

Abstract

Neuraminidase (NA) is an important target for current research on anti-influenza drugs. The acylhydrazone derivatives containing the −CONHN=CH– framework have been shown to have good NA inhibitory activity. In this paper, a series of novel acylhydrazone NA inhibitors (9a–9n) were designed and synthesized, and the inhibitory activities against NA were evaluated in vitro. The NA inhibition results showed that compound 9j has the most potent inhibitory activity (IC50 = 0.6 μM) against NA, which is significantly lower than that of the positive control oseltamivir carboxylic acid (OSC) (IC50 = 17.00 μM). Molecular docking analysis indicates that the acylhydrazone group plays an important role in compound 9j, which can bind well to the residues Arg371 and Arg292 in the S1 subsite of NA. The good potency of 9j may be also ascribed to the extending of morpholinyl ring into the 430-cavity. The results of this work may contribute to the development of more potent NA inhibitors to against mutant influenza viruses.
Keywords: Neuraminidase inhibitor, acylhydrazone, 430-cavity, bioassay
Influenza is a common viral respiratory disease caused by influenza viruses and has a strong ability to spread. Influenza virus is a negative-sense single-strand member of the Orthomyxoviridae family of RNA viruses. According to the differences in the antigenicity of nuclear proteins and matrix proteins, influenza virus are divided into three types: A, B, and C. Influenza A viruses are prone to antigenic mutations and are the main cause of seasonal or pandemic influenza worldwide.1−3 The most severe pandemic occurred in 1918, resulting in approximately 40 million deaths worldwide.4 Vaccines and antiviral drugs are currently important initiatives in the prevention and treatment of influenza. However, due to the long-lasting effect of the vaccine and its limited effectiveness in patients with low immune function, the current focus is on the development of small molecule drugs against influenza viruses.5
Neuraminidase (NA), also known as sialidase, is a glycoprotein distributed on the envelope of influenza virus. It has antigenicity and can catalyze the hydrolysis of sialic acid, which can help mature influenza virus to escape from its original host cells to infect new cells and play a crucial role in virus transmission. The active center of neuraminidase includes 19 conserved amino acids. Among them, the eight highly conserved amino acids that can directly interact with the substrate are Arg118, Arg152, Arg224, Arg292, Arg371, Asp151, Glu276, and Tyr406.6 The highly conservative structure of its catalytic active site makes it a superior target for antiviral drug development. So far, there are four approved drugs of neuraminidase inhibitors, including oseltamivir, zanamivir, peramivir, and laninamivir octanoate (Figure 1).7 Among them, oseltamivir is the only oral effective neuraminidase inhibitor. Zanamivir can only be administered by inhalation. Peramivir sodium chloride injection has been approved in China. Laninamivir octanoate has recently been authorized in Japan.8 Nevertheless, emergence and rapid spreading of resistance to these drugs poses an urgent threat to public health.9,10 Therefore, it is imperative to develop novel drug-resistant neuraminidase inhibitors against resistant virus strains.
Figure 1.

NA inhibitors had been approved for the treatment of influenza viruses.
With the further study of the crystal structure of neuraminidase, based on the phylogenetic tree, neuraminidase (NA) is divided into nine subtypes (N1–N9), in which N1, N4, N5, and N8 are the first group and N2, N3, N6, N7, and N9 are the second group. In 2006, Russell and co-workers declared that the difference between the two groups was that the first group contains a 150-cavity (residues 147−152), while the second group does not.6,11 Recently, an additional hydrophobic binding site adjacent to the active site had been discovered and named as the 430-cavity. The 430-cavity connects directly to the active site and has a large molecular volume. Interestingly, different from the 150-cavity, the 430-cavity widely and stably exists in various subtypes of neuraminidase, including group-1 and group-2.12,13 This important discovery provides a great opportunity for inhibitor design. For example, Liu’s team discovered a series of C-1 modified oseltamivir derivatives targeting the 430-cavity as potent NA inhibitors.14 A series of zanamivir analogues with C-1 and C-4 modifications had also been developed to exploit the 430-cavity.15 Therefore, as an important binding site, the 430-cavity is of great significance for the development of new NA inhibitors.
The acylhydrazones derivatives containing the −CONHN=CH– framework are the products obtained by condensation of the acylhydrazones with the corresponding aldehydes or ketones. They have strong biological activity and stable structure and have been proven to have good application prospects in biological and drug activity, bacteriostatic and antitumor.16−19 In addition, some acylhydrazone derivatives have been reported to have potential anti-influenza virus activity,20 and several of them have been evaluated for NA inhibitory activity. For instance, Xiao et al.21 synthesized a series of acylhydrazone derivatives via microwave irradiation as influenza NA inhibitors. Moreover, according to our previous reports, Zhao et al.22 discovered an acylhydrazone lead compound as a NA inhibitor through virtual screening. Yu et al.23 designed and synthesized a series of benzoylhydrazone neuraminidase inhibitors. Both series of compounds showed superior NA inhibitory activity to the positive control oseltamivir acid. Given the highly conserved active site of neuraminidase and the promising binding site of the 430-cavity, inspired by these studies, we tried to develop some new novel acylhydrazone NA inhibitors and evaluate them in vitro for inhibitory activity assay.
Based on the features derived from the pharmacophoric model in our previous study,22 a series of novel acylhydrazone neuraminidase inhibitors 9a–9n (see Scheme 1) were designed. The superimposition of the new designed compound 9j onto the pharmacophore model is shown in Figure 2. It can be seen that the two nitrogen atoms in the acylhydrazone moiety coincides with two positive nitrogen centers (NP_6 and NP_7) and the benzene ring of the acylhydrazone group is just at the position of the hydrophobic center HY_9. There are two hydrophobic centers (HY_5 and HY_10) and a hydrogen bond acceptor center (AA_3) near the substituted phenol, indicating that some hydrogen bond acceptor groups and hydrophobic groups introduced into this position may be beneficial to improve biological activity. Hence, we introduced some hydrogen bond acceptor atoms or groups containing N, O, and F (9a, 9b, and 9n). Some hydrophobic groups were also introduced, such as halogen atoms (9c, 9d, 9e, and 9m), nitro groups (9f, 9g, and 9l), methoxy groups (9h, 9i, and 9j), and an acetamide group (9k). Therefore, a total of 14 new compounds from 9a–9n were designed.
Scheme 1. Synthesis of Compounds 9a–9n.

Regents and conditions: (i) NaBH4, C2H5OH, THF, 0 °C; (ii) PBr3, CH2Cl2, 0 °C; (iii) Cs2CO3, KI, DMF, 60 °C; (iv) NH2NH2·H2O, absolute ethanol, 0 °C; (v) H2O, rt; (vi) MeOH, 70 °C reflux.
Figure 2.

(a) Features of the pharmacophore model. (b) Superposition of compound 9j onto the pharmacophore model.
The synthesis of acylhydrazones (9a–9n) was accomplished in six steps as outlined in Scheme 1.24−26 First, isophthalaldehyde (1) was subjected to a single reduction under the action of sodium borohydride to obtain 3-(hydroxymethyl) benzaldehyde 2. Then 3-(hydroxymethyl) benzaldehyde 2 was brominated under the action of phosphorus tribromide (PBr3) to form 3-(bromomethyl) benzaldehyde 3. Next, compound 3 was etherified with substituted phenol 4 by Williamson ether synthesis to obtain compounds 5a–5n. Then diethyl oxalate 6 reacted with hydrazine hydrate to prepare monoethyl oxalate hydrazine 7, which was stirred with compounds 5a–5n at room temperature to produce compounds 8a–8n. Finally, compounds 8a–8n were refluxed with N-(3-aminopropyl) morpholine in an organic solvent to obtain target compounds 9a–9n.
The biological activity against NA for the new synthesized compounds was further tested and is listed in Table 1. It can be seen that several of the target compounds show good inhibitory potency, for example, compounds 9b, 9e, 9j, and 9m, with IC50 values of 5.29, 6.20, 0.60, and 7.31 μM, respectively, which are significantly superior to the positive control OSC (IC50 = 17.00 μM). Especially, compound 9j exerts the most potency. The activities of 9i (IC50 = 13.60 μM) and 9n (IC50 = 17.82 μM) are comparable to that of OSC. Meanwhile, as shown in Table 1, the activity of compounds with the same substituents at the 4-position of the phenol ring is generally higher than that at the 2-position. This was supported by the comparison of inhibitory activity of compound 9a (IC50 = 21.57 μM) with 9b (IC50 = 5.29 μM), compound 9c (IC50 = 20.40 μM) with 9e (IC50 = 6.20 μM), compound 9f (IC50 = 85.97 μM) with 9g (IC50 = 27.84 μM), and compound 9h (IC50 = 47.75 μM) with 9j (IC50 = 0.60 μM). Particularly, the activity of 9j is about 79-fold more active against NA compared with 9h. In addition, the introduction of groups containing N, O, and F, such as methoxy and nitro groups and F atoms, leads to easy formation of hydrogen bonds with amino acids at the active site, which may increase the activity of the inhibitors to a certain extent.
Table 1. NA Inhibition Activity of Target Compounds 9a–9n.
| compd | R group | IC50 (μM) |
|---|---|---|
| 9a | 2-F | 21.57 ± 0.43 |
| 9b | 4-F | 5.29 ± 0.14 |
| 9c | 2-Cl | 20.40 ± 0.86 |
| 9d | 3-Cl | 22.13 ± 0.68 |
| 9e | 4-Cl | 6.20 ± 0.47 |
| 9f | 2-NO2 | 85.97 ± 2.19 |
| 9g | 4-NO2 | 27.84 ± 2.36 |
| 9h | 2-OCH3 | 47.75 ± 0.05 |
| 9i | 3-OCH3 | 13.60 ± 0.54 |
| 9j | 4-OCH3 | 0.60 ± 0.16 |
| 9k | 4-NHAc | 73.39 ± 2.93 |
| 9l | 2-OCH3-4-NO2 | 28.38 ± 2.86 |
| 9m | 2-OCH3-5-Br | 7.31 ± 0.23 |
| 9n | 5-F-2-NO2 | 17.82 ± 3.58 |
| OSC | 17.00 ± 1.41 |
To better understand the discrepancy in activity between compounds, molecular docking was performed to explore the binding affinities between inhibitors and NA. The Surflex-Dock module of SYBYL-X 2.1 was used for molecular docking.27 The crystal structure of the neuraminidase protein complex was taken from the RCSB protein database (PDB ID: 2HU0). The neuraminidase activity site is highly polar. It was suggested that the active site of NA could be divided into five regions empirically, termed subsites S1–S5, which are critical for the interactions with different NA inhibitors (Figure 3).28Figure 4 shows the binding patterns of compounds OSC, 9j, and 9f with NA. The docking results of the remaining compounds with NA are shown in Figure S1. As seen from Figure 4, for OSC, its whole skeleton is completely embedded in the active site of neuraminidase, which can form multiple hydrogen bonding interactions with some key amino acids, such as Arg118, Arg371, Arg292, Arg152, and Asp151. Among them, the C-1 carboxylate can form strong electrostatic interactions and three hydrogen bonds with the three positively charged arginine residues (Arg118, Arg292, Arg371) in the S1 site, which are essential for the NA inhibitory activity.29 The N atom of the C-5 amino group forms a hydrogen bond with the Asp151 residues of the S2 site. The carbonyl oxygen atom of acetamido may form a hydrogen bond with the Arg152 residue of the S3 site and a strong hydrophobic interaction with the Trp178 residue. For the most potent 9j, the acylhydrazone (−CONHN=CH−) group and its adjacent carbonyl group form four hydrogen bonds with some key residues Arg118, Arg371, and Arg292 of the S1 site, in a similar pattern with that of the 1-carboxyl group of OSC. In addition, the carbonyl oxygen atom of the acylhydrazone group forms a hydrogen bond with the Tyr406 residue. The amide nitrogen atom forms a hydrogen bond with the Tyr347 residue. The benzene ring of the acylhydrazone moeity forms hydrophobic interaction with the hydrophobic faces of residues Arg224 and Glu276 of the S4 and S5 sites. The 4-OCH3 substituted phenol may form strong hydrophobic interactions with the hydrophobic faces of residues Asp151, Glu119, and Glu227 of the S2 site. It is worth noting that the morpholine ring of compound 9j can extend into the 430-cavity and form strong hydrophobic interactions with residues Lys432, Pro431, and Ser370, which may explain its higher activity than the positive control OSC. Meanwhile, as shown in Figure 4, compound 9f can also well occupy the active site. The nitro group of the phenol ring forms two hydrogen bonds with the Arg292 residue of the S1 site, and two hydrogen bonds with the Tyr347 and Tyr406 residues, respectively. The phenol ring forms hydrophobic interactions with the hydrophobic face of Arg151 of the S2 site. The acylhydrazone moiety forms a hydrogen bond with Arg152 of the S3 site. The morpholine ring forms strong hydrophobic interaction with the residues Trp178 and Ile222 of the S3 site as well as the hydrophobic face of residue Arg224 of the S4 site. A total of five hydrogen bonds were formed. Although 9f forms as many hydrogen bonds in the active pocket of NA as OSC, its activity is not better than that of OSC, perhaps because the binding affinity of the inhibitor with residues Arg152 and Tyr406 is not as strong as that with residues Arg118 and Arg292. In addition, compared with 9j, compound 9f could occupy the active site but it has no group to extend into the 430-cavity. Therefore, the higher NA inhibitory potency of 9j may be mainly ascribed to the elongation of the molecule to the 430-cavity to form strong hydrophobic interactions with NA.
Figure 3.
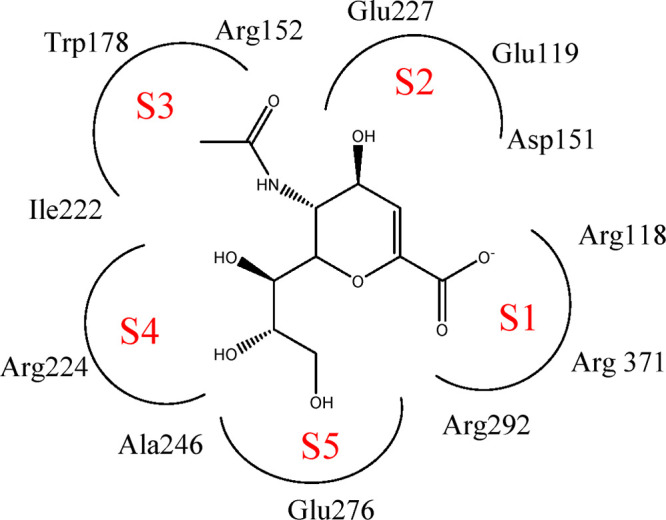
Schematic diagram of the S1–S5 region division and the combination of sialic acid (Neu5Ac) and NA active sites.
Figure 4.

Binding models of compounds OSC, 9j, and 9f with NA (PDB: 2HU0).
To clarify the dynamic interaction patterns between the inhibitors and NA protein and to verify the reliability of molecular docking results, molecular dynamics (MD) simulations were conducted for NA–inhibitor complexes by using the Amber 12.0 software package.30 The RMSD value (Å) with respect to simulation time (ns) is shown in Figure 5. It can be seen that most of the complexes are stable after 10 ns of simulation runtime, and the fluctuation of the average RMSD value during the whole simulation is between 2.0 and 4.5 Å, indicating that the complexes keep the proximate native state. Figure 6 depicts the superposition of the previously docked conformation (in green) and the conformations after the stabilization of RMSD values (in magenta) of the 9j–2HU0 complex. It can be seen that, except for a slight drift and rotation of bonds, the two conformers are in a nearly parallel poses and there seems to be no significant difference. This confirms that the binding pocket and the poses of the ligand are stable and the docking results are reliable.
Figure 5.

Diagram of RMSD value (Å) with respect to simulation time (ns).
Figure 6.

Superposition of the previously docked conformation (in green) and the conformations after the stabilization of RMSD values (in magenta) of 9j–2HU0 complex.
Based on MD simulations, the binding free energies (ΔGbind) of the 14 new inhibitors with NA were calculated and are listed in Table 2. In this research, ΔGbind was calculated by MM/PBSA and MM/GBSA methods. It is worth mentioning that MM/PBSA is generally considered superior to MM/GBSA in calculating binding free energy.31 As shown in Table 2, the smaller the ΔGbind of inhibitor-NA is, the stronger the activity of the inhibitor is for most inhibitors. For instance, the binding free energies of compounds 9e, 9j, and 9m were predicted by MM/PBSA methods to be −24.31, −22.66, and −26.40 kcal·mol–1, respectively. They are all significantly lower than that of OSC (−10.34 kcal·mol–1). Correspondingly, these three compounds have better experimental inhibitory activity, with IC50 values of 6.20 μM, 0.60 μM and 7.31 μM, which are significantly lower than that of OSC (IC50 = 17.00 μM). Moreover, the binding free energy of compound 9i (−11.23 kcal·mol–1) is similar to that of OSC. They also have comparable inhibitory activity, with IC50 values of 13.60 and 17.00 μM, respectively. The same rule also applies to compounds 9a and 9g, and compounds 9f and 9k. Hence one can see that the theoretical calculation results from MD simulation are well consistent with the experimental results.
Table 2. Calculated Binding Free Energies (ΔGbind, kcal·mol–1) of Ligand–2HU0 Complexes along with the Different Energy (kcal·mol–1) Contributions.
| compd | VDW | EEL | ΔGgas | ΔGGB | ΔGSA | ΔGsolv(GB) | ΔGbind(GB) | ΔGPB | ΔGSA | ΔGsolv(PB) | ΔGbind(PB) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OSC | –24.43 | –13.22 | –37.65 | 26.42 | –3.43 | 22.99 | –14.66 | 30.26 | –2.96 | 27.31 | –10.34 |
| 9a | –29.60 | –11.23 | –40.83 | 22.50 | –4.33 | 18.17 | –22.66 | 24.37 | –3.38 | 20.99 | –19.84 |
| 9b | –25.42 | –16.36 | –41.78 | 30.50 | –3.52 | 26.98 | –14.80 | 29.46 | –3.42 | 26.04 | –15.74 |
| 9c | –43.81 | –24.61 | –68.42 | 46.30 | –5.99 | 40.31 | –28.11 | 51.79 | –4.89 | 46.89 | –21.53 |
| 9d | –32.83 | –9.27 | –42.10 | 29.07 | –4.76 | 24.31 | –17.79 | 32.98 | –4.36 | 28.62 | –13.48 |
| 9e | –45.83 | –21.40 | –67.23 | 38.18 | –5.53 | 32.65 | –34.57 | 47.08 | –4.16 | 42.92 | –24.31 |
| 9f | –40.20 | –29.39 | –69.59 | 49.43 | –5.32 | 44.10 | –25.49 | 51.91 | –4.24 | 47.67 | –21.92 |
| 9g | –37.88 | –18.82 | –56.70 | 34.28 | –5.03 | 29.26 | –27.45 | 40.77 | –3.91 | 36.85 | –19.85 |
| 9h | –24.27 | –8.47 | –32.74 | 20.43 | –3.13 | 17.31 | –15.43 | 20.97 | –3.03 | 17.93 | –14.81 |
| 9i | –33.06 | –4.97 | –38.02 | 26.23 | –4.19 | 22.04 | –15.98 | 30.47 | –3.68 | 26.79 | –11.23 |
| 9j | –46.65 | –36.40 | –83.05 | 48.25 | –6.57 | 41.69 | –41.36 | 65.40 | –5.02 | 60.39 | –22.66 |
| 9k | –35.30 | –18.43 | – 53.73 | 38.42 | –4.36 | 34.06 | –19.67 | 36.80 | –3.99 | 32.81 | –20.92 |
| 9l | –30.91 | –11.20 | –42.11 | 29.62 | –4.33 | 25.29 | –16.82 | 29.64 | –4.09 | 25.55 | –16.56 |
| 9m | –51.77 | –15.84 | –67.61 | 38.74 | –6.15 | 32.59 | –35.02 | 46.31 | –5.10 | 41.21 | –26.40 |
| 9n | –23.46 | –48.26 | –71.72 | 54.39 | –3.37 | 51.02 | –20.70 | 54.63 | –3.25 | 51.38 | –20.33 |
In this research, a series of novel acylhydrazone neuraminidase inhibitors 9a–9n were designed and synthesized, and the inhibitory activities against NA were evaluated in vitro. Most inhibitors exhibit good inhibition activity in the range of potency of U.S. Food and Drug Administration approved oseltamivir carboxylate, which can be used for further research and development of improved inhibitors against NA. Among them, compound 9j exerts the most potency, with an IC50 value of 0.60 μM, which is significantly lower than that of the positive control OSC (IC50 = 17.00 μM). Molecular docking analysis indicates that the acylhydrazone group plays an important role, which can bind well to the key residues Arg371 and Arg292 in the S1 subsite of NA. The good potency of 9j may also be ascribed to the extending of morpholinyl ring into the 430-cavity. The result of this work may be helpful for the design of NA inhibitors with stronger activities against mutational influenza virus.
Glossary
Abbreviations
- NA
neuraminidase
- RMSD
root-mean-square deviation
- MM/GBSA
Molecular Mechanics/Generalized Born Surface Area
- MM-PBSA
Molecular Mechanics/Poisson–Boltzmann Surface Area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00313.
Methods and materials, experimental section, in vitro neuraminidase inhibition assay, 1H NMR, 13C NMR, and HRMS spectra for the target compounds (PDF)
Author Contributions
The manuscript was finished through contributions of all authors.
The authors acknowledge financial support by Natural Science Foundation of Shanghai (No. 15ZR1440400), Collaborative Innovation Fund (No. XTCX2016-14), Middle and Youth Teachers Scientific and Technological Talents Developing Fund (No. ZQ 2018-20), and Shanghai Municipal Education Commission (Plateau Discipline Construction Program).
The authors declare no competing financial interest.
Supplementary Material
References
- Wu X. A.; Wu X. L.; Sun Q. Z.; Zhang C. H.; Yang S. Y.; Li L.; Jia Z. Y. Progress of small molecular inhibitors in the development of anti-influenza virus agents. Theranostics 2017, 7, 826–845. 10.7150/thno.17071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Anjum N.; Chandra R. Combating influenza: natural products as neuraminidase inhibitors. Phytochem. Rev. 2019, 18, 69–107. 10.1007/s11101-018-9581-1. [DOI] [Google Scholar]
- Chairat K.; Tarnging J.; White N. J.; Lindegardh N. Pharmacokinetic Properties of Anti-Influenza Neuraminidase Inhibitors. J. Clin. Pharmacol. 2013, 53, 119–139. 10.1177/0091270012440280. [DOI] [PubMed] [Google Scholar]
- Das K.; Aramini J. M.; Ma L. C.; Krug R. M.; Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z. Y.; Lou K. Y.; Wang W. New small-molecule drug design strategies for fighting resistant influenza A. Acta Pharm. Sin. B 2015, 5, 419–430. 10.1016/j.apsb.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. J.; Haire L. F.; Stevens D. J.; Collins P. J.; Lin Y. P.; Blackburn G. M.; Hay A. J.; Gamblin S. J.; Skehel J. J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- Ju H.; Zhang J.; Huang B. S.; Kang D. W.; Huang B.; Liu X. Y.; Zhan P. Inhibitors of Influenza Virus Polymerase Acidic (PA) Endonuclease: Contemporary Developments and Perspectives. J. Med. Chem. 2017, 60, 3533–3551. 10.1021/acs.jmedchem.6b01227. [DOI] [PubMed] [Google Scholar]
- Chen C.-L.; Lin T.-C.; Wang S.-Y.; Shie J.-J.; Tsai K.-C.; Cheng Y.-S. E.; Jan J.-T.; Lin C.-J.; Fang J.-M.; Wong C.-H. Tamiphosphor monoesters as effective anti-influenza agents. Eur. J. Med. Chem. 2014, 81, 106–118. 10.1016/j.ejmech.2014.04.082. [DOI] [PubMed] [Google Scholar]
- Bright R. A; Medina M.-j.; Xu X.; Perez-Oronoz G.; Wallis T. R; Davis X. M; Povinelli L.; Cox N. J; Klimov A. I Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 2005, 366, 1175–1181. 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- Le Q. M.; Kiso M.; Someya K.; Sakai Y. T.; Nguyen T. H.; Nguyen K. H.; Pham N. D.; Ngyen H. H.; Yamada S.; Muramoto Y.; Horimoto T.; Takada A.; Goto H.; Suzuki T.; Suzuki Y.; Kawaoka Y. Isolation of drug-resistant H5N1 virus. Nature 2005, 437, 1108. 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- Adabala P. J. P.; LeGresley E. B.; Bance N.; Niikura M.; Pinto B. M. Exploitation of the Catalytic Site and 150 Cavity for Design of Influenza A Neuraminidase Inhibitors. J. Org. Chem. 2013, 78, 10867–10877. 10.1021/jo401854w. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Minh D. D. L.; Cheng L. S.; Lindstrom W. M.; Olson A. J.; Lin J. H.; Li W. W.; McCammon J. A. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. J. Am. Chem. Soc. 2007, 129, 7764–7765. 10.1021/ja0723535. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Cheng X. L.; Ivanov I.; Xu D.; McCammon J. A. Characterizing loop dynamics and ligand recognition in human- and avian-type influenza neuraminidases via generalized born molecular dynamics and end-point free energy calculations. J. Am. Chem. Soc. 2009, 131, 4702–4709. 10.1021/ja8085643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H.; Zhang J.; Sun Z. S.; Huang Z.; Qi W. B.; Huang B.; Zhan P.; Liu X. Y. Discovery of C-1 modified oseltamivir derivatives as potent influenza neuraminidase inhibitors. Eur. J. Med. Chem. 2018, 146, 220–231. 10.1016/j.ejmech.2018.01.050. [DOI] [PubMed] [Google Scholar]
- Feng E. G.; Shin W. J.; Zhu X. L.; Li J.; Ye D. J.; Wang J.; Zheng M. Y.; Zuo J. P.; No K. T.; Liu X.; Zhu W. L.; Tang Wei; Seong B. L.; Jiang H. L.; Liu H. Structure-Based Design and Synthesis of C-1- and C-4-Modified Analogs of Zanamivir as Neuraminidase Inhibitors. J. Med. Chem. 2013, 56, 671–684. 10.1021/jm3009713. [DOI] [PubMed] [Google Scholar]
- Rollas S.; Küçükgüzel S. G. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. 10.3390/12081910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarini C.; Haranahalli K.; Rieger R.; Ananthula H. K.; Desai P. B.; Ashbaugh A.; Linke M. J.; Cushion M. T.; Ruzsicska B.; Haley J.; Ojima I.; Del Poeta M. Acylhydrazones as antifungal agents targeting the synthesis of fungal sphingolipids. Antimicrob. Agents Chemother. 2018, 62, e00156-18. 10.1128/AAC.00156-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Yan W.; Yang J. H.; Yang Z.; Hu M. S.; Bai B.; Tang M. H.; Chen L. J. Discovery of novel β-carboline/acylhydrazone hybrids as potent antitumor agents and overcome drug resistance. Eur. J. Med. Chem. 2018, 152, 516–526. 10.1016/j.ejmech.2018.05.003. [DOI] [PubMed] [Google Scholar]
- Angelova V. T.; Valcheva V.; Pencheva T.; Voynikov Y.; Vassilev N.; Mihaylova R.; Momekov G.; Shivachev B. Synthesis, antimycobacterial activity and docking study of 2-aroyl-[1] benzopyrano[4,3-c]pyrazol-4(1H)-one derivatives and related hydrazide-hydrazones. Bioorg. Med. Chem. Lett. 2017, 27, 2996–3002. 10.1016/j.bmcl.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Carcelli M.; Rogolino D.; Gatti A.; De Luca L.; Sechi M.; Kumar G.; White S. W.; Stevaert A.; Naesens L. N-acylhydrazone inhibitors of influenza virus PA endonuclease with versatile metal binding modes. Sci. Rep. 2016, 6, 31500. 10.1038/srep31500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M. W.; Ye J.; Lian W. W.; Zhang M.; Li B. B.; Liu A. L.; Hu A. X. Microwave-assisted synthesis, characterization and bioassay of acylhydrazone derivatives as influenza neuraminidase inhibitors. Med. Chem. Res. 2017, 26, 3216–3227. 10.1007/s00044-017-2015-6. [DOI] [Google Scholar]
- Zhao Z. X.; Cheng L. P.; Li M.; Pang W.; Wu F. H. Discovery of novel acylhydrazone neuraminidase inhibitors. Eur. J. Med. Chem. 2019, 173, 305–313. 10.1016/j.ejmech.2019.04.006. [DOI] [PubMed] [Google Scholar]
- Yu R.; Cheng L. P.; Li M.; Pang W. Discovery of Novel Neuraminidase Inhibitors by Structure-Based Virtual Screening, Structural Optimization, and Bioassay. ACS Med. Chem. Lett. 2019, 10, 1667–1673. 10.1021/acsmedchemlett.9b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. T.; Fan S. Y.; Li Z. P.; Yu F. L.; Hu X. Q.; Cheng J. C.; Zhang P.; Zhong B. H.; Shi W. G. Synthesis and evaluation of novel a-aminoamides with substituted benzene scaffold for the treatment of neuropathic pain. Chin. Chem. Lett. 2016, 27, 1630–1634. 10.1016/j.cclet.2016.06.034. [DOI] [Google Scholar]
- Zhao Z. X.; Cheng L. P.; Pang W. Green synthesis of ethyl oxalate benzylidinyl hydrazides. Tetrahedron Lett. 2018, 59, 2079–2081. 10.1016/j.tetlet.2018.04.047. [DOI] [Google Scholar]
- Hao X. Q.; Wang Y. N.; Liu J. R.; Wang K. L.; Gong J. F.; Song M. P. Unsymmetrical, oxazolinyl-containing achiral and chiral NCN pincer ligand precursors and their complexes with palladium(II). J. Organomet. Chem. 2010, 695, 82–89. 10.1016/j.jorganchem.2009.09.031. [DOI] [Google Scholar]
- Duan H. X.; Zhang W. W.; Zhao J.; Liang D. S.; Yang X. L.; Jin S. H. A novel halogen bond and a better-known hydrogen bond cooperation of neonicotinoid and insect nicotinic acetylcholine receptor recognition. J. Mol. Model. 2012, 18, 3867–3875. 10.1007/s00894-012-1393-4. [DOI] [PubMed] [Google Scholar]
- Stoll V.; Stewart K. D.; Maring C. J.; Muchmore S.; Giranda V.; Gu Y. Y.; Wang G.; Chen Y. W.; Sun M. H.; Zhao C.; Kennedy A. L.; Madigan D. L.; Xu Y. B.; Saldivar A.; Kati W.; Laver G.; Sowin T.; Sham H. L.; Greer J.; Kempf D. Influenza neuraminidase inhibitors: structure-based design of a novel inhibitor series. Biochemistry 2003, 42, 718–727. 10.1021/bi0205449. [DOI] [PubMed] [Google Scholar]
- Varghese J. N.; Epa V. C.; Colman P. M. Three-dimensional structure of the complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase. Protein Sci. 1995, 4, 1081–1087. 10.1002/pro.5560040606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vay J. L.; Fawley W.. AMBER User’s Manual, 2000. [Google Scholar]
- Hou T. J.; Wang J. M.; Li Y. Y.; Wang W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.