

Abstract

Small GTPase protein Rho-kinase (ROCK) plays an important role in the pathogenesis of hypertension. Inhibition of ROCK II brings about the biochemical changes leading to vascular smooth muscles relaxation, finally resulting into potent antihypertensive activity. In the quest for potent ROCK-II inhibitors, a ligand-based pharmacophore containing four essential chemical features, namely two hydrogen bond acceptor (HBA), one hydrogen bond donor (HBD), and one hydrophobe (HY), was developed and rigorously validated. The pharmacophore was used for virtual screening, and hits retrieved from the National Cancer Institute (NCI) database were sorted on the basis of fit value, estimate value, and Lipinski’s violation. Potential feature interaction of hits was also observed during docking studies with the amino acids present in the active site of Rho-kinase. Based on the above screening, three hits (NSC 2488, NSC 2888, and NSC 4231) were chosen and subjected to in vitro Rho-kinase enzyme-based assay, followed by ex vivo rat aortic vasodilatory assay. All three compounds showed good biological activity as predicted by the model and confirmed by the docking studies.
Keywords: Rho-kinase, in vitro enzyme assay-based evaluation, hypertension, in silico drug designing
Rho-kinase (p164 protein) is the first discovered downstream effector of small GTPase-binding protein RhoA. The Rho-kinase (ROCK) belongs to serine-threonine protein kinases, and Rho gets activated by guanine nucleotide exchange factors (Figure 1). GTP-bound RhoA phosphorylates several important substrates such as the myosin-binding subunit of myosin light chain phosphatase (MLCP).1 ROCK inhibits the dephosphorylation of myosin light chain (MLC) and regulates the organization of actin cytoskeleton, along with various cellular activities, such as cell adhesion, secretion, migration, proliferation, and gene expression. Thus, the roles of ROCK inhibitors are to normalize the abnormal activation of the ROCK/Rho pathway. From the aforementioned discussion, it looks very clear that ROCK inhibitors can be of immense utility in the treatment of cardiovascular-related diseases.2
Figure 1.

Overview of Rho-kinase (ROCK) signaling and its activation and the factors leading to vascular smooth muscle cell contraction. GTP-bound RhoA leads to phosphorylation of myosin light chain phosphatase (MLCP); this inhibits the MLCP activity and increased myosin II phosphorylation. (Chem Draw Ultra Version 12.0, http://www.CambridgeSoft.com).
Rho-kinase consists of two isoforms: ROCK-I and ROCK-II. Expressions of human genes (ROCK-I and ROCK-II) are situated on chromosome 18 (18q11.1) and chromosome 2 (2p24), respectively. Both the isoforms share 60% overall and 92% identical distinctive amino acid sequences in their kinase domain. The enzyme constitutes two amino terminals, named as N-terminal kinase domain and C-pleckstrin homology (PH) domain, along with Cysteine-rich Zn finger motif and the complex coiled-coil region, which is the most active ATP-competitive binding site domain of Rho-kinase (Figure 2), where all the ROCK inhibitors would probably bind.3 ROCK-I is ubiquitously found in the lung, liver, kidney, spleen, and testes, while an abundance of ROCK-II is found in the brain, heart, and skeletal muscle. ROCK-I plays a significant role in various malignant disorders whereas ROCK-II is involved in hypertension and associated vascular smooth-muscle contraction. Upon the cleavage of caspase-3, ROCK-I gets activated whereas cleavage of granzyme-B leads to ROCK-II activation.4 Previous research has clearly shown that ROCK tends to facilitate smooth muscle contraction in tunica media of arterial wall,65 which revealed therapeutic potential of the ROCK pathway in the treatment of cardiovascular ailments. Fasudil/HA1077 (ROCK inhibitor) is the only marketed drug used in Japan for cerebral vasospasm. Initially, Fasudil was used as an intracellular calcium channel antagonist and protein kinase C inhibitor; later its vasodilatory property was observed.5 The studies have also revealed the crucial role of the Rho/ROCK pathway in diabetes-induced retinal microvasculopathy. Rho/ROCK inhibitor Fasudil protects the vascular endothelium by inhibiting neutrophil adhesion and reducing neutrophil-induced endothelial injury.
Figure 2.

Molecular structure of Rho-kinase isoform and its sequences. It consists of three domains, N-terminus entailed catalytic site of an enzyme, followed by a coiled-coil region comprising the Rho-binding domain. C-terminal is a Pleckstrin-homology (PH) with an internal cysteine-rich domain (CRD). (Chem Draw Ultra Version 12.0, http://www.CambridgeSoft.com).
Isoform selectivity is a major challenge in the development of ROCK inhibitors because of the high level of similarity between ROCK-I and II. Noticeably, the ATP binding pockets of the two isoforms are 100% identical. Putting together this poses a challenge and limitation for design of isoform specific inhibitors. Even the only clinically used ROCK inhibitor Fasudil is a nonspecific ROCK inhibitor producing multiple effects due to nonspecificity with respect to ROCK-I and II. Thus, there is an urgent need to develop ROCK-II isoform-selective inhibitors for the treatment of hypertension and related diseases.
In light of the proof of concept regarding the potent vasodilatory effect of ROCK-II inhibitors, we have attempted to develop a validated pharmacophore model using a series of isoform specific inhibitors reported in the literature.6 The model was used for virtual screening, and the identified hits were subjected to biological evaluation to experimentally validate them.
Pharmacophore Generation
As a starting point, a data set of 41 ROCK-II inhibitors6 were selected and subjected to conformation generation (Table S1) using the Best flexible conformation option available in Discovery Studio (v2.0). A total of 255 conformers were generated for each molecule of the selected training set using an energy threshold of 20.0 kcal/mol above the global minima energy in both torsional and cartesia.
With an aim to identify most crucial and common pharmacophoric features, Hypogen and HipHop (common-feature pharmacophore) models were generated.
The features obtained from common feature pharmacophore modeling (Hip Hop) were used to generate quantitative pharmacophore models (Hypogen). Three compounds (8a, 12j, and 14c) showed a high error ratio; hence, they were removed from the data set. The probable reason for these three compounds to behave as outliers could be due to inappropriate experimentation or due to different mechanism of action. Out of 10 hypotheses, the best hypothesis comprising 2HBA, 1HY, and 1HBD was selected. Pharmacophoric features along with their interfeature distances are shown in Figure 3. The chosen pharmacophore model was evaluated for statistical significance and it showed root-mean-square deviation (RMSD) of 0.98, correlation coefficient of 0.90, weight of 2.56, and configuration of 14.93 (Table S2) (Figure 4). Statistically fit model was able to differentiate between active and inactive ROCK-II inhibitors and the results of pharmacophore mapping signified this fact. For instance, most active compound of the training set (12a) showed full chemical feature mapping (Figure 5a) whereas the least active compound missed the 1HBD feature (Figure 5b).
Figure 3.
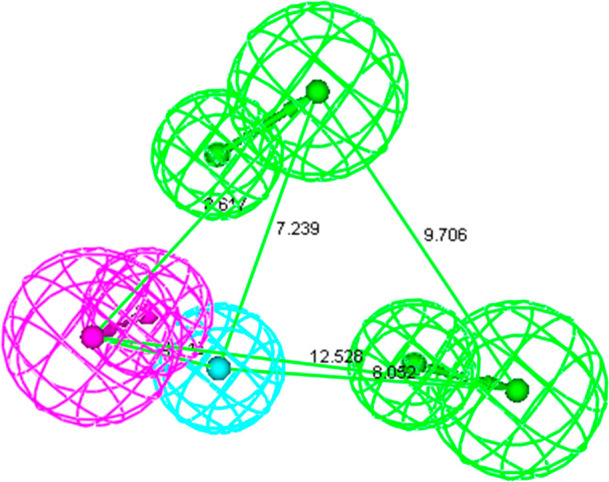
Two HBA, one HY, and one HBD pharmacophoric feature along with interfeature distances.
Figure 4.
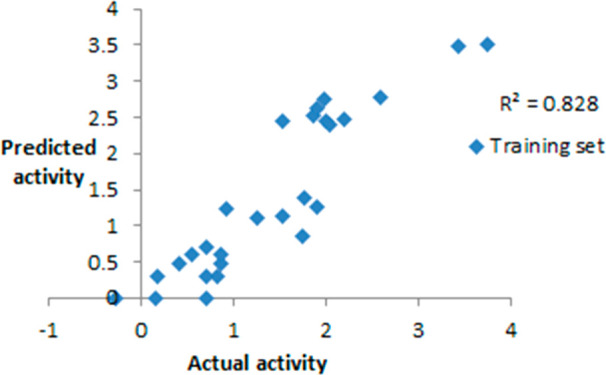
Correlation coefficient values of training set compounds.
Figure 5.
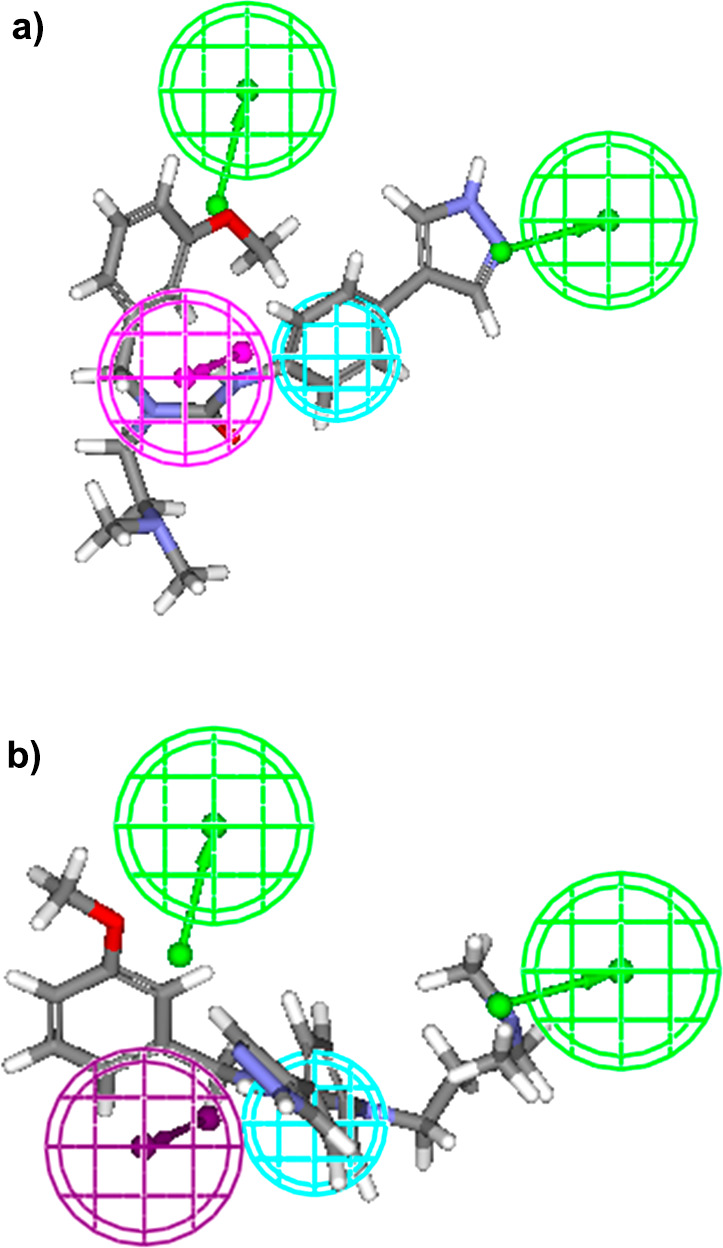
(a) Most active compound of the training set exhibiting full four feature mapping with the ligand-based pharmacophore. (b) Least active compound of the training set exhibiting three feature mapping with the ligand-based pharmacophore. (Accelrys Discovery Studio Version 2.0, http://www.rcsb.org.)
Pharmacophore Model Validation
Cat Scramble or Fischer’s Randomization Test
Using the same chemical features and parameters that were used for model development, 99 spreadsheets were generated at a 99% confidence level.7 Statistical data of the selected pharmacophore model was better than randomly generated models, which means that the selected pharmacophore is not generated by chance correlation (Figure 6).
Figure 6.

Graph of 99% Cat Scramble/Fischer’s randomization test. (Accelrys Discovery Studio Version 2.0, http://www.rcsb.org).
Internal and External Test Set Validation
The prediction capability of the model was assessed using 11 internal test set compounds and 18 external test set compounds comprising a series of indoles and 7-azaindole derivatives (Figure 7).
Figure 7.
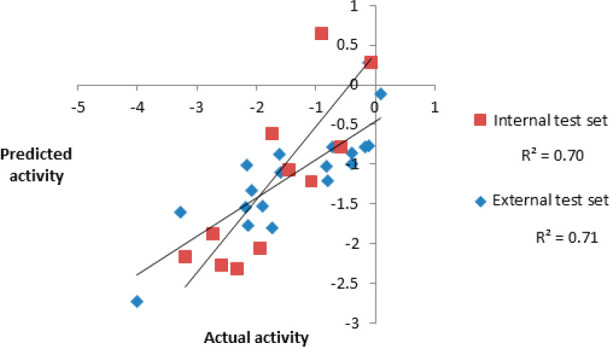
Actual activity versus predicted activity for the internal and external test set compounds.
The correlation coefficient values of 0.70 and 0.71 obtained for internal and external test sets undoubtedly indicated the high predictability and universality of the model. Figure 8 illustrates the full feature mapping of the most active compound of the external test set.
Figure 8.
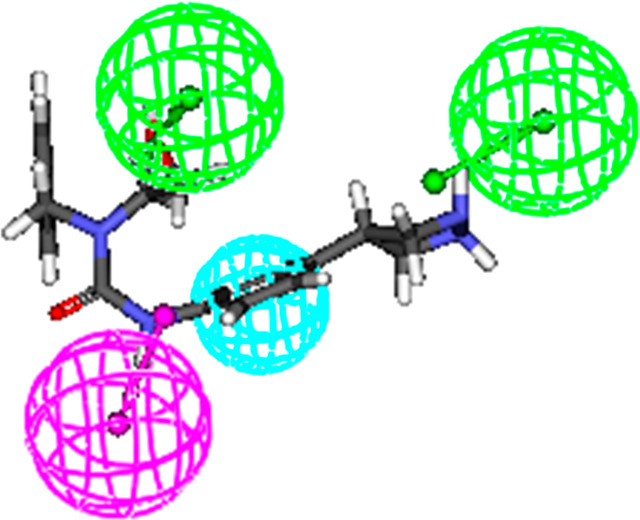
Most active compound of external set showing full feature mapping with ligand-based pharmacophore. (Accelrys Discovery Studio Version 2.0, http://www.rcsb.org.)
Rm2 matrices, developed by Roy et al., were used to compute the Rm2 matrices (average rm2 and delta rm2) for checking the proximity between estimated and actual activities. The recommended values of average and delta rm2 are >0.5 and <0.2. The average and delta rm2 for the training set was found to be 0.90 and 0.08, respectively, which clearly shows the soundness of the model.
Güner–Henry Scoring Method
The virtual screening capability of the ligand-based pharmacophore model was checked by the Güner–Henry Scoring technique. The parameters like % yield of actives, enrichment factor, false positives, and goodness of hit score (GH scoring method) were calculated and evaluated. 360 compounds appeared as actives from a pool of 440 ROCK-II inhibitors. The model showed 94% yield with an enrichment factor of 0.77. Twenty % of compounds appeared as false positives. The overall GH score of the model was 0.68, which is better than the recommended GH score (≥0.60). The result of the GH score signifies the capability of the model to estimate correctly the activity of compounds (Table S3).
Virtual Screening
The validated pharmacophore model was used as a query to search the compounds from NCI and Maybridge database comprising 2, 38, 819, and 53,000 chemical structures, respectively.8 295 NCI and 205 Maybridge hits were retrieved and sorted on the basis of the Lipinski rule of five. The remaining hits were further screened on the basis of fit value (greater than 7.5) and estimated value (less than 21 nM). This process led to the identification of seven hits NSC 2888, NSC 4231, NSC 2488, NSC 3597, NSC 2101, NSC 3837, and NSC 3596 (Table S4). Out of seven hits, three hits [NSC 2488: 1-(((6-methoxyquinolin-4-yl)methyl)amino)butan-2-ol, NSC 4231: methyl 4-(methyl(2-oxo-1,2-diphenylethyl)amino)benzoate, and NSC 2888: 2-((3-((methoxymethyl)thio)butyl)thio) ethanol] showed good fit values (greater than 8.0) and high predicted activity (less than 13 nM); hence, they were subjected to molecular docking and biological evaluation studies.
Molecular Docking
Selected NSC hits and the standard compound (Y-27632) were subjected to molecular docking studies, to understand the possible molecular interaction between the functional groups of the hits and active site amino acids of ROCK-II protein structure (4L6Q). NSC 2488 with LibDock score of 86.33 (Figure 9) showed one hydrogen bond interaction, three van der Waals, and five hydrophobic interactions. The nitro group of 6-methoxyquinolin-4-yl showed hydrogen bonding with GLY 104. On the other hand, LYS 121 and PHE 103 exhibited van der Waals interactions with the methyl group. GLY 234 showed van der Waals interaction with the hydroxyl group of 2-butanol. ASN 219, ASP 232, MET 169, ALA 119, and VAL 106 created a hydrophobic environment around NSC 2488.
Figure 9.
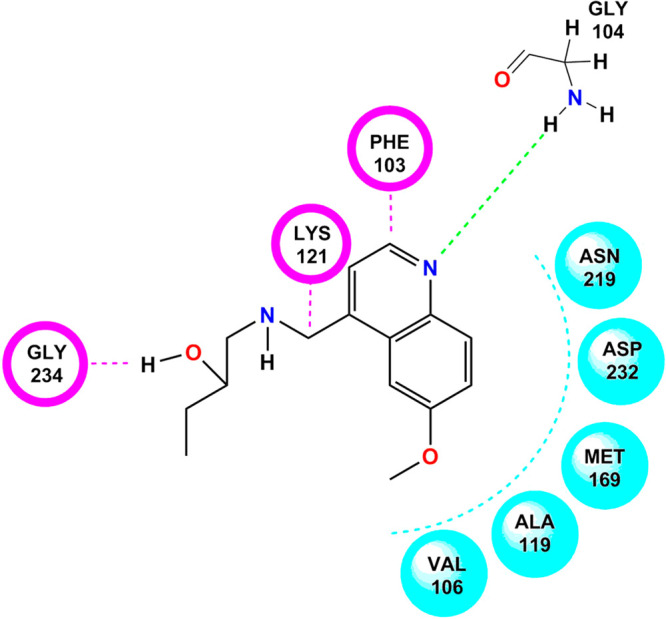
Docking pose of NSC 2488 on the active domain of Rho-kinase. Colorful dotted lines indicate the bonding interaction with the particular functional group of the amino acid. (Chem Draw Ultra Version 12.0 http://www.CambridgeSoft.com.)
With the observed LibDock score of 63.43, NSC 2888 exhibited three hydrogen bond interactions, two van der Waals interactions, and seven hydrophobic interactions. The oxygen of the ethanol group present in NSC 2888 showed hydrogen bonding with GLY 104 and PHE 103, while the other hydrogen bonding interaction was observed at the methoxy group with ALA 231. VAL 106 and LYS 121 exhibited van der Waals interactions. TYR 171, VAL 153, ILE98, ASN 219, ASP 218, MET 172, and VAL 106 created a hydrophobic pocket around the structure as shown in Figure 10.
Figure 10.
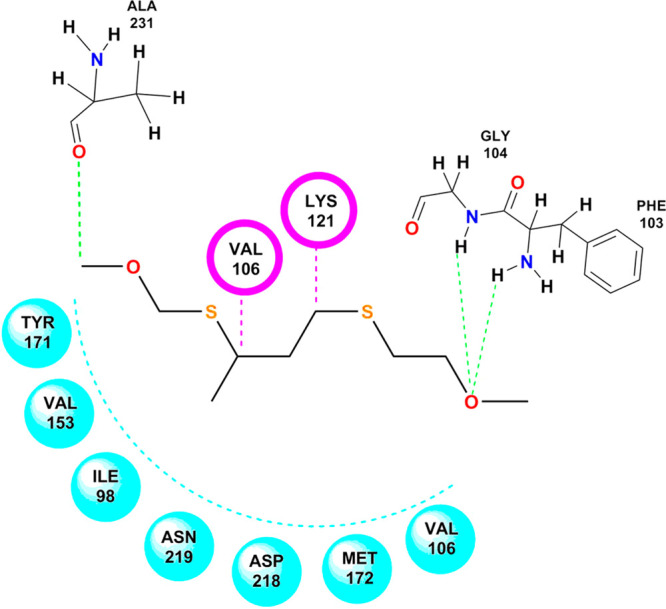
Docking pose of NSC 2888 on the active domain of Rho-kinase. (Chem Draw Ultra Version 12.0 http://www.CambridgeSoft.com.)
In NSC 4231, two hydrogen bonds, three van der Waals interactions, and five hydrophobic interactions were seen along with a good LibDock score of 95.92. The hydrogen bonding appeared between the oxygen of 1,2-diphenylethyl and GLY 104 and PHE 103. Whereas ASP 218, ASN 219, and ALA 231 showed Van der Waals interactions with the benzoic acid functionality, LEU 221, ASP 232, VAL 106, PHE 233, and LYS 121 were observed to be responsible for the hydrophobic environment around the structure of NSC 4231 (Figure 11).
Figure 11.
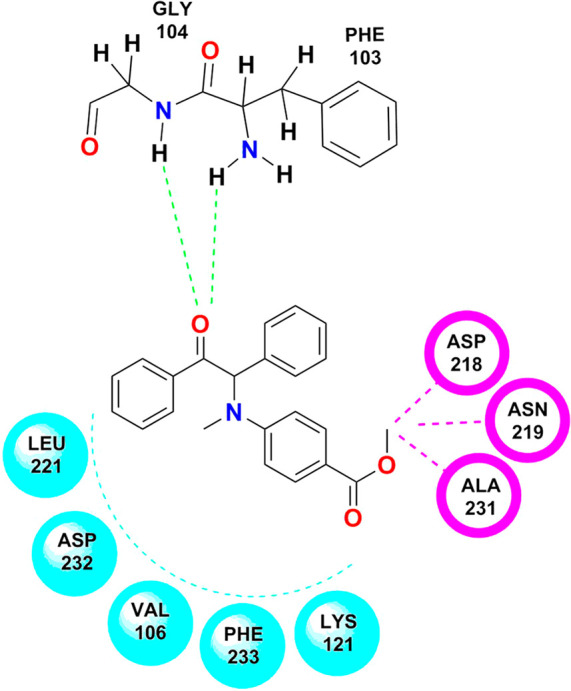
Docking pose of NSC 4231 on the active domain of Rho-kinase. (Chem Draw Ultra Version 12.0 http://www.CambridgeSoft.com.)
The reference compound Y-27632 showed interactions similar to test compounds. For instance, NSC 2488, NSC 2888, and NSC 4231 exhibited the same hydrogen bonding interactions as shown by Y-27632. GLY 104 and PHE 103 showed hydrogen bonding interactions with the oxygen group present in Y-27632. Van der Waal’s interactions were observed between the structure of Y-27632 and PHE 136, THR 235, and LYS121. ALA 119, GLY 234, VAL 106, and ILE 98 showed hydrophobic interactions with the structure of Y-27632 (Figure 12).
Figure 12.
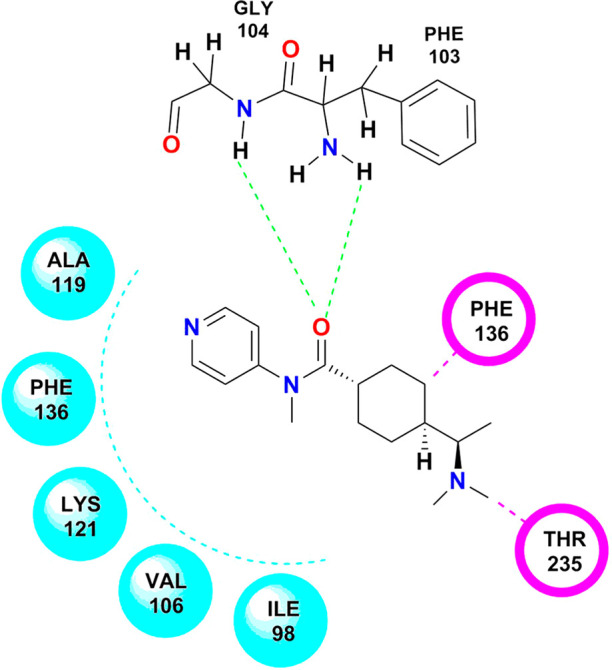
Docking pose of Y-27632 on the active domain of Rho-kinase. (Chem Draw Ultra Version 12.0 http://www.CambridgeSoft.com.)
Biological Evaluation
Enzyme-Based Rho-Kinase Inhibitory Assay
Test compounds were evaluated for Rho-kinase inhibitory activity using a microplate based Rho-associated kinase (ROCK) activity assay. Results of the study indicated that all three hits (NSC 2488, NSC 2888, and NSC 4231) are active with IC50 values of 8.02 nM, 8.4 nM, and 9.01 nM, respectively, when compared to the IC50 value of standard drug Y-27632, which was observed to be 8.3 nM (Figure 13).
Figure 13.
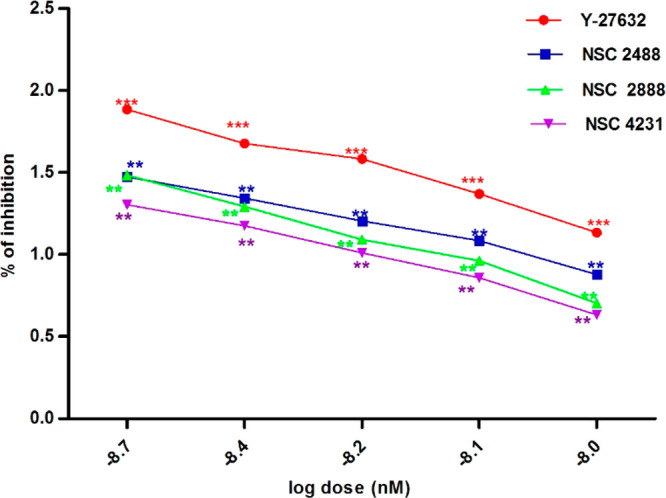
Rho-kinase inhibition at different concentrations of compounds: (a) Y-27632, (b) NSC 2488, (c) NSC 2888, and (d) NSC 4231.
Ex Vivo Isometric Contraction-Based Vasodilatory Assay
After obtaining promising results in the Rho-kinase enzyme-based inhibitory assay, all the test compounds and standard were examined for vasodilatory activity using precontracted rat aortic strips. Exposure of test compounds exhibited concentration-dependent relaxation and produced vasodilatory activity similar to standard Y-27632 (Figures 14a, b, and c). The EC50 of each test compound NSC 2888, NSC 2488, and NSC 4231 and standard was found to be 8.5 nM, 8.4 nM, 9.0 nM, and, 9.5 nM respectively.
Figure 14.

(A) Percentage of relaxation of the precontracted aortic strip at different concentrations of the compound (NSC 2488) and standard drug (Y-27632). (B) Percentage of relaxation of the precontracted aortic strip at different concentrations of the compound (NSC 2888) and standard drug (Y-27632). (C) Percentage of relaxation of the precontracted aortic strip at different concentrations of the compound (NSC 4231) and standard drug (Y-27632).
Repercussions of irregular RhoA/ROCK activity seem to be the up-regulation of renin-angiotensin aldosterone system and subsequent increase in arterial pressure coupled with increased production of reactive oxygen species (ROS). It has been well established that the inhibition of this pathway leads to vasoprotection and vasorelaxation.9
In order to get selective and structurally diverse ROCK-II inhibitors, an in silico approach was laid down along with in vitro and ex vivo studies to identify new ROCK-II inhibitors. During in silico studies, the developed pharmacophore exhibited the importance of HBA, HY, and HBD features in eliciting the RhoA/ROCK inhibitory activity. Earlier study reported10 that the pharmacophore model having 1HBA and 2HY chemical features have the ability to interact and inhibit RhoA/ROCK. In contrast to this, we observed that in addition to HBA and HY, one HBD if present can substantiate the inhibitory activity by interacting within the hinge region of ATP binding site of the enzyme. The validated four feature (2HBA, 1HY, and 1HBD) quantitative pharmacophore model was used as a query for virtual screening of large database. The retrieved hits were sorted on the basis of fit value, estimated activity, and Lipinski’s violation. Three potent virtual hits (NSC 2488, NSC 2888, and NSC 4231) were chosen for biological evaluation (Rho-kinase inhibitory assay and rat aortic ring based vasodilatory assay), but prior to this molecular docking studies were performed.
Interestingly all the three selected hits docked perfectly onto the Rho-kinase II active site and exhibited significant hydrogen bond interaction between ligand and the bottom region of the kinase domain. Previous studies revealed the importance of methionine, valine, and isoleucine in the hydrophobic interaction and tyrosine in π–π interaction. The present study also exhibited a similar kind of interaction between NSC 2488, NSC 2888, and NSC 4231 and active site amino acids (Figure S1). The type of interactions observed during docking clearly shows agreement between standard drug Y-27632 and the identified hits, which reflects the structural analogy between standard Rho-kinase inhibitor and the hits.
Recently reported studies revealed that the irregular progression of Rho-kinase enzyme leads to metabolic disorders,11 and its obvious fact that hypertension is the major component of metabolic syndrome. It is noteworthy that the up-regulation of Rho-kinase has been considered as a key factor in the pathogenesis of hypertension. Thus, inhibition of Rho-kinase would be a rational strategy in combating hypertension and associated risk factors.
In light of this and motivation received from pharmacophore and docking results, in vitro RhoA/ROCK enzyme inhibitory assay and the ex-vivo vasodilatory assay were performed.
In vitro enzyme, inhibitory assay was executed using microplate reader Synergy HI Biotek Gen 5.0. Out of three hits, NSC 2488 exhibited the highest inhibitory potential (IC50 = 8.02 nM) and showed equal potency to that of Y-27632. Rho-kinase inhibitory activity of NSC 2488 was also observed to be in agreement with high LibDock score of 122 and good estimated activity (0.98 nM). The other two hits also showed good enzyme inhibitory activity but slightly less than NSC 2488. In light of good in vitro activity, all three hits along with standard ROCK inhibitor were subjected to ex-vivo vasodilatory assay using precontracted aortic tissue. Interestingly, here also NSC 2488 produced remarkable vasodilator activity (EC50 = 8.4 nM) similar to standard. EC50 values of the other two hits were also observed to be in the nanomolar range, exhibiting their good Rho-kinase enzyme inhibitory potential.
The Tanimoto index is often used to determine the structural similarity of molecules between a set of established ligands and helps to provide information regarding specific substructures within the molecule. In the present study, the Tanimoto index protocol available in DS was utilized in order to check the novelty of the obtained hits and the Tanimoto index should be less than 0.5 for a hit to be considered as novel.
All the three identified hits NSC 2488, NSC 2888, and NSC 4231were found novel as their Tanimoto similarity index was 0.08, 0.12, and 0.13 (<0.5).
In the present study we have identified three potent inhibitors of ROCK-II through application of a battery of in-silico tools. The identified hits appear to be equally potent to that of standard drug Y-27632 in in vitro and ex-vivo studies. The potent vasodilatory activity of the identified ROCK-II inhibitors supports the predominant role of the ROCK-II isoform in the vascular smooth muscle contractility. The observed limitation of the present study is that it does not exclude the possibility of binding of the identified compounds to ROCK-I. Nevertheless, the promising results of the reported study warrant further preclinical and clinical studies.
Pharmacophore Generation and Validation
For pharmacophore model generation, structurally diverse urea-based Rho-kinase inhibitors,6 with wide activity range (1 nM to 3324 nM) were selected and sketched using Chem Draw (12.0). All the chemical structures were split into training set and test set comprising 27 and 11 compounds (Table S5), respectively, following the established ratio of 70 and 30% of compounds in each set. While splitting the compounds, care was exercised to include the most active, moderately active, and least active compounds in the training set for assuring the chemical diversity and activity range. All the sketched structures were imported into the 3D window of Discovery Studio version 2.0 for energy minimization using the CHARMm force field. Conformers were generated for all the inhibitors using the best conformation generation module.12 Pharmacophore models were generated using functional features like hydrophobic (HY), ring aromatic (RA), hydrogen bond donors (HBD), and hydrogen bond acceptors (HBA). Out of various hypotheses, the best model was selected on the basis of correlation coefficient (r) value, cost function parameters, RMSD value, and cost difference values.
Validation of Generated Model
Validation of the statistically fit model was carried out using tools such as internal and external test set prediction, Cat Scramble or Fischer’s randomization, Rm2 metrics, and GH scoring.
Internal and External Test Set Prediction
An internal test set comprising 11 compounds belonging to the same series was selected for pharmacophore modeling13 and used to check the prediction abilities of the model. In order to double-check the prognostic nature of the model, an external test containing 18 known Rho-kinase inhibitors was used, and predictions were analyzed.
Cat Scramble or Fischer’s Randomization
This validation test is often used to eliminate the possibility of chance correlation in the chosen hypothesis. Using the structures and their corresponding biological activities Cat scrambling was carried out at the 99% confidence level. The statistical results of the models constructed from the randomized data were evaluated.
Rm2 Metrics Validation
Rm2 metrics test was carried out on Rho-kinase inhibitors to predict the differences between their actual and predicted activities. A proposed value of “Average rm2” and Delta rm2 is >0.5 and <0.2, respectively.14
Güner–Henry (GH) Scoring Method
This method ascertains the virtual screening capability ofthe developed pharmacophore model and also checks the ability of the model to distinguish between the active and inactive molecules.15 GH scoring includes the calculation of percent yield of actives in a database (% Y, recall), the percent ratio of actives in the hit list (% A, precision), the enrichment factor (E), and the GH score.16 These parameters were computed using eqs 1–4.
| 1 |
| 2 |
| 3 |
| 4 |
% A is the percentage of known active compounds retrieved from the database (precision); Ha, the number of actives in the hit list (true positives); A, the number of active compounds in the database; % Y, the percentage of known actives in the hit list (recall); Ht, the number of hits retrieved; D, the number of compounds in the database; E, the enrichment of active compounds in the virtual screening hit list in comparison to the nonfiltered database; and GH is the Güner–Henry score. To calculate GH score, a data set of 440 compounds was taken from six different available series,17−22 of structurally diverse Rho-kinase inhibitors.
Virtual Screening
Database search is an important aspect of pharmacophore modeling. Hence the validated pharmacophore model was used to retrieve potential hits from the two large database libraries namely NCI and Maybridge. Parameters such as estimated value, fit value, and Lipinski rule of five were used to sort the potential hits obtained from the virtual screening for further consideration.
Molecular Docking
The Lib Dock module of Discovery Studio 2.0 was used to analyze enzyme-ligand binding interaction with minimum computational time.23 During the process, the ligand was kept movable and the target protein was kept rigid. While preparing the receptor, all water molecules were removed and hydrogen atoms were included, and the radius 10 Å was set from the geometric centroid of the ligand as defined in the active site. Top 10 docking poses of all the compounds were considered for analyzing polar and nonpolar interactions in the active site.24
Molecular Fingerprint and Structural Similarity Indices
The Tanimoto index is considered as a good measure to establish the structural similarity of molecules. The Tanimoto index protocol was implemented to check the novelty of the obtained hits using “Find Similar Molecules by Fingerprint” option available in Discovery Studio.
Biological Evaluation
Enzyme-Based Rho-Kinase Inhibitory Assay
Using microplate reader (Synergy H1 Biotek), Rho-kinase inhibitor screening assay was performed to evaluate enzyme inhibition ability of selected hits by phosphorylated MYPT1 (myosin phosphatase target subunit 1). The precoated plates with primary antibody MYPT1 were added with the enzyme, which started phosphorylation of MYPT1 at Thr 696 through the conversion of ATP to ADP using tetramethylbenzidine (TMB) as a substrate. TMB acts as a hydrogen donor, and reduction of hydrogen peroxidase to water takes place by horseradish peroxidase enzyme. Resulting diimine (carbon–nitrogen double bond) turned solution colorless to blue after addition of chromogenic substrate TMB into the wells, and the wavelength was measured at 450 nm. All the test compounds (NSC 2488, NSC 2888, and NSC 4238) were examined at five different concentrations to determine their respective IC50 values and compared with the standard (Y-27632).
Ex-Vivo Isometric Contraction-Based Vasodilation Assay
The regulations and guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals were strictly followed during the entire animal study. All the experiments and procedures were approved by the Institutional animal ethical committee of Banasthali Vidyapith, Rajasthan, India (ref No. BV/3542/17-18). Two healthy male Wistar albino rats (150–230 g) were housed in polypropylene cages and maintained at temperature of 23 ± 0.5 °C with 55 ± 5.0% relative humidity for 12 h in a light/dark atmospheric cycle.
Rats were sacrificed under high doses of anesthesia, and the thoracic aorta was quickly removed and placed in a petri dish filled with PSS (physiological salt solution) of the following composition (mM: NaCl 122; KCl 4.7; NaHCO3 15.5; KH2PO4 1.2; MgCl2 1.2; CaCl2 2.0; glucose 11.5; EDTA 0.026; pH 7.4). After removing all the fatty connective tissues, the aorta was cut into 2–3 mm rings which were used for each set of individual triplicate experiments.25 At pH 7.4, a 25 mL jacketed organ bath was filled with PSS buffer with a continuous supply of 95% O2 and 5% CO2 gas mixture at 37 °C. The rings were mounted in between two hooks. One end of the hook was connected to the aerator tube and another end of the hook was connected to a transducer where the isometric changes were recorded. Each aortic ring was progressively stretched under a passive tension of 2 g before adding any drug and the aortic ring was stabilized for 90 min.
Viability and precontraction of the rings were checked with 70 mM KCl solution and rings were allowed to return to the basal tension by washing with PSS every 10 min. The stable and sustained contraction was achieved by allowing the cumulative concentration of phenylephrine (10–6 to 10–3 M) to the aortic rings.26 The effect of each concentration was recorded on IWORX organ bath assembly and before the addition of the new concentration; the effect of previous concentration was removed with an aim to stabilize the model. After this, the cumulative concentration of test compounds and reference drug Y-27632 was added in tissue assembly and the vasodilatory effect was recorded and analyzed.
Acknowledgments
The authors are thankful to Aditya Shastri, Vice Chancellor, Banasthali Vidyapith, Rajasthan, India, for providing the necessary facility to carry out the present work.
Glossary
Abbreviations
- ROCK
Rho-associated protein kinase
- MLCP
myosin light chain phosphatase
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- HY
hydrophobe
- NCI
National Cancer Institute
- GEF
guanine nucleotide exchange factors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00126.
Data set used for pharmacophore model development, table of 10 pharmacophore hypotheses, detailing of Güner–Henry scoring, list of selected hits generated from NCI database, experimental and predicted activity values of data set compounds, and the docking poses of all three hits and standard drug captured from the Discovery Studio 2.0 itself (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Deng Ti. J.; Bhaidani S.; Sutherland C.; MacDonald A. J.; Walsh P. M. Rho-associated kinase and zipper-interacting protein kinase, but not myosin light chain kinase, are involved in the regulation of myosin phosphorylation in serum-stimulated human arterial smooth muscle cells. PLoS One 2019, 14, 1–34. 10.1371/journal.pone.0226406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H.; Kaibuchi K.; Quilliam A. L. The Insert Region of RhoA Is Essential for Rho Kinase Activation and Cellular Transformation. Mol. Cell. Biol. 2001, 21, 5287–5298. 10.1128/MCB.21.16.5287-5298.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma M.; Wei L.; Shi J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011, 7, 657–671. 10.2217/fca.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa H.; Rashid M. Development of Rho-kinase inhibitors for cardiovascular medicine. Trends Pharmacol. Sci. 2007, 28, 296–302. 10.1016/j.tips.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Feng Y.; LoGrasso V. P.; Defert O.; Li R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 1–32. 10.1021/acs.jmedchem.5b00683. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Lin Li.; Ruiz C.; Khan S.; Cameron D. M.; Grant W.; Pocas J.; Eid N.; Park H.; Schroter T.; LoGrasso V. P.; Feng Y. Synthesis and Biological Evaluation of Urea Derivatives as Highly Potent and Selective Rho Kinase Inhibitors. J. Med. Chem. 2013, 56, 3568–3581. 10.1021/jm400062r. [DOI] [PubMed] [Google Scholar]
- Singh A.; et al. Identification of novel anti-fungal lead compounds through pharmacophore modeling, virtual screening, molecular docking, anti-microbial evaluation and gastrointestinal permeation studies. J. Biomol. Struct. Dyn. 2017, 35, 2363–2371. 10.1080/07391102.2016.1218369. [DOI] [PubMed] [Google Scholar]
- Mittal A.; et al. Pharmacophore based virtual screening, molecular docking and biological evaluation to identify novel PDE5 inhibitors with vasodilatory activity. Bioorg. Med. Chem. Lett. 2014, 24, 3137–3141. 10.1016/j.bmcl.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Shah S.; Savjani J. A review on ROCK-II inhibitors: from molecular modelling to synthesis. Bioorg. Med. Chem. Lett. 2016, 26, 2383–2391. 10.1016/j.bmcl.2016.03.113. [DOI] [PubMed] [Google Scholar]
- Wang H. D.; Qu L. W.; Shi Q. L.; Wei J. Molecular docking and pharmacophore model studies of Rho-kinase inhibitors. Mol. Simul. 2011, 37, 488–494. 10.1080/08927022.2011.554548. [DOI] [Google Scholar]
- Jahani V.; Kavousi A.; Mehri S.; Karimi S. Rho kinase, a potential target in the treatment of metabolic syndrome. Biomed. Biomed. Pharmacother. 2018, 106, 1024–1030. 10.1016/j.biopha.2018.07.060. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr; Kuczera J. W.; Karplus M. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 1995, 117, 11946–11975. 10.1021/ja00153a017. [DOI] [Google Scholar]
- Iwakubo M.; Takami A.; Okada Y.; Kawata T.; Tagami Y.; Sato M.; Sugiyama T.; Fukushima K.; Taya S.; Amano M.; Kaibuchi K.; Iijima H. Design and synthesis of Rho-kinase inhibitors (III). Bioorg. Med. Chem. 2007, 15, 1022–1033. 10.1016/j.bmc.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Roy K.; et al. Some case studies on application of “(rm) 2” metrics for judging quality of quantitative structure-activity relationship predictions: emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. 10.1002/jcc.23231. [DOI] [PubMed] [Google Scholar]
- Vaghefinezhad N.; Farsani F. S.; Gharaghani S.. In silico drug-designing studies on sulforaphane analogues: pharmacophore mapping molecular docking and QSAR modeling. Curr. Drug Discovery Technol., published online November 12, 2019; 10.2174/1570163816666191112122047. [DOI] [PubMed] [Google Scholar]
- Güner O. F.; Henry D. R.. Metric for analyzing hit lists and pharmacophores. In Pharmacophore perception, development, and use in drug design; International University Line: San Diego, 2000; Vol. 1, pp 193–212. [Google Scholar]
- Bandarage K. U.; Cao J.; Come H. J.; Court J. J.; Gao H.; Jacobs D. M.; Marhefka C.; Nanthakumar S.; Green J. ROCK inhibitors 3: Design, synthesis and structure-activity relationships of 7-azaindole-based Rho kinase (ROCK) inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 2622–2626. 10.1016/j.bmcl.2018.06.040. [DOI] [PubMed] [Google Scholar]
- Chowdhury S.; Sessions E. H.; Pocas J. R.; Grant W.; Schroter T.; Lin L.; Ruiz C.; Cameron M. D.; Schurer S.; LoGrasso P.; Bannister T. D.; Feng Y. Discovery and optimization of indole and 7-azaindoles as Rho kinase (ROCK) inhibitors (Part-I). Bioorg. Med. Chem. Lett. 2011, 21, 7107–7112. 10.1016/j.bmcl.2011.09.083. [DOI] [PubMed] [Google Scholar]
- Sessions H. E.; Chowdhury S.; Yin Y.; Pocas R. J.; Grant W.; Schröter T.; Lin Li.; Ruiz C.; Cameron D. M.; LoGrasso P.; Bannister D. T.; Feng Y. Discovery and optimization of indole and 7-azaindoles as Rho kinase (ROCK) inhibitors (Part-II). Bioorg. Med. Chem. Lett. 2011, 21, 7113–7118. 10.1016/j.bmcl.2011.09.084. [DOI] [PubMed] [Google Scholar]
- Green J.; Cao J.; Bandarage K. U.; Gao H.; Court J.; Marhefka C.; Jacobs M.; Taslimi P.; Newsome D.; Nakayama T.; Shah S.; Rodems S. Design, Synthesis, and Structure–Activity Relationships of Pyridine-Based Rho Kinase (ROCK) Inhibitors. J. Med. Chem. 2015, 58, 5028–5037. 10.1021/acs.jmedchem.5b00424. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Zheng Ke.; Eid N.; Howard S.; Jeong H. J.; Yi F.; Guo J.; Park M. C.; Bibian M.; Wu W.; Hernandez P.; Park H.; Wu Y.; Luo Li. J.; LoGrasso V. P.; Feng Y. Bis-aryl Urea Derivatives as Potent and Selective LIM Kinase (Limk) Inhibitors. J. Med. Chem. 2015, 58, 1846–1861. 10.1021/jm501680m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturdivant M. J.; Royalty M. S.; Lin W. C.; Moore A. L.; Yingling D. J.; Laethem L. C.; Sherman B.; Heintzelman R. G.; Kopczynski C. C.; deLong A. M. Discovery of the ROCK inhibitor netarsudil for the treatment of open-angle glaucoma. Bioorg. Med. Chem. Lett. 2016, 26, 2475–2480. 10.1016/j.bmcl.2016.03.104. [DOI] [PubMed] [Google Scholar]
- Mei D.; Yin Y.; Wu F.; Cui J.; Zhou H.; Sun G.; Jiang Y.; Feng Y. Discovery of potent and selective urea-based ROCK inhibitors: Exploring the inhibitor’s potency and ROCK2/PKA selectivity by 3D-QSAR, molecular docking and molecular dynamics simulations. Bioorg. Med. Chem. 2015, 23, 2505–2517. 10.1016/j.bmc.2015.03.047. [DOI] [PubMed] [Google Scholar]
- Mishra P.; Kesar S.; Paliwal K. S.; Chauhan M.; Madan K. In-Silico Screening of Ligand Based Pharmacophore, Database Mining and Molecular Docking on 2, 5-Diaminopyrimidines Azapurines as Potential Inhibitors of Glycogen Synthase Kinase-3β. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 1–9. 10.2174/1871524918666180530074116. [DOI] [PubMed] [Google Scholar]
- Cordaillat M.; et al. Nitric oxide pathway counteracts enhanced contraction to membrane depolarization in aortic rings of rats on high-sodium diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, 1557–1562. 10.1152/ajpregu.00624.2006. [DOI] [PubMed] [Google Scholar]
- Mojiminiyi F. B. O.; Anigbogu C. N.; Sofola O. A.; Adigun S. A. Endothelium-dependent and -independent relaxations in Aortic rings obtained from hypertensive hooded (AGUTI). Rats. Niger. J. Physiol. Sci. 2010, 22, 109–116. 10.4314/njps.v22i1-2.54880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.