

Abstract

Proteolysis targeting chimeras (PROTACs) are catalytic heterobifunctional molecules that can selectively degrade a protein of interest by recruiting a ubiquitin E3 ligase to the target, leading to its ubiquitylation and degradation by the proteasome. Most degraders lie outside the chemical space associated with most membrane-permeable drugs. Although many PROTACs have been described with potent activity in cells, our understanding of the relationship between structure and permeability in these compounds remains limited. Here, we describe a label-free method for assessing the permeability of several VH032-based PROTACs and their components by combining a parallel artificial membrane permeability assay (PAMPA) and a lipophilic permeability efficiency (LPE) metric. Our results show that the combination of these two cell-free membrane permeability assays provides new insight into PROTAC structure–permeability relationships and offers a conceptual framework for predicting the physicochemical properties of PROTACs in order to better inform the design of more permeable and more effective degraders.
Keywords: Selective degradation, PAMPA, LPE, permeability, structure−permeability relationships
Proteolysis targeting chimeras (PROTACs) enhance our ability to drug biologically relevant targets through selective degradation.1−3 These heterobifunctional compounds include an E3 ligase-binding ligand and a protein-targeting ligand connected by a linker. PROTACs facilitate proteasomal degradation by recruiting the target protein to an E3 ligase, leading to ubiquitylation and subsequent degradation of the targeted protein.4−6 Unlike traditional inhibitors, PROTACs are catalytic and have increased target-specificity derived largely from ternary complex protein–protein contacts.7−9 While our understanding of the bioactivity of PROTACs is rapidly increasing, the physicochemical properties of these molecules have received relatively little attention.10,11
Due to the interest in PROTAC therapeutics, there is a clear need to better understand their physicochemical properties. Given their high molecular weight (MW > 800) and the presence of multiple hydrogen bond donors (HBDs) and acceptors (HBAs), PROTACs are expected to have low membrane permeability.12−15 A recent study that used the label-based chloroalkane penetration assay (CAPA)16 showed very low permeabilities for PROTACs relative to their individual components.17 While this assay provides relative cell permeabilities across a large dynamic range, it does not provide permeability coefficients that can be compared to other data sets. Also, CAPA requires a chloroalkane tag and therefore does not directly measure the permeability of the parent compound. Establishing a label-free method to quantify the permeability of PROTACs provides greater flexibility in compound design without needing to synthesize a second set of CAPA tag-containing molecules. While there are some mass spectrometry approaches to quantify the intracellular concentration of unlabeled compounds, these indirect studies do not inform on oral bioavailability and some do not differentiate between membrane-trapped compounds and those free for target binding.17−20 The VHL-NanoLuc Fusion assay21 offers label-free assessment of cell permeability, but results are confounded by their dependence on variable VHL-binding affinities. Here we report a label-free approach for studying the passive permeability of von Hippel–Lindau (VHL)-based PROTAC molecules using the parallel artificial membrane permeability assay (PAMPA) and lipophilic permeability efficiency (LPE).22 These simple, high-throughput assays correlate strongly with cell-based permeabilities and oral bioavailability while being relatively inexpensive.23 PAMPA quantifies orders-of-magnitude differences in PROTAC permeabilities with a low limit of quantitation. LPE provides insight as to how structural changes affect permeability.
We tested the membrane permeabilities of JQ-1 (1), four model compounds (SL-X series) (3–6), and 11 previously published VHL-PROTACs.24,25 These PROTACs include four series: MZ (7–9),6,7,26 AT (15–17),7 CM/CMP (12–14),27 and MZP (10–11),26 grouped according to the target-binding ligand and attachment to the VHL-recruiting ligand (Figures 2 and 5). Most previously published PROTACs have MWs ranging from 900 to 1200 and between four and six HBDs. Based on traditional criteria of drug-likeness, these compounds are expected to have low membrane permeability. This is indeed what we found. The highest PAMPA permeability measured for this set was Pe = 0.6 × 10–6 cm/s, slightly below the standard for “modest” permeability (Pe = 1 × 10–6 cm/s). Notably, we were able to quantify permeabilities for all our compounds with coefficients as low as 0.002 × 10–6 cm/s.
Figure 2.

Physicochemical properties of “AT” and “MZ” PROTACs. Cmpd = compound; PAMPA units: × 10–6 cm/s; LogD(dec/w): 1,9-decadiene and PBS pH 7.4 shake flask partition coefficient; LPE = LogD(dec/w) – 1.06(ALogP) + 5.47.
Figure 5.

Physicochemical properties of “MZP” and “CM/CMP” PROTACs. Cmpd = compound; PAMPA units: × 10–6 cm/s; LogD(dec/w): 1,9-decadiene and PBS pH 7.4 shake flask partition coefficient; LPE = LogD(dec/w) – 1.06(ALogP) + 5.47.
From our initial set of amide-containing compounds, the most permeable compound was 4 (Pe = 8.6 × 10–6 cm/s, Figure 1), an N-terminally capped VH032 analog with a phenylacetamide acting as a simple protein-targeting model. Compound 4 was 43-fold more permeable than a similar compound, 6, with a 3-unit PEG linker between the VH032 and the phenylacetamide. Strikingly, 4 was 4000-fold more permeable than the two least permeable compounds, 17 and 14 (Figures 2 and 5, respectively). Furthermore, among all 11 PROTACs tested, there was a 300-fold difference between the most permeable compound, 7, and the least permeable compounds, 14 and 17. In the MZ series alone (7–9, Figure 2), there was a 100-fold difference between the most (7, Pe = 0.6 × 10–6 cm/s) and least (9, Pe = 0.006 × 10–6 cm/s) permeable derivatives. Combined, these data demonstrate the large dynamic range of PAMPA and support its use for unlabeled, quantitative measurements.
Figure 1.

Physicochemical properties of protein-targeting small molecules and model compounds. Cmpd = compound; PAMPA units: × 10–6 cm/s; LogD(dec/w): 1,9-decadiene and PBS pH 7.4 shake flask partition coefficient; LPE = LogD(dec/w) – 1.06(ALogP) + 5.47; “--” = not determined.
MW and solvent-exposed HBDs can significantly affect membrane permeability.12 Permeability generally decreases as MW increases,28 leading to a significant reduction in permeability beyond MW = 1000.13,29 All else being equal, the relatively high MWs (900–1200 Da) of the PROTACs represent a predicted size-dependent permeability cost of approximately one log unit compared to typical small molecules of the same lipophilicity (MW < 600).13,22 However, because the PROTACs in this study are all in a similar MW range, comparisons between them reflect differences in their physical properties separate from the size penalty. Recent reviews argue that MW effects should not be considered alone because factors like hydrophobicity and HBDs affect permeability more prominently than MW.30,31 Supporting this conclusion, our two least permeable PROTACs, 14 and 17, had the highest and lowest MWs, respectively. Furthermore, 16 and 8 have nearly the same MW (1005 and 1003, respectively), the same calculated octanol–water partition coefficients (ALogP), and the same number of HBDs and HBAs, yet their permeabilities differ by 10-fold (Figure 2). Likewise, 15 and 7 are similar in terms of MW, ALogP, and HBAs/HBDs, but 7 is 120-fold more permeable than 15 (Figure 2). As expected, the compounds that had lower MW and fewer HBDs/HBAs, including 1, 3, and 4, were significantly more permeable (Pe ≥ 5 × 10–6 cm/s, Figure 1) than the PROTACs.
Permeability data alone provide little information on how structural features affect permeability. Therefore, we measured lipophilic permeability efficiency (LPE).22 Originated by our group, LPE quantifies the efficiency with which a compound achieves passive membrane permeability at a given lipophilicity based on the experimental hydrocarbon–water partition coefficient (LogD(dec/w)) and ALogP. Combining PAMPA and LPE represents a powerful method for assessing how structural features contribute to compound permeability.
This is most evident when comparing the two matched pairs from the AT and MZ series: 15 vs 7 and 16 vs 8. These compounds have the same ALogP, the same number of HBDs/HBAs, and MWs within 2 Da. Yet, the MZ compounds, 7 and 8, are significantly more permeable than their counterparts from the AT series, 15 and 16, respectively. These AT and MZ compounds differ only in the connection between their linker and VH032 ligand. In 7 and 8, the VH032 ligand has an N-terminal tert-Leu connected to a linker through an amide bond. Alternatively, 15 and 16 have a penicillamine group in place of the tert-Leu which is attached to the linker through a thioether in place of the amide bond (Figure 2).
Clearly, the chemical environment surrounding HBDs affects the PAMPA permeability of these PROTACs, similar to the effects observed in other compounds in this MW range.32,33 The LPE values of these compounds provide insight into the potential for these flexible molecules to adopt conformations capable of shielding HBDs. Typically, the addition of a solvent-exposed HBD reduces LPE by 1.8.22 The tert-Leu-containing 7 has an LPE of 0.4, and its penicillamine counterpart, 15, has an LPE of −2.3, suggesting that 15 has at least one additional exposed HBD compared to 7. The same pattern is seen with 8 and 16 that have LPE values of 0.1 and −2.6, respectively. These LPE data show that switching the tert-Leu for a penicillamine group exposes an -NH to solvent which likely contributes to the lower permeability of these AT compounds.
The crystal structure of 8 in a ternary complex with VHL and Brd4 further supports the presence of a shielded -NH in the MZ compound series.7 Inspection of this structure shows that the tert-Leu amide -NH of 8 is in a position to be shielded from solvent by the tert-Leu side chain and is within a short contact distance to the PEG oxygen, likely participating in an intramolecular hydrogen bond (IMHB) capable of shielding the -NH polarity from solvent (Figure 3). Co-crystal structures of binary complexes of VHL with bound ligands provide additional evidence for this phenomenon showing an oxygen (in a similar position to the PEG ether in 8) that points in toward the tert-Leu-NH, potentially close enough to form an IMHB.34 While the membrane permeating conformation is not necessarily the same as the target-bound conformation, these crystal structures provide a possible explanation for the difference in solvent-exposed HBDs between the MZ and AT compounds.
Figure 3.

MZ1 ternary complex with VHL and Brd4 (PDB:5T35).7 Crystal structure showing the ternary complex of MZ1 (colored by element) with Brd4 (pink) and VHL (orange). The VHL ligand tert-Leu -NH (blue arrow) is shielded by the tert-Leu side chain and is within hydrogen bonding distance of the VHL ligand PEG oxygen (red arrow).
This relationship between the MZ and AT compounds supports reducing the number of exposed HBDs to increase permeability. The extensive structural information on VHL ligand cocrystal structures has shown that the tert-Leu amide does not form a direct hydrogen bond with the VHL protein.24,25,34 Hence, we hypothesized that removing an HBD by substituting an amide for an ester would lead to increased permeability, without detrimentally comprising VHL binding affinity. To test this, we synthesized 3 and 5, ester derivatives of 4 and 6, respectively, in which the N-terminal tert-Leu amide was replaced by an ester (Figure 1). As predicted, the ester derivatives were more permeable than their amide counterparts. Compound 3 was 2-fold more permeable than 4, and 5 was 1.5-fold more permeable than 6. Thus, the amide-to-ester substitution provides a viable option to increase the permeability of these types of compounds, though with the caveat of the ester’s potential susceptibility to intracellular esterase hydrolysis.
The LPE of the amide compounds (4 and 6) is nearly the same as the LPE of their ester compound counterparts (3 and 5, respectively), suggesting that the tert-Leu is likely shielding the polarity of the HBD in the amide-containing compounds as has been observed with beta-branched amino acids.32,33 The relatively modest increase in permeability observed with these amide-to-ester substitutions reflects the unusually low desolvation penalty for the shielded amide NH—consistent with what was observed in the MZ series. Therefore, it is possible that substituting a more exposed amide with an ester could lead to even greater improvement of membrane permeability. Using a competitive fluorescence polarization (FP) assay, we found that the ester-containing 3 was still capable of binding its target protein, VHL, with a Kd only 1.7-fold higher than that of the amide-containing 4, albeit >10-fold higher than the potent VHL inhibitor VH298 (18, Figure 4).34 The Kd increase in the ester compound further advocates for trying similar substitutions farther away from the VHL-binding ligand to maintain binding capacity while improving permeability.
Figure 4.
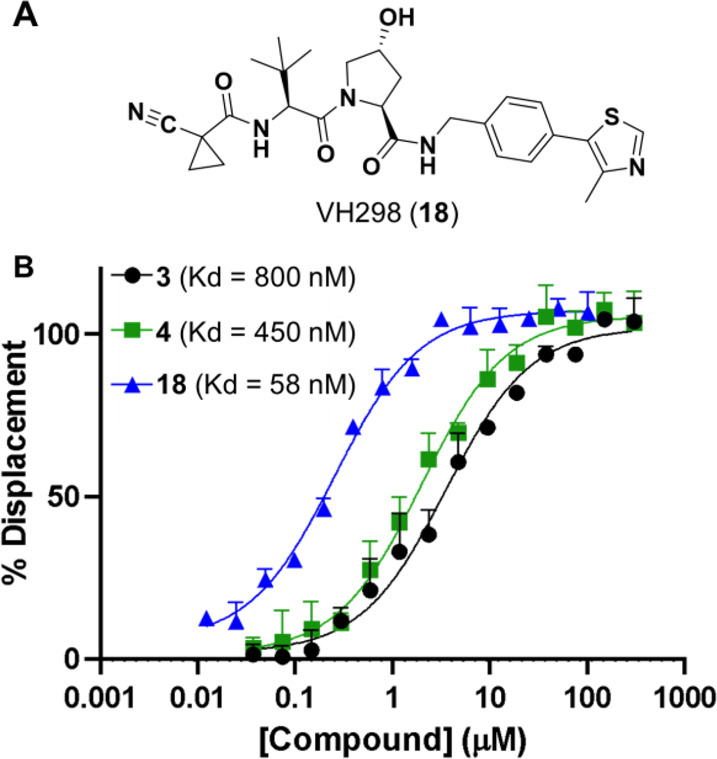
Fluorescence polarization (FP)-derived Kd of amide to ester substitution in SLX compounds: (A) VH298, a small molecule inhibitor of the E3 ubiquitin ligase VHL, used as a positive control for high-affinity binding. (B) FP data for compounds 3, 4, and 18.
Consistent with Foley et al.,17 we found that permeability increased with decreasing linker length. This was expected, as increasing the length of the linker usually results in an increase in one or more of the MW, HBDs, or HBAs. For the AT and CM/CMP series, compound permeability was reduced by half with one or two additional PEG units in the linker, respectively (cf. 15 vs 16, and 12 vs 13, Figures 2 and 5). This effect was more prominent in the MZ series as 7 (2-unit PEG linker) was 20-fold more permeable than 8 (3-unit PEG linker). A 2-fold difference in permeability was also seen in the MZP compounds (11, 4-unit PEG linker, and 10, 2-unit PEG linker, Figure 5). These results indicate that shorter linkers typically produce more permeable compounds. Moreover, for all but the MZP series, the compounds with shorter PEG linkers had higher LPE values, suggesting that compounds with shortened linkers were more efficient at permeating the membrane for their given lipophilicity.
Previous studies have advocated for the use of short alkyl linkers over PEG linkers to reduce total polar surface area to improve permeability.17 Our results diverge in this respect, as we found that our only compound bearing an alkyl linker, 17, was the least permeable (Pe = 0.002 × 10–6 cm/s). This compound was 2.5-fold less permeable than 15, which has a 1-unit PEG linker. Compound 17 has one fewer HBA in this linker than 15 which could reduce solubility and therefore affect permeability. While PAMPA allows us to quantify the differences in permeabilities directly, analyzing LPE enable us to predict which structural features cause the permeability changes. Increasing the number of PEG units in the PROTAC linker reduces the LPE of that compound (cf. 15 and 16). If the HBAs in these PEG linkers were not contributing to IMHB, substituting the PEG linker in 15 with an alkyl linker as in 17 (removing HBAs) should have little effect on LPE. However, this is not what we observed. Instead, the LPE of alkyl-linked 17 is 0.8 lower than its PEG counterpart 15, suggesting that the ether oxygen in the PEG linker of 15 is capable of shielding HBD, possibly the linker amide bond -NH (adjacent to JQ-1) in a manner similar to that observed for MZ1 (Figure 3). As the ΔLPE between 15 and 17 is less than the 1.8-unit difference expected for a fully exposed HBD, it is likely that the PEG ether provides only partial shielding by way of IMHB formation.
The same phenomenon is present in the SL-X series (3–6, Figure 1). Compounds 5 and 6 have an additional amide and 3-unit PEG linker compared to 3 and 4, respectively. If no additional IMHBs were present in 5 and 6, the inclusion of these additional HBAs and HBD should cause a decrease in LPE of at least 1.8, compared to 3 and 4. Yet, the LPE values of 5 and 6 are only moderately lower than 3 (ΔLPE = 0.8) and 4 (ΔLPE = 0.4), respectively. Thus, the PEG linker is likely involved in IMHBs responsible for shielding some polarity. Moreover, using a linker capable of forming IMHB could shield the polarity of important HBDs responsible for target engagement, a feature that would not be possible with an alkyl linker. Therefore, the best linker type for a given PROTAC is likely scaffold dependent, further highlighting the need to examine the overall lipophilicity of the molecule when designing a PROTAC.
Comparing PAMPA and LogD(dec/w) to ALogP allows us to analyze permeability trends and predict permeability improvements. For compounds with ALogPs up to ∼4, there is a positive linear correlation between ALogP and permeability.22 As lipophilicity increases beyond ALogP ∼ 4–5, compounds become insoluble or membrane-retained, and their effective membrane permeabilities diminish (Figure 6A). Therefore, designing PROTACs to have an ALogP below 5.0 could bias these compounds toward higher permeabilities. The CM/CMP compounds have low permeabilities and lower ALogPs (<1) than the other PROTACs. As permeability typically increases with ALogP from 0–4, a lipophilicity increase, such as increasing the number of −CH2– groups relative to oxygens in the linker, could greatly improve CM/CMP permeability.35
Figure 6.

PROTAC permeability and LPE. Graphs showing the (A) permeability vs ALogP and the (B) LogD(dec/w) vs ALogP for compounds 1–17. Dashed line on (A) shows the linear correlation between PAMPA and ALogP for ALogP from 0–4 (R2 = 0.9581). Dashed lines on (B) represent LPE classes, m = 1.06. LPE values (gray) are LPE averages for compounds that fall on or near the line.
Plotting LogD(dec/w) vs ALogP creates a visualization of the LPE metric which offers potential strategies to improve permeability (Figure 6B). For example, in the MZ series, 7 and 8 have low permeabilities (>0.6 × 10–6 cm/s) and moderately low LPE values (>0.5). As 7 and 8 already have ALogP values close to 4.0, further increasing lipophilicity would likely push these compounds into the insoluble region and cause a further decrease in their membrane permeability (Figure 6A). Also, the addition of a Phe residue to 8 to generate 9 leads to a 1.2-unit decrease in LPE due to the addition of an amide NH (which is less than the 1.8-unit cost expected for the addition of an amide group, indicating partial IMHB). This decrease in LPE between 8 and 9 is partially offset by an increase in ALogP of 1.1 units, leading to a 5-fold decrease in permeability and putting 9 over the edge of the solubility cliff. This analysis suggests that the decreased degrader activity observed in cells for 9 compared to 8(6) could be, in part, due to these poor physicochemical properties. Both the decrease in LPE and a significant increase in ALogP contribute to the very poor permeability of 9. An alternative solution to improving the permeability of 7 and 8 would be to replace the amide linkage to the bromodomain warhead with a group (such as an ester) that does not contribute an HBD.
The effect of structural features on permeability and bioactivity can be significant. Generally, the bromodomain-targeting compounds (MZ, AT, MZP), with extremely low permeabilities (≤0.006 × 10–6 cm/s), were less active in relevant cellular antiproliferation assays than compounds with higher permeabilities (≥0.03 × 10–6 cm/s, SI Table 2).26 Specifically, 9 was both less permeable and less bioactive than 7 and 8. This decreased bioactivity is likely attributed to the decreased permeability (Figure 2, SI Table 2), as binding affinities with the target proteins were broadly comparable.6,24 Similarly, the AT compounds were the least active compounds tested, consistent with their much lower permeability (Figure 2, SI Table 2). However, the related PROTAC, AT1, exhibited a 5-fold lower bind affinity for the VHL protein and formed less stable ternary complexes compared to 8,7,36 which could also contribute to the significant loss of cellular potency in the AT series. Conversely, the formation of a cooperative and stable ternary complex can override the impact of permeability.36 For example, 8 forms a more stable complex with its targets, Brd4 and VHL, than compound 7, leading 8 to be one log unit more active, despite being 20-fold less permeable than 7 (Figure 2, SI Table 2).26 Similarly, in the CM/CMP series, 13 is two log units more active than 12 in a cellular protein degradation assay despite being slightly less permeable (0.005 cm/s vs 0.009 cm/s, Figure 5).27 This suggests that differences in efficacy between these two compounds are likely due to the relative stability of their respective ternary complexes27 rather than differences in their extremely low permeabilities. These results suggest that efforts to improve the permeability should be monitored in conjunction with effects on ternary complex formation.
In this study, we have demonstrated that combining PAMPA and LPE provides insight into PROTAC structure–permeability relationships. These label-free assays model only passive permeability without the confounding effects of active transport. PAMPA and LogD(dec/w) are established methods; therefore, comparisons can be made to data previously gathered using these methods. With this simple method for measuring the permeability of PROTACs in hand, a more systematic study on PROTAC permeability and pharmacokinetics is required. While this study provides some evidence, assessing the permeability of PROTACs over a complete range of ALogP values would allow us to develop a more detailed lipophilicity window to guide the design of PROTACs biased toward higher permeability. As esters are generally more prone to hydrolysis than amides, additional studies are required to assess the viability of amide-to-ester substitutions. Finally, VH032-based PROTACs have a high number of HBDs and HBAs often present on both protein-binding domains of the molecule that are typically connected by a long flexible linker. This arrangement of HBDs and HBAs lends itself to the formation of IMHBs capable of shielding some of the PROTACs’ polarity, enhancing permeability. The recently reported macrocyclization of PROTACs37 could also prove beneficial in this regard by taking advantage of the IMHBs and HBD-shielding often achieved by cyclic peptides. Future studies on the permeability of these compounds, and expansion of these studies to include other PROTAC classes such as those based on cereblon-binding ligands, are warranted as they could create opportunities to model and predict a network of IMHBs and fine-tune these interactions to produce more permeable and more bioactive PROTACs.
Experimental Procedures
For methods, see the Supporting Information.
Glossary
Abbreviations
- CAPA
chloroalkane penetration assay
- FP
fluorescence polarization
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- IMHB
intramolecular hydrogen bond;
- LPE
lipophilic permeability efficiency
- MW
molecular weight
- PAMPA
parallel artificial membrane permeability assay
- PROTAC
proteolysis targeting chimera
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00265.
Experimental procedures, synthetic methods, LCMS analysis, and bioactivity data (PDF)
Author Present Address
§ A.T. and S.J.H.: Amphista Therapeutics Ltd, Bo’Ness Road, Newhouse ML1 5UH, United Kingdom
Author Present Address
∥ C.M.: School of Natural and Environmental Sciences, Chemistry, Bedson Building, Kings Road, Newcastle University, Newcastle Upon Tyne NE1 7RU, United Kingdom
Author Present Address
⊥ K.-H.C.: GlaxoSmithKline R&D, Gunnels Wood Rd, Stevenage SG1 2NY, United Kingdom
Author Contributions
V.G.K., R.S.L, and A.C. designed the project and wrote the manuscript. Permeability experiments were performed by V.G.K. Mass spectrometry data was processed by C.E.T. FP data was collected and processed by S.J.H. Compounds were synthesized by R.S.L., A.T., M.Z., and C.M with design support from A.C. Cell proliferation data was collected by K.-H.C. The manuscript was edited by all.
The R.S.L. group was funded by the National Institute of General Medicine Studies of the National Institutes of Health (NIH R01GM131135) and the National Science Foundation GRFP (NSF DGE 1339067 to V.G.K). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NIH or the NSF. The A.C. lab was funded by awards from the UK Biotechnology and Biological Sciences Research Council (BBSRC, grant BB/J001201/2), the European Research Council (ERC, Starting Grant ERC-2012-StG-311460 DrugE3CRLs), the Italian Ministry of Education, University and Research (Miur, Ph.D. Studentship to C.M.), and the European Commission (Marie Skłodowska-Curie Actions Individual Fellowship H2020-MSCA-IF-2014–655516 to K.-H.C.).
The authors declare the following competing financial interest(s): The A.C. laboratory receives or has received sponsored research support from Boehringer Ingelheim, Eisai Co., Nurix, Ono Pharmaceuticals, and Amphista Therapeutics. A.C. is a scientific founder, shareholder, nonexecutive director, and consultant of Amphista Therapeutics, a company that is developing targeted protein degradation therapeutic platforms.
While this manuscript was under review, a study was published by Scott, et al., (ACS Med. Chem. Lett.; doi.org/10.1021/acsmedchemlett.0c00194) that investigated various ADME properties, including PAMPA and cell permeabilities, of a similar (though not identical) set of PROTAC compounds.
Supplementary Material
References
- Pettersson M.; Crews C. M. Proteolysis Targeting Chimeras (Protacs) — Past, Present and Future. Drug Discovery Today: Technol. 2019, 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Ciulli A. Bifunctional Chemical Probes Inducing Protein–Protein Interactions. Curr. Opin. Chem. Biol. 2019, 52, 145–156. 10.1016/j.cbpa.2019.07.003. [DOI] [PubMed] [Google Scholar]
- Verma R.; Mohl D.; Deshaies R. J. Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cell 2020, 77 (3), 446–460. 10.1016/j.molcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 ubiquitin Ligase Cereblon to Efficiently Target Brd4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein Brd4. ACS Chem. Biol. 2015, 10 (8), 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13 (5), 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25 (1), 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10 (1), 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt G. F.; Scott-Stevens P.; Gaohua L. Targeted Protein Degradation in Vivo with Proteolysis Targeting Chimeras: Current Status and Future Considerations. Drug Discovery Today: Technol. 2019, 31, 69–80. 10.1016/j.ddtec.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Cantrill C.; Chaturvedi P.; Rynn C.; Petrig Schaffland J.; Walter I.; Wittwer M. B. Fundamental Aspects of DMPK Optimization of Targeted Protein Degraders. Drug Discovery Today 2020, 25 (6), 969–982. 10.1016/j.drudis.2020.03.012. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lead Profiling Lead- and Drug-Like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Pye C. R.; Hewitt W. M.; Schwochert J.; Haddad T. D.; Townsend C. E.; Etienne L.; Lao Y.; Limberakis C.; Furukawa A.; Mathiowetz A. M.; Price D. A.; Liras S.; Lokey R. S. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem. 2017, 60 (5), 1665–1672. 10.1021/acs.jmedchem.6b01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson S. D.; Yang B.; Fallan C. Proteolysis Targeting Chimeras (Protacs) in ‘Beyond Rule-of-Five’ Chemical Space: Recent Progress and Future Challenges. Bioorg. Med. Chem. Lett. 2019, 29 (13), 1555–1564. 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- Maple H. J.; Clayden N.; Baron A.; Stacey C.; Felix R. Developing Degraders: Principles and Perspectives on Design and Chemical Space. MedChemComm 2019, 10 (10), 1755–1764. 10.1039/C9MD00272C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraro L.; Deprey K. L.; Moser M. K.; Zou Z.; Ball H. L.; Levine B.; Kritzer J. A. Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J. Am. Chem. Soc. 2018, 140 (36), 11360–11369. 10.1021/jacs.8b06144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley C. A.; Potjewyd F.; Lamb K. N.; James L. I.; Frye S. V. Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. ACS Chem. Biol. 2020, 15 (1), 290–295. 10.1021/acschembio.9b00972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletti L. M.; Liu Y.; Koev G.; Richardson P. L.; Chen C.-M.; Kati W. Methods to Measure the Intracellular Concentration of Unlabeled Compounds within Cultured Cells Using Liquid Chromatography/Tandem Mass Spectrometry. Anal. Biochem. 2008, 383 (2), 186–193. 10.1016/j.ab.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Gordon L. J.; Allen M.; Artursson P.; Hann M. M.; Leavens B. J.; Mateus A.; Readshaw S.; Valko K.; Wayne G. J.; West A. Direct Measurement of Intracellular Compound Concentration by Rapidfire Mass Spectrometry Offers Insights into Cell Permeability. J. Biomol. Screening 2016, 21 (2), 156–164. 10.1177/1087057115604141. [DOI] [PubMed] [Google Scholar]
- Mateus A.; Gordon L. J.; Wayne G. J.; Almqvist H.; Axelsson H.; Seashore-Ludlow B.; Treyer A.; Matsson P.; Lundbäck T.; West A.; Hann M. M.; Artursson P. Prediction of Intracellular Exposure Bridges the Gap between Target- and Cell-Based Drug Discovery. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (30), E6231–E6239. 10.1073/pnas.1701848114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Naylor M. R.; Ly A. M.; Handford M. J.; Ramos D. P.; Pye C. R.; Furukawa A.; Klein V. G.; Noland R. P.; Edmondson Q.; Turmon A. C.; Hewitt W. M.; Schwochert J.; Townsend C. E.; Kelly C. N.; Blanco M.-J.; Lokey R. S. Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem. 2018, 61 (24), 11169–11182. 10.1021/acs.jmedchem.8b01259. [DOI] [PubMed] [Google Scholar]
- Avdeef A. The Rise of Pampa. Expert Opin. Drug Metab. Toxicol. 2005, 1 (2), 325–342. 10.1517/17425255.1.2.325. [DOI] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57 (20), 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J.; Galdeano C.; Soares P.; Gadd M. S.; Grzes K. M.; Ellis L.; Epemolu O.; Shimamura S.; Bantscheff M.; Grandi P.; Read K. D.; Cantrell D. A.; Rocha S.; Ciulli A. Potent and Selective Chemical Probe of Hypoxic Signalling Downstream of HIF-Α Hydroxylation Via VHL Inhibition. Nat. Commun. 2016, 7 (1), 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.-H.; Zengerle M.; Testa A.; Ciulli A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (Bet) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) Bet Inhibitor Scaffolds. J. Med. Chem. 2018, 61 (2), 504–513. 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Hughes S. J.; Testa A.; Chen W.; Lamont D. J.; Rocha S.; Alessi D. R.; Romeo R.; Ciulli A. Homo-Protacs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 2017, 8 (1), 830. 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. The Relationship between Permeant Size and Permeability in Lipid Bilayer Membranes. J. Membr. Biol. 1994, 140 (2), 111–122. 10.1007/BF00232899. [DOI] [PubMed] [Google Scholar]
- Doak B. C.; Over B.or.; Giordanetto F.; Kihlberg J. Oral Druggable Space Beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21 (9), 1115–1142. 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting Physical in Drug Discovery II: The Impact of Chromatographic Hydrophobicity Measurements and Aromaticity. Drug Discovery Today 2011, 16 (17–18), 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62 (4), 1701–1714. 10.1021/acs.jmedchem.8b00686. [DOI] [PubMed] [Google Scholar]
- Nielsen D. S.; Hoang H. N.; Lohman R.-J.; Hill T. A.; Lucke A. J.; Craik D. J.; Edmonds D. J.; Griffith D. A.; Rotter C. J.; Ruggeri R. B.; Price D. A.; Liras S.; Fairlie D. P. Improving on Nature: Making a Cyclic Heptapeptide Orally Bioavailable. Angew. Chem., Int. Ed. 2014, 53 (45), 12059–12063. 10.1002/anie.201405364. [DOI] [PubMed] [Google Scholar]
- Bockus A. T.; Schwochert J. A.; Pye C. R.; Townsend C. E.; Sok V.; Bednarek M. A.; Lokey R. S. Going out on a Limb: Delineating the Effects of Β-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide a Analogues. J. Med. Chem. 2015, 58 (18), 7409–7418. 10.1021/acs.jmedchem.5b00919. [DOI] [PubMed] [Google Scholar]
- Soares P.; Gadd M. S.; Frost J.; Galdeano C.; Ellis L.; Epemolu O.; Rocha S.; Read K. D.; Ciulli A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase: Structure–Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61 (2), 599–618. 10.1021/acs.jmedchem.7b00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulli A.; Maniaci C.; Hughes S. J.; Testa A.. Small Molecules. WO 2018/189554, October 18, 2018.
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14 (3), 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa A.; Hughes S. J.; Lucas X.; Wright J. E.; Ciulli A. Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem., Int. Ed. 2020, 59 (4), 1727–1734. 10.1002/anie.201914396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.