

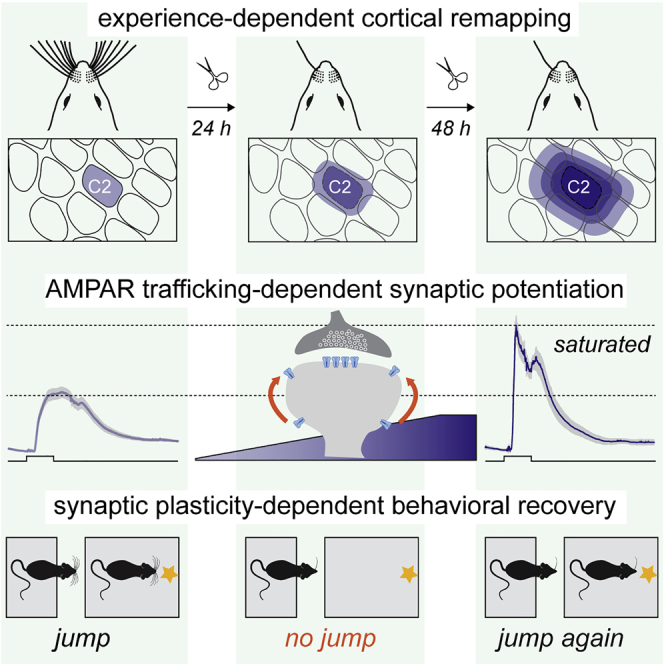
Summary
Cortical plasticity improves behaviors and helps recover lost functions after injury. However, the underlying synaptic mechanisms remain unclear. In mice, we show that trimming all but one whisker enhances sensory responses from the spared whisker in the barrel cortex and occludes whisker-mediated synaptic potentiation (w-Pot) in vivo. In addition, whisker-dependent behaviors that are initially impaired by single-whisker experience (SWE) rapidly recover when associated cortical regions remap. Cross-linking the surface GluA2 subunit of AMPA receptors (AMPARs) suppresses the expression of w-Pot, presumably by blocking AMPAR surface diffusion, in mice with all whiskers intact, indicating that synaptic potentiation in vivo requires AMPAR trafficking. We use this approach to demonstrate that w-Pot is required for SWE-mediated strengthening of synaptic inputs and initiates the recovery of previously learned skills during the early phases of SWE. Taken together, our data reveal that w-Pot mediates cortical remapping and behavioral improvement upon partial sensory deafferentation.
Keywords: synaptic plasticity, somatosensory, barrel cortex, cortical remapping, whisker trimming, behavioral recovery, AMPA receptor, AMPAR, cross-linking, surface diffusion
Graphical Abstract
Highlights
-
•
AMPAR trafficking mediates synaptic potentiation in vivo in mice with intact whiskers
-
•
Whisker trimming rapidly saturates spared-whisker responses
-
•
Synaptic potentiation causes the enhancement of spared-whisker-evoked response
-
•
Synaptic potentiation facilitates the behavioral recovery during cortical remapping
By manipulating surface AMPARs in vivo, Campelo et al. study the function of synaptic potentiation in the whisker-to-cortex system of mice upon whisker trimming. They reveal that the remapping of somatosensory regions and the recovery of altered skills depend on the potentiation of glutamatergic synapses onto layer 2/3 pyramidal neurons.
Introduction
Functional sensory maps in the cerebral cortex reorganize in response to brain trauma or peripheral injury, with active modalities gaining cortical space at the expense of less active ones (Merzenich et al., 1983). Map expansion has been proposed to adapt behaviors by optimizing neuronal circuits. Most of the evidence that map expansion is required for skills adaptation comes from studies that correlated behavioral changes with use-dependent map reorganization (Bieszczad and Weinberger, 2010; Molina-Luna et al., 2008; Reed et al., 2011). Despite these worthwhile contributions, the underlying circuit and synaptic mechanisms remain poorly understood. Here, we exploited the mouse whisker-to-barrel cortex system to explore the relationship between the synaptic mechanisms of sensory map plasticity and correlated adaptive behaviors.
Rodents use their whiskers to explore their immediate tactile environment. Under normal conditions, neurons in each barrel column have receptive fields in the primary somatosensory cortex (S1) that are strongly tuned toward one principal whisker (PW). Nevertheless, trimming all but one whisker (SWE; single-whisker experience) causes layer (L)2/3 pyramidal neurons located in the deprived and spared-related columns to respond stronger to the spared-whiskers stimulation (Feldman, 2009; Fox, 2002; Glazewski and Fox, 1996; Glazewski et al., 1996; Margolis et al., 2014), thereby resulting in the strengthening and expansion of the spared-whisker representations within the map (Feldman, 2009; Fox, 2002; Margolis et al., 2014). Synaptic plasticity—notably, long-term synaptic potentiation (LTP)—has been postulated as a synaptic mechanism for such response strengthening during learning and deprivation-induced plasticity (Clem et al., 2008; Feldman, 2009; Finnerty et al., 1999; Fox, 2002; Glazewski and Fox, 1996; Glazewski et al., 1996; Margolis et al., 2014). In support of this hypothesis, initial studies reported that activation of N-methyl-D-aspartate receptors (NMDARs) (Clem et al., 2008; Rema et al., 1998), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Clem and Barth, 2006; Dachtler et al., 2011), α/δ CREB, α-CaMKII (Ca2+/calmodulin-dependent protein kinase-II), and α-CaMKII autophosphorylation (Glazewski et al., 1996, 2000) are all involved in neuronal response potentiation in L2/3 neurons. SWE increases the synaptic strength of vertical and horizontal connections within and across cortical columns and occludes electrically induced, NMDAR-dependent LTP in acute brain slices obtained from young rodents (Clem and Barth, 2006; Clem et al., 2008; Finnerty et al., 1999), thereby providing indirect evidence for a requirement of LTP during whisker map plasticity. However, other homeostatic mechanisms, such as disinhibition, synaptic scaling, or regulation of intrinsic excitability might also be engaged upon sensory deprivation (Gainey and Feldman, 2017). It thus remained unclear whether and when synaptic plasticity is causally inducing the potentiation of spared whisker-evoked response upon whisker trimming and more particularly in adult living animals.
Therefore, although several paradigms to induce sensory-mediated LTP have been characterized in vivo (Gambino and Holtmaat, 2012; Gambino et al., 2014; Zhang et al., 2015), a direct demonstration that synaptic plasticity is required for cortical remapping and the adaptation of sensorimotor skills is still lacking in adult animals. This likely results from (1) the technical difficulty to induce and monitor synaptic plasticity in living animals with behaviorally relevant stimuli; (2) the relatively limited knowledge of the molecular mechanisms underlying this form of plasticity in vivo; and (3) the lack of specific tools to block synaptic plasticity and spare the basal circuit functions while cortical regions remap. Recently, manipulating the mobility of AMPAR has provided a specific tool to block synaptic plasticity during behavior without altering basal synaptic transmission and circuit functions (Humeau and Choquet, 2019; Penn et al., 2017). Here, we show that cross-linking the GluA2 subunit in vivo inhibits synaptic potentiation of sensory inputs induced by physiological and behaviorally relevant stimuli (whisker-mediated synaptic potentiation; w-Pot), presumably by blocking GluA2 surface diffusion and preventing the increase in synaptic AMPAR content. This demonstrates a direct link between AMPAR trafficking and physiologically induced synaptic plasticity in vivo and reaffirms decades of in vitro work. Using this strategy, we demonstrate that w-Pot is causally inducing the potentiation of whisker-evoked responses and initiates behavioral recovery in the early phase of cortical remapping following whisker trimming.
Results
SWE Reshapes Cortical Sensory Map and Rapidly Saturates Whisker-Evoked Responses in L2/3 Pyramidal Neurons
Previous studies have suggested that spared whiskers gain cortical space through LTP-mediated changes in the efficacy of existing synapses (Barth et al., 2000; Clem and Barth, 2006; Clem et al., 2008; Feldman, 2009; Feldman and Brecht, 2005; Glazewski and Fox, 1996; Glazewski et al., 1996). To explore this question in vivo, we first investigated the impact of the SWE on whisker-evoked neuronal and synaptic responses. We exposed mice to a brief period of SWE (2–3 days) by clipping all but one C2 whisker and quantified the spatial representation of the spared whisker in contralateral S1 during anesthesia using intrinsic optical signal imaging (Figures 1A–1C). For each mouse, intrinsic optical signals were acquired before, during, and after a 1-s long train (8 Hz) of C2 whisker deflection, for 3 consecutive days, in full-whisker-experience (FWE; n = 6) and SWE (n = 7) animals. For SWE mice, whiskers were trimmed after the first imaging session (SWE0; Figure 1B). The whisker deflection-evoked response area was determined as previously described (Schubert et al., 2013). In contrast to FWE mice in which signals remained stable across sessions (p = 0.3; Figure 1C), the intrinsic optical signals evoked by the deflection of the spared whisker increased gradually within the spared-whisker barrel column of SWE mice and became significantly different 48 h after clipping (SWE1 versus SWE0, p = 0.246; SWE2 versus SWE0, p = 0.004; Figure 1C). Importantly, it occurred at a time at which no alterations in the activity of L4 granular neurons have been observed in L2/3 (Benedetti et al., 2009; Glazewski and Fox, 1996; Glazewski et al., 1996), suggesting that SWE-induced map plasticity is fast and originates primarily from changes in neural activity within L2/3.
Figure 1.

SWE Increases Whisker-Evoked Supra- and Sub-threshold Synaptic Responses in L2/3 Pyramidal Neurons
(A) Schematic of intrinsic optical signals and whole-cell recordings of L2/3 pyramidal neurons in full-whisker-experience (FWE) and single-whisker-experience (SWE) mice. ns, not significant.
(B) Statistical t-maps over 3 successive days. Scale bar, 500 μm.
(C) Averaged (± SEM) PW-evoked response area (normalized to the first session at day 0). Light/dark blue lines indicate individual FWE (n = 7)/SWE (n = 6) mice, respectively. Circle indicates mean. For SWE mice, whiskers are clipped after the first imaging session (dotted arrow).
(D) Left: single-cell examples of whisker-evoked responses (gray, single trials; dark and light blue, averaged traces from SWE and FWE mice, respectively). Square pulse lines, C2 whisker deflection (100 ms). Right: fraction of spiking neurons (top: SWE2 and SWE3 pooled together) and number of spikes per whisker deflection (bottom: spiking probability; median ± interquartile range).
(E) PW-evoked PSP grand average (all recorded cells averaged) ± SEM. Cells are recorded from FWE mice and SWE mice 48 h (SWE2) and 72 h (SWE3) after trimming. Square pulse lines, C2 whisker deflection (100 ms).
(F) Median (± interquartile range) resting membrane potential (Vrest), PSP peak amplitude, and integral. FWE, n = 20 cells; SWE2, n = 4 cells; SWE3, n = 9 cells.
Thus, we next performed whole-cell recordings of L2/3 pyramidal neurons in vivo in the C2 barrel while deflecting the PW, in FWE mice and after 2 and 3 days of SWE (SWE2 and SWE3, respectively; Figures 1A and 1D–1F). In agreement with the aforementioned results, the fraction of spiking neurons in L2/3 and the number of spikes per PW deflection (i.e., the spiking probability) were increased in SWE2 mice as compared to FWE mice (spiking probability: FWE, 0; SWE2, 0.37 ± 0.18; p < 0.01) (Figure 1D). To understand the mechanisms that alter the output of L2/3 neurons during SWE, we next examined whisker-evoked neuronal responses that did not generate spikes (Figure 1E). As compared to FWE, the mean PW-evoked postsynaptic potential (PSP) peak amplitudes were significantly increased at SWE2 (FWE: 9.88 ± 0.86 mV; SWE2: 17.48 ± 2.9 mV; p < 0.01) (Figures 1E and 1F). Although we cannot exclude an effect of SWE-mediated disinhibition on PSP peak amplitude (but see Figure S1), the concurrent increase in spiking probability and strengthening of synaptic transmission suggests that, despite a moderate increase in intrinsic excitability (Figure S1F), the change of L2/3 neuronal spiking after SWE likely resulted from an increase in peak amplitude of whisker-evoked subthreshold PSP (Figure S1G). Importantly, the spiking probability (SWE2: 0.37 ± 0.18; SWE3: 0.54 ± 0.1; p = 0.186) and the mean evoked PSP peak amplitudes (SWE2: 17.48 ± 2.9 mV; SWE3: 18.41 ± 2.1 mV; p = 0.75) were similar at SWE2 and SWE3, indicating that the spared-whisker-evoked neuronal responses no longer increased after SWE2 (Figures 1D–1F).
Our data are consistent with the increase of sensory-driven spikes in vivo (Barth et al., 2000; Clem and Barth, 2006; Clem et al., 2008; Glazewski and Fox, 1996; Glazewski et al., 1996). They further suggest that plasticity in L2/3 occurs rapidly and saturates both the sub- and supra-threshold neuronal responses evoked by the spared whisker between trimming and SWE2.
w-Pot In Vivo Is Occluded after 2 Days of SWE
In vitro slice recordings suggested that LTP-like mechanisms are prime candidates for enhancing synaptic transmission during SWE in young animals (Clem and Barth, 2006; Clem et al., 2008), but in vivo evidence remains rare in older mice. Thus, given that spared-whisker neuronal responses no longer increased after SWE2, we next compared in vivo the potentiation of sensory synaptic inputs induced by whisker stimulation (w-Pot) in FWE mice and after SWE2 (Figure 2A). Indeed, LTP can be induced in vivo by rhythmic whisker stimulation (RWS) (Gambino et al., 2014; Mégevand et al., 2009; Williams and Holtmaat, 2019; Zhang et al., 2015). This form of postsynaptic LTP does not depend on back-propagating action potentials but relies instead on dendritic NMDAR-dependent plateau potentials driven by the coordinated activation of segregated leminiscal and paralemniscal thalamo-cortical circuits (Gambino et al., 2014; Williams and Holtmaat, 2019) (Figure 2B).
Figure 2.

SWE Occludes w-Pot
(A) Schematic of whisker deprivation.
(B) Schematic of thalamo-cortical circuits. L2/3 pyramidal neurons are recorded in the principal barrel-related column upon deflection of the PW. Depth of recorded cells is indicated.
(C) Time course of averaged PSP peak amplitude before and after RWS in FEW (n = 7 cells) and SWE (n = 7 cells) mice.
(D) Time course of averaged PSP amplitude normalized to baseline.
(E) Left: mean (± SEM) amplitude before (baseline) and after RWS. Error bars, sem; black lines between bars, pairs. Right: mean (± SEM) amplitude normalized to baseline. Triangles, individual cells. ns, not significant.
(F) Median (± interquartile range) of plateau strength (FWE, n = 20 cells; SWE, n = 13 cells).
(G) Correlation between normalized plateau strength and the level of RWS-induced PSP peak potentiation in FWE (light blue) and SWE (dark blue) mice.
In FWE animals, a significant potentiation of whisker-evoked PSP was elicited by stimulating the PW for 1 min at a frequency of 8 Hz (RWS) (baseline: 8.18 ± 1.17 mV; RWS: 9.77 ± 1.11 mV; n = 7; p = 0.002) (Figures 2C–2E, light blue traces; see also Figure 4A). In stark contrast, RWS failed to strengthen whisker-evoked PSP after SWE2 (baseline: 20.45 ± 2.26 mV; RWS: 19.9 ± 2.12 mV; n = 7; p = 0.264) (Figures 2C–2E, dark blue traces). To investigate the possibility that the absence of w-Pot after SWE was the consequence of the alteration of its induction mechanism, we extracted and compared NMDAR-dependent plateau potentials, as previously described (Gambino et al., 2014), in both FWE and SWE2 conditions. Indeed, suppressing these plateau potentials prevents RWS-LTP in vivo (Gambino et al., 2014). Compared to the control FWE condition, plateau potentials evoked by single-whisker stimulation were not significantly changed in SWE (plateau strength: FWE, 0.99 ± 0.03 mV∗s, n = 20; SWE, 1.35 ± 0.08, n = 13; p = 0.3) (Figure 2F). The strength of plateau potentials (i.e., the product of the probability for a whisker deflection to elicit a plateau potential and the average integrated depolarization) correlated with the magnitude of RWS-induced LTP in FWE mice (Figure 2G). However, L2/3 neurons of SWE2 mice bearing high plateau potentials could not be potentiated, indicating that SWE has altered the positive correlation between plateau strength and the level of w-Pot observed in FWE mice (Figure 2G; see also Figures S3A–S3C). This demonstrates that a key component of the NMDAR-dependent induction mechanism of w-Pot was not suppressed during SWE and could, thus, not account for the lower success rate of w-Pot. Instead, our results indicate that SWE rapidly enhances synaptic response to the spared whisker and occludes w-Pot (FWE: 123.5% ± 5.9%, n = 7; SWE: 97.6% ± 2.1%, n = 7; p = 0.001) (Figures 2D–2F), suggesting that LTP-like mechanisms have taken place in L2/3 before SWE2 and validating RWS as a tool for addressing in vivo the potentiation state of synapses activated by the spared whisker.
Figure 4.

Cross-linking GluA2 Subunit Suppresses the Expression of w-Pot in FWE Mice
(A) Time course of averaged PSP peak amplitude upon RWS (RWS, left) and when RWS is not induced (no RWS, right) in anti-GFP-injected mice.
(B) Mean (± SEM) peak amplitude before (baseline) and after RWS (n = 9 cells) or no RWS (n = 8 cells). Black lines between bars indicate pairs. ns, not significant.
(C and D) Same as in (A) and (B) but for anti-GluA2-IgG-injected mice (RWS, n = 8 cells; no RWS, n = 8 cells).
(E) Mean (± SEM) peak amplitude normalized to baseline (percentage of LTP). Triangles, individual cells.
(F) Correlation between normalized plateau strength and the level of RWS-induced w-Pot in anti-GFP- (turquoise) and anti-GluA2 (khaki) IgG-injected mice.
Cross-linking AMPAR Blocks the Expression of w-Pot in FWE Mice but Preserves Basal Synaptic Transmission
Blocking the induction of LTP with NMDAR antagonists provided the most direct evidence that synaptic plasticity at appropriate synapses is required for both potentiation of spared-whisker responses and learning (Clem et al., 2008; Rema et al., 1998; Takeuchi et al., 2013). However, NMDAR antagonists might obstruct normal sensory cortical transmission in vivo, which also relies on NMDAR conductances (Armstrong-James et al., 1993; Salt, 1986). Instead, we used an antibody cross-linking approach that has been shown to limit the surface diffusion of postsynaptic GluA2 (Constals et al., 2015; Giannone et al., 2010; Haas et al., 2018; Nair et al., 2013; Penn et al., 2017) and, thus, block the expression of synaptic plasticity while sparing NMDAR conductances (Humeau and Choquet, 2019; Penn et al., 2017). We targeted this subunit of AMPAR, as it is predominantly expressed in the neocortex (Schwenk et al., 2014), its expression in S1 is dynamically regulated upon partial sensory deafferentation (Gierdalski et al., 1999; He et al., 2004), and its immobilization has been shown to block LTP in acute slices and in vivo (Penn et al., 2017).
To investigate the role of AMPAR surface diffusion in vivo during w-Pot, we identified the C2 barrel by using intrinsic optical imaging, injected the immunoglobulins G (IgGs) against GluA2 (or anti-GFP for control) in L2/3 of the right C2 cortical column, and performed whole-cell recordings in FWE mice (Figures 3A and 3B; Figure S2). We used the non-pathogenic and highly specific 15F1 clone of anti-GluA2 IgG that does not modify the biophysical properties of GluA2-containing AMPAR GluA2 in vitro and in vivo (Constals et al., 2015; Giannone et al., 2010; Haas et al., 2018; Nair et al., 2013; Penn et al., 2017; Zhao et al., 2019). The injections, which were performed blind to the experimental conditions, resulted in diffusion of IgG across multiple columns but remained restricted to L2/3 with no visible signal in L4 (Figure 3A; Figures S2A and S2B).
Figure 3.

Cross-linking GluA2 Does Not Affect Short- and Long-Latency Whisker Subthreshold Responses
(A) Left: IgG is stereotaxically injected in FWE mice, in the superficial layers of the PW column identified by intrinsic optical imaging. Right: DAB immunostaining. White line, maximum spread of IgG. Scale bars, 100 μm.
(B) Schematic of the excitatory/inhibitory feed-forward circuit in a barrel-related column. L2/3 pyramidal neurons are recorded in the PW barrel-related column after IgG injection. Depth of recorded cells is indicated.
(C) Average (± SEM) number of action potentials (APs) triggered by incremental current injections. Insert: example of spiking pattern in anti-GFP- and anti-GluA2-injected mice upon 400-pA current injection.
(D) Single-cell example of PW-evoked PSP. Individual trials are represented with gray lines. Square pulse line, whisker deflections (100 ms).
(E) Median (± interquartile range) PSP peak amplitude, integral, onset, and onset jitter (anti-GFP, n = 34 cells; anti-GluA2, n = 31 cells).
(F) Example of NMDAR plateau strength extraction.
(G) Top: grand average of PW-evoked extracted plateau potential (all recorded cells averaged ± SEM). Black square pulse line, C2 whisker deflection (100 ms). Bottom: median (± interquartile range) onset of plateau potentials.
(H) Median (± interquartile range) plateau probability, strength (mV∗s), and full width at half maximum (FWHM, in milliseconds) in anti-GFP-injected (n = 26 cells) and anti-GluA2-injected (n = 24 cells) mice.
First, to evaluate whether IgGs have an effect on basic cell and S1 circuit electrophysiological properties, we recorded the firing properties (Figure 3C) and whisker-evoked subthreshold responses (Figures 3D–3H). No statistical differences in these parameters were detected between anti-GluA2 and anti-GFP IgGs in FWE mice. Specifically, we found no difference in the peak, onset, and jitter of short-latency PSP (Figures 3D and 3E), indicating that cortico-cortical (L4-to-L2/3 and L2/3-to-L2/3) basal synaptic transmission was not affected by anti-GluA2 IgG. Likewise, anti-GluA2 IgG did not significantly change long-latency plateau potentials evoked by single-whisker stimulation as compared to control IgG (plateau probability: anti-GFP, 0.70 ± 0.04, n = 26; anti-GluA2, 0.68 ± 0.05, n = 24; p = 0.770; plateau strength: anti-GFP, 0.71 ± 0.09 mV∗s, n = 26; anti-GluA2, 0.77 ± 0.11 mV∗s, n = 24; p = 0.828) (Figures 3F–3H). These delayed plateau potentials, which were extracted as previously described (Gambino et al., 2014), are generated through the cooperativity of lemniscal and paralemniscal circuits (see Figure 2B), suggesting that neither direct thalamo-cortical synaptic transmission nor recurrent activity was affected by the cortical infusion of IgG. Taken together, our results confirm previous studies performed in different systems (Constals et al., 2015; Giannone et al., 2010; Haas et al., 2018; Nair et al., 2013; Penn et al., 2017) and demonstrate that the cortical injection of anti-GluA2 IgG in S1 does not affect intrinsic neuronal excitability or AMPAR- and NMDAR-mediated whisker-evoked synaptic transmission in basal conditions. In addition, IgG did not modify spontaneous slow oscillations (i.e., the up and down states) of membrane potential (Figure S2C) or the whisker-evoked responses recorded at different holding membrane potentials (Figures S2D–S2F), suggesting that the net inhibitory drive onto L2/3 pyramidal neurons was also likely not affected (Haider et al., 2006; Sanchez-Vives and McCormick, 2000; Steriade et al., 1993).
RWS induced a significant potentiation of PW-evoked PSP in the presence of anti-GFP IgG, similar to that observed in control non-injected FWE mice (baseline: 8 ± 1.9 mV; RWS: 9.7 ± 2 mV; n = 9; p = 0.002) (Figures 4A and 4B; Figure S3). On average, the change in PSP amplitude when RWS was applied (RWS: 123.9% ± 1.7%; n = 9) was significantly higher than when RWS was not (no RWS: 101.6% ± 0.71%; n = 8; p < 0.001) (Figure 4B) and positively correlated with the strength of plateau potentials (Figure 4F). In contrast, GluA2 cross-linking prevented w-Pot (baseline: 10.6 ± 1.2 mV; RWS: 11.3 ± 1.3 mV; n = 8; p = 0.102; RWS+ versus RWS−: 107.1% ± 3.6% versus 97.5% ± 3.1%; p > 0.05) (Figures 4C–4E). Compared to control IgG, NMDAR-dependent plateau potentials (see Figures 3F–3H) and short-term dynamics of membrane potential during RWS (Figure S4) were not affected by anti-GluA2 IgG. On the contrary, S1 pyramidal neurons bearing high plateau strength could not be potentiated in the presence of anti-GluA2 IgG (plateau strength > 0.5; anti-GFP: 138.6% ± 5%, n = 4; anti-GluA2: 108% ± 4%; n = 7; p < 0.001) (Figure 4F; Figures S3A–S3C). This indicates that cross-linking surface GluA2 in vivo prevents the expression of w-Pot but not the NMDAR-dependent plateau potentials responsible for the induction of w-Pot.
w-Pot Mediates Neuronal Enhancement during SWE-Induced Cortical Remapping
Our data indicate that, as opposed to the pharmacological blockade of NMDAR that affects cortical transmission in vivo (Armstrong-James et al., 1993; Salt, 1986), cross-linking GluA2 subunits might represent a more specific way to prevent the expression of synaptic potentiation in vivo without modifying its induction mechanisms or basal synaptic transmission (Humeau and Choquet, 2019). Thus, we used this approach to question whether w-Pot was causally inducing the potentiation of whisker-evoked response during SWE by performing a “de-occlusion” experiment (Figure 5). Indeed, we reasoned that, if w-Pot increases synaptic responses between trimming and SWE2, then the chronic cross-linking of GluA2 subunits during this period would block this mechanism, thereby allowing RWS to potentiate whisker-evoked PSP in SWE mice when GluA2 mobility is restored.
Figure 5.

w-Pot Mediates Neuronal Responses Potentiation during SWE-Induced Cortical Remapping
(A) Schematic of experimental strategy. IgG is injected during SWE, followed by washout before recordings.
(B) Left: single-cell examples of whisker-evoked responses (gray, single traces; green and purple, averaged traces from anti-GFP- and anti-GluA2-injected mice, respectively). Square pulse line, C2 whisker deflection (100 ms). Right: fraction of spiking neurons triggered by whisker deflection.
(C) PW-evoked PSP grand average (all recorded cells averaged ± SEM). Square pulse line, C2 whisker deflection (100 ms).
(D) Left: number of spikes per whisker deflection (spiking probability; median ± interquartile range). Right: median (± interquartile range) PSP peak amplitude. FWE, n = 20 cells; SWE, n = 13 cells; GFP, n = 8 cells; GluA2, n = 9 cells. ns, not significant.
(E) Time course of averaged PSP peak amplitude upon RWS in anti-GFP-injected (green) and anti-GluA2-injected (purple) mice (after washout).
(F) Left: mean (± SEM) PSP peak amplitude before (baseline) and after RWS. Black lines between bars indicate pairs. Right: mean (± SEM) amplitude normalized to baseline. Triangles, individual cells. FWE, n = 7 cells; SWE, n = 7 cells; GFP, n = 6 cells; GluA2, n = 6 cells.
To test these predictions, anti-GluA2 IgG (or anti-GFP for controls) was injected in S1 twice a day for 2 consecutive days during SWE when LTP-like mechanisms are supposed to occur to saturate whisker-evoked neuronal responses. L2/3 pyramidal neurons were then recorded, and the potentiation state of their whisker-activated synapses was tested after a 12-h clearance period to wash out IgG (SWE2-wash; Figure 5A). This washout period was determined to remove most of IgG while preventing cortical remapping from resuming. The average number of spikes per PW deflection decreased in SWE2-wash/GluA2 as compared to SWE2-wash/GFP (anti-GluA2: 0.08 ± 0.03, n = 9; anti-GFP: 0.35 ± 0.14, n = 8; p = 0.026) (Figures 5B–5D), although the fraction of spiking neurons was not modified (SWE2-wash/GluA2: 6/9; SWE: 11/13; p > 0.05) (Figure 5B). The average PW-evoked PSP peak amplitude after the washout of anti-GluA2 IgG (7 ± 1.3 mV, n = 9, p < 0.001), but not anti-GFP IgG (14.2 ± 1.6 mV, n = 8, p = 0.126), was significantly decreased as compared to SWE (17.9 ± 2.3 mV, n = 13) and were now similar to those observed in naive FWE mice (Figures 5C and 5D). RWS potentiated PW-evoked PSP only when anti-GluA2 IgG, but not anti-GFP IgG, was washed out (SWE2-wash/GluA2: baseline, 7.6 ± 1.7 mV; RWS, 10.4 ± 2.6 mV; n = 6; p = 0.04; SWE2-wash/GFP: baseline, 12.6 ± 1.6 mV; RWS, 12.2 ± 1.9 mV; n = 6; p = 0.436) (Figures 5E and 5F). Thus, SWE2-wash with anti-GluA2 IgG preserved the expression of w-Pot in SWE mice (SWE2-wash/GluA2: 129.8% ± 7.8%, n = 6; versus SWE: 97.6% ± 2.1%, n = 7; p = 0.001) to levels similar to those in FWE mice (123.5% ± 5.9%, n = 7; p = 0.4) (Figure 5F; Figure S3). This indicates that chronically blocking AMPAR trafficking until SWE2 prevents sensory-evoked synaptic potentiation from occurring during this period.
Importantly, intrinsic excitability (Figure S3D), whisker-evoked short-latency PSP onset and jitter (Figure S3F), whisker-evoked long-latency NMDAR-plateau potentials (Figure S3B), and RWS-mediated depolarization (Figure S4) were not altered in any of our experimental conditions, suggesting that the rules governing w-Pot were likely not modified. In addition, there were no differences in basal synaptic transmission between SWE and SWE2-wash in the presence of the control anti-GFP IgG (Figure 5D; Figure S3). Taken together, our data suggest that the chronic presence of the 15F1 anti-GluA2 IgG did not alter AMPAR function as previously reported with human-disease-related anti-GluA2 auto-antibodies (Haselmann et al., 2018; Peng et al., 2015).
To exclude the possibility that chronic exposure to AMPAR cross-linking antibodies caused excessive inflammation that could have affected cell and circuit properties, we performed immunohistochemical detection of astrocyte and microglia cell markers (Figure S5). We injected anti-GluA2 and anti-GFP IgGs in the right and left hemispheres, respectively, twice a day for 2 consecutive days and assessed the number of astrocytes and microglia using anti-GFAP (glial fibrillary acidic protein) and anti-Iba1 immunostaining as previously described (Holtmaat et al., 2009) (Figure S5). While cortical injections produced inflammation in the superficial layers of the injected area as compared to the non-injected area (Figure S5), we found no quantitative differences between anti-GFP- and anti-GluA2-treated groups, as the numbers of immunopositive cells were similar between the two hemispheres, indicating that the astrocytic and microglial responses post-injection were not exacerbated in the presence of anti-GluA2 IgG compared to anti-GFP IgG. In addition, the global locomotor activity (Figures S6H–S6K) and whisker-related perception (Figures S6D–S6G) appeared unaffected in mice chronically injected with IgG in S1, suggesting that the prolonged cross-linking of AMPAR has no global cytotoxic effect.
w-Pot Facilitates the Recovery of Altered Whisker-Dependent Behaviors during the Early Phases of SWE
We demonstrated earlier that the chronic cross-linking of AMPAR prevented potentiation of whisker-evoked responses during SWE, supporting the idea that w-Pot contributes to SWE-induced cortical remapping. Because SWE alters various whisker-mediated behavioral tasks (Barnéoud et al., 1991; Celikel and Sakmann, 2007; Clem et al., 2008; Xerri, 2012), we reasoned that, if cortical remapping improves tactile perception, then blocking w-Pot during SWE should affect whisker-mediated behavioral performance.
To test this hypothesis, we monitored freely behaving mice performing a binary gap-crossing task under infrared light (Figure 6A; Figure S6A). Food-restricted mice were trained to reach a platform containing a pellet distributor delivering a calibrated food reward. The home and rewarding platforms were separated by a randomized distance between 40 and 65 mm from the home platform (Figure 6A). At a distance of 65 mm, mice used preferentially their whiskers to locate the target platform and jump onto it to receive the reward (Barnéoud et al., 1991; Celikel and Sakmann, 2007) (Figure S6B). To test the effect of cortical remapping on acquired skills while minimizing the confounding effect of learning, whiskers were trimmed after mice learned the task and attained expertise (i.e., after 4 days of training) (Figure 6B; Figure S6C). Gap-crossing performance at a distance of 65 mm dropped immediately after SWE by 28% ± 14% (fraction of success; session 4: 0.96 ± 0.04; session 5: 0.68 ± 13; n = 6; p = 0.006) but recovered quickly before SWE2 (session 6: 0.87 ± 0.09; p = 0.372) (Figure 6C). Importantly, mice that were not tested during the first sessions of SWE (sessions 5–7) had similar final success rates (session 8; 0.95 ± 0.03, n = 6; and 0.89 ± 0.08, n = 5; p > 0.05) (Figures 6C and 6D), suggesting that behavioral recovery was likely not caused by a new learning phase but, instead, resulted from the restoration of accurate perception. Interestingly, it occurred in a timescale (SWE2–SWE3) at which whisker-evoked responses saturated (Figure 1) and w-Pot has been fully occluded (Figure 2). This concurrent timing raises the possibility that LTP-like mechanisms might participate in the recovery of altered gap-crossing skills upon SWE (see Figure 6B).
Figure 6.

Cross-linking AMPAR during SWE Alters the Recovery of Gap-Crossing Skills
(A) Overview of the gap-crossing task.
(B) Schematic of the experimental strategy. Mice learn the task before SWE is induced, during which IgG is injected. The supposed effects of SWE on whisker-evoked PSP in the presence of IgG are indicated below.
(C) Top: averaged (± SEM) fraction of gap-crossing success for different gap distances in non-injected mice. Bottom: tests in sessions 5 to 7 were omitted to assess the role of learning during SWE. ns, not significant.
(D) Mean (± SEM) fraction of success in the final session (normalized to session 4 before SWE) at 65 mm for mice that are tested every day (test) and for mice that were not tested in sessions 5 to 7 (no test). Triangles, individual mice (test, n = 6; no test, n = 5).
(E) Averaged (± SEM) fraction of gap-crossing success for different gap distances in anti-GFP-injected (left) and anti-GluA2-injected (right) mice.
(F) Averaged (± SEM) fraction of gap-crossing success at 65 mm in non-injected (orange), anti-GFP-injected (green), and anti-GluA2-injected (purple) mice.
(G) Mean (± SEM) fraction of success at 65 mm after expertise in FWE mice and during SWE. Triangles, individual mice (anti-GFP, n = 7; anti-GluA2, n = 7).
To test this hypothesis, we trimmed whiskers in expert mice and injected in S1 anti-GFP or anti-GluA2 IgG twice per day, before and after each behavioral session (Figure 6B). None of the IgGs, which were injected blind after whisker trimming (sessions 5–8), altered exploration and decision latency (Figure S6). In contrast, the gap-crossing performance of anti-GluA2-IgG-injected mice decreased more (session 5: −61.8% ± 12% versus −22.6% ± 6.7%, p = 0.016) and recovered significantly more slowly compared to that of anti-GFP-injected mice (Figures 6E–6G). Success rates were, however, similar between both groups 3 days after SWE (session 7: 0.82 ± 0.09 versus 0.72 ± 0.14; p = 0.805) (Figure 6G), which might reflect the existence of additional synaptic mechanisms that preserve a slow capacity for cortical remapping (Clem et al., 2008) and/or barrel-cortex-independent behavioral strategies (Celikel and Sakmann, 2007; Hong et al., 2018). Altogether, our data indicate that cross-linking GluA2 subunits similarly affects neuronal response potentiation in vivo and behavioral output at early phases of SWE (i.e., before SWE3), thereby providing additional evidence for a critical role of w-Pot in S1 in facilitating the recovery of lost perceptual skills.
Discussion
Our study provides evidence that the rules governing synaptic plasticity in vitro through the synaptic regulation of AMPAR subunits (Granger et al., 2013; Penn et al., 2017; Shi et al., 2001) are effectively used in vivo and mediate the potentiation of spared inputs during cortical remapping. The surface diffusion and insertion of AMPAR into the postsynaptic membrane has become a well-recognized hallmark of NMDAR-dependent synaptic potentiation in vitro (Diering and Huganir, 2018; Humeau and Choquet, 2019), although a presynaptically expressed form of neocortical LTP altering release probability might also exist (Markram and Tsodyks, 1996). In the barrel cortex, while LTP, induced in vitro at L4-to-L2/3 synapses by electrical stimulation, exhibits both postsynaptic and presynaptic components (Bender et al., 2006; Feldman, 2009; Hardingham and Fox, 2006), the molecular mechanisms of w-Pot remained elusive. Here, we found that anti-GluA2 IgG did not alter whisker-evoked short-term synaptic plasticity (Figure S4). In contrast, w-Pot could not readily be produced in S1 pyramidal neurons when RWS was applied in the presence of anti-GluA2 IgG (Figure 4). This form of synaptic plasticity has been shown to depend on postsynaptic NMDAR-dependent mechanisms (Gambino and Holtmaat, 2012; Gambino et al., 2014). This is in agreement with earlier studies showing that RWS triggers the NMDAR-dependent increase of AMPAR at dendritic spines in the barrel cortex in vivo (Zhang et al., 2015) and suggests that w-Pot is most likely expressed postsynaptically in adult animals.
Together, our data indicate that cross-linking GluA2-containing AMPAR with the 15F1 anti-GluA2 IgG is sufficient to inhibit the expression of w-Pot while leaving its induction mechanism unaltered (i.e., the NMDAR-dependent plateau potentials; Figure 3; Figure S3). This specific and non-pathogenic anti-GluA2 IgG clone binds to the N-terminal domain of the GluA2 subunit, away from any functionally important domains such as the glutamate binding site (Zhao et al., 2019). Accordingly, we found that spontaneous slow-wave fluctuation of membrane potential (Figure S3) and whisker-evoked, short- and long-latency PSPs (Figure 3; Figure S3) were similar between anti-GFP- and anti-GluA2-injected mice. This supports the fact that the 15F1 antibody is not an AMPAR loss-of-function IgG and does not modify the biophysical properties of GluA2-containing AMPAR (Constals et al., 2015; Giannone et al., 2010; Haas et al., 2018; Nair et al., 2013; Penn et al., 2017). Rather, it impairs the surface diffusion of GluA2-containing AMPAR without modifying any other trafficking pathways such as endocytosis (Penn et al., 2017). This supports the possibility that, in S1 too, 15F1 anti-GluA2 IgG prevents AMPAR synaptic recruitment during w-Pot. Nevertheless, anti-GluA2 IgG can bind to all AMPARs around the injection area, including those located at synapses onto and from interneurons. While the net inhibitory drive onto L2/3 pyramidal neurons does not seem to be altered (Figure S2), we cannot exclude the possibility that IgG affected the plasticity of inhibitory or dis-inhibitory circuits. Nonetheless, RWS LTP does not appear to be caused by plasticity of interneurons (Williams and Holtmaat, 2019), suggesting that IgG at these sites only had a marginal functional impact on L2/3 pyramidal neuron plasticity.
We exploited this result to describe the relationship between synaptic plasticity and cortical remapping following SWE. In young rodents, SWE enhances synaptic communication between L4-to-L2/3 and L2/3-to-L2/3 neurons in vitro (Cheetham et al., 2007; Clem and Barth, 2006; Clem et al., 2008; Finnerty et al., 1999). In addition, 24 h of SWE has been shown to occlude electrically induced, NMDAR-dependent LTP in acute slices obtained from ∼P (post-natal day)15 mice (Clem et al., 2008), suggesting that synaptic plasticity might occur rapidly upon changes in sensory experience (Takeuchi et al., 2013). However, whether and when LTP happens in vivo in adult mice during SWE remain poorly understood. Previous results indicated that 7 days of SWE were necessary to increase whisker-evoked firing in L2/3 pyramidal neurons of 5- to 8-week-old mice, suggesting that potentiation of L2/3 pyramidal neurons might, in fact, require a longer period of SWE to occur in adult animals (Benedetti et al., 2009). In contrast, we found here that sensory inputs saturated within 2 days of SWE (Figure 1) and could not be further potentiated by RWS, indicating that 2 days of SWE is sufficient to occlude RWS-mediated LTP (Figure 2). This demonstrates that synaptic plasticity induced in vivo by behaviorally relevant stimuli occurs rapidly in the adult S1 after whisker trimming.
While LTP-like synaptic plasticity might rapidly drive the spared-whisker representation in S1 to strengthen (Figure 1B), other mechanisms, such as homeostatic disinhibition, synaptic scaling, or regulation of intrinsic excitability, might also be engaged upon sensory deprivation (Gainey and Feldman, 2017). It thus remained unclear whether synaptic plasticity is causally inducing the potentiation of spared-whisker-evoked response following SWE. To address this question, we cross-linked AMPAR for 2 days after whisker trimming and tested the effect of anti-GFP and anti-GluA2 IgGs on w-Pot and spared-whisker-evoked neuronal responses 12 h after the last IgG injection (SWE2-wash; Figure 5A). Although we cannot exclude that residual IgGs are still present after 12 h, our “de-occlusion” protocol was designed to prevent cortical remapping from blocking w-Pot after IgG removal, which would have occurred with longer washout periods. In contrast, we found that, as compared to anti-GFP, SWE-wash/GluA2 reduces the short-latency, AMPAR component of whisker-evoked PSP to levels similar to those in naive FWE mice (Figures 5B–5D) and restores the capacity to undergo w-Pot (Figures 5E and 5F), suggesting that most antibodies have been washed out. Importantly, it indicates that cross-linking the endogenous GluA2 subunit of AMPAR immediately after whisker trimming prevents the potentiation of spared-whisker-evoked PSP that would have otherwise occurred during SWE.
Overall, our data suggest that w-Pot, through the tight modulation of GluA2-containing AMPARs, is one of the major mechanisms that rapidly potentiates neuronal response during cortical remapping induced by SWE. SWE is associated with modest alterations of L2/3 pyramidal neuron excitability and spared-whisker-evoked inhibition (Figure S1), and whether it could drive synaptic scaling is currently not known. The effect of SWE on spared-whisker-evoked neuronal responses could also be sculpted by the regulation of local inhibitory interneurons. For example, complete removal of sensory inputs increases the pruning of inhibitory synapses (Keck et al., 2011). When an entire whisker row is plucked, feed-forward inhibition in S1 L2/3 pyramidal neurons is decreased (House et al., 2011; Jiao et al., 2006). Disinhibition occurs in the deprived columns to transiently increase deprived-whisker neuronal spiking before LTD-like processes take place (Li et al., 2014). However, this homeostatic mechanism has been observed in the deprived columns after 3 days of deprivation (Gainey and Feldman, 2017; Li et al., 2014), and it is currently not certain whether they might also occur in the spared columns. Nevertheless, it remains possible, due to the broad receptive field of interneurons (Swadlow and Gusev, 2002), that disinhibition within the deprived barrel further affects nearby non-deprived columns. Such disinhibition could leverage spared-whisker-evoked excitation and facilitate neuronal spiking but also might open the gate for synaptic plasticity (Williams and Holtmaat, 2019).
We verified that our results were not caused by the alteration of receptor function and basal transmission, which might occur in the prolonged presence of certain types of anti-AMPAR antibodies. Indeed, human pathogenic anti-GluA2 autoantibodies induce receptor internalization (Haselmann et al., 2018; Peng et al., 2015). In addition to our controls of basal synaptic transmission (Figure 3; Figure S3), we measured and compared the astrocytic/microglia response, NMDARs-dependent plateau potentials, neuronal excitability, and RWS-mediated depolarization, as well as the global motor activity in the acute and prolonged presence of anti-GluA2 and anti-GFP IgGs. None of these parameters were different between conditions, indicating that basic cellular, circuit, and behavioral properties as well as the rules governing RWS-mediated synaptic plasticity were likely preserved during SWE-wash. However, although the onset and jitter of short-latency whisker-evoked PSP were not different between anti-GluA2- and anti-GFP-injected mice (Figure S3), we cannot exclude the possibility that SWE-wash/GluA2 drives subunit switch by increasing the synaptic incorporation of inwardly rectifying, non-GluA2-containing AMPARs (Haselmann et al., 2018). Given that the non-pathogenic 15F1 antibodies do not induce receptor internalization but only alter GluA2 surface diffusion (Penn et al., 2017), these changes are unlikely to exist in S1. Nonetheless, if they were to occur, our overall conclusion would still remain valid, as these changes have been shown to impair synaptic LTP and behavior as well (Haselmann et al., 2018).
Our results are in line with the dynamic regulation of GluA2 expression in S1 upon partial sensory deafferentation (Gierdalski et al., 1999; He et al., 2004). However, they stand in contrast to the traditional view of AMPAR trafficking during cortical remapping, which states that the GluA2 subunit is not required for experience-dependent plasticity in the barrel cortex (Feldman, 2009; Makino and Malinow, 2011). Consistent with this model, previous in vitro studies in the barrel cortex of ∼P15 animals concluded that SWE drives LTP at L4–L2/3 synapses by inserting homomeric GluA1/GluA1 (GluA2-lacking) AMPARs (Clem and Barth, 2006; Clem et al., 2008; Miyazaki et al., 2012; Takahashi et al., 2003). Nevertheless, the GluA1 dependence of cortical and hippocampal plasticity has been shown to be developmentally regulated and decreases with age (Grosshans et al., 2002; Jensen et al., 2003). In addition, neuronal response potentiation is only partially blocked in older GluA1 knockout animals undergoing SWE (Hardingham and Fox, 2006). When GluA1 is overexpressed together with GluA2, RWS drives the incorporation of GluA1 subunit (Zhang et al., 2015), supporting the hypothesis that heteromic GluA2/GluA1 rather than homomeric GluA1/GluA1-dependent LTP likely operates during neuronal response potentiation in adult animals.
Although our results demonstrate a tight relation between w-Pot and cortical remapping, how such local synaptic changes contribute to overall behavioral adaptation upon partial sensory deafferentation remains unknown. A first attempt in addressing this issue comes from pharmacological experiments where NMDAR antagonists were used during SWE, assuming that they were selective for plasticity and not normal information processing in the brain (Clem et al., 2008; Rema et al., 1998). However, NMDAR antagonists have strong attenuating effects on long-latency spikes in supragranular layers as well as in granule cell layers (Armstrong-James et al., 1993; Salt, 1986). Instead, blocking AMPAR trafficking with cross-linking antibodies has great potential to address this question in vivo (Humeau and Choquet, 2019). The content of AMPARs at synapses in vivo has be shown to correlate with motor performance (Roth et al., 2020), and blocking AMPAR trafficking impairs learning (Penn et al., 2017). Here, by manipulating the mobility of GluA2, we found that chronic cross-linking during SWE significantly alters gap-crossing performance immediately after whisker trimming and slows down behavioral recovery after SWE (Figures 6E–6G), while it did not alter sensory processing (Figure S6). Strikingly, success rates were not different between anti-GFP- and anti-GluA2-injected mice 3 days after SWE, when whisker-evoked PSPs are saturated and w-Pot fully occluded in non-injected mice (Figure 6B). This suggests that w-Pot is associated with a fast adaptation of cortical processing and related behaviors to the change of perceptual condition. While this might rapidly take place in S1 between trimming and SWE2, other adaptation mechanisms, which are not impaired by AMPAR cross-linking, likely occur after SWE2. These mechanisms might include mGluR-mediated synaptic plasticity (Clem et al., 2008) and/or homeostatic processes (Gainey and Feldman, 2017). Interestingly, disinhibition has been proposed to briefly stabilize the deprived column in response to the loss of sensory inputs (Li et al., 2014). While these mechanisms could facilitate behavioral compensation—for example, by increasing sensory gain in the spared column or facilitating GluA2-independent form of plasticity—whether they occur in parallel and/or in response to the absence of LTP-like mechanisms remains to be elucidated.
Taken together, our results indicate that AMPAR trafficking-mediated synaptic plasticity initiates cortical remapping and occurs nearly immediately following partial sensory deafferentation, thereby providing important processing resources for spared inputs that compensate for the loss of surroundings inputs. In support of this hypothesis, training-related increases in cortical representations correlate with perceptual learning (Bieszczad and Weinberger, 2010; Molina-Luna et al., 2008; Reed et al., 2011), suggesting that sensory deafferentation could cause behavioral gains by promoting cortical remapping.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GluA2, mouse monoclonal IgG1κ | Gift by Eric Gouaux, PhD | Clone 15F1 |
| GFP, mouse monoclocnal IgG1κ | Roche | Cat#: 11814460001; RRID:AB_390913 |
| Mouse specific HRP/DAB (ABC) Detection IHC Kit | Abcam | Cat#: ab64259; RRID:AB_2818952 |
| GFAP, chicken polyclonal | USBiological Life Sciences | Cat#: G2032-25F; RRID:AB_1616563 |
| IBa1, rabbit polyclonal | WAKO | Cat#: 019-19741; RRID:AB_839504 |
| Anti-Chicken, Goat Alexa 647 | Thermofisher | Cat#: A32933; RRID:AB_2762845 |
| Anti-Rabbit, Goat Alexa 647 | Thermofisher | Cat#: A27040; RRID:AB_2536101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dustless Precision Pellets (20 mg), Rodent Grain-Based Diet | Bio-Serv | Cat#: F0163 |
| Norland Optical Adhesive 61 | Thorlabs | Cat#: NOA61 |
| https://www.thorlabs.com/thorproduct.cfm?partnumber=NOA61 | ||
| ABC Peroxidase Staining Kit | Thermofischer | Cat#: 32020 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6-Elite (SOPF) | Charles River | https://www.criver.com/products-services/find-model/c57bl6-elite-sopf-mouse?region=29 |
| Software and Algorithms | ||
| Clampfit 10 | Molecular Devices, LLC | Version 10.8.01 |
| NeuroMatic | http://www.neuromatic.thinkrandom.com | Version 2.001 |
| Igor | https://www.wavemetrics.com | Version 6.2.2.2 |
| Fiji | The NIH | https://imagej.net/Fiji |
| Poly Behavior Software | Imetronic | Version 4.5.2 |
| VDAQ | Optical Imaging, Inc. | Version 2.7.1 |
| NDP view | Hamamatsu | https://www.hamamatsu.com/eu/en/product/type/U12388-01/index.html |
| Python | Anaconda Sotfware Distribution | http://www.anaconda.com |
| MATLAB | MathWorks | https://www.mathworks.com |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frédéric Gambino (frederic.gambino@u-bordeaux.fr).
Materials Availability Statement
This study did not generate new unique reagents.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. This study did not generate any unique code.
Experimental Model and Subject Details
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council Committee (2011): Guide for the Care and Use of Laboratory Animals, 8th ed. Washington, DC: The National Academic Press.) and the European Communities Council Directive of September 22th 2010 (2010/63/EU, 74). Experimental protocols were approved by the institutional ethical committee guidelines for animal research (N°50DIR_15-A) and by the French Ministry of Research (agreement N°18892). We used male C57BL6/J 5- and 6-weeks old mice from Charles River that were housed with littermates (3 mice per cage) in a 12-h light-dark cycle. Cages were enriched with tunnels. Food and water were provided ad libitum, except during behavioral experiments (see below).
Method Details
Cranial window implantation for chronic Intrinsic Optical Imaging
Anesthesia was induced using isoflurane (4% containing ∼0.5 l/min O2) and then continued using an intraperitoneal (i.p.) injection of a MMB mixture (5 μl/g) composed of medetomidine (Sededorm, 0.2 mg/kg), midazolam (Dormicum, 5 mg/kg), and buprenorphine (Buprecare, 0.2 mg/kg) in sterile NaCl 0.9%. A heating-pad was positioned underneath the animal to keep the body temperature at 37°C. Eye dehydration was prevented by topical application of ophthalmic gel. Analgesia was achieved by local application of 100 μL of lidocaine (Lurocaine, 1%) and subcutaneous (s.c.) injection of buprenorphine (Buprecare, 0.05 mg/kg). To prevent risks of inflammation and brain swelling 40 μL of dexamethasone (Dexadreson, 0.1 mg/mL) were injected intramuscularly (i.m.) before the surgery. After disinfection of the skin (with modified ethanol 70% and betadine), the skull was exposed and a ∼5mm plastic chamber was attached to it above the relative stereotaxic location of the C2 barrel column (−1.5 mm from bregma, + 3.3 mm mideline) using a combination of super glue (Loctite) and dental cement (Jet Repair Acrylic, Lang Dental Manufacturing). The chamber was filled with saline (0.9% NaCl) and sealed with a glass coverslip.
Intrinsic Optical Imaging for barrel column targeting
To locate the cortical barrel column computing the whisker C2 (wC2), intrinsic optical signals were imaged as previously described, through the intact skull using a light guide system with a 700 nm (bandwidth of 20 nm) interference filter and stable 100-W halogen light source (Gambino and Holtmaat, 2012; Schubert et al., 2013). Briefly, the head of the animal was stabilized using a small stereotaxic frame and the body temperature kept constant with a heating pad. An image of the surface vascular pattern was taken using a green light (546 nm interference filter) at the end of each imaging session. Images were acquired using the Imager 3001F (Optical Imaging, Mountainside, NJ) equipped with a large spatial 602 × 804 array, fast readout, and low read noise charge-coupled device (CCD) camera. The size of the imaged area was adjusted by using a combination of two lenses with different focal distances (upper lens: Nikon 135 mm, f2.0; bottom lens: Nikon 50 mm, f1.2). The CCD camera was focused on a plane 300 μm below the skull surface. Images were recorded at 10 Hz for 5 s, with a spatial resolution of 4.65 μm/pixel comprising a total area of 2.9 × 3.7 mm2. wC2 was deflected back and forth (20 stimulations at 8 Hz for 1 s.) using a glass-capillary attached to a piezoelectric actuator (PL-140.11 bender controlled by an E-650 driver; Physik Instrumente) triggered by a pulse stimulator (Master-8, A.M.P.I.). Each trial consisted of a 1 s. of baseline period (frames 1-10), followed by a response period (frames 11-22) and a post-stimulus period (frames 23-50). Inter-trial intervals lasted 20 s to avoid contamination of the current intrinsic optical signal by prior stimulations. Intrinsic signals were computed by subtracting each individual frame of the response period by the average baseline signal. The obtained intrinsic signal was overlapped with the vasculature image using ImageJ software to precisely identify the cortical region computing wC2.
Craniotomy and cranial window implantation
After intrinsic optical imaging, adequate anesthesia was assessed (absence of toe pinch reflexes, corneal reflexes, and vibrissae movement) and prolonged using supplementary isoflurane if necessary. Dehydration was prevented by injecting sterile saline by s.c. injection. A 3 mm diameter craniotomy was then made over the maximum intrinsic optical signal using a pneumatic dental drill. The craniotomy was covered with sterile saline and sealed with a 3 mm glass coverslip. The coverslip was sealed to the skull using dental acrylic and dental cement (Jet Repair Acrylic, Lang Dental Manufacturing). Anesthesia was reverted by a sub-cutaneous injection of an AFB mixture (AFB) containing atipamezole (Revertor, 2.5 mg/kg), flumazenil (0.5 mg/kg), and buprenorphine (Buprecare, 0.1mg/kg) in sterile NaCl 0.9%. A delay of 2-3 weeks for surgery recovery was respected before all imaging experiments, during which the body weight of mice was daily checked.
Chronic Intrinsic Optical Imaging
MMB-anaesthetized mice were daily-imaged during 1 session with all their whiskers (baseline), followed by 2 sessions with all their whiskers trimmed but the wC2 (SWE 1-2). A cohort group was additionally recorded for 3 days with all their whiskers (FWE 1-3) as a control for barrel expansion. During each session, wC2 was deflected back and forth (20 stimulations at 8Hz for 1 s) and intrinsic optical signal recorded through a cranial window.
Spatiotemporal analysis of intrinsic optical signal
An average of 200 trials were recorded per session to quantify intrinsic optical signal as previously described (Schubert et al., 2013). The intrinsic optical signals of different sessions from the same animal were spatially aligned using the animal’s brain surface vasculature and spatially binned (6x6, final resolution: 27.9 μm/pixel or 3x3, final resolution: 13.95 μm/pixel). A high pass-filter was then applied by subtracting from each image-frame the same image-frame that was convolved using a 1270 μm full-width at half maximum (FWHM) Gaussian kernel. The whisker-evoked intrinsic optical signals were then simulated using a pixel-by-pixel paired t test, comparing the baseline period and the response period of all trials within a session. The t maps for each individual trial were low pass-filtered with a 340 μm FWHM Gaussian kernel and averaged into a final t map response. A threshold was set to t < −2.0 and any signal below this value was considered to belong to the stimulus-evoked response area. If the pixel value was t ≥ −2.0 it was considered background noise and discarded for quantification. This usually resulted in an image with a clear minimum, representing the response maximum and the barrel’s center of mass. Changes on intrinsic optical signal pixel area caused by whisker trimming were computed as the ratio between the whisker-evoked intrinsic response of the baseline and SWE sessions. All data analysis was performed using a custom software written in MATLAB (MathWorks).
In vivo whole-cell recordings
Acute AMPAR X-Linking Surgery. Anesthesia was induced using isoflurane (4% with 0.5 l/min O2) and then continue using i.p. injection of urethane (1.5 g/kg). Surgery preparation and intrinsic optical imaging were performed as aforementioned. After imaging, adequate anesthesia was assessed and prolonged by supplementary urethane (0.15 g/kg) if necessary. A small ∼1 × 1 mm craniotomy (centered above the C2 whisker maximum intrinsic optical response) was made using a pneumatic dental drill. The injections of either an anti-GluA2 antibody (clone 15F1, gift from E. Gouaux) or a monoclonal anti-GFP IgG1-K (Roche, 11814460001) were targeted to the L2/3 of S1 (−0.1 to 0.3 mm dorsoventral). A 30 nL solution containing antibody diluted in sterile saline (0.05 mg/mL) was injected at maximum rate of 15 nl/min, with 30 s intervals between injection sites as described before. All the experiments were performed blind for the antibody injected.
Chronic AMPAR X-Linking Surgery. Anesthesia was induced using isoflurane (4% containing ∼0.5 l/min O2) and continued using an i.p. injection of MB to perform intrinsic imaging targeting the wC2 cortical barrel. Adequate anesthesia was assessed and prolonged using isoflurane if necessary. Dehydration was also prevented by s.c. injection of sterile saline. A small ∼1 mm diameter craniotomy above the maximum intrinsic optical signal was made using a pneumatic dental drill. The dura was left intact and a stereotaxic injection of either anti-GluA2 or anti-GFP antibody was performed as mentioned above for acute injection. After stereotaxic injection, the craniotomy was covered with sterile saline and protected with a 3 mm polydimethylsiloxane (PDMS) coverslip. PDMS was attached to the skull using an ultraviolet (UV) curing optical adhesive (NOA61, Norland) cured with a 50 mW UV laser (3755B-150-ELL-PP, Oxxius). Before reverting anesthesia using AFB, all the whisker except C2 were trimmed (SWE 1). Antibodies were re-injected twice on the day after (SWE 2), with a 12h interval between injections using isoflurane anesthesia (4% for induction, then 2% for injection with ∼0.5 l/min O2). Stereotaxic injections were performed through the PDMS coverslip with the same injection protocol than before. After 12h of antibody washout (SWE 3), mice were finally anesthetized with isoflurane (4% with 0.5 l/min O2) and an i.p. injection of urethane (1.5 g/kg). Before the patch-clamp recordings, the PDMS cranial window was removed and the cortex protected with sterile saline. All the experiments were performed blind for the antibody injected.
Recordings
Whole-cell patch-clamp recordings of L2/3 pyramidal neurons were obtained as described previously (Gambino et al., 2014). Current-clamp recordings were made using a potassium-based internal solution in mM: 135 potassium gluconate, 4 KCl, 10 HEPES, 10 Na2-phosphocreatine, 4 Mg-ATP and 0.3 Na-GTP), pH adjusted to 7.25 with KOH, 285 mOsM). High positive pressure (200–300 mbar) was applied to the pipette (5–8 MΩ) to prevent tip occlusion. After passing the pia the positive pressure was immediately reduced to prevent cortical damage. The pipette was then advanced in 1-μm steps, and pipette resistance was monitored in the conventional voltage clamp configuration. When the pipette resistance suddenly increased, positive pressure was relieved to obtain a 3–5 GΩ seal. After break-in, membrane potential (Vm) was measured, and dialysis could occur for at least 5 min before deflecting the whisker.
Spiking pattern of patched cells was analyzed to identify pyramidal neurons. Action potentials were obtained by a step-increment of injected current. Spontaneous slow-have fluctuations of the resting membrane potentials were recorded as previously described (Petersen et al., 2003). PSPs were evoked by back and forth deflection of the whisker (100 ms, 0.133 Hz) and long-latency plateau potentials were extracted as previously described (Gambino et al., 2014). Briefly, for each whisker deflection, the relationship between the PSP half-peak amplitude and the average membrane potential between 50 and 100 ms after the onset reveals two distinct clusters of sensory-evoked PSP. Cluster 1 was defined by an index < 0, which consisted of short latency PSPs that quickly returned to the resting membrane potential. Cluster 2 was defined by an index > 0, which consisted of compound PSPs containing both short and long-latency components. The long-latency component of the PSPs in cluster 2 was obtained by subtracting the peak-scaled PSP average of cluster 1 from the PSP average of cluster 2. It was previously shown that these late components disappears when NMDAR conductances are blocked, and thus represent dendritic plateau potentials (Gambino et al., 2014).
The voltage applied to the actuator was set to evoke a displacement of 0.6 mm with a ramp of 7-8 ms of the wC2. Different frequencies of stimulation were used accordingly to the experiment (RWS-Pot: 8Hz, 1 min; cumulative PSPs: 8Hz, 2.5 s). Series and input resistance were monitored with a 100-ms long-lasting hyperpolarizing square pulse 400 ms before each single-deflection and extracted offline by using a double-exponential fit. Recordings were discarded if the change in these parameters were larger than 30%. The bridge was usually not balanced, and liquid junction potential not corrected. All the data were acquired using a Multiclamp 700B Amplifier (Molecular Devices) and digitized at 10 kHz (National Instruments) using software. Offline analysis was performed using routines with IGOR Pro (WaveMetrics).
Gap crossing apparatus
The custom-made gap crossing apparatus (Imetronic, France) consists of two individual moveable platforms: (1) a starting platform containing an automated door to precisely control the start of a trial; (2) a reward platform containing a pellet distributor to deliver a calibrated food reward. Both platforms (10x20 cm) were elevated 37.4 cm from the surface and surrounded on the three sides with a 20-cm-high Plexiglas walls. The two platforms were placed facing each other with an infra-red pad at the bottom. The edges of the platforms that face each other were made of a metal grid (10 × 10 cm) to allow a better grip where the animals should jump. A ruler placed at the bottom and between the platforms was used to precisely define the gap distances at a given trial. The apparatus was placed into a light- and soundproof cage containing ventilation, and surrounding speakers with a continuous white noise background. This ensures that mice do not have neither visual nor auditory cues regarding the reward platform. Food pellet odor was saturated inside the box to avoid any olfactory-related cues.
Behavioral protocol
At least 5 days before starting behavior, mice were food restricted and handled to decrease stress. After a 15 – 20% reduction of the initial body weight, habituation was performed during 3 days: at day 1 (maze habituation), mice were placed on the apparatus with a gap distance of 0 mm for 10 min where the pellet distributor was randomly presented for multiple times without food reward. At day 2 (reward habituation), mice were placed on the start platform and trained for 3 blocks (16 trials each block, gap distance = 0 mm) to the distribution of a food pellet in the reward platform. A given trial was defined as success if the animal reached the reward platform and ate the food pellet or as a failure if it took more than 2 min to do so. At the end of a trial, the animal was placed back in the starting platform to beginning the next one. At day 3 (jump habituation), the same protocol than day 2 was used but with a gap distance of 30 mm to habituate the animal for a distance between platforms. Habituation is considered successful if the success rate was > 95% at day 3, and the test sessions started. The test protocol had 1 session per day during 4 days where each session was composed of 16 trials containing gap distances of 40, 50, 60, and 65 mm. Individual blocks started with the minimal gap distance, had random gap distance sequences, and finished with a catch trial (gap distance of 100 mm) where the reward platform was removed. This allowed to rule out habit to jump. When addressing the effect of whisker trimming on expert mice, test sessions were performed before and after whisker trimming.
Cannula implantation for chronic AMPAR X-linking
Anesthesia was induced using isoflurane and continued by an i.p. injection of MMB to perform intrinsic optical imaging targeting of the wC2 cortical barrel as aforementioned. A small craniotomy above the maximum intrinsic optical signal was made using a pneumatic dental drill, preventing any cortical damage. After drilling, a guide cannula (62001, RWD Life Science Co., LTD) was stereotaxically inserted in the brain using a cannula holder through the craniotomy previously made. The size of the cannula (0.6 mm) was adjusted to target L1 of the somatosensory cortex. The guide cannula was fixed to the skull using two stainless steel screws and a mix of super glue (Loctite), dental acrylic and dental cement. Anesthesia was reverted by a s.c. injection of AFB and mice left to recover over 2 weeks before starting food restriction. During food restriction, mice were additionally habituated to be restrained by a different experimenter to avoid stress during antibody injection. Mice were tested during 4 sessions with FWE followed by 4 SWE sessions, during which either an anti-GluA2 or an anti-GFP antibody (0.05 mg/mL) was injected. Antibodies were injected twice per day, before and after each test session, using a pump (D404, RWD Life Science CO.) with an injection speed of 6 nL/min for the first 120 nL and 3 nL/min for the remaining 30 nL of antibody. Mice were freely moving in their home cage during injection. All experiments and analysis were performed blind for the antibody injected.
Histology
To evaluate the antibody injection profiles in S1, animals were intracardially perfused with PBS (1%) and PFA (4%). Fixed brains were sliced with a vibratome and sections incubated with PBS.H202 (0.3%) during 30 min to block endogenous peroxide. Brain slices were then incubated with a secondary anti-mouse biotinylated antibody from donkey (1/200), during 2h at room temperature (RT). To finally reveal the injected primary antibody, slices were first incubated with an avidin-biotin complex (1/200 in PBS (1x) – Triton 0.1%), and then with DAB (ab64259, Abcam). Brain slices were finally mounted between slide and coverslip and imaged post hoc using a Nanozoomer (S360, Hamamatsu). Illumination was set such that the full dynamic range of the 16-bit images was utilized. 16-bit image’s brightness was processed and a mask was registered to the corresponding coronal plates (ranging from −0.26 to −1.94 mm) of the mouse brain atlas using Illustrator (Adobe), at the various distances posterior to the bregma. To determine the horizontal and vertical spread of anti-GluA2 staining, images were converted to 8-bit images on ImageJ, and background noise was removed by using an entropy threshold algorithm (https://imagej.net/Maximum_Entropy_Threshold, ImageJ). The number of foreground pixels were then counted in ROI spanning the entire cortical column near injection site. The same analysis was performed in surrounding cortical columns.
To evaluate the astrocytic and microglial reactivity, anesthesia was induced using isoflurane (4% containing ∼0.5 l/min O2) and continued by adjusting its percentage to 1.5%–3%. One small ∼1 × 1 mm craniotomy was performed in each hemisphere, targeting the barrel field (Fronto-caudal: −1.0 / Mediolateral: ± 3.3). The dura was left intact to then perform stereotaxic injection of either an anti-GluA2 (left hemisphere) or anti-GFP antibody (right hemisphere). After stereotaxic injection the craniotomy was covered with sterile saline and protected with a 3 mm polydimethylsiloxane (PDMS) coverslip as previously described. Antibodies were re-injected twice the day after surgery, with a 12h interval between injections using isoflurane anesthesia (4% for induction, then 2% for injection with ∼0.5 l/min O2). After 12h of antibody washout, mice were finally anesthetized with isoflurane (4% with 0.5 l/min O2) and a mix of pentobarbital sodium and lidocaine (300 mg/kg; 30 mg/kg; i.p.) for animal’s intracardiac perfusion with PBS (1%) and PFA (4%). Fixed brains were sliced with a vibratome and sections were incubated afterward in PBS with Triton X-100 0.3% for 2h. A subset of brain slices were incubated either with an anti-GFAP antibody (chicken anti-GFAP, G2032-25F-100ul, USBiological Life Sciences) or an anti-Iba1 (rabbit anti-Iba1, 019-19741, WAKO) antibody (1/500 in PBS-Tween 0.05%) overnight at 4C°. Primary antibodies were washed-out 3 times with PBS, and secondary antibodies (Alexa 647 anti-chicken: A32933, Alexa 647 anti-rabbit: A27040, Thermofisher, 1/500 in PBS-Tween 0.05%) incubated at RT for 3h. Brain slices were finally mounted between slide and coverslip and imaged post hoc using a Nanozoomer (S360, Hamamatsu). Importantly, the same settings of imaging were kept for all the recorded samples. Raw data were converted to 8-bit images on ImageJ and segmented with ROI’s with similar width and height corresponding to 100 μm to determine the staining across all the superficial cortical layers. An average of pixel intensity for each ROI was calculated to determine staining intensity. LUT’s were then inverted and fitted with a Gaussian blur (5 pixels) to smooth the image and reduce noise. Finally, an entropy threshold (https://imagej.net/Maximum_Entropy_Threshold, ImageJ) was applied and the resulting number of particles for each individual ROI calculated. This allowed us to calculate the number of GFAP- or Iba1-positive cells across cortical layers.
Quantification and Statistical Analysis
All data generated or analyzed during this study are included in the manuscript, and provided in the Table S1 (data related to the main figures) and the Table S2 (data related to the supplemental figures). Data are presented as the mean ± interquartile range, except where stated differently. All statistics were performed using MATLAB (Mathworks) with an α significant level set at 0.05. Normality of all value distributions and the equality of variance between different distributions were first assessed by the Shapiro-Wilk and Levene median tests, respectively. Standard parametric tests were only used when data passed the normality and equal variance tests (p > 0.05). Non-parametric tests were used otherwise. Only two-sided tests were used. When applicable, pairwise multiple post hoc comparisons were done by using the Holm-Sidak method. Randomization and blinding methods were not used. No statistical methods were used to estimate sample size, but β-power values were calculated for parametric tests.
Acknowledgments
We thank E. Normand and the IINS in vivo core facility for animal husbandry. We thank the IINS cell biology core facilities (LABEX BRAIN [ANR-10-LABX-43]) and, in particular, C. Breillat for antibody handling. We thank H. El Oussini and B. Darracq (Imetronic) for their technical expertise and support and all the members of the Gambino and Choquet laboratories for technical assistance and helpful discussions. We thank E. Gouaux for generous gift of the 15F1 anti-GluA2 antibody.
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (NEUROGOAL, grant agreement no. 677878); the FP7 Marie-Curie Career Integration Program (grant agreement no. 631044); the ANR JCJC (grant agreement no. 14-CE13-0012-01); the University of Bordeaux (Initiative of Excellence senior chair 2014) to F.G.; from the ERC under the European Union’s Horizon 2020 Research and Innovation Program (ADOS, grant agreement no. 339541; and DynSynMem, grant agreement no. 787340) to D.C.; and the Region Nouvelle-Aquitaine to D.C. and F.G.
Author Contributions
T.C., E.A., A.d.M., V.K., and C.C. performed the experiments. T.C., D.C., and F.G. conceived the studies and analyzed the data, with the help of E.A., N.C., A.d.M., and V.K. D.C. and F.G. supervised the research and wrote the manuscript, with help from co-authors.
Declaration of Interests
The authors declare no competing interests.
Published: September 1, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108097.
Contributor Information
Daniel Choquet, Email: daniel.choquet@u-bordeaux.fr.
Frédéric Gambino, Email: frederic.gambino@u-bordeaux.fr.
Supplemental Information
References
- Armstrong-James M., Welker E., Callahan C.A. The contribution of NMDA and non-NMDA receptors to fast and slow transmission of sensory information in the rat SI barrel cortex. J. Neurosci. 1993;13:2149–2160. doi: 10.1523/JNEUROSCI.13-05-02149.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnéoud P., Gyger M., Andrés F., van der Loos H. Vibrissa-related behavior in mice: transient effect of ablation of the barrel cortex. Behav. Brain Res. 1991;44:87–99. doi: 10.1016/s0166-4328(05)80242-6. [DOI] [PubMed] [Google Scholar]
- Barth A.L., McKenna M., Glazewski S., Hill P., Impey S., Storm D., Fox K. Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J. Neurosci. 2000;20:4206–4216. doi: 10.1523/JNEUROSCI.20-11-04206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender V.A., Bender K.J., Brasier D.J., Feldman D.E. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti B.L., Glazewski S., Barth A.L. Reliable and precise neuronal firing during sensory plasticity in superficial layers of primary somatosensory cortex. J. Neurosci. 2009;29:11817–11827. doi: 10.1523/JNEUROSCI.3431-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieszczad K.M., Weinberger N.M. Remodeling the cortex in memory: Increased use of a learning strategy increases the representational area of relevant acoustic cues. Neurobiol. Learn. Mem. 2010;94:127–144. doi: 10.1016/j.nlm.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikel T., Sakmann B. Sensory integration across space and in time for decision making in the somatosensory system of rodents. Proc. Natl. Acad. Sci. USA. 2007;104:1395–1400. doi: 10.1073/pnas.0610267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham C.E.J., Hammond M.S.L., Edwards C.E.J., Finnerty G.T. Sensory experience alters cortical connectivity and synaptic function site specifically. J. Neurosci. 2007;27:3456–3465. doi: 10.1523/JNEUROSCI.5143-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem R.L., Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron. 2006;49:663–670. doi: 10.1016/j.neuron.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Clem R.L., Celikel T., Barth A.L. Ongoing in Vivo Experience Triggers Synaptic Metaplasticity in the Neocortex. Science. 2008;319:101–104. doi: 10.1126/science.1143808. [DOI] [PubMed] [Google Scholar]
- Constals A., Penn A.C., Compans B., Toulmé E., Phillipat A., Marais S., Retailleau N., Hafner A.-S., Coussen F., Hosy E., Choquet D. Glutamate-induced AMPA receptor desensitization increases their mobility and modulates short-term plasticity through unbinding from Stargazin. Neuron. 2015;85:787–803. doi: 10.1016/j.neuron.2015.01.012. [DOI] [PubMed] [Google Scholar]
- Dachtler J., Hardingham N.R., Glazewski S., Wright N.F., Blain E.J., Fox K. Experience-dependent plasticity acts via GluR1 and a novel neuronal nitric oxide synthase-dependent synaptic mechanism in adult cortex. J. Neurosci. 2011;31:11220–11230. doi: 10.1523/JNEUROSCI.1590-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering G.H., Huganir R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron. 2018;100:314–329. doi: 10.1016/j.neuron.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D.E. Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D.E., Brecht M. Map plasticity in somatosensory cortex. Science. 2005;310:810–815. doi: 10.1126/science.1115807. [DOI] [PubMed] [Google Scholar]
- Finnerty G.T., Roberts L.S., Connors B.W. Sensory experience modifies the short-term dynamics of neocortical synapses. Nature. 1999;400:367–371. doi: 10.1038/22553. [DOI] [PubMed] [Google Scholar]
- Fox K. Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience. 2002;111:799–814. doi: 10.1016/s0306-4522(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Gainey M.A., Feldman D.E. Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philos. Trans. R. Soc. B Biol. Sci. 2017;372:20160157. doi: 10.1098/rstb.2016.0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambino F., Holtmaat A. Spike-timing-dependent potentiation of sensory surround in the somatosensory cortex is facilitated by deprivation-mediated disinhibition. Neuron. 2012;75:490–502. doi: 10.1016/j.neuron.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Gambino F., Pagès S., Kehayas V., Baptista D., Tatti R., Carleton A., Holtmaat A. Sensory-evoked LTP driven by dendritic plateau potentials in vivo. Nature. 2014;515:116–119. doi: 10.1038/nature13664. [DOI] [PubMed] [Google Scholar]
- Giannone G., Hosy E., Levet F., Constals A., Schulze K., Sobolevsky A.I., Rosconi M.P., Gouaux E., Tampé R., Choquet D., Cognet L. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 2010;99:1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierdalski M., Jablonska B., Smith A., Skangiel-Kramska J., Kossut M. Deafferentation induced changes in GAD67 and GluR2 mRNA expression in mouse somatosensory cortex. Brain Res. Mol. Brain Res. 1999;71:111–119. doi: 10.1016/s0169-328x(99)00153-9. [DOI] [PubMed] [Google Scholar]
- Glazewski S., Fox K. Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J. Neurophysiol. 1996;75:1714–1729. doi: 10.1152/jn.1996.75.4.1714. [DOI] [PubMed] [Google Scholar]
- Glazewski S., Chen C.M., Silva A., Fox K. Requirement for alpha-CaMKII in experience-dependent plasticity of the barrel cortex. Science. 1996;272:421–423. doi: 10.1126/science.272.5260.421. [DOI] [PubMed] [Google Scholar]
- Glazewski S., Giese K.P., Silva A., Fox K. The role of alpha-CaMKII autophosphorylation in neocortical experience-dependent plasticity. Nat. Neurosci. 2000;3:911–918. doi: 10.1038/78820. [DOI] [PubMed] [Google Scholar]
- Granger A.J., Shi Y., Lu W., Cerpas M., Nicoll R.A. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493:495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans D.R., Clayton D.A., Coultrap S.J., Browning M.D. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Haas K.T., Compans B., Letellier M., Bartol T.M., Grillo-Bosch D., Sejnowski T.J., Sainlos M., Choquet D., Thoumine O., Hosy E. Pre-post synaptic alignment through neuroligin-1 tunes synaptic transmission efficiency. eLife. 2018;7:1–22. doi: 10.7554/eLife.31755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B., Duque A., Hasenstaub A.R., McCormick D.A. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J. Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham N., Fox K. The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J. Neurosci. 2006;26:7395–7404. doi: 10.1523/JNEUROSCI.0652-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselmann H., Mannara F., Werner C., Planagumà J., Miguez-Cabello F., Schmidl L., Grünewald B., Petit-Pedrol M., Kirmse K., Classen J. Human Autoantibodies against the AMPA Receptor Subunit GluA2 Induce Receptor Reorganization and Memory Dysfunction. Neuron. 2018;100:91–105.e9. doi: 10.1016/j.neuron.2018.07.048. [DOI] [PubMed] [Google Scholar]
- He H.-Y., Rasmusson D.D., Quinlan E.M. Progressive elevations in AMPA and GABAA receptor levels in deafferented somatosensory cortex. J. Neurochem. 2004;90:1186–1193. doi: 10.1111/j.1471-4159.2004.02590.x. [DOI] [PubMed] [Google Scholar]
- Holtmaat A., Bonhoeffer T., Chow D.K., Chuckowree J., De Paola V., Hofer S.B., Hübener M., Keck T., Knott G., Lee W.-C. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y.K., Lacefield C.O., Rodgers C.C., Bruno R.M. Sensation, movement and learning in the absence of barrel cortex. Nature. 2018;561:542–546. doi: 10.1038/s41586-018-0527-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House D.R.C., Elstrott J., Koh E., Chung J., Feldman D.E. Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron. 2011;72:819–831. doi: 10.1016/j.neuron.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y., Choquet D. The next generation of approaches to investigate the link between synaptic plasticity and learning. Nat. Neurosci. 2019;22:1536–1543. doi: 10.1038/s41593-019-0480-6. [DOI] [PubMed] [Google Scholar]
- Jensen V., Kaiser K.M.M., Borchardt T., Adelmann G., Rozov A., Burnashev N., Brix C., Frotscher M., Andersen P., Hvalby Ø. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J. Physiol. 2003;553:843–856. doi: 10.1113/jphysiol.2003.053637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Zhang C., Yanagawa Y., Sun Q.-Q. Major effects of sensory experiences on the neocortical inhibitory circuits. J. Neurosci. 2006;26:8691–8701. doi: 10.1523/JNEUROSCI.2478-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck T., Scheuss V., Jacobsen R.I., Wierenga C.J., Eysel U.T., Bonhoeffer T., Hübener M. Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron. 2011;71:869–882. doi: 10.1016/j.neuron.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Li L., Gainey M.A., Goldbeck J.E., Feldman D.E. Rapid homeostasis by disinhibition during whisker map plasticity. Proc. Natl. Acad. Sci. USA. 2014;111:1616–1621. doi: 10.1073/pnas.1312455111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H., Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis D.J., Lütcke H., Helmchen F. Microcircuit dynamics of map plasticity in barrel cortex. Curr. Opin. Neurobiol. 2014;24:76–81. doi: 10.1016/j.conb.2013.08.019. [DOI] [PubMed] [Google Scholar]
- Markram H., Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- Mégevand P., Troncoso E., Quairiaux C., Muller D., Michel C.M., Kiss J.Z. Long-term plasticity in mouse sensorimotor circuits after rhythmic whisker stimulation. J. Neurosci. 2009;29:5326–5335. doi: 10.1523/JNEUROSCI.5965-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzenich M.M., Kaas J.H., Wall J., Nelson R.J., Sur M., Felleman D. Topographic reorganization of somatosensory cortical areas 3b and 1 in adult monkeys following restricted deafferentation. Neuroscience. 1983;8:33–55. doi: 10.1016/0306-4522(83)90024-6. [DOI] [PubMed] [Google Scholar]
- Miyazaki T., Kunii M., Tada H., Sano A., Kuroiwa Y., Goto T., Malinow R., Takahashi T. Developmental AMPA receptor subunit specificity during experience-driven synaptic plasticity in the rat barrel cortex. Brain Res. 2012;1435:1–7. doi: 10.1016/j.brainres.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Luna K., Hertler B., Buitrago M.M., Luft A.R. Motor learning transiently changes cortical somatotopy. Neuroimage. 2008;40:1748–1754. doi: 10.1016/j.neuroimage.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Nair D., Hosy E., Petersen J.D., Constals A., Giannone G., Choquet D., Sibarita J.-B. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 2013;33:13204–13224. doi: 10.1523/JNEUROSCI.2381-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X., Hughes E.G., Moscato E.H., Parsons T.D., Dalmau J., Balice-Gordon R.J. Cellular plasticity induced by anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann. Neurol. 2015;77:381–398. doi: 10.1002/ana.24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn A.C., Zhang C.L., Georges F., Royer L., Breillat C., Hosy E., Petersen J.D., Humeau Y., Choquet D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature. 2017;549:384–388. doi: 10.1038/nature23658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen C.C.H., Hahn T.T.G., Mehta M., Grinvald A., Sakmann B. Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc. Natl. Acad. Sci. USA. 2003;100:13638–13643. doi: 10.1073/pnas.2235811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed A., Riley J., Carraway R., Carrasco A., Perez C., Jakkamsetti V., Kilgard M.P. Cortical map plasticity improves learning but is not necessary for improved performance. Neuron. 2011;70:121–131. doi: 10.1016/j.neuron.2011.02.038. [DOI] [PubMed] [Google Scholar]
- Rema V., Armstrong-James M., Ebner F.F. Experience-dependent plasticity of adult rat S1 cortex requires local NMDA receptor activation. J. Neurosci. 1998;18:10196–10206. doi: 10.1523/JNEUROSCI.18-23-10196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth R.H., Cudmore R.H., Tan H.L., Hong I., Zhang Y., Huganir R.L. Cortical Synaptic AMPA Receptor Plasticity during Motor Learning. Neuron. 2020;105:895–908.e5. doi: 10.1016/j.neuron.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt T.E. Mediation of thalamic sensory input by both NMDA receptors and non-NMDA receptors. Nature. 1986;322:263–265. doi: 10.1038/322263a0. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives M.V., McCormick D.A. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Schubert V., Lebrecht D., Holtmaat A. Peripheral deafferentation-driven functional somatosensory map shifts are associated with local, not large-scale dendritic structural plasticity. J. Neurosci. 2013;33:9474–9487. doi: 10.1523/JNEUROSCI.1032-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J., Baehrens D., Haupt A., Bildl W., Boudkkazi S., Roeper J., Fakler B., Schulte U. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron. 2014;84:41–54. doi: 10.1016/j.neuron.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Shi S., Hayashi Y., Esteban J.A., Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Steriade M., McCormick D.A., Sejnowski T.J. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Swadlow H.A., Gusev A.G. Receptive-field construction in cortical inhibitory interneurons. Nat. Neurosci. 2002;5:403–404. doi: 10.1038/nn847. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Svoboda K., Malinow R. Experience Strengthening Transmission by Driving AMPA Receptors into Synapses. Science. 2003;1585:1585–1589. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Takeuchi T., Duszkiewicz A.J., Morris R.G.M. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;369:20130288. doi: 10.1098/rstb.2013.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L.E., Holtmaat A. Higher-Order Thalamocortical Inputs Gate Synaptic Long-Term Potentiation via Disinhibition. Neuron. 2019;101:91–102.e4. doi: 10.1016/j.neuron.2018.10.049. [DOI] [PubMed] [Google Scholar]
- Xerri C. Plasticity of cortical maps: multiple triggers for adaptive reorganization following brain damage and spinal cord injury. Neuroscientist. 2012;18:133–148. doi: 10.1177/1073858410397894. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Cudmore R.H., Lin D.-T., Linden D.J., Huganir R.L. Visualization of NMDA receptor-dependent AMPA receptor synaptic plasticity in vivo. Nat. Neurosci. 2015;18:402–407. doi: 10.1038/nn.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Chen S., Swensen A.C., Qian W.J., Gouaux E. Architecture and subunit arrangement of native AMPA receptors elucidated by cryo-EM. Science. 2019;364:355–362. doi: 10.1126/science.aaw8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. This study did not generate any unique code.