

RT‐qPCR quantification analysis of TRECs and KRECs in gDNA of patients with many diverse molecularly confirmed Primary Immunodeficiency Diseases was performed. Our data suggest that in addition to classical SCID and agammaglobulinemia, the TREC/KREC assay may identify secondary targets including some patients with ZAP70 deficiency, DOCK8 deficiency, ataxia telangiectasia, Wiskott‐Aldrich and bare lymphocyte syndromes.

Keywords: agammaglobulinemia, ataxia telangiectasia, chronic granulomatous disease, Hyper IgE, severe combined immunodeficiency
Summary
Primary immune deficiency (PID) disorders are clinically and molecularly heterogeneous diseases. T cell receptor excision circles (TRECs) and κ (kappa)‐deleting excision circles (KRECs) are markers of T and B cell development, respectively. They are useful tools to assess T and B cell function and immune reconstitution and have been used for newborn screening for severe combined immunodeficiency disease (SCID) and agammaglobulinemia, respectively. Their profiles in several genetically confirmed PIDs are still lacking. The objective of this study was to determine TREC and KREC genomic profiling among various molecularly confirmed PIDs. We used real‐time–quantitative polymerase chain reaction (RT–qPCR)‐based triplex analysis of TRECs, KRECs and β‐actin (ACTB) in whole blood genomic DNA isolated from 108 patients with molecularly confirmed PIDs. All agammaglobulinemia patients had low KREC counts. All SCIDs and Omenn syndrome patients secondary to mutations in RAG1, RAG2, DCLRE1C and NHEJ1 had low TREC and KREC counts. JAK3‐deficient patients had normal KREC and the TREC count was influenced by the type of mutation. Early‐onset ADA patients had low TREC and KREC counts. Four patients with zeta‐chain‐associated protein kinase 70 (ZAP70) had low TREC. All purine nucleoside phosphorylase (PNP) patients had low TREC. Combined immunodeficiency (CID) patients secondary to AK2, PTPRC, CD247, DCLREC1 and STAT1 had normal TREC and KREC counts. Most patients with ataxia–telangiectasia (AT) patients had low TREC and KREC, while most DOCK8‐deficient patients had low TRECs only. Two of five patients with Wiskott–Aldrich syndrome (WAS) had low TREC counts as well as one patient each with bare lymphocyte syndrome (BLS) and chronic granulomatous disease. All patients with Griscelli disease, Chediak–Higashi syndrome, hyper‐immunoglobulin (Ig)M syndrome and IFNGR2 had normal TREC and KREC counts. These data suggest that, in addition to classical SCID and agammaglobulinemia, TREC/KREC assay may identify ZAP70 patients and secondary target PIDs, including dedicator of cytokinesis 8 (DOCK8) deficiency, AT and some individuals with WAS and BLS.
Abbreviations
- CNV
copy number variation
- CGD
chronic granulomatous disease
- DBS
dried blood spot
- DOCK8
dedicator of cytokinesis 8
- HIES
hyper‐IgE syndrome
- KREC
kappa‐deleting recombination excision circles
- NBS
newborn screening
- PID
primary immunodeficiency disease
- qPCR
quantitative polymerase chain reaction
- SCID
severe combined immunodeficiency disease
- SNV
single nucleotide variant
- STAT3
signal transducer and activator of transcription 3
- TREC
T cell receptor excision circle
- WAS
Wiskott‐Aldrich syndrome
Introduction
Primary immunodeficiency diseases (PIDs) are a phenotypically and molecularly heterogeneous group of diseases with more than 300 known genetic defects that affect the development or the function of the immune system [1, 2]. Patients affected with these genetic defects present with varying clinical presentations that range from mild to life‐threatening infections, dysregulated immunity, inflammation and malignancy [1, 2, 3]. PIDs are classified based on the affected immunological system and clinical presentations [2]. These include defects in antibody production by B cells (humoral immune deficiencies) and/or T cell functions (cellular immunity), impaired antigen presentation, defects in natural killer (NK) cell cytotoxicity, Toll‐like receptor (TLR) activation, phagocytosis, macrophage activation and complement.
During T and B cell development, premature T and B cells undergo the process of recombination of receptor gene segments. This occurs at the T cell (TCR) and B cell receptor (BCR) loci to generate the diversity of T and B cell repertoires, respectively, that are required to develop effective immunity against a wide range of antigens, such as bacteria, viruses and others [4, 5]. These recombination processes result in the formation of extra‐chromosomal non‐replicating excision circles. Quantitative measurements of these excision circles can be used clinically to assess the degree of T and/or B cell defects. Multiple types of TRECs are generated during the process of TCR rearrangement, and the TCR delta deletion TREC (delta rec psi‐J‐alpha signal joint TREC) appears to be the most accurate TREC for measuring thymic output. Conversely, BCR rearrangement results in the formation of κ (kappa)‐deleting excision circles (KRECs). Each (B/T) lymphocyte is believed to produce a single copy of TREC or KREC which have been used to develop newborn screening (NBS) methods for T or B cell lymphopenia, such as severe combined immunodeficiency disease (SCID) and X‐linked agammaglobulinemia (XLA), respectively [6, 7, 8, 9]. TRECs are generated during the latest stages of T cell development just before they exit to the periphery. Hence, their levels in the peripheral circulation represent thymic activity [10, 11, 12, 13]. Also, the longevity of naive T cells in the peripheral circulation, as well as TRECs intracellular degradation and dilution by peripheral T cell division, represent limitations that should be kept in mind. Conversely, KRECs are formed in the early stages of development up to mature B lymphocytes. Hence, KREC numbers in the periphery represent fresh and functional B cells [14].The utility of TREC and KREC in NBS aims to provide early diagnosis of severe forms of PID which have a significant impact on patient morbidity and mortality [14].
TREC measurements have not only been used for NBS: this method was also used as a tool to assess T cell immune reconstitution after thymic or hematopoietic stem cell transplantation (HSC) transplantation [15, 16, 17, 18]. Thymic output has been studied using TREC measurements, with the effect of aging showing an inverse correlation where TREC numbers decreased by more than 95% between the ages of 25 and 60 years [19]. Also, TREC measurements have been used in the studies of T cell generation in neonates [20]. T cell homeostasis was also evaluated in patients with rheumatoid arthritis [21], HIV and in response to highly active anti‐retroviral therapy (HAART) [22]. In such studies, genomic DNA (gDNA) extracted from whole blood was used.
The combined triplex assay of the TREC, KREC and β‐actin (ACTB) screening approach is useful for the detection of severe forms of PID that present with a low number of TREC, KREC or both [23, 24]. These assays have been validated by screening large numbers of newborn babies. Babies with low TREC or KREC numbers were then recalled for second‐tier testing [25]. Conversely, using a duplex (TREC, ACTB) assay, we recently conducted a SCID NBS pilot study that showed a high incidence of SCID in the Saudi newborn population (1 : 2906 live births) [26].
In this study, we sought to assess TREC and KREC profiling using genomic DNA extracted from whole blood samples from a large and diverse group of patients with molecularly confirmed diagnoses of PIDs.
Materials and methods
This study was approved by the institutional review board at King Faisal Specialist Hospital and Research Center (KFSHRC), RAC no. 2130‐027. The study was also supported by the Kingdom of Saudi Arabia National Science, Technology and Innovation Plan’s Strategic Technologies Grant KACST: 13‐BIO 755‐20 under the King Abdulaziz City for Science and Technology (Riyadh, Kingdom of Saudi Arabia).
Sample collection
All patients identified and reported in this study received comprehensive clinical and laboratory evaluations as part of their routine clinical care at the Pediatric Immunology and Allergy Clinics at King Faisal Hospital and Research Center. Previously consented, archived extracted DNA samples from this collection were used for this study. Written informed consent to obtain blood samples or to use the stored DNA was obtained from each patient or his/her parents if available; otherwise, the sample was anonymized. Data of interest collected from the patients’ medical records were secured as governed by the institutional policies on patient confidentiality and privacy.
DNA extraction
Genomic DNA of children known to have various PIDs (and their families, when available) was isolated from peripheral blood samples obtained by venipuncture as described previously using standard protocols. DNA was diluted to a working concentration of 10 ng/μl [27].
Sequence analysis and copy number analysis of PID genes
Bi‐directional Sanger sequencing was performed in all gDNA samples. Sequencing primers were designed to amplify the region of interest, which was sequenced as previously described [28]. Sequence data were aligned against the reference GenBank sequences and examined for variation. For novel mutation validation, anonymized 250 DNA samples derived from normal Saudi blood donors were sequenced to rule out potential novel Saudi normal single nucleotide polymorphisms (SNPs). The Affymetrix high‐definition scan SNP‐microarray platform (Affymetrix, Santa Clara, CA, USA) was used to evaluate patients with dedicator of cytokinesis 8 (DOCK8) deficiency for possible copy number abnormalities.
Real‐time–quantitative triplex polymerase chain reaction (RT–qPCR)
RT–qPCR of gDNA of individuals with known diagnoses of primary immune deficiency was used to quantify TRECs, KRECs and ACTB, which served as an internal control for determining the efficiency of the RT–qPCR reaction. Reagents for this assay (SCREEN‐ID neonatal screening kit) were purchased from Mabtech Diagnostics (Nacka Strand, Sweden) (https://www.mabtech.com/), and the assay was performed as per the manufacturer’s recommendations which have been previously described [25, 26]. Briefly, the RT–qPCR reactions were performed in a final volume of 20 μl containing 100 ng (10 ng/μl) gDNA, 1× TaqMan Gene Expression Master Mix, 20 μM TREC primers, 25 μM KREC primers, 7·5 μM ACTB primers, 15 μM 6‐carboxyfluorescein (6FAM)‐labeled minor groove‐binding (MGB) TREC‐ and NED‐labeled MGB ACTB probes, 17·5 μM VIC‐labeled MGB KREC probe and 0·8 μl of 10 mg/ml bovine serum albumin (BSA). The 96‐well plate reactions were carried out on the ABI QuantStudio 7 real‐time PCR System (Applied Biosystems, Foster City, CA, USA). PCR conditions were: initial cycle at 50°C for 2 min and a heating cycle at 95°C for 10 min, followed by 45 cycles of 30 s at 95°C and 30 s at 60°C. An individual cycle threshold for TREC, KREC or ACTB was fixed for automated data collection and analysis of the amplification during the exponential phase. Calibration curves were generated by 10‐fold serial dilution using TREC‐KREC‐ACTB. All analyzed RT–qPCR assays met the quality requirements of similar slopes and R 2 values > 0·97. While amplification of ACTB was used to assess PCR efficiency, this was disregarded if TREC and KREC copy numbers were above the cut‐off level.
Ethical approval
This study was submitted to the Institutional Review Board of King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia, and was approved by the Research Advisory Committee through established procedures.
Results
Patient demographics and T/B lymphocyte subset flow cytometry analysis
This large group of patients represents a wide array of inborn errors of immunity. In 100 patients, the median age at sample collection was 39 ± 72 months (0·25–372) and prehematopoietic stem cell transplantation lymphocyte subset analysis results were available on 90 subjects (Supporting information, Table S1). The median count for CD3+ cells was 1672·5 ± 1932 (reference: 1600–2700), CD4+ cells 706 ± 1322 (reference: 1000–1700), CD8+ cells 480 ± 843·9 (reference: 600–1000), CD19 cells 275 ± 806 (reference: 400–800), NK CD56+ and 16+ cells 220 ± 556·9 (reference: 200–400). Lymphocyte subpopulations counts were low in 21 (CD3+), 58 (CD4+), 50 (CD8+), 45 (CD19+) and 65 (NK CD56+, 16+) samples. However, naive and memory T cell counts were not available for many samples. TREC and KREC counts were low in 50 and 34 samples, respectively, while 29 samples were low for both markers, as shown in Supporting information, Table S1.
Table 1.
Combined TREC/KREC counts and DNA mutation analysis in 108 patients with various inborn errors of immunity
| Sample Name | Clinical phenotype | Gene | gDNA variant | Protein change | Reference | TRECs/μl | KRECs/μl |
|---|---|---|---|---|---|---|---|
| 08R‐01806 | Agammaglobulinemia | BTK | NM_000061:exon8:c.C763T | p.R255X | [45] | 1494 | 1·3 |
| 12R‐01584 | BTK | 894 | 1·2 | ||||
| 08R‐01805 | BTK | 861 | 1·7 | ||||
| 11R‐00514 | BTK | NM_000061:exon20:c.1909‐9T>C | n.a. | [46] | 1.2 | 631 | |
| 11R‐00404 | CID | AK2 | NM_001625:exon6:c.C545A | p.A182D | [46] | 24 | 19 |
| 09R‐00412 | AK2 | 64 | 250 | ||||
| 09R‐00413 | AK2 | 235 | 1932 | ||||
| 11R‐01877 | PTPRC | NM_002838:exon32:c.T3545C | p.L1182S | 273 | 656 | ||
| 12R‐03326 | CD247 (CD3Z) | NM_198053:exon2:c.58+8C>T | n.a. | 318 | 219 | ||
| 11R‐00457 | DCLRE1C | NM_022487:exon10:c.C614G | p.S205C | 47 | 154 | ||
| 13R‐01205 | ZAP70 | NM_001079:exon12:c.G1606A | p.G536S | 0.8 | 8 | ||
| 13R‐01204 | ZAP70 | 12 | 23 | ||||
| 09R‐00001 | STAT1 | NM_007315: c.1220T>G | L407R | 15 | 26 | ||
| 06R‐00778 | SCID | RAG1 | NM_000448:exon2:c.T1201C | p.S401P | [47] | 0.1 | 0·7 |
| 12R‐02223 | RAG1 | NM_000448:exon2:c.555delG | p.R186fsX15 | [46] | 0 | 0 | |
| 11R‐00776 | RAG1 | NM_000448:exon2:c.G2210A | p.R737H | [48] | 0 | 0 | |
| 13R‐00820 | RAG1 | NM_000448:exon2:c.C1180T | p.R394W | [49] | 0 | 0·1 | |
| 12R‐02253 | RAG1 | NM_000448:exon2:c.G2041T | p.E681X | [46] | 0 | 0 | |
| 06R‐00454 | JAK3 | NM_000215:exon 8: c.1012 delG | A338LfsX14 | This report | 0 | 350 | |
| 08R‐01741 | JAK3 | NM_000215:exon7:c.C913T | p.Q305X | [46] | 0 | 23 | |
| 11R‐00138 | JAK3 | NM_000215:exon3:c.G308A | p.R103H | 67 | 270 | ||
| 11R‐00199 | RAG2 | NM_000536:exon2:c.374_375delCA | p.T125RfsX10 | 0 | 0 | ||
| 08R‐00802 | RAG2 | NM_000536:exon2:c.G686C | p.R229P | [50] | 0 | 0 | |
| 09R‐00477 | RAG2 | NM_000536:exon2:c.T547C | p.F183L | [46] | 0 | 0 | |
| 09R‐00114 | RAG2 | NM_000536:exon2:c.442C>T | p.R148X | [47] | 0 | 0 | |
| 10R‐00041 | RAG2 | NM_000536:exon2:c.1403_1406delATCT | p.H468RfxX16 | [46] | 0 | 0 | |
| 12R‐03041 | ADA | NM_000022: c.385G>A | p.V129M | [51] | 3 | 2 | |
| 07R‐01680 | ADA | NM_000022:exon10:c.956_960delAAGAG | p.E319GfsX3 | [52] | 56 | 65 | |
| 06R‐00782 | ADA | NM_000022:exon 10:c.956_960delAAGAG | p. E319GfsX3 | 0 | 0 | ||
| 07R‐00376 | DCLRE1C | (artemis) deletion exon 1 – 3 | n.a. | [53] | 0 | 0 | |
| 08R‐00625 | DCLRE1C | NM_001033855:exon6:c.G403A | p.G135R | [46] | 0 | 0 | |
| 07R‐01595 | ZAP70 | NM_001079.3: exon 12:c.1606G>A | p.G536S | 12 | 6 | ||
| 10R‐00846 | ZAP70 | 319 | 1242 | ||||
| 09R‐00066 | ZAP70 | NM_001079:exon5:c.569G>C | p.R190T | 5 | 632 | ||
| 08R‐00795 | PNP | NM_000270:exon5:c.487T>C | p.S163P | [54] | 0 | 82 | |
| 11R‐02131 | PNP | 0 | 3 | ||||
| 09R‐00329 | PNP | Splice: NM_000270:exon2:c.12‐1G>C | n.a. | [46] | 0 | 75 | |
| 10R‐00194 | SCID with microcephaly, growth retardation and sensitivity to ionizing radiation | NHEJ1 | NM_024782:exon4:c.390+1G>C | 0.4 | 0 | ||
| 10R‐00190 | NHEJ1 | 2 | 6 | ||||
| 11R‐00581 | MSMD | IFNGR2 | NM_005534:exon5:c.C705A | p.Y235X | [55] | 508 | 1159 |
| 08R‐00113 | Omenn syndrome | RAG1 | NM_000448:exon2:c.555delG | R186fsX15 | [46] | 0 | 0 |
| 11R‐00236 | RAG2 | NM_000536:exon2:c.1332C>G | p.I444M | [56] | 0 | 0 | |
| 10R‐00808 | AT | ATM | NM_000051: c.C8732A | p.T2911N | This report | 1.7 | 4·4 |
| 08R‐01809 | ATM | NM_000051:exon61:c.8833_8834delCT | L2945VfsX10 | [57] | 5.5 | 4·5 | |
| 11R‐01857 | ATM | NM_000051:exon63:c.9066delA | p.G3023AfsX10 | [46] | 2.4 | 3·9 | |
| 07R‐01996 | ATM | NM_000051:exon8:c.903_904insT | p.A302CfsX3 | 1 | 1 | ||
| 08R‐1863 | ATM | ATM:NM_000051:exon5:c.381delA | p.V128X | [58] | 0.1 | 0·3 | |
| 08R‐01896 | ATM | NM_000051:exon63:c.9066delA | p.G3023AfsX10 | [46] | 34 | 3 | |
| 08R‐01634 | HIES | DOCK8 | NM_203447:exon7:c.827+6T>C | n.a. | [28] | 19 | 146 |
| 12R‐00998 | DOCK8 | NM_001190458:exon7:c.647delT | p.L216RfsX24 | This report | 2.2 | 171 | |
| 12R‐03319 | DOCK8 | DOCK8 deletion of exons 6 to 8 | n.a. | This report | 12 | 81 | |
| 11R‐02472 | DOCK8 | n.a. | This report | 8.2 | 479 | ||
| 12R‐03038 | DOCK8 | n.a. | This report | 0 | 0 | ||
| 09R‐00002 | DOCK8 | NM_203447:exon44:c.T5625G | p.Y1875X | [28] | 24 | 390 | |
| 10R‐00007 | DOCK8 | NM_203447:exon40:c.C5132A | p.S1711X | 3 | 192 | ||
| 06R‐01268 | DOCK8 | NM_203447:exon44:c.T5625G | p.Y1875X | 26 | 1277 | ||
| 07R‐02055 | DOCK8 | 23 | 980 | ||||
| 11R‐00115 | DOCK8 | DOCK8 deletion up to exon 19 | n.a. | 0.8 | 20 | ||
| 11R‐00061 | DOCK8 | NM_203447:c.4626+5G>A | [46] | 20 | 339 | ||
| 11R‐00422 | DOCK8 | NM_203447:exon40:c.C5132A | p.S1711X | [28] | 11 | 128 | |
| 11R‐00761 | DOCK8 | NM_203447: c.4626+5 G>A | n.a. | [46] | 0.1 | 839 | |
| 11R‐00753 | DOCK8 | DOCK8 deletion of exons 1–13 | This report | 2 | 67 | ||
| 13R‐00808 | DOCK8 | NM_203447:exon44:c.T5625G | p.Y1875X | [28] | 42 | 2617 | |
| 08R‐00244 | DOCK8 | NM_203447:exon40:c.C5132A | p.S1711X | 0 | 123 | ||
| 11R‐00140 | DOCK8 | NM_203447:exon44:c.T5625G | p.Y1875X | 7 | 354 | ||
| 07R‐00097 | DOCK8 | Whole DOCK8 deletion up to intron 1 of KANK1 | n.a. | 0.3 | 0·7 | ||
| 10R‐00180 | DOCK8 | Deletion up to exon 19 | This report | 7 | 418 | ||
| 11R‐02144 | DOCK8 | NM_203447:exon44:c.T5625G | p.Y1875X | [28] | 19 | 90 | |
| 09R‐00179 | STAT3 | NM_003150:exon20:c.G1868T (hetero) | p.W623L | [46] | 139 | 162 | |
| 11R‐00298 | STAT3 | NM_003150:exon13:c.G1145A (hetero) | p.R382Q | [59] | 50 | 60 | |
| 07R‐00795 | STAT3 | NM_003150:exon13:c.1144C>T (hetero) | p.R382W | 107 | 64 | ||
| 09R‐00184 | STAT3 | 153 | 31 | ||||
| 09R‐00182 | STAT3 | 675 | 748 | ||||
| 11R‐01442 | WAS | WAS | NM_000377:exon4:c.G397A | p.E133K | [60] | 14 | 110 |
| 12R‐02926 | WAS | M_000377: c.11dupG | M6NfsX32 | [61] | 31 | 80 | |
| 11R‐00333 | WAS | NM_000377:exon2:c.G257A | p.R86H | [62] | 119 | 28 | |
| 11R‐02389 | WAS | 0 | 88 | ||||
| 12R‐02269 | WAS | NM_000377:exon1:c.G91A | p.E31K | [63] | 144 | 611 | |
| 11R‐02714 | BLS | RFXANK | NM_003721:c.C477A | p.S159R | This report | 29 | 333 |
| 12R‐02302 | RFXANK | NM_003721:exon6:c.A362T | p.D121V | [64] | 20 | 172 | |
| 12R‐02646 | RFXANK | 20 | 386 | ||||
| 13R‐00818 | RFXANK | NM_134440:exon4:c.271+1G>C | n.a. | [46] | 1.2 | 2·4 | |
| 09R‐00132 | RFXANK | NM_003721:exon3:c.8_11dupTTAC | Q5YfsX22 | 84 | 86 | ||
| 10R‐00303 | CIITA | NM‐000246: c.G2551C | p.A851P | This report | 133 | 912 | |
| 11R‐00156 | CGD | CYBB | NM_000397:exon6:c.G618A | p.W206X | [65] | 104 | 322 |
| 08R‐00975 | CYBB | NM_000397:exon5:c.C388T | p.R130X | [66] | 0 | 581 | |
| 11R‐00186 | CYBB | NM_000397:exon13:c.1618delG | p.E540KfsX7 | [67] | 323 | 363 | |
| 07R‐01787 | CYBB | NM_000397:exon10:c.1217T>A | p.L406X | [46] | 247 | 646 | |
| 08R‐01338 | NCF2 | NM_000433:exon2:c.C229T | p.R77X | [68] | 213 | 481 | |
| 08R‐00833 | GS | MLPH | NM_001042467:exon2:c.G104A | p.R35Q | [46] | 49 | 65 |
| 07R‐00416 | MLPH | 28 | 70 | ||||
| 10R‐00003 | MLPH | NM_001042467: exon8: c.987delC | p.S330fsX15 | 159 | 290 | ||
| 10R‐00558 | RAB27A | NM_004580:c.148_149delAGinsC | p.R50QfsX35 | [69] | 541 | 372 | |
| 11R‐00810A | RAB27A | NM_004580:exon5:c.A400C | p.K134Q | [46] | 90 | 161 | |
| 07R‐00817 | RAB27A | NM_004580:exon6:c.598C>T | p.R200X | [70] | 151 | 239 | |
| 07R‐00962 | RAB27A | 888 | 631 | ||||
| 11R‐02385 | RAB27A | NM_004580:exon2:c.53_54delCT | p.S18WfsX15 | [71] | 122 | 87 | |
| 12R‐02807 | CHS | LYST | NM_000081:exon23:c.G6694T | p.G2232X | [46] | 45 | 77 |
| 13R‐01455 | LYST | NM_000081:exon37:c.9044+1G>T | n.a. | 659 | 712 | ||
| 11R‐00696 | HIGM | CD40 | NM_001250:exon3:c.256+2T>C | [72] | 240 | 323 | |
| 12R‐02921 | CD40 | 84 | 18 | ||||
| 07R‐00087 | CD40 | 705 | 1551 | ||||
| 11R‐00167 | CD40LG | NM_000074:exon5:c.719_720delAT | p.N240SfsX3 | [46] | 1417 | 674 | |
| 11R‐02335 | CD40LG | NM_000074:exon5:c.A506G | p.Y169C | [73] | 115 | 1175 | |
| 10R‐00971 | AICDA | NM_020661:exon3:c.G169A | p.V57M | [46] | 53 | 218 | |
| 10R‐00970 | AICDA | 16 | 63 | ||||
| 10R‐00972 | AICDA | 137 | 340 | ||||
| 07R‐00662 | AICDA | NM_020661:exon2:c.152A>T | p.N51I | 109 | 234 |
All variants (mutations) in this table are homozygous except for STAT3 (heterozygous) and BTK (X‐linked hemizygous, ♣).
TREC = T cell receptor excision circle; n.a. = not applicable; KREC = kappa deleting recombinant excision circle; PNP = purine nucleoside phosphorylase; CID = combined immunodeficiency; SCID = severe combined immunodeficiency; MSMD = Mendelian susceptibility to mycobacterial disease; AT = ataxia telangiectasia; HIES = hyper‐immunoglobulin (Ig)E syndrome; WAS = Wiskott‐Aldrich syndrome; BLS = bare lymphocyte syndrome; DOCK8 = dedicator of cytokinesis 8; CGD = chronic granulomatous disease; GS = Griscelli syndrome; CHS = Chediak–Higashi syndrome; HIGM = hyper‐IgM.
PID genetic mutations detected by Sanger sequencing and SNP microarray analyses
In this large cohort of 108 patients with a wide spectrum of PIDs (Table 1), missense mutations were the most common (n = 44) followed by stop–gain mutations (n = 24), frameshift mutations (n = 19) and splicing mutations (n = 13), which were all identified via Sanger sequencing. Four of the frameshift mutations were identified in four of the six patients with ataxia‐telangiectasia (AT). Large intragenic deletions detectable by SNP‐microarray analysis were confirmed in eight patients, seven of which involved DOCK8, and one deletion spanned exons 1–3 of DCLRE1C (ARTEMIS). In 11 patients, we detected eight unique and novel mutations (Table 1), six of which involved DOCK8 (including five multi‐exon deletions). While STAT3 and BTK mutations are heterozygous and hemizygous (X‐linked), respectively, all remaining mutations reported here are homozygous. Clinically and biochemically, all patients with novel mutations had typical presentation except for cases 5, 6 and 7, who had atypical AK2 deficiency which was recently reported to cause hypogammaglobulinemia and limited T cell dysfunction [29], and cases 11 and 12 with zeta‐chain‐associated protein kinase 70 (ZAP70) deficiency who presented with CID phenotype without CD8 deficiency.
Triplex (TREC, KREC, ACTB) assay
Using a cut‐off of ≤ 15 TRECs/μl and < 10 KRECs/μl, all agammaglobulinemia patients [4] secondary to BTK defects had low KREC and normal TREC counts. All SCIDs and Omenn syndrome patients secondary to RAG1 [6], RAG2 [6], DCLRE1C [2] and NHEJ1 [2] had low TREC and KREC counts. All SCIDs secondary to JAK3 deficiency had normal KRECs and two of three had low TRECs. Two of the three ADA‐deficient patients had low TRECs and KRECs. Interestingly, two patients of three with ZAP70 deficiency had low TRECs and low KREC in one patient. All purine nucleoside phosphorylase (PNPP)‐deficient patients [3] had low TRECs, and one of them had low KREC. CID patients secondary to AK2 [3], protein tyrosine phosphatase receptor type C (PTPRC) [1], CD247 [1], DNA cross‐linking repair protein 1 (DCLREC1) [1] and signal transducer and activator of transcription 1 (STAT‐1) [1] had normal TRECs and KRECs, while the CID patients with atypical ZAP70 (no CD8 deficiency) had a low TREC count. Five of six AT patients had low TREC and all had low KREC counts. Thirteen of 20 DOCK8‐deficient patients had low TRECs and two had low KRECs. Two of five patients with Wiskott–Aldrich syndrome (WAS) had low TRECs. One of six patients with bare lymphocyte syndrome (BLS) had low TRECs and KRECs. One of the five chronic granulomatous disease patients had a low TREC count. All patients with Griscelli disease [8], Chediak–Higashi syndrome [2], hyper‐immunoglobulin (Ig)M syndrome [9] and interferon gamma‐receptor receptor 2 (IFN‐γR2) [1] had normal TREC and KREC counts.
Discussion
TRECs and KRECs are considered markers of new lymphocyte output. They are used in research studies, but not routinely used in the routine clinical evaluation of patients with PIDs. TREC levels are extremely low or undetectable in all patients with typical SCID, and have been used effectively for routine NBS for SCID [7, 30]. KRECs can be used to screen for agammaglobulinemia [6, 8]. Currently, there is no universally validated NBS method for other PIDs.
TREC levels have been extensively assayed in children with PIDs in whom the decreased number and function of T and/or B cells result in severe impairment of immunity. Limited KREC studies were performed for various types of PIDs. Roifman et al. studied TRECs among 51 patients with T cell dysfunction. All classical SCIDs patients had low TREC numbers while partial ADA, interleukin (IL)‐2γR (p. R222C mutation), CD40L, forkhead box protein 3 (FoxP3) and IL‐10R had normal TREC results [31]. Using the same multiplex assay reported here, Borte et al. applied this approach to study 49 original stored Guthrie cards from patients diagnosed with SCID (n = 18), XLA (n = 4), AT (n = 4), Nijmegen breakage syndrome (n = 2), common variable immunodeficiency (CVID) (n = 4), IgA deficiency (n = 15) and X‐linked hyper‐IgM syndrome (HIGM) (n = 2) and demonstrated its ability to identify patients with XLA, AT, NBS and WAS [23]. Separately, in two of three young children with DOCK8‐related HIES, we found low dried blood spot (DBS) TREC copy numbers [32]. Subsequently, a few additional reports demonstrated low TREC counts in DBS samples from patients with various secondary (non‐SCID) target disorders such as AT [33], Nijmegen breakage syndrome [34, 35], cartilage hair hypoplasia (CHH) [36], WAS [36, 37] and DNA ligase 4 (LIG4) deficiency [38]. Table 2 summarizes the primary immunodeficiency diseases in which TREC and KREC numbers have been evaluated both in the literature as well as in our current study.
Table 2.
Primary immunodeficiency diseases in which T cell receptor excision circles (TREC) and kappa deleting recombinant excision circles (KREC) have been evaluated in the literature and this study
| Disease | Genetic defect | TREC | KREC | References |
|---|---|---|---|---|
| Severe combined immunodeficiency disease | ADA | Low | Not done (n.d.) | [31, 74, 75] |
| Partial ADA | Normal (N) | n.d. | [31] | |
| ADA | Low | Low | This report | |
| Partial ADA | N | N | This report | |
| AK2 | Low | Low | [23], this report | |
| AK2 (p.A182D mutation) | N | N | This report | |
| CD3G | Low | n.d. | [31] | |
| CD247 (CD3Z) | N | N | This report | |
| DCLRE1C | Low | Low | This report | |
| IL2RG | Low | N | [7, 23, 31, 75] | |
| IL2RG (p.R222C mutation) | N | N | [31] | |
| JAK3 | Low | N | [7, 23, 75], this report | |
| JAK3 (p.R103H mutation) | N | N | This report | |
| LIG4 | Low | n.d. | [31, 75] | |
| PNP | Low | n.d. | [74] | |
| Low | Low | This report | ||
| PTPRC | N | N | This report | |
| NHEJ1 | Low | Low | This report | |
| RAG1 | Low | Low | [23, 31, 75], this report | |
| RAG2 | Low | Low | [31, 75], this report | |
| RMRP | Low | n.d. | [31] | |
| ZAP70 | Low | n.d. | [31] | |
| Low or N | Low or N | This report | ||
| Agammaglobulinemia | BTK | N | Low | [8, 23], this report |
| Ataxia‐telangiectasia | ATM | Borderline | Low | [23] |
| Low (5 of 6) | Low | This report | ||
| Hyper‐immunoglobulin (Ig)E syndromes | DOCK8 | Low | n.d. | [32] |
| Low (13 of 20) | N in 18 and low in 2 of 20 | This report | ||
| STAT3 | N | N | This report | |
| Wiskott–Aldrich syndrome | WAS | Low | Low | [76] |
| Low (2 of 6) | Low (1 of 6) | This report | ||
| Bare lymphocyte syndrome | Not reported | Low | n.d. | [31] |
| RFX5 | Low | n.d. | [41] | |
| RFXANK | N (1 of 5 low) | N (1 of 5 low) | This report | |
| CIITA | N | N | This report | |
| Hyper‐IgM syndrome | CD40L | N | N | [23, 31, 76], this report |
| CD40 | N | N | This report | |
| AICDA | N | N | This report | |
| Griscelli syndrome | RAB27 | N | N | This report |
| MLPH | N | N | This report | |
| Chediak–Higashi syndrome | LYST | N | N | This report |
| Nijmegen breakage syndrome | NBN | N | Low | [23, 77] |
| Immune dysregulation | FOXP3 | N | n.d. | [31] |
| IL2RA | Low | n.d. | [31] | |
| IL10RA | N | n.d. | [31] | |
| Mendelian susceptibility to mycobacterial diseases | IFNGR2 | N | N | This report |
| STAT1 | Borderline | N | This report | |
| Chronic granulomatous disease | CYBB | Low | n.d. | [43] |
| CYBB | N (low in 1 of 4) | N | This report | |
| NCF2 | N | N | This report |
In this report, we confirm that a subset of patients with AT, WAS and the majority of DOCK8‐deficient individuals had low TREC and/or KREC counts (Table 1, Fig. 1), thus potentially allowing early NBS for these disorders. It is interesting to note that one of our three cases with ADA deficiency had normal TREC counts and therefore would have been missed on NBS. Either late‐onset or partial ADA deficiency may explain such phenomena, as a significant subset (15–20%) of ADA‐deficient patients exhibit a ‘delayed clinical onset’ and present with less severe, but slowly progressive, combined immune deficiency later in life [39]. It was believed that some PID patients with residual T cells, such as ZAP70 deficiency, will be missed by TREC‐based NBS. ZAP70 deficiency, a TCR signaling defect, is characterized by normal/elevated number of circulating lymphocytes, absent CD8+ T cells, abundant CD4+ T cells, normal number and activity of natural killer cells and normal number with reduced function of B cells. In this cohort, four of five cases with ZAP70 deficiency had low TREC counts, a finding that was previously reported by Roifman et al. [31], thus making some patients with this disorder potentially amenable to TREC‐based detection. In contrast, a recent publication reported that the TREC count was normal at birth and undetectable at age of 9 months in a patient with ZAP70 defect [40], reflecting possible variations that could be observed at different ages.
Fig. 1.
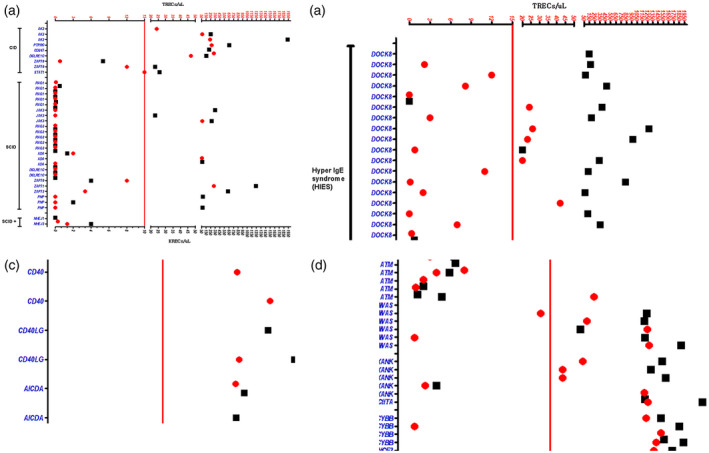
T cell receptor excision circle (TREC) and κ (kappa)‐deleting excision circle (KREC) counts in a cohort of 108 patients with various primary immune deficiencies. Primary immune deficiency (PID) categories and related genes are displayed on the y‐axis and TRECs/KRECs on the x‐axis. Vertical red lines within each group represent TREC (red circles) and KREC (black squares) count cut‐offs at 15 and 10, respectively. BLS = bare lymphocyte syndrome; CGD = chronic granulomatous disease; CHS = cartilage hair hypoplasia syndrome; CID = combined immunodeficiency; HIES = hyper‐immunoglobulin (Ig)E syndrome; HIGM = hyper‐IgM syndrome; SCID = severe combined immunodeficiency.
Major histocompatibility complex (MHC) class II deficiency, also known as bare lymphocyte syndrome (BLS) type II, is a primary immunodeficiency disorder that results from the lack of expression of MHC class II molecules on the surface of immune cells rather than an intrinsic defect in T cell development. It is another disease that is expected to be missed by TREC based newborn screening. Recently, an MHC II‐deficient patient was identified through SCID NBS [41]. Interestingly, one of our six patients had low TREC and KREC counts. The possible mechanism for this observation is not clear, but might be related to T cell depletion secondary to chronic unresolved infections. In this report, we showed for the first time, to our knowledge, that patients with SCID secondary to CD3Z (CD247) and PTPRC (CD45) deficiency have a normal TREC and KREC, confirming the notion that these rare types of SCID will unlikely to be identified by the current SCID NBS. The roles of the different CD3 chains in T cell differentiation are not well known. Mutations in those chains impair T cell development to varying degrees. Deficiency of CD3δ or CD3ε causes severe T cell lymphopenia, while CD3γ or CD247 (CD3Z) causes mild T cell lymphopenia, and this probably explains the normal TREC level in our patient with CD3Z defect [42].
We noticed unexpectedly that one of the five patients with chronic granulomatous disease (CGD), a phagocyte defect, had a low TREC count, an observation that was previously observed in some patients with CGD, which is thought to be due to T cell depletion (through unknown mechanisms) [43, 44]. This finding supports our observation, and calls for additional comprehensive T cell evaluation and monitoring in these patients.
Genomic DNA extracted from whole blood samples of individuals with fully characterized genetic mutations is the best approach to determine which PID can be detected by NBS using TREC and KREC quantification, followed by validation on DBS. In this study, we explored the genomic TREC and KREC profiles of 108 patients with diverse molecularly confirmed PIDs as representing the largest series of patients ever studied, and showed that various PID disorders are associated with low TREC and/or KREC copy numbers, not limited to SCID and agammaglobulinemia. Such findings will possibly increase the list of secondary targets for NBS. Additionally, as the majority of our patients’ DNA samples were collected beyond the neonatal period, our results suggest the potential utility of this triplex assay beyond the newborn period to evaluate the immune system during work‐up of various immunodeficiency diseases in addition to the assessment of immune reconstitution post‐HSCT. As these assays were performed well beyond the newborn period, these results should be validated by assays in the newborn period.
Disclosures
The authors declare no conflicts of interest.
Author contributions
M. D. designed the study, analyzed data and prepared the manuscript. H. A.‐M. and A. H. participated in the data analysis and manuscript preparation. G. A.‐D. and A. J. performed the triplex (TREC, KREC, ACTB) screening assay. F. E. helped with the data analysis. A. M. A., B. A., R. A., H. A.‐D. and I. A. provided the patients’ information. All authors approved the final version for submission.
Supporting information
Table S1. Flowcytometry profiles of T/B lymphocytes and corresponding TREC/KREC counts in patients with primary immune deficiencies. Highlighted cells in the table indicate low counts as defined by the reference ranges: TREC < 15, KREC < 10 and reference ranges for T/B cell subsets: CD3 1,600‐2,700; Helper T CD4 1,000‐1,700; CD8 600‐1,000; CD19 400‐800; NK CD56 &16 200‐400; CD4/CD8 Ratio 1.3‐2.0. NA not available.
Acknowledgements
This research is supported by National Science, Technology and Innovation Plan’s (NSTIP) strategic technologies program in Saudi Arabia (KACST: 13‐BIO‐755‐20).
Contributor Information
M. Dasouki, Email: madasouki@kfshrc.edu.sa.
H. Al‐Mousa, Email: hamoudalmousa@kfshrc.edu.sa.
A. Hawwari, Email: hawwariab@ngha.med.sa.
References
- 1. Bousfiha A, Jeddane L, Picard C et al The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol 2018; 38:129–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Picard C, Bobby Gaspar H, Al‐Herz W et al International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee report on inborn errors of immunity. J Clin Immunol 2018; 38:96–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol 2017; 14:7–18. [DOI] [PubMed] [Google Scholar]
- 4. Chen J, Alt FW. Gene rearrangement and B‐cell development. Curr Opin Immunol 1993; 5:194–200. [DOI] [PubMed] [Google Scholar]
- 5. Krangel MS. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol 2009; 21:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borte S, Wang N, Oskarsdottir S, von Dobeln U, Hammarstrom L. Newborn screening for primary immunodeficiencies: beyond SCID and XLA. Ann NY Acad Sci 2011; 1246:118–30. [DOI] [PubMed] [Google Scholar]
- 7. Chan K, Puck JM. Development of population‐based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2005; 115:391–8. [DOI] [PubMed] [Google Scholar]
- 8. Nakagawa N, Imai K, Kanegane H et al Quantification of kappa‐deleting recombination excision circles in Guthrie cards for the identification of early B‐cell maturation defects. J Allergy Clin Immunol 2011; 128:223–5 e2. [DOI] [PubMed] [Google Scholar]
- 9. Serana F, Chiarini M, Zanotti C et al Use of V(D)J recombination excision circles to identify T‐ and B‐cell defects and to monitor the treatment in primary and acquired immunodeficiencies. J Transl Med 2013; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al‐Harthi L, Marchetti G, Steffens CM, Poulin J, Sékaly R, Landay A. Detection of T cell receptor circles (TRECs) as biomarkers for de novo T cell synthesis using a quantitative polymerase chain reaction‐enzyme linked immunosorbent assay (PCR‐ELISA). J Immunol Methods 2000; 237:187–97. [DOI] [PubMed] [Google Scholar]
- 11. Arellano MV, Ordonez A, Ruiz‐Mateos E et al Thymic function‐related markers within the thymus and peripheral blood: Are they comparable? J Clin Immunol 2006; 26:96–100. [DOI] [PubMed] [Google Scholar]
- 12. Dion ML, Sekaly RP, Cheynier R. Estimating thymic function through quantification of T‐cell receptor excision circles. Methods Mol Biol 2007; 380:197–213. [DOI] [PubMed] [Google Scholar]
- 13. Douek DC, McFarland RD, Keiser PH et al Changes in thymic function with age and during the treatment of HIV infection. Nature 1998; 396:690–5. [DOI] [PubMed] [Google Scholar]
- 14. van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen‐induced B cell expansion. J Exp Med 2007; 204:645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiesa R, Gilmour K, Qasim W et al Omission of in vivo T‐cell depletion promotes rapid expansion of naive CD4+ cord blood lymphocytes and restores adaptive immunity within 2 months after unrelated cord blood transplant. Br J Haematol 2012; 156:656–66. [DOI] [PubMed] [Google Scholar]
- 16. Davies EG, Cheung M, Gilmour K et al Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol 2017; 140:1660–70 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaspar HB, Cooray S, Gilmour KC et al Long‐term persistence of a polyclonal T cell repertoire after gene therapy for X‐linked severe combined immunodeficiency. Sci Transl Med 2011; 3:97ra79. [DOI] [PubMed] [Google Scholar]
- 18. Hassan A, Lee P, Maggina P et al Host natural killer immunity is a key indicator of permissiveness for donor cell engraftment in patients with severe combined immunodeficiency. J Allergy Clin Immunol 2014; 133:1660–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naylor K, Li G, Vallejo AN, et al The influence of age on T cell generation and TCR diversity. J Immunol 2005; 174:7446–52. [DOI] [PubMed] [Google Scholar]
- 20. Schonland SO, Zimmer JK, Lopez‐Benitez CM et al Homeostatic control of T‐cell generation in neonates. Blood 2003; 102:1428–34. [DOI] [PubMed] [Google Scholar]
- 21. Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA 2000; 97:9203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu IN, Meyer‐Olson D, Stoll M, Witte T, Schmidt RE, Baerlecken NT. Increased T‐cell turnover is associated with spondyloarthritis in virally suppressed patients with HIV‐1 infection. HIV Med 2015; 16:255–60. [DOI] [PubMed] [Google Scholar]
- 23. Borte S, von Dobeln U, Fasth A et al Neonatal screening for severe primary immunodeficiency diseases using high‐throughput triplex real‐time PCR. Blood 2012; 119:2552–5. [DOI] [PubMed] [Google Scholar]
- 24. Speckmann C, Neumann C, Borte S et al Delayed‐onset adenosine deaminase deficiency: strategies for an early diagnosis. J Allergy Clin Immunol 2012; 130:991–4. [DOI] [PubMed] [Google Scholar]
- 25. Barbaro M, Ohlsson A, Borte S et al Newborn screening for severe primary immunodeficiency diseases in Sweden‐a 2‐year pilot TREC and KREC screening study. J Clin Immunol 2017; 37:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Al‐Mousa H, Al‐Dakheel G, Jabr A et al High incidence of severe combined immunodeficiency disease in Saudi Arabia detected through combined T cell receptor excision circle and next generation sequencing of newborn dried blood spots. Front Immunol 2018; 9:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Al‐Saud BK, Al‐Sum Z, Alassiri H et al Clinical, immunological, and molecular characterization of hyper‐IgM syndrome due to CD40 deficiency in eleven patients. J Clin Immunol 2013; 33:1325–35. [DOI] [PubMed] [Google Scholar]
- 28. Alsum Z, Hawwari A, Alsmadi O et al Clinical, immunological and molecular characterization of DOCK8 and DOCK8‐like deficient patients: single center experience of twenty‐five patients. J Clin Immunol 2013; 33:55–67. [DOI] [PubMed] [Google Scholar]
- 29. Chou J, Alazami AM, Jaber F et al Hypomorphic variants in AK2 reveal the contribution of mitochondrial function to B cell activation. J Allergy Clin Immunol 2020; 146(1):192–202. [DOI] [PubMed] [Google Scholar]
- 30. Kwan A, Abraham RS, Currier R et al Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014; 312:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roifman CM, Somech R, Kavadas F et al Defining combined immunodeficiency. J Allergy Clin Immunol 2012; 130:177–83. [DOI] [PubMed] [Google Scholar]
- 32. Dasouki M, Okonkwo KC, Ray A et al Deficient T Cell Receptor Excision Circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening. Clin Immunol 2011; 141:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mallott J, Kwan A, Church J et al Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol 2013; 33:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patel JP, Puck JM, Srinivasan R et al Nijmegen breakage syndrome detected by newborn screening for T cell receptor excision circles (TRECs). J Clin Immunol 2015; 35:227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deripapa E, Balashov D, Rodina Y et al Prospective study of a cohort of Russian Nijmegen breakage syndrome patients demonstrating predictive value of low kappa‐deleting recombination excision circle (KREC) numbers and beneficial effect of hematopoietic stem cell transplantation (HSCT). Front Immunol 2017; 8:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jilkina O, Thompson JR, Kwan L, Van Caeseele P, Rockman‐Greenberg C, Schroeder ML. Retrospective TREC testing of newborns with severe combined immunodeficiency and other primary immunodeficiency diseases. Mol Genet Metab Rep 2014; 1:324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W, Sun X, Wang J et al Defective thymic output in WAS patients is associated with abnormal actin organization. Sci Rep 2017; 7:11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tamura S, Higuchi K, Tamaki M et al Novel compound heterozygous DNA ligase IV mutations in an adolescent with a slowly‐progressing radiosensitive‐severe combined immunodeficiency. Clin Immunol 2015; 160:255–60. [DOI] [PubMed] [Google Scholar]
- 39. Santisteban I, Arredondo‐Vega FX, Kelly S et al Novel splicing, missense, and deletion mutations in seven adenosine deaminase‐deficient patients with late/delayed onset of combined immunodeficiency disease. Contribution of genotype to phenotype. J Clin Invest 1993; 92:2291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grazioli S, Bennett M, Hildebrand KJ, Vallance H, Turvey SE, Junker AK. Limitation of TREC‐based newborn screening for ZAP70 Severe Combined Immunodeficiency. Clin Immunol 2014; 153:209–10. [DOI] [PubMed] [Google Scholar]
- 41. Marcus N, Stauber T, Lev A et al MHC II deficient infant identified by newborn screening program for SCID. Immunol Res 2018; 66:537–42. [DOI] [PubMed] [Google Scholar]
- 42. Marin AV, Jimenez‐Reinoso A, Briones AC et al Primary T‐cell immunodeficiency with functional revertant somatic mosaicism in CD247. J Allergy Clin Immunol 2017; 139:347–9 e8. [DOI] [PubMed] [Google Scholar]
- 43. Albuquerque AS, Fernandes SM, Tendeiro R et al Major CD4 T‐cell depletion and immune senescence in a patient with chronic granulomatous disease. Front Immunol 2017; 8:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heltzer M, Jawad AF, Rae J, Curnutte JT, Sullivan KE. Diminished T cell numbers in patients with chronic granulomatous disease. Clin Immunol 2002; 105:273–8. [DOI] [PubMed] [Google Scholar]
- 45. Bradley LA, Sweatman AK, Lovering RC et al Mutation detection in the X‐linked agammaglobulinemia gene, BTK, using single strand conformation polymorphism analysis. Hum Mol Genet 1994; 3:79–83. [DOI] [PubMed] [Google Scholar]
- 46. Al‐Mousa H, Abouelhoda M, Monies DM et al Unbiased targeted next‐generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol 2016; 137:1780–7. [DOI] [PubMed] [Google Scholar]
- 47. Villa A, Sobacchi C, Notarangelo LD et al V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 2001; 97:81–8. [DOI] [PubMed] [Google Scholar]
- 48. Villa A, Santagata S, Bozzi F et al Partial V(D)J recombination activity leads to Omenn syndrome. Cell 1998; 93:885–96. [DOI] [PubMed] [Google Scholar]
- 49. Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG‐dependent primary immunodeficiencies. Hum Mutat 2006; 27:1174–84. [DOI] [PubMed] [Google Scholar]
- 50. Schuetz C, Neven B, Dvorak CC et al SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS‐deficient SCID. Blood 2014; 123:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arrendondo‐Vega FX, Santisteban I, Notarangelo LD et al Seven novel mutations in the adenosine deaminase (ADA) gene in patients with severe and delayed onset combined immunodeficiency: G74C, V129M, G140E, R149W, Q199P, 462delG, and E337del. Mutations in brief no. 142. Humn Mut 1998; 11:482. Online. [DOI] [PubMed] [Google Scholar]
- 52. Hirschhorn R, Chen AS, Israni A, Yang DR, Huie ML. Two new mutations at the adenosine deaminase (ADA) locus (Q254X and del nt1050‐54) unusual for not being missense mutations. Hum Mutat 1993; 2:320–3. [DOI] [PubMed] [Google Scholar]
- 53. Al‐Herz W, Massaad MJ, Chou J, Notarangelo LD, Geha RS. DNA recombination defects in Kuwait: clinical, immunologic and genetic profile. Clin Immunol 2018; 187:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Al‐Saud B, Alsmadi O, Al‐Muhsen S et al A novel mutation in purine nucleoside phosphorylase in a child with normal uric acid levels. Clin Biochem 2009; 42:1725–7. [DOI] [PubMed] [Google Scholar]
- 55. Kong XF, Vogt G, Itan Y et al Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet 2013; 22:769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alsmadi O, Al‐Ghonaium A, Al‐Muhsen S et al Molecular analysis of T‐B‐NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet 2009; 10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Telatar M, Wang Z, Udar N et al Ataxia‐telangiectasia: mutations in ATM cDNA detected by protein‐truncation screening. Am J Hum Genet 1996; 59:40–4. [PMC free article] [PubMed] [Google Scholar]
- 58. Teraoka SN, Telatar M, Becker‐Catania S et al Splicing defects in the ataxia‐telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet 1999; 64:1617–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Minegishi Y, Saito M, Tsuchiya S et al Dominant‐negative mutations in the DNA‐binding domain of STAT3 cause hyper‐IgE syndrome. Nature 2007; 448:1058–62. [DOI] [PubMed] [Google Scholar]
- 60. Kwan SP, Hagemann TL, Radtke BE, Blaese RM, Rosen FS. Identification of mutations in the Wiskott–Aldrich syndrome gene and characterization of a polymorphic dinucleotide repeat at DXS6940, adjacent to the disease gene. Proc Natl Acad Sci USA 1995; 92:4706–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jin Y, Mazza C, Christie JR et al Mutations of the Wiskott–Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood 2004; 104:4010–9. [DOI] [PubMed] [Google Scholar]
- 62. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell 1994; 78:635–44. [DOI] [PubMed] [Google Scholar]
- 63. Kolluri R, Shehabeldin A, Peacocke M et al Identification of WASP mutations in patients with Wiskott–Aldrich syndrome and isolated thrombocytopenia reveals allelic heterogeneity at the WAS locus. Hum Mol Genet 1995; 4:1119–26. [DOI] [PubMed] [Google Scholar]
- 64. Wiszniewski W, Fondaneche MC, Louise‐Plence P et al Novel mutations in the RFXANK gene: RFX complex containing in‐vitro‐generated RFXANK mutant binds the promoter without transactivating MHC II. Immunogenetics 2003; 54:747–55. [DOI] [PubMed] [Google Scholar]
- 65. Gono T, Yazaki M, Agematsu K et al Adult onset X‐linked chronic granulomatous disease in a woman patient caused by a de novo mutation in paternal‐origin CYBB gene and skewed inactivation of normal maternal X chromosome. Intern Med 2008; 47:1053–6. [DOI] [PubMed] [Google Scholar]
- 66. Roos D, de Boer M, Kuribayashi F et al Mutations in the X‐linked and autosomal recessive forms of chronic granulomatous disease. Blood 1996; 87:1663–81. [PubMed] [Google Scholar]
- 67. Al‐Muhsen S, Al‐Hemidan A, Al‐Shehri A et al Ocular manifestations in chronic granulomatous disease in Saudi Arabia. J Am Assoc Pediatr Ophthalmol Strabis 2009; 13:396–9. [DOI] [PubMed] [Google Scholar]
- 68. Koker MY, Sanal O, van Leeuwen K et al Four different NCF2 mutations in six families from Turkey and an overview of NCF2 gene mutations. Eur J Clin Invest 2009; 39:942–51. [DOI] [PubMed] [Google Scholar]
- 69. Enders A, Zieger B, Schwarz K et al Lethal hemophagocytic lymphohistiocytosis in Hermansky–Pudlak syndrome type II. Blood 2006; 108:81–7. [DOI] [PubMed] [Google Scholar]
- 70. Rajadhyax M, Neti G, Crow Y, Tyagi A. Neurological presentation of Griscelli syndrome: obstructive hydrocephalus without haematological abnormalities or organomegaly. Brain Dev 2007; 29:247–50. [DOI] [PubMed] [Google Scholar]
- 71. Aksu G, Kutukculer N, Genel F, Vergin C, Omowaire B. Griscelli syndrome without hemophagocytosis in an eleven‐year‐old girl: expanding the phenotypic spectrum of Rab27A mutations in humans. Am J Med Genet A 2003; 116A:329–33. [DOI] [PubMed] [Google Scholar]
- 72. Al‐Dhekri H, Al‐Sum Z, Al‐Saud B et al Successful outcome in two patients with CD40 deficiency treated with allogeneic HCST. Clin Immunol 2012; 143:96–8. [DOI] [PubMed] [Google Scholar]
- 73. Madkaikar M, Gupta M, Chavan S et al X‐linked hyper IgM syndrome: clinical, immunological and molecular features in patients from India. Blood Cell Mol Dis 2014; 53:99–104. [DOI] [PubMed] [Google Scholar]
- 74. Gerstel‐Thompson JL, Wilkey JF, Baptiste JC et al High‐throughput multiplexed T‐cell‐receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population‐based newborn screening. Clin Chem 2010; 56:1466–74. [DOI] [PubMed] [Google Scholar]
- 75. Morinishi Y, Imai K, Nakagawa N et al Identification of severe combined immunodeficiency by T‐cell receptor excision circles quantification using neonatal Guthrie cards. J Pediatrics 2009; 155:829–33. [DOI] [PubMed] [Google Scholar]
- 76. Sottini A, Ghidini C, Zanotti C et al Simultaneous quantification of recent thymic T‐cell and bone marrow B‐cell emigrants in patients with primary immunodeficiency undergone to stem cell transplantation. Clin Immunol 2010; 136:217–27. [DOI] [PubMed] [Google Scholar]
- 77. van der Burg M, Pac M, Berkowska MA et al Loss of juxtaposition of RAG‐induced immunoglobulin DNA ends is implicated in the precursor B‐cell differentiation defect in NBS patients. Blood 2010; 115:4770–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Flowcytometry profiles of T/B lymphocytes and corresponding TREC/KREC counts in patients with primary immune deficiencies. Highlighted cells in the table indicate low counts as defined by the reference ranges: TREC < 15, KREC < 10 and reference ranges for T/B cell subsets: CD3 1,600‐2,700; Helper T CD4 1,000‐1,700; CD8 600‐1,000; CD19 400‐800; NK CD56 &16 200‐400; CD4/CD8 Ratio 1.3‐2.0. NA not available.