

Abstract
The membrane protein claudin-3 (CLDN3) is critical for the formation and maintenance of tight junction and its high expression has been implicated in dictating malignant progression in various cancers. However, the post-translational modification of CLDN3 and its biological function remains poorly understood. Here, we report that CLDN3 is positively correlated with ovarian cancer progression both in vitro and in vivo. Of interest, CLDN3 undergoes S-palmitoylation on three juxtamembrane cysteine residues, which contribute to the accurate plasma membrane localization and protein stability of CLDN3. Moreover, the deprivation of S-palmitoylation in CLDN3 significantly abolishes its tumorigenic promotion effect in ovarian cancer cells. By utilizing the co-immunoprecipitation assay, we further identify ZDHHC12 as a CLDN3-targating palmitoyltransferase from 23 ZDHHC family proteins. Furthermore, the knockdown of ZDHHC12 also significantly inhibits CLDN3 accurate membrane localization, protein stability and ovarian cancer cells tumorigenesis. Thus, our work reveals S-palmitoylation as a novel regulatory mechanism that modulates CLDN3 function, which implies that targeting ZDHHC12-mediated CLDN3 S-palmitoylation might be a potential strategy for ovarian cancer therapy.
Key words: Claudin-3, ZDHHC12, S-Palmitoylation, Ovarian cancer, Cancer progression, Membrane localization
Graphical abstract
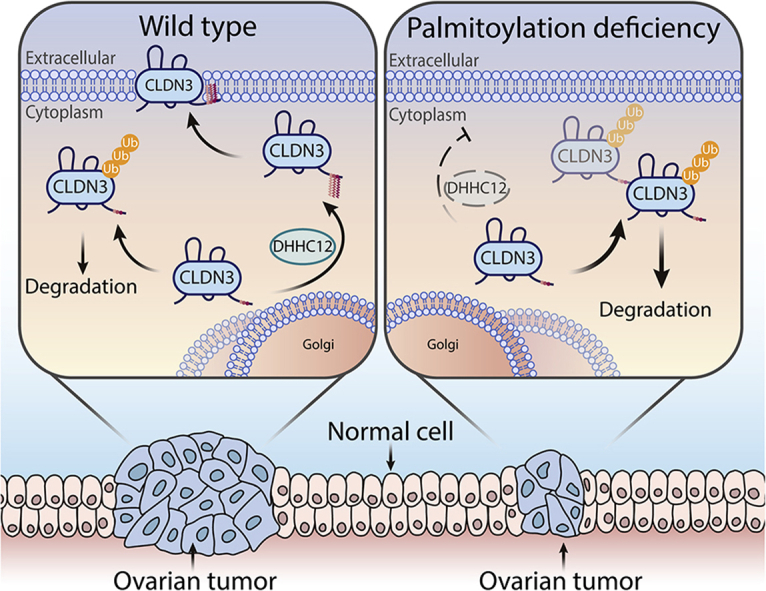
ZDHHC12-mediated S-palmitoylation promotes the cell membrane localization of CLDN3 and maintains its stability in ovarian cancer cells, contributing to the oncoprotein function of CLDN3. When ZDHHC12 was knocked down, CLDN3 was insufficiently S-palmitoylated leading to intracellular distribution and preference to degradation, which berried tumor growth.
1. Introduction
Claudins (encoded by CLDNs genes) are the most important component of tight junction1, which are critical for sealing the epithelial sheets and controlling paracellular flux of ions and small molecules2. CLDNs maintain the function of tight junction, like closing the cell gap, keeping cell polarity, regulating tissue permeability and assisting the signaling pathway on the cellular membrane of epithelial endothelial and mesothelial cells3, 4, 5. It has been reported that some claudins are overexpressed in certain types of cancer while others are downregulated in different types of cancer, thus creating differential expression patterns between tumor and normal cells6, 7, 8. Moreover, the function of individual claudin in tumorigenesis and cancer progression is also varied depending on the types of cancer3. For instance, overexpression of CLDN7 is related to metastatic potential and poor prognosis in pancreatic adenocarcinoma9, whereas low level of CLDN7 is correlated with clinicopathologic grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast10,11. Importantly, among the 23 proteins of claudin family, increasing evidences suggest that abnormal overexpression of CLDN3 is highly associated with tumor growth and cell invasion, motility, and survival in various types of cancer, such as colorectal, breast, prostate and so on12, 13, 14, 15. Therefore, CLDN3 has been recognized as a potential biomarker and therapeutic target for certain type of cancer.
Given the critical role of CLDN3 in tumorigenesis and cancer progression, several efforts on cancer therapeutics have been made. The high expression levels of CLDN3 in multiple types of cancer may provide a unique opportunity for anticancer therapy using Clostridium perfringens enterotoxin (CPE), that is because CLDN3 is one of the only transmembrane tissue-specific claudin proteins capable of mediating CPE binding and cytolysis16. However, the most important challenge is systemic toxicity, which could hinder the use of CPE for potential anti-cancer therapy. Besides, since claudins are transmembrane proteins and contain two extracellular loops, they may also offer promising targets for antibody-based or small molecular-based therapy17. Nevertheless, the antibody or small molecules designed to disrupt tight junctions may also be a double-edged sword, because an increase in cell permeability can enhance the uptake of anticancer agents. But at the same time, it may also increase the nutritional uptake to promote tumor progression. Therefore, further studies identifying the potential molecular target based on a deeper understanding of the regulatory mechanism on CLDN3 are highly desirable.
For the membrane protein, cysteine palmitoylation (S-palmitoylation), a kind of reversible fatty acylation, has been proven to play crucial roles in regulating protein function such as cell signaling, localization, trafficking and protein–protein interactions18, 19, 20, 21. The role of palmitoylation in cancer has focused on many membrane-associated oncoproteins like H-RAS, N-RAS and EGFR22, 23, 24, 25, 26, which facilitates these proteins stability and is required for activity. Most recently, Rodenburg et al.27 found that S-palmitoylations on multiple sites in CLDN3 occurs stochastically by native mass spectrometry. However, the way in which S-palmitoylation impacts CLDN3 function has not been studied. More importantly, as an enzymatic reaction, S-palmitoylation is the attachment of a 16-carborn atom fatty acid to a target protein at cysteines through a thioester linkage catalyzed by palmitoyltransferases (ZDHHCs)28,29. Accordingly, a characterization of which ZDHHC regulates CLDN3 S-palmitoylation is needed, regarding the potential application for cancer therapy by modulating CLDN3 biological function.
In the current study, the expression of CLDN3 was shown positively correlated with the ovarian cancer tumorigenesis and progression. In ovarian cancer cell, CLDN3 undergoes ZDHHC12-mediated S-palmitoylation on multiple cysteines in C-terminus region. The S-palmitoylation regulates CLDN3 protein stability and localization, and eventually determines its tumorigenesis in ovarian cancer cells. Our work provides insights into the functions of S-palmitoylation on CLDN3, and suggests that inhibiting ZDHHC12 is a potential anticancer strategy for ovarian cancer.
2. Materials and methods
2.1. Common reagents and antibodies
Anti-FLAG antibody [A00187-100, 1:4000 for immunoblotting (IB) and A00170-40, 1:4000 for IB] and the anti-FLAG resin beads (L00425) for immunoprecipitation (IP) were obtained from GenScrip Biotech Corp. (Nanjing, China). Anti-FLAG antibody [#8146, 1:500 for immunofluorescence (IF)] was purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-CLDN3 antibody [#34–1700, 1:100 for immunohistochemistry (IHC), 1:1000 for IB], Alexa Fluor 488 anti-rabbit secondary antibody (A32731, 1:1000 for IF) and Alexa Fluor 568 anti-mouse secondary (A-11004, 1:1000 for IF) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-CLDN3 antibody (ab52231, 5 μg/mL for IP) was purchased from Abcam (Shanghai, China). Anti-HA (db2603, 1:4000 for IB), β-actin (db1959, 1:5000 for IB) and GAPDH antibodies (db106, 1:5000 for IB) were purchased from Diagnostic Biosystems (Hangzhou, China). Anti-ZDHHC12 antibody (HPA059339, 1:50 for IHC), chloroquine (CQ) (C6628), cycloheximide (C7698), tris [(1-benzyl-1H-1,2,3-triazo-4-yl)methy]amine (TBTA), tris(2-carboxyethyl) phosphine (TCEP), sulforhodamine B (SRB) (#S1402) and agarose (A9414) were purchased from Sigma–Aldrich (St. Louis, MO, USA). For click chemistry assay, CY5.5-azide (B4030) was purchased from Lumiprobe (MA, USA). Alk-14 compound was synthesized according to reported procedures30. MG132 (S2619) was purchased from Selleck Chemicals (Munich, Germany). For immunofluorescence, saponin (8047-15-2) was purchased from Aladdin (Shanghai, China). DAPI (EF704) was purchased from Dojindo Molecular Technologies, Inc. (Rockville, MD, USA). HRP-conjugated secondary antibody (PV-6001) and 3,3′-diaminobenzidine tetrahydrochloride chromogen (DAB) kit (ZLI-9018) for immunohistochemistry were purchased from ZSGB-BIO company (Beijin, China).
2.2. Cell culture, transfection and transduction
293T, 293FT, OVCAR8, SKOV3, HaCaT, and A2780 cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). A2780 cell lines were grown in DMEM (#12800, Gibco, Grand Island, NY, USA) supplemented with 10% fetal calf serum (Sijiqing, China). OVCAR8 and SKOV3 cell lines were cultured in RPMI-1640 (#31800, Gibco) supplemented with 10% fetal calf serum. HaCaT, 293FT and 293T cell lines were cultured in DMEM supplemented with 10% fetal calf serum. Cells were incubated at 37 °C in a humidified space of 5% CO2. All cell lines used were authenticated by STR profiling. The cell lines were monitored for mycoplasma contamination ever six months.
To transiently overexpress the protein of interested in cells, the expression vectors were transfected into cells using jetPRIME (114–75, Polyplus-transfection®, France) according to the manufacturer's protocol. Empty vector was transfected as a negative control. To stably overexpress or knockdown the protein of interest, the lentivirus stocks were produced by co-transfection of 293FT cells with the packaging vector pCMV-R8.91, the envelope vector pMD2-VSGS and the lentiviral cloning vectors pCDH encoding the protein of interest and shRNA vectors pLKO.1. The sequences of shRNA used in this study were listed in Supporting Information Table S1. For infection, cells were grown at 20%–30% confluency, and 0.5–2 mL of each virus was added with 6 μg/mL polybrene. After 12–18 h, culture medium was replaced with fresh one.
2.3. Immunoprecipitation and immunoblotting
For co-immunoprecipitation, cells overexpressing FLAG-tagged CLDN3 and HA-tagged DHHC were lysed in lysis buffer [25 mmol/L Tris-base pH 8.0, 150 mmol/L NaCl, 1% Nonidet-40 (NP-40) and 10% glycerin] with protease inhibitor cocktail. The lysate was then centrifuged at 12,000×g for 30 min at 4 °C to remove insoluble materials. The supernatants were incubated with the anti-FLAG resin beads at 4 °C for 3 h. Resin beads were washed at least five times using wash buffer (25 mmol/L Tris-base pH 8.0, 300 mmol/L NaCl and 0.8% NP-40) and mixed with protein loading buffer, and then the mixture was boiled at 95 °C for 20 min. The samples were analyzed by immunoblotting.
For immunoblotting, proteins were resolved by SDS-PAGE and transferred to PVDF membrane. The PVDF membrane was blocked with 5% skimmed milk in T-PBS (0.1% Tween-20 in PBS) at room temperature for 1 h. For immunoblotting of p-ERK, 3% BSA in T-PBS was applied as blocking buffer. Then, the primary antibody was diluted with T-PBS and incubated the membrane for different time points according to the manual. The PVDF membrane was washed extensively in T-PBS and incubated with HRP-conjected secondary antibody (1:5000 diluted with 5% skimmed milk in T-PBS) for 1 h at room temperature. The chemiluminescence signal in membrane was recorded after developing in ECL plus Western blotting detection reagents using AI600 (GE Healthcare Inc., Boston, MA, USA).
For endogenous immunoprecipitation, the protein lysate was incubated with the anti-CLDN3 coupled to protein G-sepharose beads at 4 °C overnight and then was performed as described previously.
2.4. Labeling and detection of S-palmitoylation on CLDN3 by in-gel fluorescence
Cells overexpressing FLAG-tagged CLDN3 WT (wild type) and other mutants were treated with 50 μmol/L of Alk-14 for 4 h. Cells were collected and lysed in 4% SDS buffer [4% SDS, 150 mmol/L NaCl and 50 mmol/L triethylamine in double distilled water (ddH2O), pH 8.0] with protease inhibitor cocktail. The immunoprecipitation was performed as described previously. The click chemistry reaction was performed after washing beads step, beads were incubated in 20 μL reaction buffer (50 mmol/L Tris-base, 150 mmol/L NaCl, 1% NP-40, 0.1% SDS and 0.25% deoxycholate without EDTA, pH 8.0) followed by adding 0.5 μL CY5.5-azide (4 mmol/L in DMSO, final concentration 1 μmol/L), 1 μL TBTA (10 mmol/L in DMF, final concentration 500 μmol/L), 0.4 μL CuSO4 (100 mmol/L in ddH2O, final concentration 2 mmol/L) and 0.5 μL TCEP (40 mmol/L in ddH2O, final concentration 0.2 μmol/L). The click chemistry reaction was allowed to proceed for 30 min at room temperature. Then, the reaction mixture was mixed with protein loading buffer and boiled at 95 °C for 20 min. 10 μL of the sample was treated with 1 μL NH2OH (5 mol/L in ddH2O) or equivalent volume of water for 5 min at room temperature then at for 5 min at 95 °C. The samples were resolved by SDS-PAGE. CY5.5 fluorescence signal was recorded by Odyssey (LI-COR) with setting 700 nm channel.
2.5. Immunofluorescence
A2780 or OVCAR8 cells were plated beforehand in the fluorescent chamber slides (#4615–4534, TAB-TEKII Chamber Slide, USA Scientific Inc., Ocala, FL, USA) and cultured until confluent. When most of cells were in contact with each other to form tight junction, discarded the medium and fixed the cells with ice-cold 4% paraformaldehyde (PFA, in PBS) at room temperature for 15 min. The slide then was washed with PBS for 5 min three times. Cells were permeabilized with blocking buffer (0.1% Saponin, 5% BSA in PBS) at room temperature for 30 min. Then, cells were incubated with the anti-FLAG antibody (1:500 diluted in blocking buffer) and the anit-ZO-1 antibody (1:400 diluted in blocking buffer) at 4 °C overnight. After washing three times by PBS, cells were incubated with Alexa 488-conjugated anti-mouse (1:1000 diluted in blocking buffer) and Alexa 568-conjugated anti-rabbit (1:1000 diluted in blocking buffer) secondary antibodies in a humidified light-protected chamber for 1 h at room temperature. Cells were washed with PBS extensively followed by incubating DAPI (1:5000 diluted in PBS) for nuclei stain for 5 min at room temperature. The slides were coverslipped and sealed with anti-fade reagent (0100–01, SouthernBiotech, Birmingham, AL, USA). Confocal images were observed under a confocal microscope (FV1000, OLYMPUS, Tokyo, Japan).
2.6. Cell proliferation assay
For each assay, cells were seeded in 6 wells centered on a 24-well plate (100 cells per well), and then maintained in medium with 10% FBS for 10 days. After washing twice with ice-cold PBS, cells were fixed by ice-cold 10% trichloroacetic acid at 4 °C for 1 h. Then, washed the plate with water and applied 200 μL SRB (1.5 g in 1% acetic acid) was added and incubated for 25 min at room temperature. The stained cells were washed with 1% acetic acid and all excess dye was removed, air-dried and solubilized in 200 μL of 1% Tris-base. Absorbance of the resulting solution was measured at 540 nm.
2.7. Soft agar colony formation assay
Melt 5% agarose solution, prepare the bottom medium (0.6% agarose gel, 10% FBS in DMEM or RPMI-1640) and keep it in 42 °C to prevent from solidifying. For bottom layer, gently add 2 mL of the bottom medium into each well of the 6-well culture plate and place the plate horizontally at 4 °C for least 15 min to solidification. For the upper layer, cell suspension and the bottom medium were mixed in a 1:1 dilution to a cell/agar mixture (0.3% agarose gel, 1.5 × 103 A2780 cells/mL or 3.0 × 103 OVCAR8 cells/mL). Gently add 1 mL of the cell/agar mixture onto the bottom layer to a final cell number of 1.5 × 103 A2780 cells or 3.0 × 103 OVCAR8 cells per well. After the upper layer solidifies at room temperature, place the plate into a 37 °C incubator. Add 100 μL of cell culture medium twice weekly to prevent desiccation. The time required for adequate colony formation varies for each cell line, typically around 21 days. Stain cells by adding 100 μL of 0.5% MTT solution (5 mg/mL MTT in PBS) per well and incubating plate at 37 °C for at least 1 h. Take photographs of each well and count colonies using ImageJ Fiji software (Bethesda, MA, USA).
2.8. Real-time PCR
Total RNA isolation was carried out with RNAiso plus kit (#9109, TaKaRa) according to the manufacturer's instructions. And then first-strand complementary DNA synthesis was obtained using the Reverse Transcription System Kit (3AT331-03, TransGen Biotech) according to the manufacturer's instructions. The cDNA was used to perform the real-time PCR using SYBR green reagent (#172–5124, Bio-Rad, Hercules, CA, USA). The CT values were obtained using a real-time PCR platform (QuantStudio 6 Flex Real-Time PCR System, Thermo Fisher Scientific). The primers used for real-time PCR were listed in Supporting Information Table S2.
2.9. Immunohistochemistry
The ovarian cancer paraffin-embedded tissue slides (OV2161 and T112b, Alenabio, China) were deparaffinized. Antigen retrieval was performed using EDTA repair fluid (pH 8.0), microwaved for 8–15 min until boiling. After boiling, allow the slides to cool to room temperature. Wash the slides with PBS for 5 min three times. Tissues were covered with peroxidase block buffer (3% hydrogen peroxide in methanol) for 15 min and then washed with PBS for 5 min three times. Cover the tissue with block reagent (5% normal goat serum, 0.1% Triton X-100 in PBS) for 30 min at room temperature. Tissues were incubated with the anti-CLDN3 antibody (1:100 diluted in block reagent) or anti-ZDHHC12 (1:50 diluted in block reagent) in a humidified light-protected chamber overnight at 4 °C. Gently wash the tissue three times in PBS and incubated with HRP-conjugated secondary antibody (PV-6001, ZSGB-BIO) in a humidified light-protected chamber for 1 h at room temperature. Tissues were washed extensively in PBS followed by the colorimetric detection using 3,3′-diaminobenzidine tetrahydrochloride chromogen (DAB) kit (ZLI-9018, ZSGB-BIO). Tissues were counterstained with hematoxylin and dehydrated. Then, mount and coverslip the slides. Additionally, the proportion of stain was estimated based on a combined evaluation of the extent and intensity of staining. Scores 0–3 were assigned according to the percentage of positive tumor cells (0, = 0%; 1, ≤40%; 2, = 41%–70%; 3, ≥70%) and the intensity of tumor staining. Two scores were added up to give an overall score of 0–6: –, no expression (score 0, complete negative staining); +, weak expression (score 2–3); ++, median expression (score 4); +++ indicates high expression (score 5–6).
2.10. Animal study
Two individual in vivo experiments were executed as follow describing.
BALB/c-nu mice (4–5 weeks old) were obtained from National Rodent Laboratory Animal Resource (Shanghai, China). 1.8 × 106 OVCAR8 cells transfected with shCtrl or two shCLDN3 lentivirus were suspended in the culture medium and injected subcutaneously into either left or right flanks of each nude mouse, respectively. Each group contains 8 mice in the start. The body weight and tumor size of mice were measured every two days before sacrificed. After 37 days, all the mice were sacrificed. Tumor tissues was harvested and weighted.
BALB/c-nu mice were brought from the same situation. 1.0 × 106 OVCAR8 cells transfected with shCtrl or two shZDHHC12 lentivirus were suspended in the culture medium and injected subcutaneously into either left or right flanks of each mouse, respectively. Each group contains 8 mice (n = 8) in the start. The body weight and tumor size of mice were measured every two days before sacrificed. After 37 days, all mice were sacrificed. Tumor tissues was harvested and weighted.
Maintenance and experimental procedures for the mice studies were approved by Zhejiang University's IACUC.
2.11. Statistical analysis
Statistical analysis was performed and plotted using GraphPad Prism 7.0 software. Grayscale analysis, cell counting and colocalization analysis were performed using ImageJ Fiji software and the colocalization coefficients (Manders' colocalization coefficients, tM1 and tM2) were analyzed by Plugin Colocalization Finder. Results expressed as mean ± standard deviation (SD) or mean ± standard error (SEM) of mean determined from a minimum of three independent experiments and were analyzed using the Student's t-test. Differences were considered significant for P values less than 0.05.
3. Results
3.1. Clinicopathologic characteristics of CLDN3 in ovarian cancer
It has been well documented that CLDNs are abnormally expressed in different types of cancer. However, most of the clinical pathological studies only focused on certain CLDN in specific cancer type. Given that the function of certain CLDN in tumorigenesis is largely depends on the tissue specificity, we reasoned that finding a suitable cancer type is important for studying modulatory mechanism of CLDN3. Accordingly, we analyzed the relation between human CLDNs expression and the cancer progression among the different types of cancer from public microarray or RNA-seq database (PROgeneV2 database, http://genomics.jefferson.edu/proggene/). The heat map showed the hazard ratio of 23 CLDNs in different types of cancer from different database (Fig. 1A, and the raw data can be found in Supporting Information Table S3). From which, we found that high expression of CLDN3 had a significantly high hazard ratio in ovarian cancer (Fig. 1A and B), indicating that expression level of CLDN3 is positively correlated with poor progression in ovarian cancer. To confirm it, we next examined the expression of CLDN3 by immunohistochemistry in tissue microarray containing 240 ovarian cancer tissues and normal ovarian tissues. As shown in Fig. 1C and D, the protein expression level of CLDN3 is markedly lower in normal ovarian tissues than that in tumor tissues. Furthermore, there is a positive correlation between the protein expression of CLDN3 and ovarian cancer malignancy, as the higher-grade classification of pathology diagnosis responds to the smaller ratio of CLDN3 negative expression and higher proportion of +++ expression level of CLDN3.
Figure 1.

Clinicopathologic characteristics of CLDN3 in ovarian cancer. (A) The HR (hazard ratio) value of each human CLDN family protein (data of 22 CLDNs are available) expression in different types of cancers. All data is obtained from TCGA and GEO dataset. The threshold of scale bar is from 0 to 32 and white color indicates the baseline value of 2. Shades of gray indicate no available data. (B) Kaplan–Meier curves for overall survival rate of patients with ovarian cancer from the GEO dataset (GSE8842 and GSE63885; green curve, patients with low expression of CLDN3; red curve, patients with high expression of CLDN3). (C) Representative images of different score of immunohistochemical stain, indicating different expression level of CLDN3 (+++ indicates high expression; ++ indicates median expression; + indicates low expression; – indicates negative expression). (D) The pie chart shows the proportion of different expression level of CLDN3 in each ovarian tumor grade (from GRADE-1 to GARDE-III and normal tissue group). Darker shades of blue indicate higher expression of CLDN3 (n = 240). (E) The knockdown efficiency of shRNA against CLDN3 in OVCAR8 cells was validated by immunoblotting. (F) Colony forming assay was performed on shCtrl, shCLDN3#1 and shCLDN3#2 OVCAR8 cells. Quantification of colony numbers relative to shCtrl group are shown on the right panel. Values with error bars indicate mean ± SD of three biological replicates (∗∗P < 0.01; ∗∗∗P < 0.001). (G) The tumorigenesis of shCtrl, shCLDN3#1 and shCLDN3#2 groups according to time course (37 days). The first time that tumor was found in each group was on day 5, day 15 and day 18. In shCtrl group, 8 tumors formed on day 18 which remained to the end of in vivo assay. In shCLDN3#1 group, 4 tumors formed on day 23 which remained to the end of in vivo assay. In shCLDN3#2 group, 6 tumors formed on day 35 which remained to the end of in vivo assay. (H) Tumor growth curves of shCtrl, shCLDN3#1 and shCLDN3#2 groups in vivo animal assay. Error bars represent SEM. (∗P < 0.05). (I) Tumor formed in the nude mice at the end of in vivo assay (n = 8 for each group). At the endpoint of the in vivo animal assay, the tumorigenesis ratio (the number of tumorigenic individuals/the number of total individuals) of each group: 8/8 in shCtrl group, 5/8 in shCLDN3#1 group, 6/8 in shCLDN3#2. The tumors were arranged from left to right by weight. (J) The nude mice were sacrificed, and the tumors were removed and measured in weight. Dot plots represent the weight of tumors (n = 8 for each group in all statistical analysis; ∗P < 0.05).
To further confirm the critical role of CLDN3 in ovarian cancer, we carried out the tumorigenic growth assays in OVCAR8 cells whose CLDN3 was highly expressed (Supporting Information Fig. S1A). Stably knocking down CLDN3 cells were generated by shCLDN3 lentivirus with two different sequences in OVCAR8 cells (Fig. 1E). We observed a significant decrease in proliferation in shCLDN3 OVCAR8 cells in vitro (Fig. 1F). Next, we used these stably knocking down CLDN3 cells for the in vivo experiment. Tumor formation in nude mouse was tested by subcutaneous injection of shCLDN3 OVCAR8 cells (n = 8 in each group), and shCtrl OVCAR8 cells (n = 8) were used as controls. As shown in Fig. 1G and H, the tumorigenesis and tumor growth rate of shCLDN3 groups were also slower than the shCtrl group. Moreover, the tumor size of shCLDN3 group was remarkably smaller than that of shCtrl group (Fig. 1I), and the mean tumor weight in shCLDN3 groups was significantly less than those in shCtrl group inoculated nude mice (0.023 ± 0.008 g and 0.016 ± 0.008 vs. 0.293 ± 0.122 g, Fig. 1J). Collectively, these results demonstrated that CLDN3 is positively correlated with the malignant degree of ovarian tumor, and contributes to tumorigenesis and progression in ovarian cancer both in vitro and in vivo.
3.2. CLDN3 is palmitoylated on multiple cysteine residues in C-terminus
To explore the regulatory mechanism of CLDN3, we mainly focus on S-palmitoylation here. Because S-palmitoylation has been detected in CLDN3 and this post-translational modification is tightly determined by certain enzyme, which could be potentially regulated by small molecule compounds. To verify that CLDN3 is S-palmitoylated, we used a metabolic labeling method with the Alk-14 probe (Fig. 2A). Alk-14 is a palmitic acid analogy and the C-terminal structure of which is an alkenyl group rather than an alkyl in natural palmitoyl acid. The intracellular palmitoylated proteins will be metabolically incorporated with Alk-14. Since Alk-14 contains the alkyne group, palmitoylated protein can be further conjugated to a fluorescence probe conjected with an azide group like CY5.5-azide by click chemistry reaction31 (Fig. 2A). This modification can be visualized by in-gel fluorescence. The property of S-palmitoylation can be verified by the treatment of hydroxylamine (NH2OH), because hydroxylamine (NH2OH) as a nucleophile removes fluorescent-labeled palmitoylation on cysteine residues. As shown in Fig. 2B, FLAG-tagged wild type of CLDN3 overexpressed in 293T cells has strong fluorescent labeling and the fluorescence signal on CLDN3 was totally removed with the treatment of hydroxylamine, suggesting that CLDN3 contains S-palmitoylation. Similarly, we also detected the constitutively S-palmitoylation of endogenous CLDN3 in ovarian cancer cell line OVCAR8, SKOV3 and normal immortal keratinocyte cell line HaCaT (Fig. 2C and Figs. S1B and S1C).
Figure 2.

CLDN3 is S-palmitoylated at multiple cysteine sites. (A) The schematic overview of the Alk-14 metabolic labeling method to study fatty acylation of CLDN3. (B) In-gel fluorescence detection of the S-palmitoylation level of exogenous FLAG-tagged CLDN3 overexpressing in 293T cells by Alk-14 metabolic labeling method with or without NH2OH treatment. Black triangle indicates fluorescent band of S-palmitoylated CLDN3. (C) In-gel fluorescence detection of the S-palmitoylation level of endogenous CLDN3 in OVCAR8 cells by Alk-14 metabolic labeling method with or without NH2OH treatment. Black triangle indicates fluorescent band of S-palmitoylated CLDN3. (D) Schematic representation of CLDN3 structure (top panel) and comparison of amino acid sequences of CLDN3 at the C-terminus in different species (bottom panel). The model depicts the conserved amino acid sequence of CLDN3. CLDN3 alignments for the parts of the intracellular C-terminus domain where the potential palmitoylations occur, including multiple CLDN3 homologs. Shown are human CLDN3 (O15551), rat CLDN3 (Q63400), mouse CLDN3 (Q9Z0G9), dog CLDN3 (Q95KM5), cow CLDN3 (Q765N9) and zebrafish CLDN3 (E7EXG8). Amino acid positions of the potential palmitoylation are indicated above the alignment. (E) In-gel fluorescence shows the S-palmitoylation level of CLDN3 WT, C181/182S, C181/184S and C182/184S overexpressed in 293T cells. Quantification of the fluorescence intensity relative to CLDN3 WT is shown on the right panel. Values with error bars indicate mean ± SD of three biological replicates. (∗∗P < 0.01; ∗∗∗P < 0.001).
Next, we tried to identify which cysteine residues of CLDN3 were S-palmitoylated. We got the clue from mass spectrometry data27 and online in silico analysis (https://swisspalm.org/), and then determined candidates that are cysteine residues 103, 106, 181, 182 and 184 of CLDN3. Firstly, we tested cysteine residues of Cys103 and Cys106. Delusively, palmitoylation level of CLDN3 did not recede by mutating both cysteine 103 and 106 into serine (Fig. S1D). Then, we focused on other three cysteine residues (Cys181, Cys182, and Cys184) at C-terminus of CLDN3, which of them are mostly conserved across different species (Fig. 2D). Although single cysteine mutation slightly decreased in-gel fluorescent intensity, no statistic difference was observed (Fig. S1E). Therefore, we mutated two out of three cysteines and tested S-palmitoylation levels of these double mutants. As shown in Fig. 2E, we observed a remarkable fluorescence signal decrease for the three double cysteine mutants (C181/182S, C181/184S, and C182/184S). These results indicate that CLDN3 is palmitoylated in multi-cysteine residues which are closed to the CLDN3 intracellular C-terminus.
3.3. S-palmitoylation of CLDN3 is required for its protein stability
Given a consistent observation of some left-over fluorescence signal for three double cysteine mutants, we further constructed the triple cysteine residues mutated CLDN3 (refer to CLDN3 3CS). Interestingly, after transfecting CLDN3 3CS in 293T cells, we could not detect the accumulation of it by Western blot unless with the treatment of MG132. And chloroquine did not have the effect as MG132. This evidence gave us an assumption that an accelerate degradation by ubiquitin–proteasome occurs to CLDN3 3CS compared to CLDN3 WT (Fig. 3A and B). Co-immunoprecipitation result showed that more ubiquitin proteins conjugated to CLDN3 3CS compared to CLDN3 WT (Fig. S1F), which validated our assumption. To further prove this, we tested the protein degradation rate of CLDN3 3CS. Cells stably expressing FLAG-tagged CLDN3 WT and CLDN3 3CS were treated with MG132 for 8 h firstly, and then treated with cycloheximide (CHX) after refreshing the culture medium. The data showed that while less than 20% of CLDN3 WT degraded after 12 h of CHX treatment, more than 70% of CLDN3 3CS degraded after 12 h of CHX treatment in the same time period (Fig. 3C). This result suggests that CLDN3 3CS is much less stable than its WT counterpart. Simultaneously, we checked the fluorescence intensity of CLDN3 3CS in previous immunoprecipitation system. Compared to the fluorescence intensity of CLDN3 WT, fluorescence intensity of CLDN3 3CS was barely detected, which indicates that CLDN3 3CS might be S-palmitoylated-deficient (Fig. 3D). Collectively, our data implied that the S-palmitoylation of CLDN3 is required for its protein stability preventing from ubiquitination-mediated degradation.
Figure 3.

The S-palmitoylation of CLDN3 is required for its protein stability. (A) Expression of FLAG-tagged CLDN3 wildtype, double cysteine mutants and triple cysteine mutant that transfected in 293T cells was validated by immunoblotting with anti-FLAG antibody. (B) Validation of the degradation pathway for CLDN3 WT and CLDN3 3CS. 293T cells were transfected with FLAG-tagged CLDN3 WT or FLAG-tagged CLDN3 3CS. Each group of cells were treated with MG132 for 8 h or with CQ for 24 h or with DMSO before lysed and tested by immunoblotting. (C) Degradation kinetics curves of CLDN3 WT and CLDN3 3CS. OVCAR8 cells stably expressing FLAG-tagged CLDN3 WT or 3CS mutant were exposed to MG132 for 8 h, followed by the treatment of CHX for 3, 6, 9 and 12 h. Lysates were resolved by immunoblotting. The protein level of CLDN3 WT and CLDN3 3CS with CHX treatment for 0 h is set to 1. Quantification of relative protein level in each group at the indicated time point is shown on the right. Values with error bars indicate mean ± SD of three independent replicates (∗∗∗P < 0.001). (D) In-gel fluorescence shows the S-palmitoylation level of CLDN3 WT and CLDN3 3CS. 293T cells were transfected with FLAG-tagged CLDN3 WT or FLAG-tagged CLDN3 3CS. After the treatment of MG132 and Alk-14, cells were lysed and submitted to immunoprecipitation and immunoblotting. Quantification of fluorescent intensities relative to CLDN3 WT is shown on the right panel. Values with error bars indicate mean ± SD of three independent experiments (∗∗∗P < 0.001).
3.4. S-palmitoylation facilitates an accurate subcellular localization and cell proliferation for CLDN3
Of note is the fact that though the S-palmitoylation level is significantly decreased in three double cysteines mutants (C181/182S, C181/184S, and C182/184S), there is no difference in protein expression compared to CLDN3 WT (Fig. 3A), suggesting other biological functions might be affected by S-palmitoylation besides the protein stability. CLDN3 is the plasma membrane protein and plays a central role in the assembly and maintenance of tight junction complex, directly determining paracellular permeability32,33. And ZO-1 helps it keep general function by tethering it to the actin cytoskeleton. We performed the immunofluorescence assay to investigate whether S-palmitoylation is required for its transport to the cytomembranes. FLAG-tagged wildtype, double mutants and triple mutants were stably expressed in A2780 cells (whose CLDN3 expression is low in vitro, Fig. S1A) and shCLDN3#2 OVCAR8 cells. Immunofluorescence was performed with anti-FLAG and anti-ZO-1 antibodies to visualize the intracellular localization of FLAG-tagged CLDN3 and endogenous ZO-1. A serial of stacked confocal images might help illustrating the co-localization of ZO-1 and CLDN3 in spatial manner. We introduced the thresholded Manders’ colocalization coefficients (tM1 and tM2)34 as the quantitative colocalization evaluation of CLDN3 and ZO-1 on plasma membrane. As shown in Fig. 4A and Supporting Information Fig. S2A, CLDN3 WT was almost distributed on plasma membrane. But CLDN3 3CS was predominantly localized to cytoplasm. For the three double cysteines mutants, though these were still predominantly localized on plasma membrane (weaker than wild type), the plasma membrane located mutated proteins were more evenly distributed compared to CLDN3 WT (Fig. 4A and Fig. S2A), implying that S-palmitoylation might be required for the accurate plasma membrane localization of CLDN3. Furthermore, the pairs of tM1 and tM2 value of each CLDN3 mutant were less than that of CLDN3 WT (Fig. 4B, the mean value of tM1 and tM2 was displayed above each group of scatter plot), which means less CLDN3 mutants colocalized with ZO-1. These evidences revealed an expected hypothesis that S-palmitoylation of CLDN3 contributes its plasma membrane targeting and colocalization with ZO-1.
Figure 4.

S-Palmitoylation facilitates an accurate intracellular localization and cell proliferation for CLDN3. (A) Confocal images show subcellular localization of endogenous ZO-1, FLAG-tagged CLDN3 WT or mutants in A2780 cell line. Cells expressing FLAG-tagged CLDN3 3CS were treated with MG132 for 4 h before fixed and immunofluorescence staining with indicated antibodies. The y–z axis shows the location of ZO-1 and CLDN3 in basal and apical regions. (B) Quantitative analysis of correlated co-localization of CLDN3 (green) and ZO-1 (red) in (A), representing by the mean of Manders' coefficient with threshold (tM1 and tM2) (n = 7 for each group; ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001). (C) Validation of the expression of FLAG-tagged CLDN3 WT and mutants in shCLDN3 OVCAR8 cells by immunoblotting. (D) Representative data from soft agar colony formation of shCLDN3 OVCAR8 cells expressing FLAG-tagged CLDN3 WT or mutants. 3.0 × 103 cells were cultured in RPMI-1640 medium contained 10% FBS and 0.35% agarose gel for 21 days. Cell colonies were stained by MTT. (E) The quantification of the colony numbers in soft agar colony formation assay (D). Values with error bars indicate mean ± SD of three independent replicates (∗P < 0.05; ∗∗P < 0.01).
Based on the positive correlation between CLDN3 protein level and ovarian cancer progression, we wondered if S-palmitoylation encourages CLDN3 possessing the pathological biofunction in ovarian cancer. Then, we performed the soft agar colony formation assay, which mimics in vivo environment in some degree, on groups of shCLDN3 OVCAR8 cells and A2780 cells which express CLDN3 WT and mutants. As shown in Fig. 4C–E, the colony number of CLDN3 WT group increased by 1.64-fold compared to the control group, while any other group of CLDN3 mutants performed almost indistinctive proliferation compared to the control group. The similar experimental results (Figs. S2B and S2C) also could be observed in A2780 cells. Taken together, our data suggests that CLDN3 is more likely to promote tumorigenesis ability of ovarian cancer cells when it is equipped with a full set of S-palmitoylation on three cysteine sites at the C-terminus, which might be attributing to multiple effects of S-palmitoylation on CLDN3.
3.5. ZDHHC12 is identified as a CLDN3-palmitoylating enzyme
If S-palmitoylation probably determines the fate of CLDN3 as an oncoprotein, it will be profound and prospective to get access to diminishing S-palmitoylation of CLDN3. It has been known that S-palmitoylation is a reversible modification that is mediated by a family of Asp-His-His-Cys (DHHC)-containing protein acyltransferases encoded by ZDHHC genes. To date, the ZDHHC family of genes in mammals consists of 23 members35. We thus ask which palmitoyltransferases is responsible for the S-palmitoylation of CLDN3. As there are conserved domains between human and mouse-derived DHHC family proteins, we utilized the expression vectors of 23 mouse-derived DHHCs to identify the specific DHHC that determines S-palmitoylation of CLDN3. Each HA-tagged DHHC (from DHHC1 to DHHC23) was transfected in 293T cells which stably expresses FLAG-tagged CLDN3. Then the cell lysate was submitted to anti-FLAG immunoprecipitation. Co-precipitated HA-tagged DHHC was detected by Western blot with anti-HA antibody. After the several rounds of screening, we confirmed that DHHC12 and DHHC1 had a strong interaction with CLDN3 (Fig. 5A, Supporting Information Figs. S3A and S3B). Subsequently, we wiped off DHHC1, since no significant effect of DHHC1 on accumulated S-palmitoylation level of CLDN3 (Fig. S3C).
Figure 5.

ZDHHC12 is a CLDN3-palmitoylaiting enzyme. (A) Identification of the ZDHHC12 palmitoyltransferase by co-immunoprecipitation screening of CLDN3 and DHHC enzymes. 293T cells stably expressing FLAG-tagged CLDN3 were transfected with HA-tagged DHHCs for 24 h. Cell lysate was submitted to anti-FLAG immunoprecipitation and immunoblotting. The specific immunoblot band of HA-tagged DHHC12 co-immunoprecipitated with FLAG-tagged CLDN3 was indicated by yellow triangle. (∗represents the heavy chain of anti-FLAG antibody). (B) In-gel fluorescence shows the S-palmitoylation level of CLDN3 WT was affected by vector, DHHC12 WT and DHHC12 C127S. Right histogram shows the quantification of the fluorescence intensity relative to CLDN3 WT. Values with error bars indicate mean ± SD of four independent replicates (n.s. indicates no statistic difference; ∗∗P < 0.01). (C) The knockdown efficiency of shRNA against human ZDHHC12 in A2780 cells was validated by real-time PCR. Values with error bars indicate mean ± SD of three independent replicates (∗∗∗P < 0.001). (D) In-gel fluorescence shows the weaken S-palmitoylation level of shZDHHC12 group compared to shCtrl group. CLDN3 expressed A2780 cells with knocking down endogenous ZDHHC12 were submitted to anti-FLAG immunoprecipitation and immunoblotting. Right histogram shows the quantification of the fluorescence intensity of shZDHHC12 group relative to shCtrl group. Values with error bars indicate mean ± SD of three independent replicates (∗P < 0.05).
Meanwhile, as shown in Fig. 5B, S-palmitoylation level of CLDN3 in 293T cells co-expressing with wild type DHHC12 was increased by 1.40-fold compared to 293T cells expressing CLDN3 only. Furthermore, we introduced a catalytic dead mutant of DHHC12 (DHHC12 C127S) to verify the palmitoylation catalysis of DHHC12 on CLDN3. Indeed, deficiency of catalytic form (DHHC12 C127S) failed to elevate S-palmitoylation of CLDN3 (Fig. 5B), even if it kept the interaction with CLDN3. Since we had demonstrated that Cys181, Cys182 and Cys184 of CLDN3 were the palmitoylation residues, we performed the co-expression of DHHC12 with CLDN3 WT or mutants in 293T cells, and then detected S-palmitoylation level of each CLDN3 mutant. The result showed that CLDN3 WT carried an increased S-palmitoylation level with co-expression of DHHC12 WT, while other CLDN3 mutants had no much difference in the S-palmitoylation level (Fig. S3D). Thus, the result suggested that ZDHHC12 could catalyze three cysteine residues of CLDN3 in S-palmitoylation in an enzymatic dependent manner.
Additionally, we validated CLDN3 S-palmitoylation level in A2780 cells expressing FLAG-tagged CLDN3 by knocking down the expression of endogenous human ZDHHC12 by shRNA lentivirus. As expectedly, knockdown of ZDHHC12 brought about approximately 50% decrease in S-palmitoylation level of CLDN3 and expression level of CLDN3 also decreased slightly in A2780 cells (Fig. 5C and D). Taken together, we could profile ZDHHC12 as a dominant CLDN3-palmitoylation enzyme, which manipulates S-palmitoylation of CLDN3 in most case.
3.6. ZDHHC12 affects CLDN3-related ovarian tumorigenesis
Next, we asked whether manipulating the ZDHHC12 will affect the biological function of CLDN3 in ovarian cancer progression. First, we tested the membrane localization of CLDN3. A serial of stacked confocal images showed that knockdown of ZDHHC12 hindered CLDN3 targeting cytomembrane which resulted in its intracellular mis-localization partly (Fig. 6A). And the quantitative analysis of colocalization of CLDN3 and ZO-1 in immunofluorescence assay further described the insufficient colocalization of them (Fig. 6B), which is similar to the result of CLDN3 with C-terminus S-palmitoylation deficiency as previous data. Multiple signaling pathways including MAPK/ERK pathway have been described as the vehicle for the role of CLDN3 in various tumorigenesis13,15,36, we thus examined the effect of silencing ZDHHC12 on MAPK/ERK signaling pathway. As expected, we observed the diminution of phosphorylation level of EKR1/2 in ZDHHC12-mediated CLDN3 downregulation manners (Fig. 6C, Supporting Information Figs. S4A and S4B).
Figure 6.

ZDHHC12 affects CLDN3 related ovarian tumorigenesis. (A) Confocal images show subcellular localization of endogenous ZO-1and FLAG-tagged CLDN3 in shCtrl and shZDHHC12 A2780 cells. The y–z axis shows the location of ZO-1 and CLDN3 in basal and apical regions. (B) Quantification of correlated colocalization of CLDN3 and ZO-1, representing by the mean of Manders’ coefficient with threshold (tM1 and tM2) (n = 7 for each group; ∗∗∗P < 0.001). (C) Downregulation of phosphorylated ERK1/2 in shZDHHC12 OVCAR8 cells, compared to shCtrl OVCAR8 cells, was indicated by immunoblotting. (D) The tumorigenesis of shCtrl, shZDHHC12#1 and shZDHHC12#2 groups in vivo animal assay according to time course (37 days). The first time that tumor was found in each group was on day 5, day 33 and day 20. In shCtrl group, 7 tumors formed on day 15 which remained to the end of in vivo assay. In shZDHHC12#1 group, 2 tumors formed on day 37 which remained to the end of in vivo assay. In shZDHHC12#2 group, 4 tumors formed on day 35 which remained to the end of in vivo assay. (E) Tumor growth curves of shCtrl, shZDHHC12#1 and shZDHHC12#2 groups in vivo animal assay. Error bars represent SEM (∗∗P < 0.01). (F) Tumor formed in nude mice at the endpoint of in vivo animal assay (n = 8 for each group). At the endpoint of the in vivo animal assay, the tumorigenesis ratio (the number of tumorigenic individuals/the number of total individuals) of each group: 7/8 in shCtrl group, 2/8 in shZDHHC12#1 group, 4/8 in shZDHHC12#2. The tumors were arranged from left to right by weight. (G) The nude mice were sacrificed, and the tumors were removed and measured in weight. Dot plots represent the weight of tumors (n = 8 for each group in all statistical analysis; n.s. indicates no statistic difference; ∗P < 0.05).
Since ZDHHC12-mediated CLDN3 S-palmitoylation dictates its biological function, it will be interesting to test whether silencing ZDHHC12 could phenocopy the effect of CLDN3 knockdown on ovarian cancer tumorigenesis inhibition. To further confirm this, we subsequently performed in vivo experiment. 1 × 106 OVCAR8 cells with shCtrl or shZDHHC12 were vaccinated subcutaneously on each side of BALB/c-nu mice body (n = 8). The mice were executed at the termination of assay. Expectedly, the tumorigenesis and tumor growth rate of both shZDHHC12 groups were much slower than shCtrl group (Fig. 6D–G). Moreover, we also established the relationship of ZDHHC12 and CLDN3 with more clinical evidence. As shown in Figs. S4C–S4F, ZDHHC12 has a positive correlation with CLDN3 in protein level or mRNA level in ovarian cancer.
4. Discussion
We found a positive correlation between CLDN3 expression and the pathological grade of ovarian cancer, and proved that targeting CLDN3 S-palmitoylation might be a potent therapeutic strategy for malignant ovarian cancer. What's more important, we initially introduce ZDHHC12 as a novel target for CLDN3. A growing body of evidence indicates that dysfunction of tight junctions is responsible for the tumor pathology in carcinogenesis. In this regard, emerging evidence further suggests that tight junction harbors and recruits evolutionarily conserved protein complexes and signaling cascades to help maintain normal homeostasis. Therefore, decreasing claudin expression might lead to the compromised tight junction function.
In this study, we decipher that the S-palmitoylation of CLDN3 is important for its biological function especially in regulating cell proliferation. Here, we focus on cysteine 181, 182 and 184 sites, which are close to the C-terminus of CLDN3. Importantly, the C-terminus of CLDN3 interacts with many signaling pathways because this domain has a number of serine, tyrosine and phosphorylated sites that can potentially enable various molecular interactions15,37,38. Therefore, it provides the conditions for regulating the biological function of CLDN3 that the possible interplay of multiple post-translational modifications.
Profoundly, we found that palmitoylation impacted the localization of CLDN3 to the cell membrane and facilitates its stability. When lacking palmitoylation, CLDN3 was likely to be distributed among the cytoplasm. Similarly, some researches referred that phosphorylation could also affect the intracellular localization of CLDN3 and affect TJ function37. It will be interesting to test if there is a possible crosstalk between palmitoylation and phosphorylation in the future studies, as the palmitoylation sites we founded are close to the phosphorylation site in mammalian CLDN3 amino acid sequence. Interestingly, in our finding, CLDN3 3CS was not able to localize at cell membrane normatively, which resulted in its lack of stability. We hypothesize that the correct intracellular localization is a prerequisite for maintaining CLDN3 protein stability and its biological function, which mechanism we might consider as a potential strategy for downregulation of CLDN3. A similar situation can be observed in some members of the tetraspanin family of proteins of which the structures are analogous to the claudin. For example, the palmitoylation deficient mutant of CD15139 and CCR540 showed diffusely distribution and markedly diminished stability during biosynthesis, compared to its wild type. These facts give us more credibility in our experiment and highlight our point of view. Parallelly, the uncovered results hint to our further research.
On the other view, in recent years, the mechanism of a peculiar form of internalization of claudins was uncovered gradually, which was associated with the dynamic remodeling of TJs, followed by epithelial cells moving against adjacent cells, intercellular TJs are remodeled. When TJs were endocytosed, claudins appeared to be dissociated from other TJ components and underwent the individual endocytosis, that is TJ-incorporated claudins are recognized for ubiquitylation and internalization followed by lysosome targeting by unknown mechanisms. Our finding that CLDN3 3CS is more likely to be ubiquitinated for degradation by proteasome is partly consistent with these views41,42. Collectively, we speculate that stable S-palmitoylation of CLDN3 as a sign for ovarian cancer cells keeping barrier function while loss of S-palmitoylation may trigger ubiquitylation-mediated degradation of CLDN3 followed by internalization.
Although knocking down CLDN3 by lentiviral virus is not equivalent to that caused by internalization of CLDN3, the function of TJs is disrupted in both cases. In our in vitro and in vivo tests, loss of CLDN3 dramatically hampered the proliferation of ovarian cancer. Besides, our in vitro study showed that ovarian cells expressing CLDN3 S-palmitoylation deficient mutants did not promote cell tumorigenesis compared to cells expressing wild type CLDN3, which hinted that S-palmitoylation of CLDN3 contributes to its oncogenic character and make it more trustworthy if animal study is carried out.
Importantly, by screening the 23 ZDHHCs, we found that ZDHHC12 had a strong interaction with CLDN3. More evidence supported that CLDN3 is a direct palmitoylation target of the endogenous ZDHHC12. Nevertheless, there is still approximately a half level of S-palmitoylation in A2780 cell compared to control after knocking down about 80% of endogenous ZDHHC12, which can be attributed to incomplete ZDHHC12 knockdown or to additional minor contribution from other ZDHHC proteins. Some studies showed that palmitoylated proteins are likely to assemble with their cognate ZDHHC during the palmitoylation process43,44. The mammalian ZDHHC12, ZDHHC21 and ZDHHC23 harbor analogous amino acid alignments, which might explain the previous result.
Accumulated evidence has shown that subcellular localization of claudins is critical to determine their oncogenic role in cancer. In this study, we demonstrate that CLDN3 is S-palmitoylated by ZDHHC12, which PTM facilitates cytomembrane targeting of CLDN3 and maintains the tight junction. Moreover, we profiled the positive correlation of CLDN3 and ZDHHC12 in ovarian cancer from TCGA database. Researches continue to explore different ZDHHC protein structures, binding patterns to substrates, and enzymatic catalytic mechanism in order to develop selective inhibitors of ZDHHC. With the demonstration and discovery of assorted inhibitors45,46, it can be expected that palmitoylation modification is gradually becoming a spotlight of oncoprotein therapy, such as impelling degradation and blocking downstream signal activation. Although in our research, we have not fully elucidated the mechanisms responsible for the oncogenic S-palmitoylation role, ZDHHC12 may become a potential in therapeutic intervention in ovarian cancer patients with high expression of CLDN3.
5. Conclusions
In this paper, we confirmed the role of CLDN3 in promoting tumorigenesis and progression in ovarian cancer progress based on database. As digging the biochemical role of CLDN3, we found that CLDN3 is palmitoylated on multiple cysteine residues in C-terminus, which contributes to the stability and subcellular localization of CLDN3. Importantly, the observation and analysis of immunofluorescence results bring us to a speculation that a full set of three cysteine residues is required for accurate cytomembrane targeting and ZO-1 binding. By screening the potential interaction between CLDN3 and 23 ZDHHC family proteins and the metabolic labeling method, we found that ZDHHC12 is a dominant CLDN3-palmitoylation enzyme, which benefits the appropriated anchoring and stability of CLDN3. What's nascent and important is the potential role of ZDHHC12 as a manipulator in ovarian cancer progression which promoted by CLDN3.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 91753114 to Hong Jiang; No. 81872885 to Ji Cao), Zhejiang Provincial Natural Science Foundation (No. Y18H310005 to Ji Cao, China), and the Talent Project of Zhejiang Association for Science and Technology (No.2018YCGC002 to Ji Cao, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.03.008.
Contributor Information
Ji Cao, Email: caoji88@zju.edu.cn.
Qiaojun He, Email: qiaojunhe@zju.edu.cn.
Author contributions
Conception and design: Ji Cao and Meidan Ying; Experimental operation: Meng Yuan and Xiaobing Chen; Collection and assembly of data: Meng Yuan, Yitang Sun and Li Jiang; Manuscript writing: Meng Yuan, Xiaobing Chen and Li Jiang; Final approval of manuscript: all authors.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Morita K., Furuse M., Fujimoto K., Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci U S A. 1999;96:511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Runkle E., Mu A.D. Tight junction proteins: from barrier to tumorigenesis. Canc Lett. 2013;337:41–48. doi: 10.1016/j.canlet.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee B., Moon K.M., Kim C.Y. Tight junction in the intestinal epithelium: its association with diseases and regulation by phytochemicals. J Immunol Res. 2018;2018:2645465. doi: 10.1155/2018/2645465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heiskala M., Peterson P.A., Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic. 2001;2:93–98. doi: 10.1034/j.1600-0854.2001.020203.x. [DOI] [PubMed] [Google Scholar]
- 5.Singh A.B., Uppada S.B., Dhawan P. Claudin proteins, outside-in signaling, and carcinogenesis. Pflügers Archiv. 2017;469:69–75. doi: 10.1007/s00424-016-1919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A.B., Sharma A., Dhawan P. Claudin family of proteins and cancer: an overview. J Oncol. 2010;2010:1–11. doi: 10.1155/2010/541957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rangel L.B., Agarwal R., D'Souza T., Pizer E.S., Alo P.L., Lancaster W.D. Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Canc Res. 2003;9:2567–2575. [PubMed] [Google Scholar]
- 8.Kojima F., Ishida M., Takikita-Suzuki M., Hotta M., Katsura K., Nagata A. Claudin expression profiles in Epstein-Barr virus-associated nasopharyngeal carcinoma. Oncol Rep. 2010;23:927–931. doi: 10.3892/or_00000716. [DOI] [PubMed] [Google Scholar]
- 9.Thuma F., Heiler S., Schnolzer M., Zoller M. Palmitoylated claudin 7 captured in glycolipid-enriched membrane microdomains promotes metastasis via associated transmembrane and cytosolic molecules. Oncotarget. 2016;7:30659–30677. doi: 10.18632/oncotarget.8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X., Li Y., Qiu H., Wang Y. Downregulation of claudin-7 potentiates cellular proliferation and invasion in endometrial cancer. Oncol Lett. 2013;6:101–105. doi: 10.3892/ol.2013.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kominsky S.L., Argani P., Korz D., Evron E., Raman V., Garrett E. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene. 2003;22:2021–2033. doi: 10.1038/sj.onc.1206199. [DOI] [PubMed] [Google Scholar]
- 12.Todd M.C., Petty H.M., King J.M., Piana Marshall B.N., Sheller R.A., Cuevas M.E. Overexpression and delocalization of claudin-3 protein in MCF-7 and MDA-MB-415 breast cancer cell lines. Oncol Lett. 2015;10:156–162. doi: 10.3892/ol.2015.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L., Wang Y., Zhang B., Zhang H., Zhou M., Wei M. Claudin-3 expression increases the malignant potential of lung adenocarcinoma cells: role of epidermal growth factor receptor activation. Oncotarget. 2017;8:23033–23047. doi: 10.18632/oncotarget.14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim N.I., Kim G.E., Lee J.S. Diagnostic usefulness of claudin-3 and claudin-4 for immunocytochemical differentiation between metastatic adenocarcinoma cells and reactive mesothelial cells in effusion cell blocks. Acta Cytol. 2016;60:232–239. doi: 10.1159/000447008. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad R., Kumar B., Chen Z., Chen X., Muller D., Lele S.M. Loss of claudin-3 expression induces IL6/gp130/Stat 3 signaling to promote colon cancer malignancy by hyperactivating Wnt/beta-catenin signaling. Oncogene. 2017;36:6592–6604. doi: 10.1038/onc.2017.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan X., Lin X., Manorek G., Kanatani I., Cheung L.H., Rosenblum M.G. Recombinant CPE fused to tumor necrosis factor targets human ovarian cancer cells expressing the claudin-3 and claudin-4 receptors. Mol Canc Therapeut. 2009;8:1906–1915. doi: 10.1158/1535-7163.MCT-09-0106. [DOI] [PubMed] [Google Scholar]
- 17.Li X., Iida M., Tada M., Watari A., Kawahigashi Y., Kimura Y. Development of an anti-claudin-3 and -4 bispecific monoclonal antibody for cancer diagnosis and therapy. J Pharmacol Exp Therapeut. 2014;351:206–213. doi: 10.1124/jpet.114.216911. [DOI] [PubMed] [Google Scholar]
- 18.Ko P.J., Dixon S.J. Protein palmitoylation and cancer. EMBO Rep. 2018;19:1–18. doi: 10.15252/embr.201846666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linder M.E., Deschenes R.J. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 20.Hornemann T. Palmitoylation and depalmitoylation defects. J Inherit Metab Dis. 2015;38:179–186. doi: 10.1007/s10545-014-9753-0. [DOI] [PubMed] [Google Scholar]
- 21.Deng H., Li W. Monoacylglycerol lipase inhibitors: modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm Sin B. 2019:1–21. doi: 10.1016/j.apsb.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Runkle K.B., harbanda A.K., Stypulkowski E., Cao X.J., Wang W., Garcia B.A. Inhibition of DHHC20-mediated EGFR palmitoylation creates a dependence on EGFR signaling. Mol Cell. 2016;62:385–396. doi: 10.1016/j.molcel.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch S.J., Snitkin H., Gumper I., Philips M.R., Sabatini D., Pellicer A. The differential palmitoylation states of N-Ras and H-Ras determine their distinct Golgi subcompartment localizations. J Cell Physiol. 2015;230:610–619. doi: 10.1002/jcp.24779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song S.P., Hennig A., Schubert K., Markwart R., Schmidt P., Prior I.A. Ras palmitoylation is necessary for N-Ras activation and signal propagation in growth factor signalling. Biochem J. 2013;454:323–332. doi: 10.1042/BJ20121799. [DOI] [PubMed] [Google Scholar]
- 25.Kharbanda A., Runkle K., Wang W., Witze E.S. Induced sensitivity to EGFR inhibitors is mediated by palmitoylated cysteine 1025 of EGFR and requires oncogenic Kras. Biochem Bioph Res. 2017;493:213–219. doi: 10.1016/j.bbrc.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P.Y., Wang Y.J., Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B. 2019;9:871–879. doi: 10.1016/j.apsb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodenburg R.N., Snijder J., van de Waterbeemd M., Schouten A., Granneman J., Heck A.J. Stochastic palmitoylation of accessible cysteines in membrane proteins revealed by native mass spectrometry. Nat Commun. 2017;8:1–9. doi: 10.1038/s41467-017-01461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korycka J., Lach A., Heger E., Boguslawska D.M., Wolny M., oporkiewicz M.T. Human DHHC proteins: a spotlight on the hidden player of palmitoylation. Eur J Cell Biol. 2012;91:107–117. doi: 10.1016/j.ejcb.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Zaballa M.E., van der Goot F.G. The molecular era of protein S-acylation: spotlight on structure, mechanisms, and dynamics. Crit Rev Biochem Mol Biol. 2018;53:420–451. doi: 10.1080/10409238.2018.1488804. [DOI] [PubMed] [Google Scholar]
- 30.Hang H.C., Wilson J.P., Charron G. Bioorthogonal chemical reporters for analyzing protein lipidation and lipid trafficking. Accounts Chem Res. 2011;44:699–708. doi: 10.1021/ar200063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao J., Sun L., Aramsangtienchai P., Spiegelman N.A., Zhang X., Huang W. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc Natl Acad Sci U S A. 2019;116:5487–5492. doi: 10.1073/pnas.1815365116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overgaard C.E., Daugherty B.L., Mitchell L.A., Koval M. Claudins: control of barrier function and regulation in response to oxidant stress. Antioxidants Redox Signal. 2011;15:1179–1193. doi: 10.1089/ars.2011.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson J.M., van Itallie C.M. Physiology and function of the tight junction. Csh Perspect Biol. 2009;1:1–17. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manders E.M., Verbeek F.J., Aten J.A. Measurement of colocalization of objects in dual-color confocal images. J Microsc Oxford. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 35.De I., Sadhukhan S. Emerging roles of DHHC-mediated protein S-palmitoylation in physiological and pathophysiological context. Eur J Cell Biol. 2018;97:319–338. doi: 10.1016/j.ejcb.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Jiang L., Yang Y.D., Fu L., Xu W.Q., Liu D.B., Liang Q.Y. CLDN3 inhibits cancer aggressiveness via Wnt-EMT signaling and is a potential prognostic biomarker for hepatocellular carcinoma. Oncotarget. 2014;5:7663–7676. doi: 10.18632/oncotarget.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Souza T., Agarwal R., Morin P.J. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J Biol Chem. 2005;280:26233–26240. doi: 10.1074/jbc.M502003200. [DOI] [PubMed] [Google Scholar]
- 38.Greaves J., Carmichael J.A., Chamberlain L.H. The palmitoyl transferase DHHC2 targets a dynamic membrane cycling pathway: regulation by a C-terminal domain. Mol Biol Cell. 2011;22:1887–1895. doi: 10.1091/mbc.E10-11-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X., Claas C., Kraeft S.K., Chen L.B., Wang Z., Kreidberg J.A. Palmitoylation of tetraspanin proteins: modulation of CD151 lateral interactions, subcellular distribution, and integrin-dependent cell morphology. Mol Biol Cell. 2002;13:767–781. doi: 10.1091/mbc.01-05-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanpain C., Wittamer V., Vanderwinden J.M., Boom A., Renneboog B., Lee B. Palmitoylation of CCR5 is critical for receptor trafficking and efficient activation of intracellular signaling pathways. J Biol Chem. 2001;276:23795–23804. doi: 10.1074/jbc.M100583200. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi S., Iwamoto N., Sasaki H., Ohashi M., Oda Y., Tsukita S. The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J Cell Sci. 2009;122:985–994. doi: 10.1242/jcs.040055. [DOI] [PubMed] [Google Scholar]
- 42.Matsuda M., Kubo A., Furuse M., Tsukita S. A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci. 2004;117:1247–1257. doi: 10.1242/jcs.00972. [DOI] [PubMed] [Google Scholar]
- 43.Abrami L., Dallavilla T., Sandoz P.A., Demir M., Kunz B., Savoglidis G. Identification and dynamics of the human ZDHHC16-ZDHHC6 palmitoylation cascade. Elife. 2017;6:1–24. doi: 10.7554/eLife.27826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lemonidis K., Salaun C., Kouskou M., Diez-Ardanuy C., Chamberlain L.H., Greaves J. Substrate selectivity in the zDHHC family of S-acyltransferases. Biochem Soc Trans. 2017;45:751–758. doi: 10.1042/BST20160309. [DOI] [PubMed] [Google Scholar]
- 45.Yang Z.H., Cappello T., Wang L. Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B. 2015;5:145–150. doi: 10.1016/j.apsb.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Jiang X., Feng F., Liu W., Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.