

Abstract
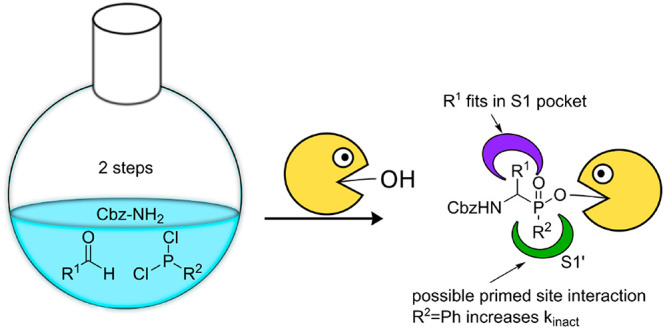
Serine proteases comprise about one-third of all proteases, and defective regulation of serine proteases is involved in numerous diseases. Therefore, serine protease inhibitors are promising drug candidates. Aminomethyl diphenyl phosphonates have been regularly used as scaffolds for covalent serine protease inhibition and the design of activity-based probes. However, they cannot make use of a protease’s primed site. Therefore, we developed a facile two-step synthesis toward a set of phenyl phosphinates, which is a related scaffold but can interact with the primed site. We tested their inhibitory activity on five different serine proteases and found that a phenyl group directly attached to the phosphorus atom leads to superior activity compared with phosphonates.
Keywords: Covalent inhibitors, phosphinates, protease inhibitor, serine proteases
Serine proteases comprise about one-third of all proteases. A multitude of them are implicated in human diseases. Neutrophil elastase, for instance, plays a role in inflammatory diseases such as inflammatory bowel disease,1 chronic obstructive pulmonary disease,2 and cystic fibrosis.3 Furthermore, it is a component of neutrophil extracellular traps (NETs), a network of chromatin and granular proteins that is secreted by neutrophils to trap and kill pathogens.4 Chymase is linked to atherosclerosis and gastric cancer, and numerous other serine proteases are involved in carcinogenesis, tumor progression, and neurodegeneration.5 Hence, serine proteases are attractive drug targets and protease inhibitors represent promising drug candidates. For example, camostat is an approved drug for treatment of chronic pancreatitis.
The development of covalent inhibitors is a re-emerging topic in drug development.6,7 Various recently approved drugs, such as ibrutinib for the treatment of leukemia, have a covalent inhibitory mechanism.7 In the past, several covalent inhibitor scaffolds for serine proteases have been developed, such as isocoumarins,8 benzoxazinones,9N-(sulfonyloxy) and N-(acyloxy) phtalimides,10 sulfonyl11 and phosphonyl fluorides,12 1,2,5-thiadiazolidin-3-one 1,1-dioxides,13 diphenyl phosphonates,14 β-lactams, and more recently oxolactams.15,16 Interestingly, these can not only be used as inhibitors but also as warheads for activity-based probes (ABPs).
α-Aminomethyl diphenyl phosphonates are of particular interest due to their ease of synthesis by a one-step three-component reaction.17 Although usually synthesized as an enantiomeric mixture, the R-configuration, which corresponds to the natural L-configuration of amino acids, is usually the active species.18,19 Because of their close resemblance to amino acid residues, peptidyl diphenyl phosphonates allow for a rational inhibitor design, using knowledge of the protease substrate specificity, which is determined by subsites neighboring the active site. The subsites interact with side chains of individual amino acid residues around the cleaved peptide bond (scissile bond).27 For S1 family proteases, the biggest family of serine proteases, the S1 pocket is the primary recognition site, but other subsites also contribute to selectivity. Therefore, extending phosphonate inhibitors beyond the P1 position can improve selectivity. For example, inclusion of non-natural amino acids in the P2–P4 position has led to highly active and selective inhibitors for the serine protease human neutrophil elastase (HNE).20,21
Aminomethyl diphenyl phosphonates make no efficient use of the primed site of the protease. Although adding substituents to the phenyl group has been shown to influence the activity,22,23 this may be a result of the leaving group capacity, possible primed site interaction, or a combination thereof. However, the leaving groups are not observed in crystal structures of the compounds due to “aging” (Figure 1, upper part). Knowledge of the interaction with the primed site, however, could lead to a potential gain in selectivity. Phosphinates are structurally similar to phosphonates but have one of the ester bonds replaced by a carbon substituent. As a result, phenyl phosphinate esters only possess one leaving group (Figure 1, lower part) but have an additional stereocenter at the phosphorus atom. With their carbon substituent, phosphinates may have the ability to interact with the primed site, thereby potentially increasing selectivity.
Figure 1.

Phosphonate esters (1) and phosphinate esters (2) bind with their side chain to the protease S1 subsite. Phosphinates can potentially make use of the primed sites with their R2 group. Diphenyl phosphonates (upper) react with the nucleophilic active site serine of the protease, thereby expelling a phenoxy leaving group. The second phenoxy group is later lost in an “aging” process, and the negatively charged oxygen occupies the oxyanion hole. Phosphinates react in the same manner but do not undergo the aging process.
A small number of studies have reported phosphinate serine protease inhibitors,24−26 but these studies only incorporated one carbon substituent, preventing a thorough analysis of their influence on activity. Additionally, the synthesis of most of these compounds requires three to five synthetic steps from commercially available starting materials.25
Here we report a facile one-pot (for benzaldehydes) and two-pot (for other aldehydes) two-step synthesis of α-amino phenyl phosphinate esters as covalent, irreversible serine protease inhibitors. We show that they display better inhibition of various S1 family serine proteases compared with diphenyl phosphonates and we present a possible binding mode of the different carbon substituents in the active site of several serine proteases.
Inspired by work on the synthesis of phosphinodepsipeptides by Meng and Xu,28 we explored a two-step one-pot synthesis of the phosphinate scaffold in a pseudo-four-component condensation reaction (Scheme 1). The procedure starts with a three-component Mannich-type reaction between commercially available aldehydes 6, dichlorophosphines 7, and benzyl carbamate (Scheme 1). From these materials, the aldehyde R1 substituent forms the eventual amino acid side chain mimic, whereas the R2 substituent on the phosphine constitutes the potential primed site binding element. The formed phosphinyl chloride intermediate 8 is then reacted with phenol and triethylamine to yield the final phosphinate ester 10. Although successful for benzaldehyde as a starting material, the one-pot procedure did not succeed with aliphatic aldehydes. As an alternative, we directed our efforts toward a two-step synthesis, using the same starting materials (Scheme 2). Here, we isolated intermediate phosphinic acids 9, which were then reacted with phenol in a separate, Steglich-type esterification using DMAP and DIC (Scheme 2). Satisfyingly, this yielded the desired compounds 10–13, as a mixture of diastereomers. Since no purification of the intermediate is necessary, this two-step synthesis is a facile way to access α-amino phosphinate esters with aliphatic as well as aromatic amino acid side chains.
Scheme 1. Two-Step, One-Pot Synthesis of Phosphinate Esters.

Reagents and conditions: (a) CbzNH2, acetonitrile, reflux; (b) TEA, phenol.
Diastereomeric ratio as determined after silica chromatography.
Mimic of amino acid: Phg = phenylglycine.
Scheme 2. Two-Step, Two-Pot Synthesis of Phosphinate Esters.

Reagents and conditions: (a) CbzNH2, acetonitrile, reflux, aqueous workup; (b) phenol, DIC, DMAP, toluene, 80 °C, 4 h.
Diastereomeric ratio as determined after silica chromatography.
Mimic of amino acid: Phg = phenylglycine.
Since one of our aims was to identify the influence of R2, we used different dichlorophosphines to vary this substituent. Unfortunately, our choice was limited by the small amount of commercially available dichlorophosphines. Eventually, we selected three different ones, each with a distinct carbon substituent: 7a with a phenyl substituent would most closely resemble the diphenyl phosphonate counterparts and was the only commercially available dichlorophosphine with an aromatic substituent (1, see Figure 1B). Besides this, 7b with n-propyl as a sterically small and 7c with t-butyl as a bulky, sterically demanding substituent were chosen. The aldehydes were selected according to the proteases to be targeted. We decided for phenylacetaldehyde, benzaldehyde, isovaleraldehyde, and isobutyraldehyde leading to analogues of Phe, Phg, Leu, and Val, respectively.
The free acid intermediates 9 were generally obtained as pure compounds after a simple aqueous workup. Unfortunately, despite several efforts, the reaction with 7b or 7c and phenylacetaldehyde did not yield the desired product. Overall, the obtained yields are generally higher for reactions with n-propyldichlorophosphine and lower for the sterically more demanding t-butyldichlorophosphine, whereas the ones for phenyldichlorophosphine are found in between.
Having the small series of phosphinates in hand, we tested their inhibitory activity on different serine proteases from the S1 family of serine proteases. We decided for neutrophil elastase (NE), porcine pancreas elastase (PPE), and proteinase 3 (PR3) which possess a preference for small hydrophobic amino acids in their S1 site as well as for chymotrypsin (ChT) and cathepsin G (CatG) which in turn prefer cleavage after large hydrophobic amino acids. In order to select active compounds, we first performed a competitive activity-based protein profiling experiment (competitive ABPP) at high inhibitor concentration. To this end, each of these enzymes was incubated with 100 μM of the compounds, and residual activity was measured by labeling with the general serine hydrolase ABP FP-rhodamine.29 As can be seen in Figure 2, NE, PR3, and PPE are best inhibited by the valine derivative 12a, in agreement with their P1 preference. The substituent on the phosphorus, however, seems crucial. Whereas the phenyl substituent results in an active compound (12a), the n-propyl substituent renders the compound inactive (NE and PPE) or dramatically reduces the inhibitory capacity (PR3) and a t-butyl inactivates it completely. For CatG, the Phe derivative 13a is the most active compound. All compounds with the exception of the t-butyl-substituted ones and 12b show some degree of inhibition of ChT, in agreement with the general preference of the digestive protease ChT for large hydrophobic residues in the P1 position. Overall, the compounds with the phenyl substituent on the phosphorus seem most active, followed by the n-propyl ones. The t-butyl leads to inactivity for all tested proteases. In addition to the general screening, we tested 12a as the best compound with a small aliphatic residue in different concentrations on NE and, for comparison, on ChT to get an insight into the compound’s activity and selectivity. Furthermore, we performed a titration of 13a, the best compound with a large hydrophobic residue on ChT as well as NE. To our delight, both of these experiments reveal high selectivity of the individual compound for their respective target protease over the off-target protease (see Figure S1).
Figure 2.

Initial testing of compounds by competitive activity-based protein profiling. Proteases were treated with the indicated compound at 100 μM or DMSO control (−), after which residually active enzyme was fluorescently labeled with the activity-based probe FP-Rh. Hence, a disappearance of a band corresponds to an active compound. NE = human neutrophil elastase, PR3 = human proteinase 3, ChT = bovine chymotrypsin, PPE = porcine pancreatic elastase, CG = human cathepsin G.
Past reports about phosphinate esters as serine protease inhibitors in comparison with phosphonate esters have been contradictory in their results. Boduszek et al. reported di- and tripeptidyl phosphinate esters to be nearly unreactive with NE and PPE, whereas similar phosphonate esters were found to be active.24 In contrast, Walker et al. found that phosphinate esters with a single amino acid in the P1 position displayed consistently higher activity than their phosphonate counterparts.25
To shed more light on the difference in activity of phosphinate and phosphonate esters, we made a comparable assessment of the potency of the here described compounds as well as their diphenyl phosphonate ester counterparts 1a and 1b. To this end, we performed inhibition kinetics experiments. Because of the covalent, irreversible nature of the inhibitors, we used a two-step binding model (Figure S2) consisting of a first, reversible, non-covalent interaction and a second, irreversible reaction between enzyme and inhibitor. Applying this binding model, we determined kinact and KI. kinact is the rate constant of the second, irreversible step in this process at infinite inhibitor concentration and as such an indicator of compound reactivity. The inhibitor concentration that results in an observed rate constant of 1/2kinact is called KI. It reflects the reversible binding process between inhibitor and enzyme before reaction. The overall inhibitory potency is reflected in the ratio of these two values with better inhibitors possessing higher kinact/KI values.
Valine derivatives 12a and 1a were tested against three enzymes with elastase-like activity (NE, PR3, and PPE). Additionally, 12b was tested against PR3. Whereas 12a showed activity with the highest kinact/KI against PR3, the diphenyl phosphonate was mostly inactive, in line with previous reports,22 except against NE (Table 1, entries 1–7). Compound 12b, which only showed activity against PR3 in the gel-based experiments (Figure 2), displayed much lower activity than 12a.
Table 1. Kinetic Values of the Synthesized Inhibitors.
| entry | enzyme | compound | AAa | kinact (10–3 s–1) | KI (μM) | kinact/KI (M–1 s–1) | DRb |
|---|---|---|---|---|---|---|---|
| 1 | NE | 12a | Val | 13.0 ± 3.0 | 51.0 ± 15.4 | 255 ± 96 | 11:89 |
| 2 | 1a (ValP) | Val | 0.59 ± 0.04 | 2.96 ± 0.85 | 200 ± 59 | ||
| 3 | PR3 | 12a | Val | 11.0 ± 1.6 | 26.5 ± 6.2 | 415 ± 115 | 11:89 |
| 4 | 12b | Val | 1.75 ± 0.20 | 83.8 ± 18.3 | 21 ± 5 | 54:46 | |
| 5 | 1a (ValP) | Val | n.d.c | n.d.c | n.i.d | ||
| 6 | PPE | 12a | Val | 7.80 ± 0.79 | 132 ± 19 | 59 ± 10 | 11:89 |
| 7 | 1a (ValP) | Val | n.d.c | n.d.c | n.i.d | ||
| 8 | ChT | 12a | Val | 3.04 ± 0.15 | 7.42 ± 0.83 | 409 ± 50 | 11:89 |
| 9 | 10a | Phg | 1.57 ± 0.07 | 0.46 ± 0.076 | (3.4 ± 0.6) × 103 | 20:80 | |
| 10 | 10b | Phg | 5.84 ± 0.9 | 14.5 ± 3.8 | 404 ± 122 | 55:45 | |
| 11 | 11a | Leu | 21.1 ± 2.0 | 21.6 ± 3.3 | 978 ± 176 | 44:56 | |
| 12 | 11b | Leu | 2.44 ± 0.31 | 13.0 ± 2.9 | 188 ± 48 | 53:47 | |
| 13 | 13a | Phe | 23.0 ± 1.7 | 1.40 ± 0.28 | (16.4 ± 3.6) × 103 | 39:61 | |
| 14 | 1b (PheP) | Phe | 5.63 ± 0.24 | 0.69 ± 0.07 | (8.1 ± 0.9) × 103 |
The tested compound mimics the indicated amino acid.
Compounds were tested as indicated mixtures of diastereomers (either as obtained in the reaction or as a mixture with a DR of approximately 50:50).
n.d.: not determined; since no inhibition was seen, these values were not determined.
n.i.: no inhibition; no good inhibition was seen in a dilution series between 200 and 26.7 μM.
Unfortunately, the kinetic experiments with cathepsin G gave no good progression curves and thus no good fitting was possible in that case. Hence, phosphinates with large hydrophobic P1 residues (Leu, Phe, Phg) were tested against chymotrypsin, as this protease was inhibited by the largest number of compounds (Figure 2) and may be able to give more insight into the primed site effect. Phe in the P1 position is clearly preferred with its kinact/KI value being 5–20-fold higher than the values of the corresponding Phg and Leu derivatives with the same primed site element (Table 1, entries 9–13). Additionally, it is twice as active as the corresponding diphenyl phosphonate inhibitor 1b, which is especially due to a higher kinact. Unfortunately, the Phe phosphinates could only be synthesized with one R2 group. Therefore, comparison of the primed site binding elements can only be made with the Leu and Phg derivatives. In both cases, the inhibitors with a phenyl R2 substituent displayed higher potency. Interestingly, for the Leu phosphinate esters, the difference was mainly caused by a higher kinact for the R2=Ph, whereas, for the Phg phosphinate esters, the underlying potency difference had its cause in a better KI for the R2=Ph compound.
For further insight into the different binding modes, we performed covalent docking with AutoDock 4.230 of some selected compounds in crystal structures of NE, PR3, and ChT (Figure 3). In these experiments, only the R-configurations at the α-carbon to the phosphorus were assessed, because these reflect the natural L-configuration of amino acids and correspond to the active species, as previously shown in structural and kinetics studies.26,31 However, we included both configurations at the phosphorus atom: the bound RR- and RS-diastereomers. Note that the R-configuration at the phosphorus bound to the enzyme originates from the S-configuration at the phosphorus in the inhibitor, because the stereochemistry inverts upon reaction with the active site serine in an SN2P-type mechanism with trigonal bipyramidal transition state (Figure 3D) Strikingly, we found the RS-diastereomer to be consistently superior to the RR-diastereomer in binding energy, with the only exception being 11a (Table S1). Additionally, a visual inspection of the top scoring poses showed that the RR-derivatives generally displayed unlikely binding modes (Figure S3). Therefore, we propose that the RS-diastereomers are probably the (most) active species. Future efforts in synthesis toward enantiomerically pure phosphinate esters may address this further and result in compounds with higher potency.
Figure 3.
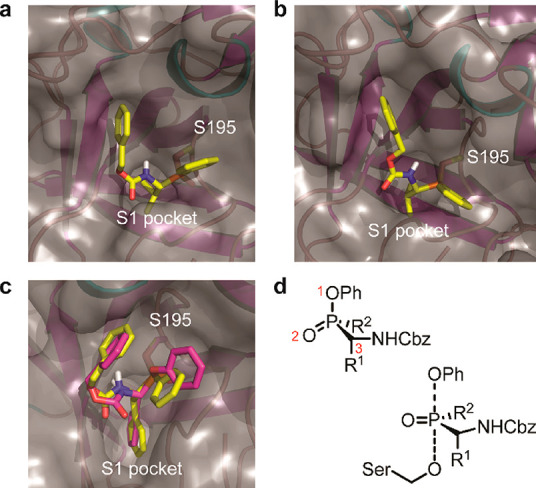
Proposed binding models by in silico covalent docking. Shown are close-ups of the active site with the protein in cartoon format (α-helices in cyan, β-sheets in magenta, loops in pink), molecular surface depicted as gray transparent, and inhibitors as stick models. All phosphinates are colored yellow. Water molecules and inhibitor molecules originally present in the structure were removed prior to docking. (a) Compound 12a covalently bound to S195 of human NE (PDB code: 4WVP). (b) Compound 12a covalently bound to S195 of human PR3 (PDB code: 1FUJ). (c) Compound 13a and diphenyl phosphonate 1b (in magenta) bound to S195 of bovine α-chymotrypsin (PDB code: 4Q2K). (d) Chirality at the phosphorus atom with priorities of the substituents (the P=O bond is treated as a normal single-bonded substituent according to IUPAC rules). Here, the S-enantiomer is depicted. Note that the chirality is inverted upon reaction with the enzyme in an SN2P-type mechanism.
For NE and PR3 we took a closer look at the interactions of 12a. In both proteases the valine side chain docks well into the S1 pocket and the Cbz group occupies the S2 pocket, whereas the phenyl substituent at the phosphorus atom reaches toward the primed site (Figure 4A,B). The better KI value seen for PR3, as measured in the kinetics experiments, may be explained by the somewhat different orientation of the Cbz and Ph groups as well as the preference of PR3 for slightly larger P1 and P2 residues, as has been shown before.20,21,31
Figure 4.
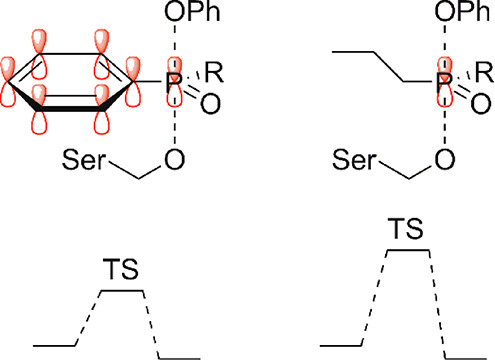
Proposed explanation for the higher reactivity of phosphinate esters with a phenyl ring attached to the phosphorus. In the SN2P reaction mechanism, the orbitals on the phosphorus that form bonds to the incoming nucleophile and outgoing leaving group have overlap with the π-orbitals of the phenyl ring (left), thereby stabilizing this structure and lowering the transition state energy. This stabilization is not present with aliphatic substituents on the phosphorus (right).
For ChT, we compared the most active compound 13a with the reference diphenyl phosphonate 1b. For both compounds, the Phe side chain reaches deep into the S1 pocket and the carboxybenzyl substituent interacts with the S2 pocket. The phenoxy as well as the phenyl group are both oriented toward the S1′ pocket. The overall very similar binding mode may explain their comparable KI values.
In conclusion, we report a straightforward synthetic procedure to synthesize phenyl phosphinates in one to two steps from commercially available aldehydes and dichlorophosphines. These compounds inhibit S1 family serine proteases depending on their P1 and primed site binding elements. As expected, for the P1 position, the compounds follow the known substrate specificity preferences of the targeted enzymes: phosphinates with a P1 valine inhibit elastase-like proteases, whereas those with large, hydrophobic P1 elements are active against proteases with chymotryptic activity.
As a primed site binding element (R2), a phenyl substituent was generally preferred. More particular, these compounds were superior to phosphonate esters, in line with a previous study by Walker et al.25 In all compounds, the phenyl was also preferred over the n-propyl substituent, and all compounds with t-Bu substituents on the phosphorus atom were inactive. The last observation may be explained by a steric clash: the bulky t-Bu group could either prevent attack of the active site serine or substantially disturb the interaction with the enzyme (i.e., raising KI). However, steric hindrance is not a logical explanation for the lower activity of the compounds with n-Pr substituents. Interestingly, the reason for the activity difference is mainly rooted in a better kinact of the phosphinates with the Ph substituent. Hence, it is not the initial binding to the enzyme’s active site but rather the reaction rate with the active site serine residue that underlies the difference in activity. Remarkably, the kinact was also higher than the one for diphenyl phosphonates. This may be counterintuitive, because the electrophilicity of the phosphorus in phosphinate esters is lower than that for phosphonate esters, indicated by a lower partial Gasteiger charge as calculated during ligand preparation for in silico docking. As a possible explanation for the increased reactivity, we here propose that the R2 phenyl ring stabilizes the transition state in the reaction with the enzyme (Figure 4).
We also found in compounds 12 (the Val series of phosphinates) that the n-Pr substituent only showed activity, albeit with low potency, against PR3 but not against NE. This result may offer future possibilities to obtain inhibitors that are selective for PR3 over the closely related NE. It also illustrates the capacity of the primed site to influence the binding to the target protease. Overall, we expect that phosphinate inhibitors with extended primed site elements as well as residues that bind the S2–S4 sites will lead to more specific inhibitors and activity-based probes. Work along these lines is currently being investigated and will be reported in due course.
Acknowledgments
We thank Luc Baudemprez for recording NMR spectra.
Glossary
Abbreviations
- ABPP
activity-based protein profiling
- CatG
cathepsin G
- Cbz
carboxybenzyl
- ChT
chymotrypsin
- DIC
N,N′-diisopropylcarbodiimide
- DMAP
4-dimethylaminopyridine
- DR
diastereomeric ratio
- HNE
human neutrophil elastase
- NET
neutrophil extracellular trap
- Phg
phenyl glycine
- PPE
porcine pancreas elastase
- PR3
proteinase 3
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00284.
Supplemental figures, supplemental tables, experimental methods, and copies of NMR spectra and LC-MS chromatograms (PDF)
Author Contributions
S.H.L.V. and J.P.K. developed the project and designed compounds, S.L. synthesized compounds, J.P.K. performed biochemical analysis, M.A.T.v.d.P. performed covalent docking, J.P.K. wrote the manuscript, and S.H.L.V. revised the manuscript. All authors have given approval to the final version of the manuscript.
We acknowledge funding by the FWO (project grant G0D8617N and PhD fellowship 1S18520N to J.P.K.), the Ministerium für Kultur and Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin–inkl. Wissenschaft and Forschung, and the Bundesministerium für Bildung and Forschung.
The authors declare no competing financial interest.
This paper was originally published ASAP on August 13, 2020. A revised Supporting Information file was uploaded, and the paper reposted on August 14, 2020.
Supplementary Material
References
- Anderson B. M.; et al. Application of a chemical probe to detect neutrophil elastase activation during inflammatory bowel disease. Sci. Rep. 2019, 9, 13295. 10.1038/s41598-019-49840-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenderdos K.; Condliffe A. The neutrophil in chronic obstructive pulmonary disease: Too little, too late or too much, too soon?. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- Roghanian A.; Sallenave J. M. Neutrophil elastase (NE) and NE inhibitors: canonical and noncanonical functions in lung chronic inflammatory diseases (cystic fibrosis and chronic obstructive pulmonary disease). J. Aerosol Med. Pulm. Drug Delivery 2008, 21, 125–144. 10.1089/jamp.2007.0653. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; et al. Neutrophil extracellular traps kill bacteria. Science (Washington, DC, U. S.) 2004, 303, 1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Rachel K. V.; Sirisha G. V. D.. Serine proteases and their inhibitors in human health and disease. In Proteases in Human Diseases; Chakraborti S., Chakraborti T., Dhalla N. S., Eds.; Springer Nature: 2017; pp 195–226. 10.1007/978-981-10-3162-5. [DOI] [Google Scholar]
- Bauer R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20, 1061–73. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Samanta I.; Mondal A.; Liu W. R. Covalent Inhibition in Drug Discovery. ChemMedChem 2019, 14, 889–906. 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. W.; Powers J. C. Reaction of serine proteases with substituted 3-alkoxy-4-chloroisocoumarins and 3-alkoxy-7-amino-4-chloroisocoumarins: new reactive mechanism-based inhibitors. Biochemistry 1985, 24, 7200–7213. 10.1021/bi00346a028. [DOI] [PubMed] [Google Scholar]
- Gütschow M.; Neumann U.; Sieler J.; Eger K. Studies on 2-benzyloxy-4H-3,1-benzoxazin-4-ones as serine protease inhibitors. Pharm. Acta Helv. 1998, 73, 95–103. 10.1016/S0031-6865(98)00003-X. [DOI] [PubMed] [Google Scholar]
- Neumann U.; Gütschow M. N-(sulfonyloxy)phthalimides and analogues are potent inactivators of serine proteases. J. Biol. Chem. 1994, 269, 21561–21567. [PubMed] [Google Scholar]
- Fahrney D. E.; Gold A. M. Sulfonyl Fluorides as Inhibitors of Esterases. I. Rates of Reaction with Acetylcholinesterase, α-Chymotrypsin, and Trypsin. J. Am. Chem. Soc. 1963, 85, 997–1000. 10.1021/ja00890a037. [DOI] [Google Scholar]
- Ni L. M.; Powers J. C. Synthesis and kinetic studies of an amidine-containing phosphonofluoridate: A novel potent inhibitor of trypsin-like enzymes. Bioorg. Med. Chem. 1998, 6, 1767–1773. 10.1016/S0968-0896(98)00109-6. [DOI] [PubMed] [Google Scholar]
- Groutas W. C.; et al. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry 1997, 36, 4739–4750. 10.1021/bi9628937. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J.; Powers J. C. Irreversible inhibition of serine proteases by peptidyl derivatives of alpha-aminoalkylphosphonate diphenyl esters. Biochem. Biophys. Res. Commun. 1989, 161, 143–149. 10.1016/0006-291X(89)91572-6. [DOI] [PubMed] [Google Scholar]
- Mulchande J.; et al. Azetidine-2,4-diones (4-oxo-β-lactams) as scaffolds for designing elastase inhibitors. J. Med. Chem. 2008, 51, 1783–1790. 10.1021/jm701257h. [DOI] [PubMed] [Google Scholar]
- Mulchande J.; et al. 4-Oxo-β-lactams (azetidine-2,4-diones) are potent and selective inhibitors of human leukocyte elastase. J. Med. Chem. 2010, 53, 241–253. 10.1021/jm901082k. [DOI] [PubMed] [Google Scholar]
- Oleksyszyn J.; Subotkowska L.; Mastalerz P. Diphenyl 1-Aminoalkanephosphonates. Synthesis 1979, 1979, 985–986. 10.1055/s-1979-28903. [DOI] [Google Scholar]
- Häußler D.; et al. Phosphono Bisbenzguanidines as Irreversible Dipeptidomimetic Inhibitors and Activity-Based Probes of Matriptase-2. Chem. - Eur. J. 2016, 22, 8525–8535. 10.1002/chem.201600206. [DOI] [PubMed] [Google Scholar]
- Schulz-Fincke A. C.; Blaut M.; Braune A.; Gütschow M. A BODIPY-Tagged Phosphono Peptide as Activity-Based Probe for Human Leukocyte Elastase. ACS Med. Chem. Lett. 2018, 9, 345–350. 10.1021/acsmedchemlett.7b00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Kasperkiewicz P.; et al. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2518–2523. 10.1073/pnas.1318548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasperkiewicz P.; Altman Y.; D’Angelo M.; Salvesen G. S.; Drag M. Toolbox of Fluorescent Probes for Parallel Imaging Reveals Uneven Location of Serine Proteases in Neutrophils. J. Am. Chem. Soc. 2017, 139, 10115–10125. 10.1021/jacs.7b04394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieńczyk M.; et al. Simple phosphonic inhibitors of human neutrophil elastase. Bioorg. Med. Chem. Lett. 2011, 21, 1310–1314. 10.1016/j.bmcl.2011.01.083. [DOI] [PubMed] [Google Scholar]
- Wang C.; Abegg D.; Dwyer B. G.; Adibekian A. Discovery and Evaluation of New Activity-Based Probes for Serine Hydrolases. ChemBioChem 2019, 20, 2212–2216. 10.1002/cbic.201900126. [DOI] [PubMed] [Google Scholar]
- Boduszek B.; Brown A. D.; Powers J. C. α-aminoalkylphosphonate di(chlorophenyl) esters as inhibitors of serine proteases. J. Enzyme Inhib. 1994, 8, 147–158. 10.3109/14756369409020197. [DOI] [PubMed] [Google Scholar]
- Hamilton R.; Wharry S.; Walker B.; Walker B. J. The synthesis of phosphinic acid based proteinase inhibitors. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 144, 761–764. 10.1080/10426509908546356. [DOI] [Google Scholar]
- Walker B.; et al. Asymmetric preference of serine proteases toward phosphonate and phosphinate esters. Biochem. Biophys. Res. Commun. 2000, 276, 1235–1239. 10.1006/bbrc.2000.3597. [DOI] [PubMed] [Google Scholar]
- Meng F.; Xu J. Synthesis of phosphinodepsipeptides via the pseudo-four-component condensation reaction. Tetrahedron 2013, 69, 4944–4952. 10.1016/j.tet.2013.04.032. [DOI] [Google Scholar]
- Patricelli M. P.; Giang D. K.; Stamp L. M.; Burbaum J. J. Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics 2001, 1, 1067–1071. . [DOI] [PubMed] [Google Scholar]
- Bianco G.; Forli S.; Goodsell D. S.; Olson A. J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. 10.1002/pro.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtenberg B. C.; Kasperkiewicz P.; Robinson H.; Drag M.; Riedl S. J. The elastase-PK101 structure: Mechanism of an ultrasensitive activity-based probe revealed. ACS Chem. Biol. 2015, 10, 945–951. 10.1021/cb500909n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.