

Abstract

The toll-like receptor (TLR) family is an evolutionarily conserved component of the innate immune system, responsible for the early detection of foreign or endogenous threat signals. In the context of autoimmunity, the unintended recognition of self-motifs as foreign promotes initiation or propagation of disease. Overactivation of TLR7 and TLR9 have been implicated as factors contributing to autoimmune disorders such as psoriasis, arthritis, and lupus. In our search for small molecule antagonists of TLR7/9, 7f was identified as possessing excellent on-target potency for human TLR7/9 as well as for TLR8, with selectivity against other representative TLR family members. Good pharmacokinetic properties and a relatively balanced potency against TLR7 and TLR9 in mouse systems (systems which lack functional TLR8) made this an excellent in vivo tool compound, and efficacy from oral dosing in preclinical models of autoimmune disease was demonstrated.
Keywords: Toll-like receptor, TLR, autoimmunity, innate immunity, psoriasis, lupus
The innate immune system, a first line of host-defense, includes several families of pattern recognition receptors (PRRs) such as the Toll-like receptor (TLR) family. PRRs recognize pathogen-associated molecular patterns (PAMPs) from invading microbes and danger-associated molecular patterns (DAMPs) released from stressed or damaged host cells.1 TLRs are transmembrane proteins composed of an extracellular (or intraendosomal) ligand-binding domain connected via a transmembrane region to a cytosolic toll/interleukin (TIR) signaling domain.2 Binding of ligands (PAMPs/DAMPs) to the ectodomain (ECD) of hetero- or homodimeric TLR pairs initiates signaling events via recruitment of adaptor proteins to the TIR domain. This activates signal transduction cascades resulting in cellular proliferation as well as production of type-I interferons (IFN) and other pro-inflammatory cytokines and chemokines. To date, 13 mammalian TLRs have been identified, including nine subtypes (TLR1–9) with defined functions in human.2 These PRRs are localized either on the plasma cell membrane (TLR1, 2, 4, 5, 6) or on endosomal membranes (TLR3, 7, 8, 9), with the endosomal TLRs responding to double-stranded RNA (TLR3), single-stranded RNA (TLR7 and 8), or hypomethylated double-stranded DNA (TLR9) as activating ligands. In autoimmune disorders, sustained activation of TLR7/9 has been shown to play a key role in immune dysfunction, leading to enhanced disease states.3 In mouse models, overexpression of TLR7 led to autoimmune disease progression, while TLR7 deficiency protected against disease development in the lupus-prone MRL/lpr background.4,5 Although specific knockout of TLR9 led to worsening of disease in lupus-prone mice, the dual knockout of TLR7 and 9 again showed enhanced protection, supporting the potential benefit of dual inhibition of these targets for the treatment of lupus.6 Among the other endosomal TLRs, TLR3 signaling has not been as strongly associated with autoimmune disorders, while deciphering the role of TLR8 has been complicated by species differences. Unlike TLR7 or TLR9, the roles of TLR8 in humans are not recapitulated in mouse—mouse TLR8 does not respond to single-stranded RNA ligands, which do stimulate TLR8 in human.4,7 As such, an understanding of the function of TLR8 in autoimmune diseases has lagged behind that of other TLRs.8
Herein, we detail the identification of potent and selective small molecule antagonists of TLR7/8/9 based on a 2-phenyl-indole chemical series. While TLR8 function is not captured in mouse models, the in vivo studies presented here support at least the potential utility of small molecule antagonists of TLR7/9 as therapeutics for the treatment of autoimmune disorders. Examples of potent small molecule inhibitors of endosomal TLR signaling were quite limited at the outset of our efforts, with none showing the desired TLR selectivity profile that we were seeking.9 Antimalarials (e.g., chloroquine, quinacrine) and structurally related compounds have been shown to inhibit TLR9 but were reported to exert their effects through binding to the nucleic acid ligands, resulting in limited selectivity against TLR3 as well as inhibition of other cytosolic DNA sensors.10,11 We initiated a screen of a proprietary compound collection utilizing cellular reporter assays to monitor for inhibition of TLR activity, focused on the identification of antagonists of TLR7 and TLR9, given the body of data connecting their dysregulation to autoimmune disorders. The screen utilized the HEK293 Blue secreted embryonic alkaline phosphatase (SEAP) assay,12 in which the cells were engineered to overexpress the TLRs of interest. The functional nature of this screen necessitated deconvolution to filter out general pathway inhibitors with alternative mechanisms of action. Counter-screening against additional endosomal (TLR3) and cell-surface (TLR4) family members helped to eliminate nonspecific pathway inhibitors or compounds with assay interference.
Given the human-to-mouse TLR8 functional disconnect discussed above, activity on that receptor was not tracked for all compounds, with only advanced examples subsequently evaluated. From these screening efforts, a 2-phenyl-indole-5-piperidine chemical series was identified, with examples demonstrating encouraging dual antagonism of TLR7/9 and an indication that selectivity against TLR3/4 could be achieved (Figure 1, 4a,b). This scaffold afforded an opportunity to readily investigate changes to several positions, including at N-1, on the C-2 phenyl, at C-3, and the nitrogen of the C-5 piperidine.
Figure 1.

Indole-5-piperidine chemical series identified from screening.
Structure–activity relationship (SAR) studies began with variation of the indole C-2 group as shown in Table 1. Mono- and disubstituted phenyl patterns were explored using both electron-donating and electron-withdrawing groups. The tolerability for substitution of the C-2 phenyl was narrow for TLR7, with unsubstituted and monosubstituted systems, regardless of substituent position or electronics, showing poor TLR7 activity. Of the single substitution patterns evaluated, a methoxy group in the meta or para positions (5f, 5g) were the most encouraging. Combination of these substitutions into the 3′,4′-dimethoxyphenyl compound 5n provided a dramatic improvement in TLR7 potency. Constraining these substituents as in dioxolane 5p reduced TLR7 activity, as did increasing the size of the alkoxy groups, as in 5o. Specific positioning of the dimethoxy groups was critical, with the 2′,4′- (5l) and 2′,3′-dimethoxy (5m) patterns yielding substantially reduced TLR7 activity.
Table 1. C-2 SAR of C-3-Methyl-indoles.


IC50 values are a mean of two or more individual determinations except those marked with *.
Activity against TLR9 was more accommodating to changes in C-2 phenyl substitution, although the SAR was flat in comparison to TLR7 activity. Across the range of changes described in Table 1, at least modest TLR9 antagonism was retained, spanning a range of approximately 300–5000 nM. Single substitutions at any position of the C-2 phenyl with a methyl group led to a slight (3-fold or less) decrease in activity, whereas introduction of a methoxy group was most advantageous when placed at the para position (5g). Nitrile substitution was also beneficial for TLR9 activity, regardless of position (5h–j). An additional electron withdrawing group, the primary carboxamide, was explored only at the para position, providing further improved activity (5k). For the disubstituted examples, the 3′4′-dimethoxy provided favorable results, as was observed for TLR7 activity (5n). The constrained (5p) and bulkier (5o) analogues were once again found to exhibit reduced activity relative to the 3′4′-dimethoxy substitution pattern.
Compound 5n was profiled for selectivity and exhibited reduced activity against TLR3 (IC50 2160 nM) and TLR4 (IC50 14 970 nM) relative to TLR7/9. On the basis of the encouraging dual 7/9 antagonist profile of 5n, the 3′,4′-dimethoxyphenyl group was retained at C-2 for subsequent studies. Efforts turned to the evaluation of C-5 piperidine capping groups (Table 2). In comparison to the relatively steep and unaccommodating TLR7 SAR noted for the substitution pattern of the C-2 phenyl, a high level of chemical diversity was tolerated off of the C-5 piperidine. For TLR9, the antagonist activity was noted to be influenced by the basicity of the region, specifically the number of basic atoms and the varying level of basicity associated with each. Removal of the piperidine capping group to give monobasic 6a was tolerated for TLR7 but not for TLR9 (>10-fold reduction in activity). Acetylation of the piperidine to afford a nonbasic C-5 group (6b) was somewhat detrimental to TLR7 activity (5-fold reduced vs 5n) but highly detrimental to TLR9 activity (IC50 > 50 000 nM). The spacing between the basic atoms and nature of their arrangement was flexible, as highlighted by examples 6c–6f. Homologated compound 6c demonstrated activity against both TLRs that was close to that of 5n, particularly for TLR9. Cyclic constraint of the terminal amine chain such as piperidines 6d and 6e were comparable in activity to homologated 6c. As compared to the piperidine (6d), constraint as an azetidine (6f) led to slightly reduced TLR7 activity (∼2-fold) but TLR9 potency was 10-fold reduced, possibly corresponding to the reduction in pKa of both amines introduced by this moiety.
Table 2. C-5 Piperidine SAR.


ACDpKa calculated by Instant JChem software (ChemAxon); showing values within a range of 0–15.
IC50 values are a mean of two or more individual determinations except those marked with *.
The remaining compounds in Table 2 describe the effects of introducing a pendant heteroaryl group off of the piperidine nitrogen via a methylene linker. In each case, the basicity of the piperidine nitrogen is attenuated by the substituent, which includes a second nitrogen of varying basicity. While 1-methyl-5-yl-imidazole 6g showed modest activity for TLR7 and TLR9, 2-methylimidazole 6h was 4-fold reduced in TLR7 potency but demonstrated very good TLR9 activity despite only a slight change in calculated basicity of these constitutional isomers. Imidazole 6i, regioisomeric to 6g, also exhibited reduced TLR7 but increased TLR9 activity despite a reduction in calculated pKa values.
Compound 5n also served as the basis for optimization at the N-1 and C-3 indole positions (Table 3). The N-1 position was found to be intolerant to substitution in regards to TLR7 activity as evidenced by methyl indole 7a (IC50 5260 nM), whereas TLR9 activity was only slightly reduced by this change (IC50 740 nM). Modification of the C-3 position proved much more rewarding, with a range of variations being well tolerated for TLR9 activity and select changes leading to dramatic improvement in TLR7 potency. While chloro (7b) or cyano (7c) replacements at C-3 reduced TLR7 activity, extension of the alkyl group to ethyl (7d) or trifluoroethyl (7e) improved TLR7 activity by 2- to 4-fold. This improvement was even more pronounced with isopropyl substituted 7f, which provided low nanomolar TLR7 potency (IC50 10 nM). Homologation to isobutyl substitution (7g) led to a precipitous decline in TLR7 potency (IC50 1290 nM), although that extension had little effect on TLR9 activity.
Table 3. SAR of Indole N-1 and C-3 Positions.

| compd | R1 | R2 | TLR7 IC50 (nM)a | TLR9 IC50 (nM)a |
|---|---|---|---|---|
| 5n | H | Me | 250 ± 160 | 430 ± 260 |
| 7a | Me | Me | 5260 ± 1540 | 740 ± 170 |
| 7b | H | Cl | 1880 ± 710 | 430 ± 170 |
| 7c | H | CN | >10 000 | 260 ± 200 |
| 7d | H | CH2CH3 | 130 ± 100 | 490 ± 370 |
| 7e | H | CH2CF3 | 70 ± 40 | 750 ± 270 |
| 7f | H | isopropyl | 10 ± 7 | 700 ± 390 |
| 7g | H | isobutyl | 1290 ± 770 | 830 ± 770 |
IC50 values are a mean of two or more individual determinations.
As described above, 7f was found to be a potent antagonist of TLR7 with modest activity against TLR9. Further evaluation against other TLRs in the HEK293 reporter system (Table 4) showed 7f to also possess potent activity against TLR8 (IC50 17 nM) but much reduced activity against endosomal TLR3 (IC50 5440 nM) and cell-surface TLR4 (IC50 15 100 nM). In agonist mode, 7f was inactive up to 50 000 nM against TLR7, 8, or 9. The antagonist activity of 7f was tested in additional cellular and whole blood assays. In a peripheral blood mononuclear cell (PBMC) assay measuring IL-6 production, inhibition of TLR7- or 9-driven responses were very similar to that observed in the HEK293 reporter assays. Inhibition of the TLR9-stimulated production of IFNα in plasmacytoid dendritic cells (pDCs) was found to be more pronounced (IC50 210 nM) than for other cellular assays. As pDCs express both TLR9 and TLR7, this cell type was also used for the evaluation of TLR7 inhibition. The TLR7 agonist employed in the pDC assay was inactivated flu virus rather than a small molecule agonist as utilized in the other assays. The excellent response of 7f in this assay (IC50 11 nM) with a more physiologically relevant TLR7 ligand highlights that the potency appears to be independent of the nature of the TLR7 agonist. This differentiates from the antimalarials, for which poor inhibitory activity was observed against non-nucleic acid-based agonists of TLR7.10 In human whole blood (hWB) assays, 7f demonstrated inhibition of IL-6 production consistent with the cellular responses for TLR7, 8, 9. In anticipation of in vivo studies in mouse, the activity of 7f was evaluated in mouse whole blood (mWB) assays. As described above, TLR8 is nonfunctional in mouse, and for that reason only TLR7 and TLR9 responses were examined in this mWB assay. The TLR9 inhibitory response of 7f in mWB (IC50 510 nM) was similar to that found in hWB, however, the TLR7 inhibition in mWB (IC50 43 nM) was ∼7-fold reduced relative to hWB, indicating a more balanced TLR7/9 dual antagonist profile in mouse.
Table 4. TLR Cellular and Whole Blood Activity Data for 7f.
| assay | receptor | IC50 (nM)a |
|---|---|---|
| HEK293; NF-kB reporter (antagonist mode) | TLR9 | 700 ± 390 |
| TLR8 | 17 ± 37 | |
| TLR7 | 10 ± 7 | |
| TLR4 | 15100 ± 16500 | |
| TLR3 | 5440 ± 5000 | |
| HEK293; NF-kB reporter (agonist mode) | TLR7,8,9 | >50000 |
| PBMCs; IL-6 | TLR9 | 510 ± 80 |
| TLR7 | 4.8 ± 3.6 | |
| pDC (ODN2216); IFNαpDC (flu virus); IFNα | TLR9 | 210 ± 390 |
| TLR7 | 11 ± 9 | |
| human whole blood; IL-6 | TLR9 | 320 ± 97 |
| TLR8 | 5.1 ± 5.6 | |
| TLR7 | 5.7 ± 6.3 | |
| mouse whole blood; IL-6 | TLR9 | 510 ± 150 |
| TLR7 | 43 ± 45 | |
IC50 values are a mean of two or more individual determinations. See Supporting Information for assay details.
Assessment of pharmacokinetic (PK) properties for 7f in mouse showed good oral bioavailability (62%F) and a flat exposure profile with a 24 h peak-to-trough ratio of ∼4 (Table 5). On the basis of exposures in blood, the time to maximal concentration (Tmax) for oral dosing was rapid (0.5 h) with maximal concentration (Cmax) of 1628 nM (15 mg/kg). The low clearance and high volume of distribution (20 L/kg) led to a long terminal half-life of 32 h. Compound 7f was found to partition preferentially into the blood (blood-to-plasma ratio 4.6:1 at 2 h and 6.9:1 at 7 h) and also into the central nervous system (7 h brain to plasma ratio 1.4:1).
Table 5. Mouse Pharmacokinetic Profile of 7fa.
| dose (mg/kg) |
||
|---|---|---|
| 3 (IV) | 15 (PO) | |
| Tmax (h) | 0.5 | |
| Cmax (nM) | 1628 | |
| Cl (mL/min/kg) | 7.7 | |
| Vss (L/kg) | 20 | |
| T1/2 (h) | 32 | |
| F (%) | 62 | |
IV vehicle: 70/30 PEG400/water. PO vehicle: 80/20 PEG400/200 mM citrate buffer (pH 3.0).
In mouse pharmacodynamic (PD) models, oral dosing with 7f was able to effectively block cytokine production elicited by treatment with agonists of TLR7 (gardiquimod) or TLR9 (ODN1585) (Figure 2). A dose-dependent decrease in agonist-induced IL-6 production was observed, with plasma EC50 of 2.4 nM (TLR7) or 25 nM (TLR9). This was noted to be 4- to 5-fold more potent than the mWB assay results when factoring in the blood-to-plasma ratio, possibly reflecting the preferential distribution of the compound into tissues.
Figure 2.
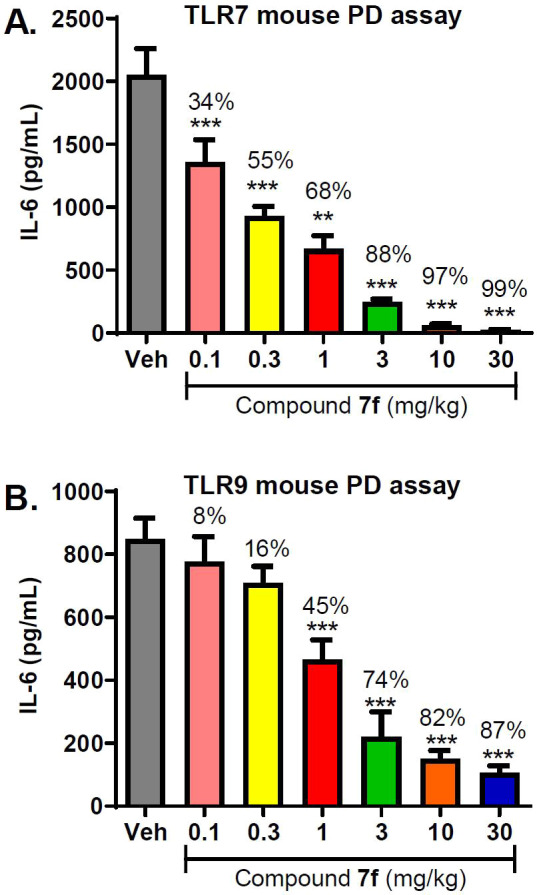
Efficacy of 7f in TLR7 (A) and TLR9 (B). PD experiments measuring suppression of agonist-induced IL-6. **p < 0.01, ***p < 0.001. One-way ANOVA with Dunnett’s test compared to the vehicle. N = 8 mice per group.
7f was then advanced to rodent models of autoimmune disorders. Topical application of imiquimod (a TLR7 agonist) is a well-precedented method of eliciting skin changes characteristic of human psoriasis.13 In this model, imiquimod cream (5%) was applied to the shaved back region of mice once a day for 6 days, and the effect of concurrent once-daily oral treatment with 7f was compared to dexamethasone (3 mg/kg qd) or vehicle control (Figure 3).
Figure 3.

Efficacy of 7f (1, 3, 10, 30 mg/kg) or dexamethasone (3 mg/kg) vs vehicle in imiquimod-induced mouse model of psoriasis showing changes to scaling **p < 0.01, ***p < 0.001. One-way ANOVA with Dunnett’s test compared to the vehicle. N = 7–10 mice per group.
Significant and dose-dependent suppression of multiple end points was observed, with maximal activity of 7f exceeding the efficacy shown by dexamethasone. As shown for the scaling end point, treatment with 7f led to a reduction of up to ∼70% relative to vehicle at the top dose. Exposures from day 5 (Supporting Information) show that achieving partial coverage above mWB TLR7 IC50 as observed for the 3 mg/kg dose appears sufficient to elicit ∼50% inhibition in scaling, whereas sustained coverage above the mWB IC50, as achieved for the 10 and 30 mg/kg doses, provided further improvement.
Compound 7f was subsequently progressed to a disease model with a more extensive duration of dosing. MRL/lpr mice spontaneously develop disease with manifestations that are reminiscent of human lupus, including proteinuria, lymphadenopathy, splenomegaly, elevated cytokine production, and increased antibody production.14Figure 4 shows the effect of 8 weeks of treatment with 7f (1, 3, 10, 30 mg/kg, qd) as compared to treatment with prednisolone (10 mg/kg, qd) on the urinary marker proteinuria (Figure 4A), antidouble-stranded DNA (dsDNA) antibody titer (Figure 4B), and the serum cytokine IL-10 (Figure 4C) in the MRL/lpr mice.
Figure 4.

Efficacy of 7f (1, 3, 10, 30 mg/kg) or prednisolone (10 mg/kg) vs vehicle in a MRL/lpr mouse model of lupus showing changes to proteinuria levels (A), anti-dsDNA antibody titer (B), and plasma IL-10 (C). *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA with Dunnett’s test compared to the vehicle. N = 12 mice per group.
For each end point, a dose-dependent decrease in severity was noted, with a maximal response comparable to that shown by the steroid positive control. Exposures of 7f at the end of the study (Supporting Information) indicated that sustained coverage above the mWB EC50 for TLR7 was achieved with a dose of 3 mg/kg, where notable impact on proteinuria (55% reduction) was observed and modest reduction (30–35%) of the other end points was seen. At the higher doses (10, 30 mg/kg), coverage above the TLR9 mWB EC50 was achieved as well, and more robust efficacy was demonstrated.
Subsequent to the identification of 7f as a potent antagonist of TLR7/8/9, structural evidence was obtained informing on the direct binding mode of inhibition for this class of compounds. The availability of hTLR8 protein enabled the generation of cocrystal structures with compound 17c, as detailed in Figure 5.
Figure 5.

X-ray crystal structure of 17c bound to TLR8. (A) Structure of 17c in complex with human TLR8 (RCSB PDB 6V9U). The two protein chains are shown as light-green and light-blue ribbons, respectively. The two ligand molecules are shown in space-filling representation, with the carbon atoms of one molecule in green and the other in cyan. (B) Detailed view of one of the two 17c binding sites. Colors are as in (A), with hydrogen bonds indicated as dashed orange lines. (C) The complex of TLR8 bound to 17c is shown aligned to agonist-bound TLR8 (RCSB PDB 3W3J, orange color), apo TLR8 (3W3G, pink), and TLR8 bound to antagonist CU-CPT9b (5WYZ, purple).
TLR8 crystallized as a dimer with two symmetric ligand binding sites located at the dimer interface, consistent with previously reported TLR8 X-ray structures15,16 (Figure 5A,C). Interestingly, 17c stabilizes an inactive conformation of TLR8 in which the C-termini of the ECD are positioned far apart; the structure aligns well with the apo conformation of the protein (Figure 5C, middle). The open, inactive conformation observed in this study is differentiated from both the closed, active agonist-bound conformation (Figure 5C, left) and from a recently reported antagonist-bound structure, which demonstrated an inactive, moderately open conformation (Figure 5C, right).17Figure 5B shows a detailed view of one of the binding sites. The 3′,4′-dimethoxyphenyl moiety forms a bidentate hydrogen bond to the Gly-351 backbone NH and stacks between Tyr-348 of one chain and Phe-494/Phe-495 of the second chain. These interactions account for the SAR favoring the 3′,4′-dimethoxy substitution pattern on the C-2 phenyl. The importance of the indole NH for activity (e.g., indole N-methyl 7a; TLR8 IC50 958 nM) can be understood in light of the hydrogen bond between the backbone carbonyl of Phe-494 and the indole NH. The protonated piperidine is able to interact with the Glu-427 side chain and is oriented toward the solvent-exposed interior of the ectodomain, consistent with the flexible SAR observed for substitution of the C-5 position. TLR7 and TLR8 share 40% sequence identity in their ectodomains, and 50% of the residues in the 17c binding site are identical. TLR7 and TLR8 activity align well for the compounds tested, and homology modeling of the TLR7 binding site supports a similar set of protein–ligand interactions. The TLR9 ECD is related more distantly to TLR8 (33% sequence identity overall and 25% in the binding site), which is reflected in the more disparate SAR for TLR7/8 vs TLR9 inhibition.
Preparation of indole-based antagonists is depicted in Scheme 1. Variation of the C-3 position was accomplished through the use of commercially available 5-bromoindoles. The C-3 methylindole (9a) was prepared via LiAlH4 reduction of 5-bromo-indole-3-carboxaldehyde (8a), while the C-3 alkyl indoles 9b–d were prepared by exposing 8b to the appropriate secondary amine in the presence of Shvo’s catalyst. These indole cores 9a–d, along with the commercially available 3-cyano-5-bromoindole (9e), were further elaborated to the general structure 10 by Suzuki–Miyaura cross-coupling and subsequent hydrogenation. Preparation of C-3 chloro intermediate 10f began with cross-coupling and reduction of 5-bromoindole (8b) in a manner similar to that described above, followed by NCS-mediated chlorination. Bromination of 10a–f with NBS occurred selectively at the C-2 position to afford intermediates 11a–f, which were transformed into the final compounds by one of several routes. Functionalization of the indole C-2 position required the removal of the piperidine Boc protecting group via treatment of 11a–c with HBr/acetic acid in DCM. Addition of TEA, followed by aldehyde 12 under reductive amination conditions, afforded scaffolds 13a–c, which were subjected to cross-coupling with phenylboronic acids 14 to afford, after Boc deprotection, analogues 5a–p and 7d,f. Alternatively, 11a,c–f were reacted with 3,4-dimethoxyphenylboronic acid under cross-coupling conditions, followed by Boc removal via treatment with HCl to give 6a or 17c–f.
Scheme 1. Synthesis of TLR Antagonists.
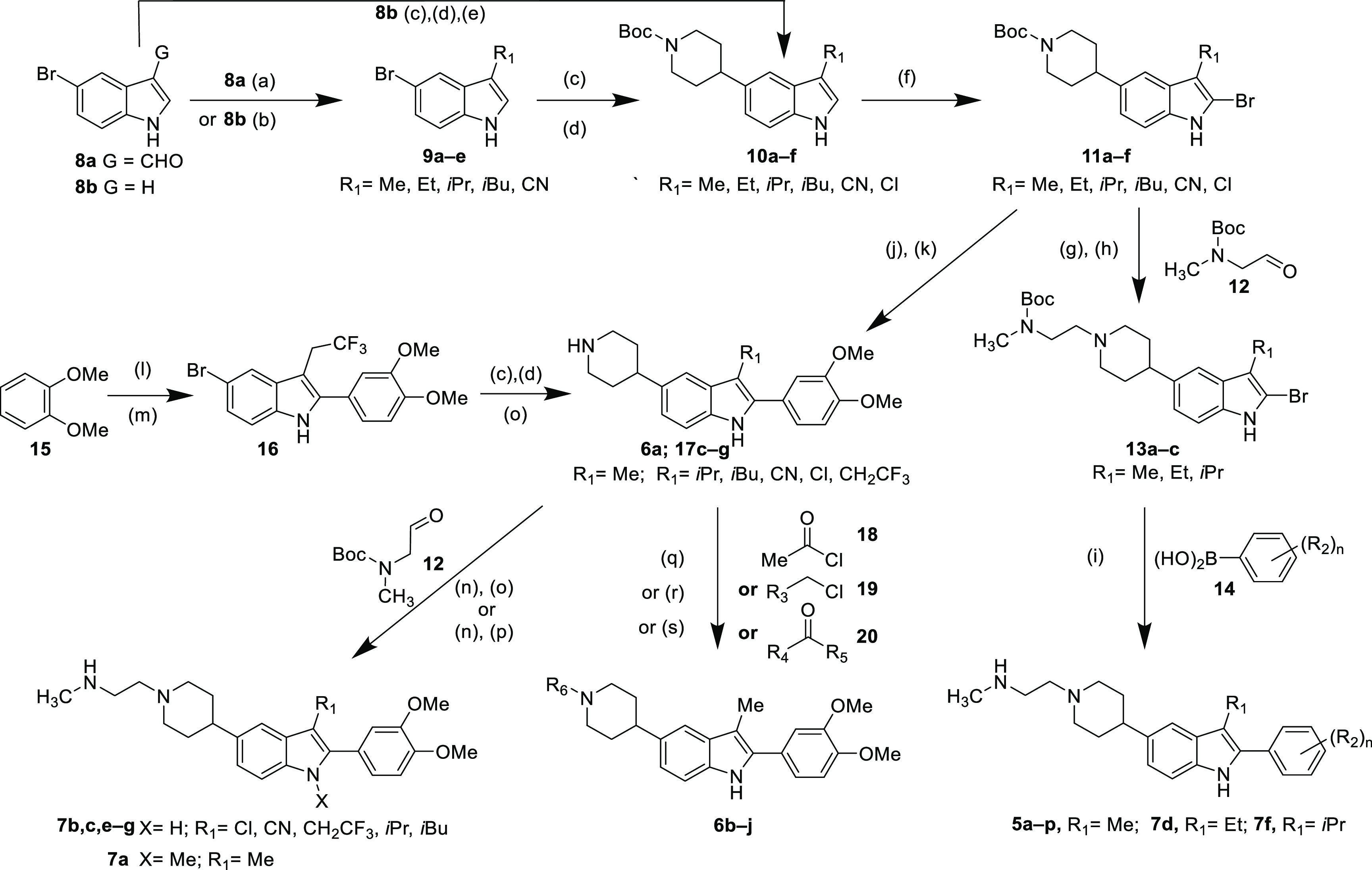
Reagents and conditions: (a) LiAlH4, THF, reflux, 45%; (b) Shvo’s catalyst, amine, K2CO3, 155 °C, 21%–72%; (c) tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate, 3.0 M K3PO4, Pd(dppf)Cl2, THF, 70–84%; (d) 10% Pd/C, H2, EtOAc, 85–97%; (e) NCS, DCM, 84%; (f) NBS, DCM then 10% aqueous Na2SO3, 20–75%; (g) HBr(AcOH), DCM; (h) TEA, AcOH, Na(OAc)3BH, DCM, 85% 2-steps; (i) 3.0 M K3PO4, Pd(dppf)Cl2, THF, then TFA, DCM, 2–66%; (j) 3.0 M K3PO4, 3,4-dimethoxyphenylboronic acid, Pd(dppf)Cl2, THF, 1 h; (k) 4.0 M HCl in dioxane, 44–100% (2 steps); (l) trifluoromethylbutanoic acid, PPA, toluene, 43%; (m) 4-bromophenylhydrazine, PPA, 155 °C, 38%; (n) TEA, AcOH, Na(OAc)3BH, DCM; (o) 4.0 M HCl in dioxane, 12–95%; (p) NaH, DMF, then MeI, then 50% TFA in DCM, 52%; (q) TEA, DCM, 25–73%; (r) DBU, DCM, 6%; (s) TEA, AcOH, Na(OAc)3BH, DCM, 11–88%.
Synthesis of C-3-trifluoroethylindole 17g began with the reaction of 3,4-dimethoxybenzene with trifluoromethylbutanoic acid and polyphosphoric acid (PPA). The resulting intermediate was heated with 4-bromophenylhydrazine and PPA to afford 16. A three-step sequence (coupling/hydrogenation/deprotection) analogous to that described for the conversion of 9 to 10 above was applied to 16 to afford 17g. Reaction of intermediates 17 with acetyl chloride (18), alkyl chlorides 19, or aldehyde or ketones 20 afforded 6b–j. Intermediates 6a/17c–g were separately subjected to reductive with a subsequent Boc deprotection to give final compounds 7b,c,e–g. N-Methylindole 7a was prepared by reductive amination of 6a and 12, followed by exposure to sodium hydride and MeI prior to Boc removal.
In conclusion, we discovered 7f as a promising lead for a small molecule TLR7/9 dual antagonist program aimed at the treatment of lupus and other autoimmune diseases. Through an HTS campaign, we identified a 2-phenyl-indole-5-piperidine series and conducted optimization studies, which led to the incorporation of dimethoxyphenyl substitution at C-2 to drive dual potency against TLR7 and TLR9. Potency was further enhanced through modifications of alkyl substituents at C-3, while changes to C-5 provide an area for tuning of potency as well as PK and off-target liabilities through modification of compound basicity and polarity. The excellent potency and oral PK properties of 7f in mouse enabled its use in multiple in vivo models for the evaluation of modulating TLR7/9 driven pharmacodynamic responses (with TLR8 not able to be evaluated in mouse systems) and also for efficacy in rodent disease models for psoriasis and lupus. These results establish small molecule inhibition of the TLR7/9 pathway as a promising approach for treatment of autoimmune disorders.
Glossary
Abbreviations
- TLR
toll-like receptor
- PRR
pattern recognition receptor
- PAMP
pathogen-associated molecular patterns
- DAMP
danger-associated molecular patterns
- ECD
extracellular domain or ectodomain
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00264.
Preparation and characterization of compounds 5–7; biologic assays. X-ray cocrystallographic data, and refinement statistics for compound 17c bound to TLR8 (PDF)
Accession Codes
Atomic coordinates for the X-ray structure of compound 17c: (PDB 6V9U) in TLR8 is available from the RCSB Protein Data Bank (www.rscb.org). Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
Supplementary Material
References
- Baccala R.; Gonzalez-Quintial R.; Lawson B.; Stern M.; Kono D.; Beutler B.; Theofilopoulos A. Sensors of the innate immune system: Their mode of action. Nat. Rev. Rheumatol. 2009, 5, 448–456. 10.1038/nrrheum.2009.136. [DOI] [PubMed] [Google Scholar]
- O’Neill L.; Golenbock D.; Bowie A. The history of Toll-like receptors — redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- Marshak-Rothstein A.; Rifkin I. Immunologically active autoantigens: The role of Toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 2007, 25, 419–441. 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- Heil F.; Hemmi H.; Hochrein H. Species specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Subramanian S.; Tus K.; Li Q. Z.; Wang A.; Tian X. H.; Zhou J.; Liang C.; Bartov G.; McDaniel L. D.; Zhou X. J.; Schultz R. A.; Wakeland E. K. A TLR7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9970–9975. 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen S.; Shupe J.; Nickerson K.; Kashgarian M.; Flavell R.; Shlomchik M. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Liu J.; Xu C.; Hsu L.; Luo Y.; Xiang R.; Chuang T. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol. Immunol. 2010, 47, 1083–1090. 10.1016/j.molimm.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes J.; Weinerman B.; Basole C.; Salazar J. TLR8: The forgotten relative revindicated. Cell. Mol. Immunol. 2012, 9, 434–438. 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patinote C.; Karroum N.; Moarbess G.; Cirnat N.; Kassab I.; Bonnet P.; Deleuze-Masquéfa C. Agonist And Antagonist Ligands Of Toll-Like Receptors 7 And 8: Ingenious Tools For Therapeutic Purposes. Eur. J. Med. Chem. 2020, 193, 112238. 10.1016/j.ejmech.2020.112238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kužnik A.; Benčina M.; Švajger U.; Jeras M.; Rozman B.; Jerala R. Mechanism Of Endosomal TLR Inhibition By Antimalarial Drugs And Imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- An J.; Woodward J.; Sasaki T.; Minie M.; Elkon K. Cutting Edge: Antimalarial Drugs Inhibit IFN-Β Production Through Blockade Of Cyclic GMP-AMP Synthase–DNA Interaction. J. Immunol. 2015, 194, 4089–4093. 10.4049/jimmunol.1402793. [DOI] [PubMed] [Google Scholar]
- HEK-Blue TLR Cells; InvivoGen, 2019; https://www.invivogen.com/hek-blue-tlr-cells (accessed 2019-03-14).
- van der Fits L.; Mourits S.; Voerman J.; Kant M.; Boon L.; Laman J.; Cornelissen F.; Mus A.; Florencia E.; Prens E.; Lubberts E. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- Perry D.; Sang A.; Yin Y.; Zheng Y.; Morel L. Murine models of systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011, 1–19. 10.1155/2011/271694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H.; Ohto U.; Shibata T.; Miyake K.; Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- Tanji H.; Ohto U.; Shibata T.; Taoka M.; Yamauchi Y.; Isobe T.; Miyake K.; Shimizu T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. 10.1038/nsmb.2943. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Hu Z.; Tanji H.; Jiang S.; Das N.; Li J.; Sakaniwa K.; Jin J.; Bian Y.; Ohto U.; Shimizu T.; Yin H. Small-molecule inhibition of TLR8 through stabilization of its resting state. Nat. Chem. Biol. 2018, 14, 58–64. 10.1038/nchembio.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.