ImmunoResearch) and goat anti-mouse Alexa Fluor 594 conjugated (1:500; Jackson ImmunoResearch), respectively.
Quantitative analysis of Aβ plaque load and that of astrocytes and microglia
The load is defined as the percentage of a cross-sectional profile covered by positively-thresholded objects. It was determined using whole-section approach, where the entire cross-sectional profile of a given anatomical structure is digitally photographed and thresholded to prevents sampling bias [27]. We quantified the load of Th-S labeled fibrillar Aβ plaques and that of HJ3.4 immunostained Aβ plaques both in the brain cortex and in the hippocampus and loads of GFAP+ astrocytes and Iba1+ and CD68+ microglia in the brain cortex. Cortical load was analyzed on 3 coronal sections per brain taken at the approximated levels of the anterior commissure, the rostral portion of the hippocampus, and the mammillary bodies, while the hippocampal load was analyzed on 4–5 serial sections spaced 800 μm apart. Load values obtained from individual cortical and hippocampal profiles were averaged for each brain. Infrequent Th-S or anti-Aβ positive vascular profiles were edited out from digitized images. Sections immunolabelled for GFAP, Iba1 and CD68 also were counterstained with Th-S and the glia cell load was indexed to that of Th-S+ plaques on the same section. All load analyses were performed using NIH ImageJ v1.52. In addition, the load of GFAP+ cells was quantified in the dentate hilus in mice aged 2 months before the onset of Aβ deposition to assess for differences resulting from variable Prdx6 expression. Numerical density of neuritic plaques was determined by taking their count on 3 cortical and 4–5 hippocampal profiles per brain on Gallyas stained sections and dividing obtained count by combined area of cross-sectional profiles. Similarly, we quantified the numerical density of nascent plaques in the brain cortex of mice aged 6 months.
Laser scanning confocal microscopy (LSCM) and segmentation analysis of Aβ plaques
Images of multi-fluorochrome labeled Aβ plaques were taken under immersion oil, 63x, and 1.4 N.A. objective and 2x digital zoom using Zeiss LSM 880 microscope and ZEN Black 2.3 SP1 acquisition software v. 14.0.18.201 (Carl Zeiss AG; Oberkochen, Germany). Z stacks of 0.5-μm-thick serial tomograms with 25% overlap (increased to 50% for TREM2 imaging) were acquired through the entire thickness of Aβ plaques. For morphometric analysis 8 serial tomograms (or 12 for TREM2 imaging) dissecting through the center of the plaque fibrillar core were taken out from the original Z-stack and collapsed into a 2D image. Monochromatic images of individual plaque labels were then converted to 8-bit format, contrast enhanced, and analyzed using NIH ImageJ v1.52 (Fig. S1a-c). The following metrics of Aβ plaques were analyzed: 1) plaque area (the cross-sectional area of Aβ plaque implicit within the outline of HJ3.4 immunolabel [Fig. S1b]), 2) plaque/core ratio (ratio between the plaque area and the area of its X-34+ fibrillar core), 3) GFAP plaque index (GFAP+ % plaque area) (Fig. S1c), 4) density of plaque-associated microglia (number of Iba1+ microglia cells implicit within the Aβ plaque outline divided by the outline area), 5) Iba1 plaque index (Iba1+ % plaque area), 6) microglia barrier robustness index (Iba1/X-34% co-localization), 7) CD68 phagosome plaque index (CD68+ % plaque area), 8) TREM2 plaque area (TREM2+ % area within the 40 μm radius from the plaque center), 9) TREM2 to Iba1 ratio in plaque-associated microglia, 10) number of NF-positive swellings per plaque, and 11) NF plaque index (NF+ % plaque area).
Statistical analysis
Normal distribution of data was confirmed first using the Kolmogorov-Smirnov and Shapiro-Wilk tests. Differences across multiple data sets were analyzed using one-way analysis of variance (ANOVA) followed by Holm-Sidak’s multiple comparison test to ascertain significance between individual data set pairs. Statistical analysis was performed using GraphPad Prism v 7.03 (GraphPad Software Inc. San Diego, CA). All data presented in the manuscript are given as the mean and the standard error of the mean (SEM). Magnitude of change across genotypes was always expressed as the percentage value for APP/Prdx6+/+ mice, or as a fold-change when direct comparison between APP/Prdx6Tg and APP/Prdx6+/− genotypes was made.
Results
Overexpression of Prdx6 reduces Aβ load while Prdx6 haplodeficiency increases it
To differentially express Prdx6 we crossed APPSWE/PS1dE9 (APP/Prdx6+/+) AD Tg model mice [12, 13] with mice transgenically over-expressing Prdx6129X1/SvJ allele on the wild-type Prdx6 background (Prdx6+/+/Tg1/1) [15] or with Prdx6 knock out (Prdx6−/−) mice [18]. Resulting lines were termed APP/Prdx6Tg and APP/Prdx6+/−, respectively (Fig. 1a-c); and exhibited no phenotypical or gross behavioral defects or precocious morbidity compared to parental APP/Prdx6+/+ mice. Levels of Prdx6 mRNA and PRDX6 protein in the brain cortex of mice aged 4–6 weeks show 2:1:0.6 ratio across APP/Prdx6Tg, APP/Prdx6+/+, and APP/Prdx6+/− genotypes, while the expression of APP and GFAP remain unchanged both by mRNA and protein analysis (Fig. 1d-f). Likewise, we found no differences in the morphology of GFAP+ astrocytes and their load in the dentate hilus, which we analyzed in mice aged 3 months (Fig. 1g, h). Soluble Aβx-40 and Aβx-42 peptides were extracted using diethyalamine [DEA] from the brain cortex of mice aged 2 months and quantified using C-terminus specific ELISAs and their levels showed no significant differences across the genotypes (Fig. 1i). These data evidence that alteration of Prdx6 gene expression does not affect resting state of astrocytes or brain Aβ levels prior to the onset of plaque formation.
The effect of Prdx6 gene dose on Aβ deposition was detailed in female mice aged 10 months. APP/Prdx6Tg line shows 21% reduction in the fibrillar (Th-S+) Aβ plaque load in the brain cortex (p < 0.0001) and 26% reduction in the hippocampus (p < 0.0001) compared to APP/Prdx6+/+ controls, while conversely APP/Prdx6+/− mice demonstrate 28% (p < 0.0001) and 22% (p < 0.001) increase in respective structures (Fig. 2a, b). The load of immunolabeled Aβ plaques shows 22% reduction in the brain cortex (p < 0.0001) and 17% reduction in the hippocampus (p < 0.01) in APP/Prdx6Tg mice compared to APP/Prdx6+/+ controls, while in APP/Prdx6+/− mice it is increased by 32% (p < 0.0001) and 80% (p < 0.0001) (Fig. 2c, d), respectively. In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 1.6-fold higher load of fibrillar Aβ plaques both in the brain cortex and in the hippocampus (p < 0.0001), while the load of immunolabeled Aβ plaque is 1.7-fold higher in the cortex (p < 0.0001) and 2.2-fold higher in the hippocampus (p < 0.0001).
Fig. 2.

Aβ load in APPSWE/PS1dE9 mice varies inversely with Prdx6 gene expression level. a and c Representative microphotographs of coronal cross-sections through the somatosensory cortex and the dorsal hippocampus from 10-month-old female mice of indicated genotypes, which were stained for fibrillar Aβ plaques with Thioflavin-S or immunolabeled with HJ3.4 clone directed against the N-terminus of Aβ peptide, respectively. Quantitative analysis of fibrillar b and immunopositive d Aβ plaque load in the brain cortex and in the hippocampus evidencing suppression of Aβ deposition in Prdx6 overexpressing mice and increased Aβ deposition in Prdx6 haplodeficient mice. Values represent mean (+SEM) from n = 9–12 female mice per genotype. e and f ELISA analysis of soluble (DEA-extractable) and total (FA-extractable) Aβx-40 and Aβx-42 levels in the brain cortex of female 10-month-old mice, respectively. Values represent mean (+SEM) from n = 7–13 animals per genotype. p < 0.0001 in b, d, e, and f (ANOVA); *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Abbreviations: CA1 – cornu Ammonis sector 1, Crtx – cortex, DG – dentate gyrus, Hip – hippocampus. Scale bars: 750 μm in a and 50 μm in c
Levels of soluble and total deposited Aβx-40 and Aβx-42 were quantified in DEA and FA brain cortex extracts, respectively. In APP/Prdx6Tg mice soluble Aβx-40 and Aβx-42 levels are reduced by 35% (p = 0.18) and 45% (p < 0.05) compared to APP/Prdx6+/+ controls, respectively; while in APP/Prdx6+/− mice they are increased by 156% (p < 0.0001) and 28% (p = 0.09), respectively (Fig. 2e). Total Aβx-40 and Aβx-42 levels are reduced by 26% (p < 0.01) and 22% (p < 0.01) in APP/Prdx6Tg mice compared to APP/Prdx6+/+ controls, respectively; while in APP/Prdx6+/− mice they are increased by 20% (p < 0.01) and 18% (p < 0.05), respectively (Fig. 2f). In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 3-fold (p < 0.0001) and 1.3-fold (p < 0.01) higher levels of soluble Aβx-40 and Aβx-42, respectively; and 1.6-fold (p < 0.0001) and 1.5-fold (p < 0.01) higher levels of total Aβx-40 and Aβx-42, respectively. Of note, Prdx6 haplodeficiency effects an increase in Aβ accumulation in AD Tg mice comparable to that caused by Trem2 haplodeficiency or Trem2 R47H mutant, conferring loss of TREM2 function [29].
To examine whether the effect of Prdx6 gene dose on Aβ deposition is gender-specific, we compared Aβ plaque load in female and male littermates at the age of 10 months (Fig. S1 a-d). Significant reduction in Aβ plaque load in APP/Prdx6Tg line and its increase in APP/Prdx6+/− line compared to APP/Prdx6+/+ controls is evident in both sexes. When matched for Prdx6 genotype, female mice show significantly higher load of plaques than male mice, irrespective of plaque type and brain structure analyzed. This latter observation is consistent with reports indicating gender-based disparity in Aβ plaque load in some of AD Tg mouse models, with propensity toward greater Aβ accumulation in females [30, 31]. All further analyses presented in this study were performed on female mice to assure gender consistence.
Prdx6 gene dose does not influence Aβ-associated astrocytic load but has a direct relationship with that of microglia
To gain insight into the mechanism(s) underlying opposing relationship between Prdx6 gene dose and Aβ deposition and inform design of subsequent experiments we quantified loads of GFAP+ astrocytes and Iba1+ and CD68+ microglia in the cortex of female mice aged 10 months and indexed them to the load of fibrillar (Th-S+) Aβ plaques. The unindexed GFAP+ load in APP/Prdx6Tg mice shows 12% (p < 0.0001) reduction, while in APP/Prdx6+/− mice 15% increase (p < 0.0001) compared to APP/Prdx6+/+ controls (Fig. 3a, b). The differences across genotypes equalize and become statistically insignificant when GFAP+ load is indexed to that of Th-S+ plaques (Fig. 3c). Unindexed loads of Iba1+ and CD68+ microglia vary only modestly across the genotypes: with 5% reduction (p < 0.05) in the Iba1+ load in APP/Prdx6+/− mice vs. APP/Prdx6+/+ controls (Fig. 3d, e), and 16% increase (p < 0.0001) in the CD68+ load in APP/Prdx6Tg mice vs. APP/Prdx6+/+ controls (Fig. 3g, h). However, when either Iba1+ or CD68+ load becomes indexed to that of Th-S+ plaques, a strong gradient of microglia activation across the three genotypes emerges: APP/Prdx6Tg > APP/Prdx6+/+ > APP/Prdx6+/− (Fig. 3f, i). The Th-S+ indexed Iba1+ load is increased by 16% (p < 0.0001) in APP/Prdx6Tg mice, while in APP/Prdx6+/− mice it is reduced by 23% (p < 0.0001) compared to APP/Prdx6+/+ controls. Analogously, the Th-S+ indexed CD68+ load is increased by 41% (p < 0.0001) in APP/Prdx6Tg mice and reduced by 15% (p < 0.0001) in APP/Prdx6+/− mice compared to APP/Prdx6+/+ controls. In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 1.5-fold (p < 0.0001) and 1.7-fold (p < 0.0001) reduction in the loads of Iba1+ and CD68+ microglia cells, when these are indexed to that of Th-S+ plaques. These findings collectively indicate that neither Prdx6 overexpression effects hyperactive astrogliosis nor Prdx6 haplodeficiency impairs ability of astrocytes to mount response to Aβ deposition since the GFAP+ load remains proportional to that of fibrillar amyloid deposits across the three genotypes. Unexpectedly, we find that activation of microglia varies directly with the Prdx6 gene dose and this includes CD68 antigen expression, which reflects phagocytic microglia activity.
Fig. 3.

Prdx6 gene dose does not affect global response of astrocytes to Aβ deposition but shows a direct relationship with that of microglia. a, d, g Representative microphotographs of anti-GFAP, −Iba1, or -CD68 immunolabeled coronal sections through the somatosensory cortex from 10-month-old female mice of indicated genotypes, respectively. Counterstaining with Thioflavin-S (Th-S) reveals fibrillar plaques to which the load of glial cells was indexed. b, e, and h Quantitative analysis of GFAP+, Iba1+, and CD68+ cell load, in the brain cortex, respectively. c, f, and i Show loads of GFAP+, Iba1+, and CD68+ cells indexed to these of Th-S+ fibrillar plaques, respectively. While Th-S+-indexed GFAP+ cell load demonstrates no statistically significant differences across the genotypes, those for Iba+ and CD68+ cells have significant direct relationship with Prdx6 gene dosage. All values represent mean (+SEM) from n = 12 mice per genotype. p < 0.0001 in b, f, h, and i, p = 0.27 in c and p = 0.0142 in e (ANOVA); *p < 0.05, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Scale bar: 50 μm in a, d, and g
Prdx6 overexpression and haplodeficiency produce opposite effects on Aβ plaque modeling by plaque-associated astrocytes and microglia
We used multi-antigen labeling and laser scanning confocal microscopy (LSCM) to analyze effects of Prdx6 gene dose on the morphology of Aβ plaques and behavior of plaque-associated glial cells (Fig. S2 a-c). In APP/Prdx6Tg mice, the average plaque area (i.e. area implicit within the outline of anti-Aβ label) is reduced by 18% (p < 0.01) compared to APP/Prdx6+/+ controls, while the ratio of plaque area to the area of its X-34+ fibrillar core (plaque/core ratio) is reduced by 17% (p < 0.01) evidencing increased plaque compactness (Fig. 4a-c). Opposite effects are seen in APP/Prdx6+/− mice, where the average plaque area and the plaque/core ratio are increased by 39% (p < 0.0001) and 16% (p < 0.01) compared to APP/Prdx6+/+ controls, respectively. In direct comparison with APP/Prdx6Tg line, in APP/Prdx6+/− mice an average Aβ plaque area is 1.7-fold bigger (p < 0.0001) and has 1.4-fold higher (p < 0.0001) plaque/core ratio. These changes in Aβ plaque morphology are closely associated with differential behavior of plaque-associated astrocytes and microglia discernible across the genotypes. In APP/Prdx6Tg mice, there is increased infiltration of Aβ plaques by GFAP+ astrocytic processes, reflected by 28% increase in the GFAP plaque index (GFAP+ % plaque area) vs. APP/Prdx6+/+ controls (Fig. 4a, d). Conversely, in APP/Prdx6+/− mice Aβ plaques are characterized by rarefaction of astrocytic processes and show 23% reduction (p < 0.05) in the GFAP plaque index vs. APP/Prdx6+/+ controls. In direct comparison with APP/Prdx6Tg line, the GFAP plaque index in APP/Prdx6+/− mice is reduced by 1.7-fold (p < 0.001). These observations evidence that Prdx6 gene dose alters the way astrocytes engage Aβ plaques, rather than produces global effect on the astrocytic activation in response to Aβ deposition. In addition, we analyzed plaque content of apolipoprotein (apo) E, which in the CNS is produced by astrocytes and co-deposited with Aβ in the plaques [32]. In APP/Prdx6Tg mice, there is 41% increase (p < 0.0001) in the apoE plaque index (ApoE+ % plaque area) compared to APP/Prdx6+/+ controls, while in APP/Prdx6+/− mice that index is reduced by 26% (p < 0.0001) (Fig. S3 a, b). In direct comparison with APP/Prdx6+/− line, APP/Prdx6Tg mice show 1.9-fold higher (p < 0.0001) apoE plaque index reflecting increased engagement of Aβ plaques by astrocytes. We also assessed expression of complement component 3 (C3) by astrocytes engaging Aβ plaques by measuring the C3+ / GFAP+ plaque index (Fig. S4 a, b). This was done because elevated C3 expression is considered a marker of the neurotoxic astrocytic phenotype also known as A1 [33–35]. In APP/Prdx6Tg mice the C3+ / GFAP+ index is reduced by 26.7% compared to APP/Prdx6+/+ controls (p < 0.0001), while in APP/Prdx6+/− mice it is increased by 31.8% (p < 0.0001). In direct comparison with the APP/Prdx6Tg line, plaque-associated astrocytes in APP/Prdx6+/− mice show 1.8-fold higher C3 expression (p < 0.0001).
Fig. 4.

Prdx6 expression alters plaque morphology and their penetrance by astrocytes. a Representative LSCM images of mature plaques triple labeled with X-34 (fibrillar core), anti-Aβ (HJ3.4 clone), and anti-GFAP antibodies from female mice of indicated genotypes demonstrating inverse relationship between Prdx6 gene dose and size of Aβ plaques and degree of plaque compactness expressed as a ratio between cross-sectional areas of Aβ plaque label and X-34 labeled fibrillar core. Prdx6 overexpression also is associated with increased penetrance of Aβ plaques by astrocytes, while in Prdx6 haplodeficient mice the number and caliber of plaque penetrating GFAP positive processes are diminished. Shown are quantitative analyses of: average Aβ plaque cross-sectional area b, plaque/core ratio c, and GFAP plaque index (GFAP+ % plaque area) d. Values in b and c represent mean (+SEM) from n = 70–90 randomly selected plaques per genotype, while those in d from n = 30 randomly selected plaques per genotype. p < 0.0001 in b, c, and d (ANOVA); *p < 0.05, **p < 0.01, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Scale bar: 10 μm in a
Along with the differences in the way how astrocytes engage Aβ plaques we also noticed significant differences in the behavior of plaque-associated microglial cells. In APP/Prdx6Tg mice, there is 65% increase (p < 0.0001) in the number of plaque-associated Iba1+ microglia cells (Fig. 5a, c), 26% increase (p < 0.01) in the plaque Iba1 index (Iba1+ % plaque area) (Fig. 5d), 32% increase (p < 0.05) in the microglia barrier robustness index (Iba1/X-34% co-localization) (Fig. 5e), and 12% increase (p = 0.16) in the CD68 phagosome plaque index (CD68+ % plaque area) (Fig. 5b. f) compared to APP/Prdx6+/+ controls. APP/Prdx6+/− mice display opposite changes in microglia activation metrics. The average number of plaque-associated Iba1+ microglia cells shows 29% reduction (p < 0.01), plaque Iba1 index 27% reduction (p < 0.01), microglia barrier robustness index 36% reduction (p < 0.05), and CD68 phagosome index 31% reduction (p < 0.01) vs. APP/Prdx6+/+ controls. In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 2.3-fold lesser number of periplaque Iba1+ cells (p < 0.0001), 1.7-fold lower plaque Iba1 index (p < 0.0001), 2.1-fold lower microglia barrier robustness index (p < 0.0001), and 1.6-fold lower CD68 phagosome index (p < 0.0001). There also is a striking effect of Prdx6 gene dose on TREM2 expression by periplaque microglia. APP/Prdx6Tg mice feature 44% increase (p < 0.0001) in the TREM2+ label of the plaque (TREM2 plaque area) (Fig. 6a, b) and 42% increase (p < 0.0001) in the TREM2/Iba1 ratio (Fig. 6c) reflecting upregulation of TREM2 expression on the individual microglial cell level compared to APP/Prdx6+/+ controls. In contrast, APP/Prdx6+/− mice show down regulation of TREM2 expression, evidenced by 23% reduction (p < 0.05) in TREM2 plaque area and 31% reduction (p < 0.001) in TREM2/Iba1 ratio compared to APP/Prdx6+/+ controls. In direct comparison with APP/Prdx6Tg line these metrics are reduced by 1.9-fold (p < 0.0001) and 2.1-fold (p < 0.0001) in APP/Prdx6+/− mice, respectively. Thus, Prdx6 gene dose shows direct relationship with phagocytic activation of periplaque microglia and inverse relationship with the size and compactness of Aβ plaques, what is consistent with function of periplaque microglia recognized for phagocytosing diffuse Aβ in the plaque brim and forming the plaque core. Prdx6 overexpression has a protective effect by enhancing Aβ proteostasis, while Prdx6 haplodeficiency to the contrary confers functional impairment of periplaque microglia in countering Aβ deposits akin to that rendered by TREM2 haplodeficiency or R47H Trem2 mutant [3, 29, 36].
Fig. 5.

Phagocytic activation of plaque-associated microglia is enhanced in mice overexpressing Prdx6 and attenuated in Prdx6 haplodeficient mice. Representative LSCM images of mature plaques co-labeled with X-34 (fibrillar core), anti-Aβ (HJ3.4 clone), anti-Iba1, and DRAQ5 (nuclear stain) in a, and with X-34, anti-Aβ, anti-CD68, and DRAQ5 in b evidencing direct relationship between Prdx6 gene dose and the number of periplaque microglia cells and expression of Iba1 and CD68 microglial activation markers. Shown are morphometric analyses of plaque-associated microglia number c, Iba1 plaque index (Iba1+ % plaque area) d, microglia barrier robustness (Iba1/X-34% co-localization) e, and CD68 phagosome plaque index (CD68+ % plaque area) f. Values in c through f represent mean (+SEM) from n = 26–35 randomly selected plaques per staining and per genotype. p < 0.0001 in c through f (ANOVA); *p < 0.05, **p < 0.01, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Scale bar: 10 μm in a and b
Fig. 6.

Expression of TREM2 in plaque-associated microglia shows direct relationship with Prdx6 gene dose. a Representative LSCM images of mature plaques co-labeled with X-34 (fibrillar core), anti-Iba1, and anti-TREM2 antibodies from female animals of indicated genotypes evidence increased periplaque expression of TREM2 in APP/Prdx6Tg line and conversely reduced expression in APP/Prdx6+/− line compared to APP/Prdx6+/+ controls. Quantification of TREM2 immunoreactivity within surrounding of the X-34+ fibrillar core (TREM2 plaque area) b and TREM2 to Iba1 ratio in plaque-associated microglia c. Values represent mean (+SEM) from n = 45 randomly selected plaques per genotype. p < 0.0001 in b and c (ANOVA); *p < 0.05, **p < 0.01, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Scale bar: 10 μm in a
PRDX6 protein is upregulated in plaque-associated astrocytes, but it is undetectable in microglia
We used multi-antigen labeling and LSCM imaging to interrogate cellular localization of PRDX6 protein in plaque-associated glial cells. PRDX6 strictly co-localizes to GFAP+ astrocytes and its expression follows APP/Prdx6Tg > APP/Prdx6+/+ > APP/Prdx6+/− gradient across the genotypes (Fig. 7a). Plaque-associated astrocytes feature marked upregulation of PRDX6 expression in comparison to astrocytes in Aβ plaque free, 3-month-old mice (Fig. S5a, b). Redistribution of PRDX6 immunoreactivity from the soma to astrocytic processes is a striking feature of periplaque astrocytic activation. This is pronounced in APP/Prdx6Tg mice, noticeable in APP/Prdx6+/+ mice, but absent in APP/Prdx6+/− animals (Fig. S5b). Activated astrocytes in APP/Prdx6Tg mice also appear to be more hypertrophied compared to those in APP/Prdx6+/− mice, especially in respect to the processes penetrating plaques (Fig. 4a, Fig. S4a, Fig. S5b). Apart from astrocytes grouped around Aβ plaques, activated, PRDX6+ astrocytes also can be found alongside blood vessels in 10-month-old mice (Fig. S5c). Similarly, expression of PRDX6 by activated, perivascular astrocytes has been described in AD autopsy material, including vessels which do not exhibit overt cerebral amyloid angiopathy [9]. Akin to plaque-associated astrocytes, perivascular astrocytes also show differences in PRDX6 expression level across the genotypes, which are commensurate with Prdx6 gene dose.
Fig. 7.
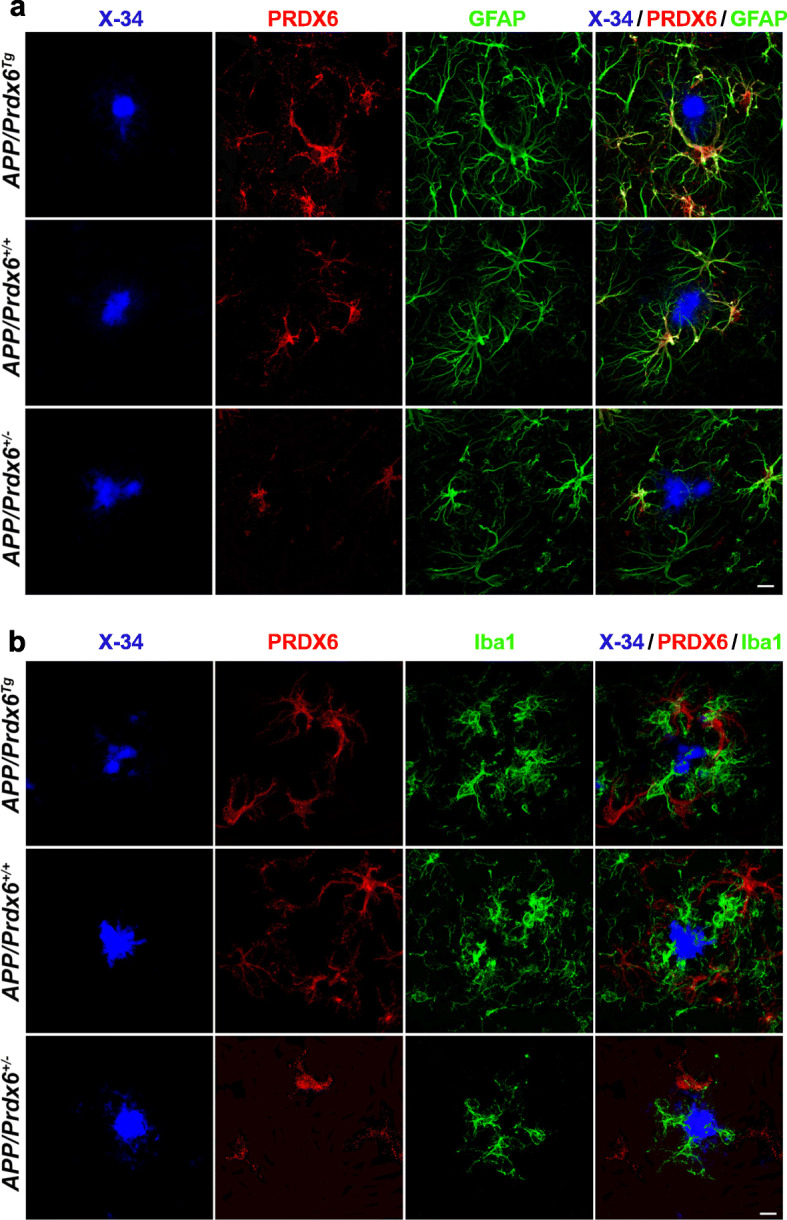
PRDX6 protein is exclusively expressed by astrocytes and absent in plaque-associated microglia. Representative LSCM images of mature plaques co-labeled with X-34 (fibrillar core), anti-PRDX6, and anti-GFAP antibodies in a, and with X-34, and anti-PRDX6, and anti-Iba1 antibodies in b evidencing colocalization of PRDX6 immunostaining with that of GFAP but not with Iba1. Co-labeling with anti-PRDX6, and anti-Iba1 antibodies in b reveals in fact two distinct cell populations localized further apart and closer to the fibrillar core, respectively. There is apparent APP/Prdx6Tg > APP/Prdx6+/+ > APP/Prdx6+/− gradient of PRDX6 immunostaining intensity across the genotypes. PRDX6 immunostaining does not co-localize with the X-34 labeled fibrillar plaque core. Scale bar: 10 μm in a and b
There is no evidence of PRDX6 expression by plaque-associated microglia. Anti-PRDX6 and anti-Iba1 immunostainings show no co-localization in any of studied genotypes. In fact, the X-34/PRDX6/Iba1 triple labeling reveals two strikingly different, non-overlapping populations of plaque-associated cells: Iba1+ activated microglia and PRDX6+ cells, which judging by their “spider-like morphology” and localization farther apart from the X-34+ fibrillar core, are likely plaque-associated astrocytes (Fig. 7b). In a supplementary experiment, we also examined Prdx6 transcript in affinity-purified microglia cultures where we find its level to be merely 1% of that in cultured astrocytes, and not increasing following lipopolysaccharide stimulation (Fig. S6). Thus, confirmatory to previous work of us and others [8, 9] we endorse that PRDX6 protein is expressed by astrocytes but not by microglia and its level becomes upregulated during astrocytic activation in Prdx6 gene dose-dependent manner. Our demonstration that manipulation of the expression level of an astrocytic inherent protein produces coextensive effects on periplaque microglia activation implies that astrocytes circuitously guide microglia dependent Aβ plaque processing.
Seeding of nascent Aβ plaque is inhibited by Prdx6 overexpression while promoted by haplodeficiency
Early stages of Aβ deposition remain only partially understood. Increasing concentration of soluble Aβ peptides is believed to drive Aβ self-assembly giving rise to precipitation of the earliest, immunodetectable deposits, which attract glial cells. To explore how variable Prdx6 expression impacts Aβ plaque seeding we quantified numerical density of nascent plaques and their subsets devoid of GFAP+ or Iba1+ cells on X-34/Aβ/GFAP or X-34/Aβ/Iba1 triple-labeled sections, respectively. We define nascent plaques as circumscribed, immunopositive Aβ deposits having no X-34+ core or having X-34+ core with cross-sectional surface ≤40μm2 (Fig. 8a, b). This analysis was done in mice aged 6 months, representing an early stage of Aβ deposition in the APPswe/PS1dE9 Tg model [13]. In APP/Prdx6Tg line the numerical density of all nascent plaques in the brain cortex shows 17% reduction (p < 0.05), while in APP/Prdx6+/− mice 22% increase (p < 0.05) compared to APP/Prdx6+/+ controls (Fig. 8a-d). In comparison with APP/Prdx6Tg line, 6-month-old APP/Prdx6+/− mice have 1.5-fold (p < 0.001) higher density of nascent plaques. Numerical densities of plaques devoid of GFAP+ or Iba1+ cells also show significant differences across the genotypes. In APP/Prdx6Tg mice numbers of astrocyte and microglia-free plaques are lesser by 33% (p < 0.01) and 43% (p < 0.001) compared to APP/Prdx6+/+ controls, respectively; while in APP/Prdx6+/− mice they are increased by 38% (p < 0.001) and 24% (p < 0.01), respectively. In direct comparison between APP/Prdx6Tg and APP/Prdx6+/− lines, the latter shows 2.1-fold (p < 0.0001) and 2.2-fold (p < 0.0001) more of astrocyte and microglia-free plaques, respectively. We also detected significant differences in the phagocytic activity of microglia associated with nascent plaques across the mouse lines. This was determined by quantification of the CD68+ / X-34+ plaque index in a subset of nascent plaques, which already developed the amyloid core (Fig. S7a, b). APP/Prdx6Tg mice exhibit 39.9% increase in the CD68+ / X-34+ plaque index compared to APP/Prdx6+/+ controls (p < 0.0001), while in APP/Prdx6+/− mice the CD68+ / X-34+ index is reduced by 20.1% (p < 0.01). In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 1.8-fold lower CD68+ / X-34+ index (p < 0.0001).
Fig. 8.

Prdx6 expression modulates plaque seeding. Shown are representative images of nascent plaques in female mice aged 6 months of indicated genotypes, which were co-labeled with X-34 (fibrillar core), and anti-Aβ (HJ3.4 clone) and anti-GFAP antibodies in a and with X-34, anti-Aβ (HJ3.4 clone), and anti-Iba1 antibodies in b evidencing an inverse relationship between Prdx6 expression level and the numerical density of nascent plaques and the numerical density of nascent plaques devoid of GFAP+ and Iba1+ cells. Quantitative analysis of nascent plaque numerical density in the brain cortex performed on X-34/Aβ/GFAP co-labeled sections in c, and on X-34/Aβ/Iba1 co-labeled sections in d. In this analysis nascent plaques were arbitrary defined as having no X-34+ core or having X-34+ core < 40 μm2. Black bars represent number of plaques devoid of GFAP+ or Iba1+ cells /mm2 and are superimposed on grey bars representing the total number of plaques /mm2 for a given genotype. Values in c and d represent mean (+SEM) from n = 6 female mice per genotype. p < 0.0001 in c and d (ANOVA); ●●p < 0.01, ●●●p < 0.001, and ●●●●p < 0.0001 denote significance for the density of GFAP and Iba1 devoid plaques across genotypes; *p < 0.05, and ***p < 0.001, denote significance for the density of all plaques (Holm-Sidak’s post-hoc test). Scale bar: 20 μm in a and b
In a complementary experiment, we quantified the load and numerical density of all Th-S+ plaques (nascent + mature) in the brain cortex and the hippocampus in mice aged 6 months and this analysis revealed APP/Prdx6Tg < APP/Prdx6+/+ < APP/Prdx6+/− rank order concerning both metrics with statistically significant differences across the genotypes for either anatomical structure (Fig. S8a-c). Taken together these findings indicate that activity level of astro- and microglia cells effectively impacts early stages of Aβ deposition and that this process is regulated by PRDX6.
Plaque-associated axonal dystrophy varies inversely with Prdx6 gene dose
Numerical density of neuritic plaques was quantified in female mice aged 10 months on section impregnated with Gallyas silver stain [25, 26, 28]. In APP/Prdx6Tg mice, neuritic plaque density in the brain cortex and the hippocampus is reduced by 38% (p < 0.0001) and 39% (p < 0.01) compared to APP/Prdx6+/+ controls, respectively; while in APP/Prdx6+/− mice conversely it shows 31% (p < 0.0001) and 29% (p < 0.05) increase, respectively (Fig. 9a, b). In comparison with APP/Prdx6Tg line, the numerical density of neuritic plaques in APP/Prdx6+/− mice is 2.1-fold higher in either the brain cortex or the hippocampus (p < 0.0001). There also are noticeable differences in the morphology of neuritic plaques across the genotypes. These concern the number of argentophilic spheroids and the distance they spread out from the plaque center following the APP/Prdx6Tg < APP/Prdx6+/+ < APP/Prdx6+/− rank order (Fig. 9a, c). Argentophilic spheroids represent focal swelling of axons, which degenerate passing through diffuse Aβ amassed around the plaque fibrillar core. They also are known to accumulate endosomal vesicles and neurofilaments [37], thus, we used double anti-Aβ/anti-neurofilament (NF) immunolabeling protocol to further characterize the impact of Prdx6 gene dose on axonal dystrophy (Fig. 9d). APP/Prdx6Tg mice show reduction in the average number of NF-positive swelling per plaque by 32% (p < 0.01) and in the NF plaque index (NF+ % plaque area) by 28% (p < 0.01) compared to APP/Prdx6+/+ controls, while in APP/Prdx6+/− mice there is 31% (p < 0.01) and 28% (p < 0.01) increase in these metrics, respectively (Fig. 9e, f). In direct comparison with APP/Prdx6Tg line, APP/Prdx6+/− mice show 1.9-fold more of NF-positive swellings per plaque (p < 0.0001) and 1.7-fold higher NF plaque index (p < 0.0001).
Fig. 9.

Neuritic degeneration is attenuated in APPSWE/PS1dE9 mice overexpressing Prdx6 and exacerbated in Prdx6 haplodeficient mice. a Shown are representative microphotographs of coronal cross-sections through the somatosensory cortex (upper panel) and the dorsal hippocampus (lower panel) from 10-month-old female mice of indicated genotypes, which were stained with Gallyas silver stain. Both the numerical density of neuritic plaques and the number and size of spheroid bodies forming the plaques increase in the rank order of APP/Prd6Tg < APP/Prd6+/+ < APP/Prd6+/−. Arrows indicate mature plaques while arrowheads indicate early stage of neuritic degeneration associated with emerging plaques, which are abundant in the APP/Prd6−/− mice. b Quantitative analysis of numerical density of Gallyas+ neuritic plaques in the brain cortex and in the hippocampus depicted as mean value (+SEM) from n = 6 female mice per genotype. c Shown are high magnification microphotographs of neuritic plaques produced by superimposing bright field picture of silver impregnated spheroids on fluorescent image of X-34 labeled fibrillar plaque core highlighting the differences in plaque composition across the genotypes, which include both the number and the distance spheroid spread away from the fibrillar core. d Representative LCMS images of plaques co-labeled with antibodies against the N-terminus of Aβ peptide (Aβ1–16) and neurofilaments (NF) from female mice of indicated genotypes demonstrating distorted trajectories of axons passing in the vicinity of the plaques and presence of axonal swellings (spheroids) in the area occupied by anti-Aβ immunolabeling (white encirclement indicated by Aβ plaque mask). Quantification of the number of NF+ axonal swellings per Aβ plaque in e and NF+ plaque index (NF+ % plaque area) in f. Values represent mean (+SEM) from n = 40 randomly selected plaques per genotype. p < 0.001 for the hippocampus in b, and p < 0.0001 for the brain cortex in b, e, and f (ANOVA); *p < 0.05, **p < 0.01, and ****p < 0.0001 (Holm-Sidak’s post-hoc test). Scale bars: 50 μm – the cortex and 200 μm the hippocampus in a, and 10 μm in c and d
Discussion
Unlike microglia, function of astrocytes in Aβ proteostasis has received limited attention and remains of rudimentary understanding [38]. Peri-plaque astrocytic activation has been classically viewed as a passive response to neuritic degeneration through formation of a glial scar [39]. Here, we provide novel evidence that astrocytes play an active role in countering Aβ deposition, and identify PRDX6 protein as a key element of this process. PRDX6 is expressed by astrocytes but no other type of CNS glial cells [8, 9]. As a prerequisite for this study, we determined that two-fold change in Prdx6 expression level does not affect astrocyte-resting state or alter soluble Aβ levels in AD Tg mice prior to appearance of Aβ plaques. Altered Prdx6 expression also does not influence mounting of Aβ-associated astrogliosis, as the load of GFAP+ astrocytes remains proportional to that of fibrillar Aβ plaques. However, we find a striking, direct dependence between Prdx6 gene dose and propensity of astrocytes to engage and penetrate Aβ plaques. We also show that Prdx6 gene overexpression is associated with reduced Aβ plaque load through suppression of nascent plaque seeding and remodeling of mature plaque by phagocytically activated microglia, while Prdx6 haplodeficiency conversely effects increased Aβ load in association with reduced periplaque microglia activation (Fig. 10 a-d). Demonstration that altering expression of an astrocytic inherent protein produces coextensive effect on periplaque microglia implies that astrocytes circuitously guide microglia dependent Aβ plaque processing.
Fig. 10.
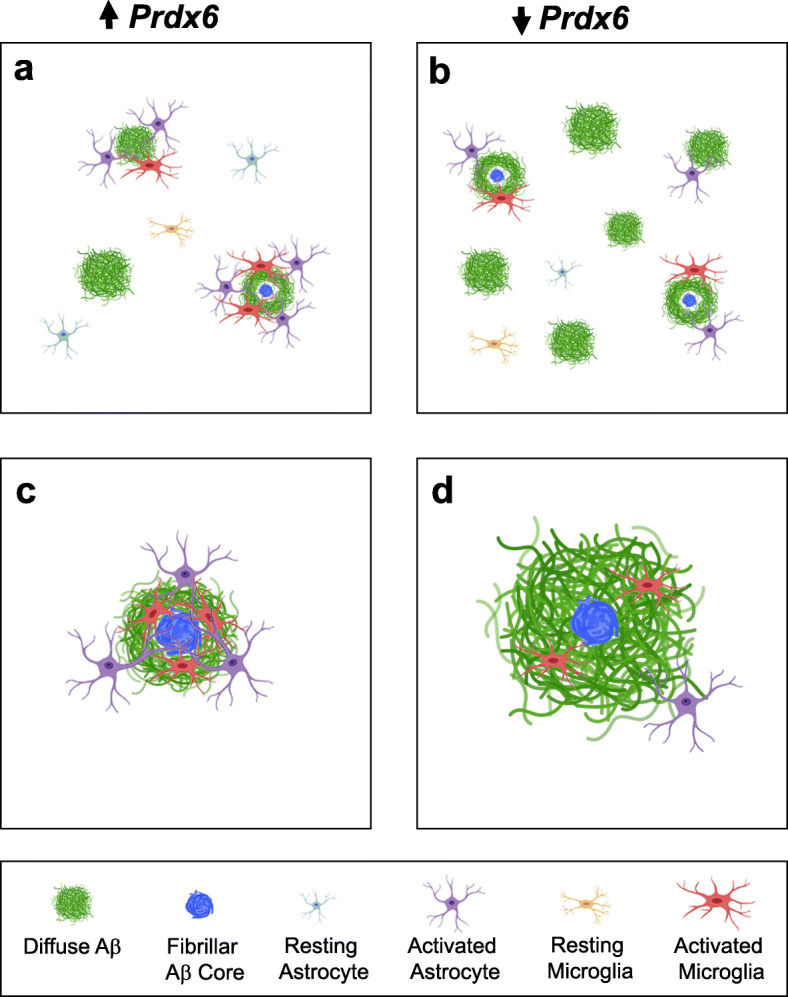
Schematic depiction of PRDX6 dependent mechanisms in Aβ homeostasis: a and b preventive effect on nascent plaque seeding and c and d remodeling of mature plaques. a Overexpression of Prdx6 is associated with reduced number of nascent plaques and specifically those, which are devoid of activated astrocytes and/or microglia cells. b In contrast, Prdx6 haplodeficiency increases number of nascent plaques including those, which are not associated with astro- and micro- glial cells. c Mature plaques in Prdx6 overexpressing mice show increased plaque compactness and diminished deposition of diffuse Aβ peptide around the plaque fibrillar core. d Conversely, Prdx6 haplodeficiency is associated with reduced plaque compactness and increased amount of diffuse Aβ in the plaque periphery. There is a direct association between the Prdx6 gene dose and periplaque activation of astrocytes and microglia. Since PRDX6 is an astrocytic protein these observations imply phagocytic function of microglia in Aβ proteostasis is regulated by astrocytes
PRDX6 is a cytoplasmic, enzymatic protein with independent Gpx and PLA2 activities [40]. Gpx activity is endowed by Cys47, while the PLA2 activity by the triad of Ser32, His26, and Asp140 brought into a single catalytic site by the tertiary PRDX6 folding [41]. PRDX6 is abundant in lung epithelium, endothelium, hepatocytes, and peripheral macrophages [5], where it is involved in antioxidant defense (with H2O2, short chain hydroperoxides, and phospholipid hydroperoxides as reduction substrates), phospholipid turnover, and cellular signaling [5]. In particular, PRDX6 is recognized for its ability to restitute peroxidized cell membrane lipids by their reduction through Gpx activity or excision through PLA2 action with subsequent replacement by PLA2-conjugated lysophosphatidylcholine acyl transferase activity. This particular functionality of PRDX6 is essential for the integrity of lung epithelium endowing resistance to constant oxidative damage [5, 6]. In AD, Aβ plaques constitute a source of locally confined yet enduring oxidative stress, which contributes importantly to disease pathogenesis by rendering protein misfolding, impairing ability of microglia to phagocytose Aβ while promoting their pro-inflammatory phenotype, and effecting neuritic degeneration [42]. Astrocytes, which penetrate Aβ plaques need to withstand consequences of chronic oxidative stress. In this study we showed that penetrance of Aβ plaques by astrocytes is associated with upregulation of PRDX6 protein expression along with redistribution of PRDX6 from the cell body to astrocytic processes. Similar reactive behavior of PRDX6 is known from extra-CNS cell lineages where oxidative stress upregulates PRDX6 level and promotes its translocation from the cytosol to the cell membrane allowing access to peroxidized lipids as the metabolic substrate [43]. In our previous work, we experimentally proved that PRDX6 renders astrocytes resistant to oxidative damage [8]. Astrocytes cultured from Prdx6 overexpressing mice exhibit increased resistance to in vitro oxidative insult while astrocytes cultured from Prdx6−/− mice, conversely show significantly reduced viability. Thus, greater penetrance of Aβ plaques by astrocytes in APP/Prdx6Tg mice and reduced penetrance in APP/Prdx6+/− mice reflect increased and decreased resistance of astrocytes to plaque-associated oxidative stress, respectively. Interestingly, we also observed that PRDX6 overexpressing astrocytes, which engage Aβ plaques have significantly reduced expression of C3, which is a marker of astrocytic neurotoxic phenotype, also known as A1 [33–35]. Conversely, in Prdx6 haplodeficient mice, which cannot mount proper PRDX6 expression, astrocytes not only poorly penetrate Aβ plaques but they also show upregulation of C3 expression. These results imply that chronic oxidative stress may contribute importantly to astrocytes assuming neurotoxic phenotypes, and that PRDX6 besides increasing endurance to oxidative insult helps in maintaining astrocytic homeostasis.
PRDX6 also is widely recognized for cellular signaling owing to its PLA2 activity, and this functionality is triggered by various cellular stressors requiring oxidative, inflammatory, and mechanical stress response [5, 7]. PRDX6/PLA2 activity can engage several signaling pathways including down-stream activation of NADPH oxidase 2 (NOX2), which is a member of the NOX/DuOX enzyme family generating O2•- or H2O2 [44, 45]. Although rapid and uncontrollable release of O2•- or H2O2 is a culprit in tissue injury during acute oxidative stress, low levels of either intracellular or extracellular O2•- or H2O2 messengers are deemed essential for many homeostatic processes, involving intracellular signaling, microbial defense and guiding immune cell chemotaxis into sites of inflammation [46–50]. A well-established example of such, is NOX2-dependent activation of toll-like receptor (TLR) pathway promoting antibacterial autophagy [51]. Other examples of PLA2-dependent signaling include arachidonic acid pathway [52] and nuclear factor kappa B (NF-κB) pathway [53], which are up and downregulated by PRDX6 level, respectively. In summary, we hypothesize that the beneficence of PRDX6 overexpression against Aβ pathology is owed both to its anti-oxidative function, which increases oxidative stress endurance and allows for a more robust penetration of Aβ deposits by astrocytes and for the signaling function of PLA2 by which astrocytes circuitously can modulate the activity of plaque-associated microglia. It also is important to conceptualize, that with their somas located outside Aβ plaques and a substantial number of their processes spreading outward into the brain parenchyma, activated astrocytes are well-poised to extent signaling message far distant from the perimeter of Aβ deposits they engage. As the role of astrocytic PRDX6/PLA2 in cellular signaling remains unexplored, ours is the first observation to suggest it may be involved in astro/microglia crosstalk. Certainly, aforementioned PRDX6/PLA2-related signaling pathways like TLR and NF-κB are regarded important to microglia function [54–56]. Thus, in the future studies it will be imperative to precisely determine, which of these pathways underlie astrocyte/microglia cooperative effort to counter Aβ deposition.
Incipient stages of Aβ deposition remain underexplored. It is unclear how nascent Aβ plaques become recognized by glial cells and what sequence of events leads to periplaque glia activation. In the CNS, astrocytes numerically surpass microglia and form a dense surveying network readily responding to changes affecting milieu of the nervous tissue. Examining early stage of Aβ deposition in 6-month-old mice, we found that the numerical density of nascent plaques and the subset of these plaques devoid of astrocytes and microglia companionship vary inversely with the Prdx6 gene dose. We also found that phagocytic activity of microglia associated with nascent plaques remains directly proportional to the Prdx6 expression. These observations merit two conclusions. Firstly, that PRDX6 is involved in engagement of nascent plaques by co-operative action of astrocytes and microglia and secondly, that a portion of newly seeded plaques is cleared by activated microglia. Though the second notion may appear speculative, it is supported by the fact that the level of APP transcript and the level of soluble Aβ prior to the onset of Aβ deposition are similar across all three analyzed transgenic mouse lines rendering them similarly capable of plaque seeding. Our histopathological analysis also endorses recently published spatio-temporal transcriptomic studies in AD Tg mice, which indicate transcriptome alterations in astrocytes and microglia occur in parallel and coincide with emergence of early Aβ deposits [57, 58]. Thus, both studies emphasize an importance of astro/microglia cooperative effort in maintaining Aβ homeostasis.
Likewise, mature plaques we examined in mice aged 10 months reveal influence of Prdx6 gene dosage on their morphology and degree of periplaque microglia activity. Aβ plaques in Prdx6 overexpressing mice feature reduced amount of diffuse Aβ in the plaque brim, increased plaque compactness, and increased number and phagocytic activity of plaque-associated microglia. Such morphological changes reflect established function of microglia, known to phagocytose Aβ peptide in the plaque brim what limits plaque diffusion and facilitates compacting Aβ within the fibrillar core [36]. In Prdx6 hemizygous mice Aβ plaques exhibit opposite features including increased amount of loose, non-fibrillar Aβ in the plaque brim and reduced plaque/core ratio indicative of reduced plaque compactness. Diminished number of periplaque microglial cells and microglia phagocytosis markers remain consistent with this more diffuse plaque morphology, resembling plaques in AD mice with Trem2 hemizygocity or those harboring Trem2 R47H mutant, both of which confer loss of TREM2 function and impair Aβ proteostatic function of microglia [29]. In fact, in our study peri-plaque microglia in Prdx6 hemizygous mice feature reduced TREM2 expression, while those in Prdx6 overexpressing mice show TREM2 upregulation. TREM2 and the TREM2/mTOR signaling pathway are recognized for their critical role in promoting phagocytic activity of microglia and other cells of myeloid linage [4]. However, how TREM2 expression is triggered during microglia activation and how it is sustained effecting microglia phagocytic phenotype remains unclear [59]. TREM2 activity is suppressed by activation of the NF-κB [59] pathway, which also is inversely regulated by PRDX6 expression level [53]. TREM2 also is stimulated by a set of specific ligands, including apoE [4, 59], which plaque content directly correlates with PRDX6 expression. Hence, through cellular signaling and increased apoE plaque content PRDX6 can exert two-prong effect on promoting TREM2 activity in periplaque microglia.
Axons passing through diffuse Aβ in the plaque periphery undergo degeneration forming spherical bodies [36, 37] and we found neuritic degeneration to be another essential feature directly associated with Prdx6 gene dose. There is reduced numerical density of neuritic plaques in Prdx6 overexpressing mice and conversely their density is increased in Prdx6 hemizygous animals. While, these differences can be explained by variable effect of Prdx6 expression on plaque seeding and resulting discrepancies in plaque density, we also found dissimilarities in the amount of degenerating axons within individual plaques. These differences likely reflect differential effect of Prdx6 gene dosage on phagocytic activity of periplaque microglia and resulting degree of plaque diffusion [36]. It is also possible that anti-oxidant activity of PRDX6 itself contributes to attenuating neuritic degeneration as this process is driven by oxidative stress [42]. However, PRDX6 is compartmentalized to the cytoplasm and its anti-oxidant activity takes place primarily intracellularly rather than it is known to exert significant extracellular effect [5].
While this study utilizes models of chronic neurodegeneration and its conclusions imply beneficence of PRDX6 overexpression in Aβ pathology, opposing findings were described using acute/subacute models of neurodegeneration. Yun et al. showed that intraventricular infusion of oligomeric Aβ42 in mice transgenically overexpressing Prdx6 paradoxically exacerbates oxidative stress and metrics of neurodegeneration compared to wild type mice [60]. Similarly, the same group showed that administration of MPTP to Prdx6 Tg mice exacerbate loss of dopaminergic neurons in the substantia nigra compared to wild type animals [61]. Results of both studies suggest that overexpression of PRDX6 may potentiate outcome of an oxidative insult in the CNS. However, studies concerning PRDX6 function in the lungs may provide insight into understanding these seemingly opposing outcomes. There is extensive literature to indicate opposing behavior of pulmonary PRDX6 during chronic vs. fulminant oxidative insult (reviewed in [5]). While PRDX6 is recognized for its essential role in maintaining the integrity of cellular membranes in the pulmonary epithelium, which is exposed to chronical low-grade oxidative stress, in acute lung injury, (e.g. in the course of sepsis), over activation of PLA2/NOX2 signaling pathway may lead to uncontrolled generation of O2•- or H2O2 and paradoxical exacerbation of lung injury. Based on the original observations made by Yun and colleagues, brain PRDX6 also may be capable of paradoxical amplification of oxidative stress related injury. However, since Aβ deposition is an inherently protracted phenomenon, associated with chronic, low-level oxidative stress, we elected to test the role of PRDX6 in this process by developing and examining novel APP/PS1 Tg mice varying in Prdx6 gene expression. In contrast to the model utilizing intraventricular infusion of preformed Aβ1–42 oligomers, we demonstrated beneficence of PRDX6 overexpression in Aβ deposition and associated neurodegeneration, which we also confirmed by showing opposing outcomes when PRDX6 expression was reduced.
Unlike other cell types, astrocytes remain strikingly under-investigated in AD-related research [38]. There are few reports tying astrocyte dysfunction to propagation of Aβ pathology. Deletion of Gfap and vimentin genes encoding intermediate filaments of astrocytic cytoskeleton was shown to impair migration of astrocytes toward Aβ plaques, which resulted in accelerated plaque pathogenesis and enhanced neuritic degeneration in AD Tg mice [62]. Also, astrocytic rarefaction of Aβ plaques was identified as one of the neuropathological hallmarks in rapidly progressing AD, which is a rare but aggressive AD variant. Proteomic analysis in rapidly progressing AD identified PRDX6 as one of top proteins, whose peri-plaque expression is significantly reduced compared to sporadic disease [63]. In the backdrop of these observations, our study provides original insights into a role of astrocytes in Aβ proteostasis by proposing that astrocytes rather than being passive by-standers actively recognize Aβ deposits and circuitously modulate microglia phagocytic activity to counter Aβ accumulation. Plaques that escape initial seeding control and continue to grow remain penetrated by astrocytes, which resistance to oxidative stress is mediated by PRDX6 antioxidant capacity. Astrocytes also propagate phagocytic activity of microglia, which limits plaque diffusion and curtails neuritic degeneration and this effect may be endowed by PLA2 signaling functionality of PRDX6. Thus, emerging evidence recognizes astrocytes as important players in Aβ proteostasis, and suggests that upregulation of PRDX6 expression can be of therapeutic benefit in AD.
Conclusions
Our work identifies PRDX6 as an important factor selectively regulating periplaque activation of astrocytes and playing a critical role in astro/microglia cross talk in maintaining Aβ proteostasis.
Supplementary information
Additional file 1: Figure S1. Comparative analysis of Aβ plaque load in female and male littermates shows significant opposing effects of Prdx6 overexpression and haplodeficiency in both sexes. Figure S2. Segmentation analysis of LSCM images of Aβ plaques. Figure S3 Overexpression of Prdx6 is associated with increased accumulation of apoE within Aβplaques, while Prdx6 haplodeficiency conversely reduces apoE plaque content. Figure S4. Expression of complement component 3 (C3) by plaque associated astrocytes varies inversely with Prdx6 gene dose. Figure S5. Development of Aβ pathology is associated with upregulation of PRDX6 protein expression in astrocytes. Figure S6. Analysis of Prdx6 RNA level in microglia cells. Figure S7. Phagocytic activity of microglia cells surrounding nascent plaques varies directly with Prdx6 gene dose. Figure S8. Prdx6 gene dose affects early stage of Aβ deposition. Supplementary methods. Table S1. Two-way ANOVA analysis of fibrillar and Aβ plaque load in the brain cortex and in the hippocampus in 10-month-old female and male littermates from APP/Prdx6Tg, APP/Prdx6+/+, and APP/Prdx6+/- transgenic mouse lines.
Acknowledgements
We would like to thank Dr. D. M. Holtzman from Washington University School of Medicine (St. Louis, MO) for providing HJ2, HJ3.4, and HJ7.4 anti-Aβ monoclonal antibodies and HJ6.3 monoclonal antibody against murine apoE and Dr. Gary E. Landreth from Indiana University School of Medicine (Indianapolis, IN) for advising us on TREM2 immunohistochemistry.
Abbreviations
- Aβ
β-amyloid
- AD
Alzheimer’s disease
- APP
Amyloid precursor protein
- CD68
Cluster of differentiation 68
- CNS
Central nervous system
- GFAP
Glial fibrillary acidic protein
- Gpx
Glutathione peroxidase
- Iba1
Ionized calcium binding adaptor molecule
- PLA2
Phospholipase A2
- Prdx6
Peroxiredoxin 6 gene
- PRDX6
Peroxiredoxin 6 protein
- PS1
Presenilin 1
- Th-S
Thioflavin-S
- TREM2
Triggering Receptor Expressed in Myeloid cells 2
Authors’ contributions
J.E.P. and M.J.S. conceived of the project and designed the experiments. J.E.P., J.R.D., M.M.A., A.M.L., and L.A.F. conducted the research. J.E.P., J.R.D., M.M.A., A.M.L., and M.J.S. analyzed the data. M.J.S. wrote the manuscript. All authors have read and approved the final version of the manuscript.
Authors’ information
N/A
Funding
This work was supported by grants from the National Institute on Aging R01 AG031221 (M.J.S.), R01 AG053990 (M.J.S.), and R01 AG067478 (M.J.S.).
Availability of data and materials
Raw images and datasets that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
All mouse care and experimental procedures were approved by Institutional Animal Care and Use Committees of the New York University School of Medicine.
Consent for publication
N/A
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Joanna E. Pankiewicz, Email: Joanna.Pankiewicz@nyulangone.org
Jenny R. Diaz, Email: Jenny.DiazRodriguez@nyulangone.org
Mitchell Martá-Ariza, Email: Mitchell.MartaAriza@nyulangone.org.
Anita M. Lizińczyk, Email: Anita.Lizinczyk@nyulangone.org
Leor A. Franco, Email: Leor.Franco@nyulangone.org
Martin J. Sadowski, Email: sadowm01@med.nyu.edu
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13024-020-00401-8.
References
- 1.Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, et al. Association of Amyloid and tau with Cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed]
- 2.Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18:759–772. doi: 10.1038/s41577-018-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90:724–739. doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol. 2018;14:667–675. doi: 10.1038/s41582-018-0072-1. [DOI] [PubMed] [Google Scholar]
- 5.Fisher AB. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch Biochem Biophys. 2017;617:68–83. doi: 10.1016/j.abb.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Benipal B, Zhou S, Dodia C, Chatterjee S, Tao JQ, Sorokina EM, Raabe T, Feinstein SI, Fisher AB. Critical role of peroxiredoxin 6 in the repair of peroxidized cell membranes following oxidative stress. Free Radic Biol Med. 2015;87:356–365. doi: 10.1016/j.freeradbiomed.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel P, Chatterjee S. Peroxiredoxin6 in Endothelial Signaling. Antioxidants (Basel). 2019:8. [DOI] [PMC free article] [PubMed]
- 8.Asuni AA, Guridi M, Sanchez S, Sadowski MJ. Antioxidant peroxiredoxin 6 protein rescues toxicity due to oxidative stress and cellular hypoxia in vitro, and attenuates prion-related pathology in vivo. Neurochem Int. 2015;90:152–165. doi: 10.1016/j.neuint.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Power JH, Asad S, Chataway TK, Chegini F, Manavis J, Temlett JA, Jensen PH, Blumbergs PC, Gai WP. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer's disease pathology. Acta Neuropathol. 2008;115:611–622. doi: 10.1007/s00401-008-0373-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, Asuni AA, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the interaction between Apolipoprotein E and Abeta reduces Intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am J Pathol. 2013;182:1750–1768. doi: 10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li YS, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol. 2004;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am J Respir Cell Mol Biol. 2006;34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelan SA, Wang X, Wallbrandt P, Forsman-Semb K, Paigen B. Overexpression of Prdx6 reduces H2O2 but does not prevent diet-induced atherosclerosis in the aortic root. Free Radic Biol Med. 2003;35:1110–1120. doi: 10.1016/s0891-5849(03)00462-3. [DOI] [PubMed] [Google Scholar]
- 16.Fisher AB, Dodia C, Yu K, Manevich Y, Feinstein SI. Lung phospholipid metabolism in transgenic mice overexpressing peroxiredoxin 6. Biochim Biophys Acta. 1761;2006:785–792. doi: 10.1016/j.bbalip.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Phelan SA, Petros C, Taylor EF, Ledinski G, Jurgens G, Forsman-Semb K, Paigen B. Peroxiredoxin 6 deficiency and atherosclerosis susceptibility in mice: significance of genetic background for assessing atherosclerosis. Atherosclerosis. 2004;177:61–70. doi: 10.1016/j.atherosclerosis.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem. 2003;278:25179–25190. doi: 10.1074/jbc.M302706200. [DOI] [PubMed] [Google Scholar]
- 19.Burkholder T, Foltz C, Karlsson E, Linton CG, Smith JM. Health evaluation of experimental laboratory mice. Curr Protoc Mouse Biol. 2012;2:145–165. doi: 10.1002/9780470942390.mo110217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asuni AA, Guridi M, Pankiewicz JE, Sanchez S, Sadowski MJ. Modulation of amyloid precursor protein expression reduces beta-amyloid deposition in a mouse model. Ann Neurol. 2014;75:684–699. doi: 10.1002/ana.24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26:4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantification. Methods Mol Biol. 2005;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- 25.Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the apoE/A-beta interaction ameliorates A-beta-related pathology in APOE epsilon 2 and epsilon 4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014;2:75. doi: 10.1186/s40478-014-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wisniewski HM, Sadowski M, Jakubowska-Sadowska K, Tarnawski M, Wegiel J. Diffuse, lake-like amyloid-beta deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:674–683. doi: 10.1097/00005072-199807000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Pankiewicz JE, Baquero-Buitrago J, Sanchez S, Lopez-Contreras J, Kim J, Sullivan PM, Holtzman DM, Sadowski MJ. APOE genotype differentially modulates effects of anti-a beta, Passive Immunization in APP Transgenic Mice. Mol Neurodegener. 2017;12:12. doi: 10.1186/s13024-017-0156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 29.Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, von Saucken VE, Williams RY, Karlo JC, Moutinho M, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer's disease. Mol Neurodegener. 2018;13:29. doi: 10.1186/s13024-018-0262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ordonez-Gutierrez L, Fernandez-Perez I, Herrera JL, Anton M, Benito-Cuesta I, Wandosell F. AbetaPP/PS1 transgenic mice show sex differences in the cerebellum associated with aging. J Alzheimers Dis. 2016;54:645–656. doi: 10.3233/JAD-160572. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Tanila H, Puolivali J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318–327. doi: 10.1016/j.nbd.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 32.Sadowski MJ, Wisniewski T: Apoliporoteins in different amyloidoses. In Protein Misfolding, Aggregation, and Conformational Diseases. Edited by Uversky VN, Fink AL. Singapore: Springer; 2006: 101–122.[Atassi MZ (Series Editor): Protein Reviews].
- 33.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24:931–938. doi: 10.1038/s41591-018-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016;213:667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, Ferguson SM. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer's disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112:E3699–E3708. doi: 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 39.Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging. 1991;12:593–600. doi: 10.1016/0197-4580(91)90091-w. [DOI] [PubMed] [Google Scholar]
- 40.Bannitz-Fernandes R, Aleixo-Silva R, Silva JP, Dodia C, Vazquez-Medina JP, Tao JQ, Fisher A, Netto L. Non-Mammalian Prdx6 Enzymes (Proteins with 1-Cys Prdx Mechanism) Display PLA(2) Activity Similar to the Human Orthologue. Antioxidants (Basel). 2019:8. [DOI] [PMC free article] [PubMed]
- 41.Manevich Y, Reddy KS, Shuvaeva T, Feinstein SI, Fisher AB. Structure and phospholipase function of peroxiredoxin 6: identification of the catalytic triad and its role in phospholipid substrate binding. J Lipid Res. 2007;48:2306–2318. doi: 10.1194/jlr.M700299-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manevich Y, Shuvaeva T, Dodia C, Kazi A, Feinstein SI, Fisher AB. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase a(2) activities. Arch Biochem Biophys. 2009;485:139–149. doi: 10.1016/j.abb.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, Fisher AB. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem. 2011;286:11696–11706. doi: 10.1074/jbc.M110.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ambruso DR, Ellison MA, Thurman GW, Leto TL. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim Biophys Acta. 1823;2012:306–315. doi: 10.1016/j.bbamcr.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 46.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Go YM, Jones DP. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic Biol Med. 2011;50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bogdan C, Rollinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12:64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 51.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitt A, Schmitz W, Hufnagel A, Schartl M, Meierjohann S. Peroxiredoxin 6 triggers melanoma cell growth by increasing arachidonic acid-dependent lipid signalling. Biochem J. 2015;471:267–279. doi: 10.1042/BJ20141204. [DOI] [PubMed] [Google Scholar]
- 53.Arevalo JA, Vazquez-Medina JP. The Role of Peroxiredoxin 6 in Cell Signaling. Antioxidants (Basel). 2018:7. [DOI] [PMC free article] [PubMed]
- 54.Su F, Bai F, Zhou H, Zhang Z. Microglial toll-like receptors and Alzheimer's disease. Brain Behav Immun. 2016;52:187–198. doi: 10.1016/j.bbi.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Gambuzza ME, Sofo V, Salmeri FM, Soraci L, Marino S, Bramanti P. Toll-like receptors in Alzheimer’s disease: a therapeutic perspective. CNS Neurol Disord Drug Targets. 2014;13:1542–1558. doi: 10.2174/1871527313666140806124850. [DOI] [PubMed] [Google Scholar]
- 56.Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Lalakova J, Kuhnemund M, Voytyuk I, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease. Cell. 2020;182:976-991 e919. [DOI] [PubMed]
- 58.Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, et al. The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. 2019;27:1293–1306. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jay TR, von Saucken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017;12:56. doi: 10.1186/s13024-017-0197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yun HM, Jin P, Han JY, Lee MS, Han SB, Oh KW, Hong SH, Jung EY, Hong JT. Acceleration of the development of Alzheimer's disease in amyloid beta-infused peroxiredoxin 6 overexpression transgenic mice. Mol Neurobiol. 2013;48:941–951. doi: 10.1007/s12035-013-8479-6. [DOI] [PubMed] [Google Scholar]
- 61.Yun HM, Choi DY, Oh KW, Hong JT. PRDX6 exacerbates dopaminergic Neurodegeneration in a MPTP mouse model of Parkinson's disease. Mol Neurobiol. 2015;52:422–431. doi: 10.1007/s12035-014-8885-4. [DOI] [PubMed] [Google Scholar]
- 62.Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27:187–198. doi: 10.1096/fj.12-208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drummond E, Nayak S, Faustin A, Pires G, Hickman RA, Askenazi M, Cohen M, Haldiman T, Kim C, Han X, et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol. 2017;133:933–954. doi: 10.1007/s00401-017-1691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Comparative analysis of Aβ plaque load in female and male littermates shows significant opposing effects of Prdx6 overexpression and haplodeficiency in both sexes. Figure S2. Segmentation analysis of LSCM images of Aβ plaques. Figure S3 Overexpression of Prdx6 is associated with increased accumulation of apoE within Aβplaques, while Prdx6 haplodeficiency conversely reduces apoE plaque content. Figure S4. Expression of complement component 3 (C3) by plaque associated astrocytes varies inversely with Prdx6 gene dose. Figure S5. Development of Aβ pathology is associated with upregulation of PRDX6 protein expression in astrocytes. Figure S6. Analysis of Prdx6 RNA level in microglia cells. Figure S7. Phagocytic activity of microglia cells surrounding nascent plaques varies directly with Prdx6 gene dose. Figure S8. Prdx6 gene dose affects early stage of Aβ deposition. Supplementary methods. Table S1. Two-way ANOVA analysis of fibrillar and Aβ plaque load in the brain cortex and in the hippocampus in 10-month-old female and male littermates from APP/Prdx6Tg, APP/Prdx6+/+, and APP/Prdx6+/- transgenic mouse lines.
Data Availability Statement
Raw images and datasets that support the findings of this study are available from the corresponding author upon reasonable request.