

Abstract

Drugs that block voltage-gated sodium channels (NaVs) have utility in treating conditions including pain, epilepsy, and cardiac arrhythmias and as anesthetics (Lancet Neurol. 2010;9:413–424. doi: 10.1016/S1474-4422(10)70059-4.; Expert Opin. Ther. Pat. 2010;20:755–779. doi: 10.1517/13543771003774118.). The identification of compounds with improved efficacy and safety is a key aim for the discovery of improved NaV blocking drugs (Comprehensive Medicinal Chemistry III; Elsevier, 2017; pp 131−175). We report the identification of a novel class of brain penetrant and voltage-gated sodium channel blockers, leading to the discovery of vixotrigine, a use-dependent sodium channel blocker with activity in in vivo models of pain. Vixotrigine has excellent physiocochemical properties for drug development, and both preclinical and clinical data support a safety profile suitable for potential use in neuropathic pain and other conditions. It has shown efficacy in a Phase II study for pain associated with trigeminal neuralgia.
Keywords: Use-dependent, state-dependent, sodium channel blocker, NaV, trigeminal neuralgia, analgesic
Introduction
The mechanism of action of several drugs in current clinical practice is, at least in part, due to nonselective blockade of voltage-gated sodium channels.1−4 For example, lamotrigine5 for epilepsy and bipolar disorder and carbamazepine6 for epilepsy and trigeminal neuralgia are characterized as nonselective sodium channel blockers. However, the utility of these drugs is limited by side effects and multiple drug–drug interactions that require careful and prolonged titration to the effective clinical dose. Hence there is a need for sodium channel blockers with improved safety profiles.
Within the mammalian genome, nine subtypes of the voltage-gated sodium channel (designated NaV1.1–NaV1.9) have been identified and cloned.7 Physiological and human genome-wide association studies have identified mutations of several NaV subtypes in a range of diseases (“channelopathies”).8 For example heteroallelic mutations in the SCN4A gene encoding the α-subunit of NaV1.4 (the skeletal muscle sodium channel) have been associated with a myasthenic (fatigue and weakness) syndrome.9 Of particular focus, various NaV mutations have been implicated in different types of pain; several mutations in the human SCN9A gene, encoding the α-subunit of the NaV1.7 channel, have been found to be causative for primary erythromelalgia,10 an autosomal dominant neurological disorder characterized by severe burning pain and erythema. Furthermore, SCN9A and SCN10A (encoding NaV1.8) are both genetically associated11 with painful small fiber neuropathy due to dorsal root ganglion neuron hyperexcitability.
Trigeminal neuralgia12,13 is a rare disease associated with intense, unilateral facial pain characterized by electric shock-like episodes linked to entrapment of the trigeminal nerve. 150,000 cases are diagnosed in the United States each year. Treatment options for this debilitating condition include microvascular decompression surgery, which risks facial numbness and paralysis, or drug treatment with carbamazepine or oxcarbazepine. These agents are nonselective voltage-gated sodium channel blockers but are poorly tolerated, and their use is hampered by multiple clinically relevant drug–drug interactions and the active pharmacology of their metabolites.14
Our goal was to identify a NaV blocker with potential utility in the treatment of neuropathic pain, including trigeminal neuralgia and other CNS disorders. Pain is associated with high frequency nerve signaling and our hypothesis is that preferential binding to the inactivated state of the sodium channel should favor high frequency blockade and give selectivity over normal physiological signaling at lower firing frequencies. Achieving a sufficient degree of inactivated-state block of sodium channels was targeted for optimal safety and efficacy. This is measured by the use-dependence and voltage dependence of the channel inhibition.15 Furthermore, based on genetic association studies, by targeting state dependence we were aiming for enhanced NaV blockade in neurons with pathologically elevated activity, while sparing channels associated with low frequency activities such as cardiac function.16 Importantly, we chose to optimize for CNS penetration on the basis that nociceptors express a variety of NaV subtypes, and blockade of brain sodium channels (NaV 1.1, 1.2, 1.3) is highly precedented17 for efficacious NaV blockers. We thereby sought to harness the proven mode of action of existing sodium channel blockers while significantly improving the therapeutic index and side effect profile.
In summary, we targeted compounds with preferential binding to the inactivated state of relevant sodium channels; selectivity over the hERG channel;18 and pharmacokinetic properties suitable to support oral dosing and efficient brain exposure with low potential for clinically relevant drug–drug interactions.
Discovery
A requirement for an improved safety profile, including avoiding the need for dose titration,19 led us to look for starting points structurally differentiated from currently marketed drugs. A high throughput FLIPR screen was conducted using cells expressing the brain sodium channel NaV1.2 to find general chemotypes with sodium channel blocking activity. Hits were assessed for their tonic and use-dependent block on an Ionworks platform using the NaV1.3 channel as a surrogate for brain sodium channels. Tonic block was assessed by measuring the effect of test compounds on the peak current elicited by the first pulse in a 10 Hz pulse train at a range of concentrations, from which a pIC50 was determined.20 The use-dependent block was assessed on the final pulse, and data presented here show the negative log of the concentration that caused a 15% inhibition of the sodium current (pUD15). The ratio of UD15 to IC50 is reported as the Tonic Selectivity (TS).
A pharmacophore for use-dependent sodium channel blockers emerged in which a lipophilic aromatic group was linked to a basic amine or polar heterocycle, which was in turn linked to an amide or amide isostere (Figure 1). This was precedented in the published literature with the compounds ralfinamide and safinamide reported to be state dependent blockers by Newron.21 In addition the Abbott compound A-803467 and Merck’s rufinamide, mapping to this pharmacophore, are reported as NaV1.8 and NaV1.7 blockers, though for rufinamide no channel selectivity is claimed.22,23
Figure 1.

NaV pharmacophore generation and selected published NaV blockers.
Following this analysis, an N-benzyl glycinamide scaffold was chosen as a minimum template with both tonic and use-dependent block of NaV1.3 to examine for optimization through substitution and derivatization. Compound 1, an unsubstituted N-benzyl glycinamide, incorporating the precedented para 2-fluorobenzyloxy group, was synthesized and showed modest tonic and reasonable use-dependent block of NaV1.3 (Table 1). We hypothesized that conformational restriction around the benzyl-amino-amide feature could be associated with enhanced state-dependency, improved tonic selectivity, and better chemical developability properties. Our strategy was to induce conformational restriction of this simple template (Figure 2) to build the targeted profile. This starting point had the additional advantage of good solubility and reasonable synthetic accessibility, favoring a lead optimization campaign.
Table 1. Selected Constrained Benzyl Glycinamides.


Figure 2.

Targeted contraints of benzyl glycinamides.
Conformationally constrained amides were selected by introducing cyclization along the benzyl glycinamide backbone: between the amide nitrogen and either the α-position or the basic nitrogen (A), from the aromatic ring on to the glycinamide backbone (B), and between positions α- to the amino nitrogen (C) (Figure 2). In order to maintain developability properties within a range suitable for a brain penetrant drug, we targeted analogs with clogP values between 2 and 3.5 and with molecular weight below 400. The 2-fluorobenzyl substituent was maintained as a 2-fluoro benzyl or benzyl group.
A. Cyclization between the amide nitrogen and the basic nitrogen gave analog 2, which showed no significant sodium channel blockade. Cyclization through the glycine α-carbon resulted in compound 3, which retained some use-dependent block, but at a significantly lower level than 1 and with very low tonic block (Table 1).
B. Constraint of the benzylic position was investigated by cyclization onto the central aromatic ring, either through nitrogen (4–10) or benzylic carbon (11–13), while retaining an unsubstituted amide. Ring systems incorporating the amino nitrogen and branching at nitrogen (4–8) gave markedly reduced tonic block at NaV1.3. Similar compounds in which the amide group was attached α to the nitrogen (9, 10) still showed significantly reduced use-dependent block relative to benzyl glycine 1. Exocyclic amines in which the benzylic position alone was constrained (11, 12) were relatively potent tonic blockers of NaV1.3 and showed significant use-dependent block. Interestingly, however, an oxygen atom in the heterocyclic constraint, forming a benzopyran system (13), led to a loss of sodium channel blockade.
C. A further constraint incorporated the amino nitrogen into a discrete pyrrolidine ring. Isomers of structure 14 maintained blockade of NaV1.3 with good tonic selectivity. Isomers of compound 15, incorporating a tertiary nitrogen, had higher use-dependent block, while the proline derivative 16 gave the highest use-dependent block in the series with a tonic selectivity of 64.
Compounds 11, 12, and 14–16 were selected for further profiling and series optimization. Individual isomers were prepared, their ADME properties investigated, and hERG inhibition profile examined to evaluate series properties and liabilities. Specific indanes and tetrahydroquinoline isomers related to 11 and 12 had a low potential for hERG blockade and showed low microsomal clearance; however, they proved to be inhibitors of cytochrome P450 isoforms. Modification with additional alkyl substituents typically led to much higher hERG inhibition or increased rat microsomal clearance. Analogs of 15 or 16 possessing a tertiary pyrrolidine ring were associated with high clearance in rat microsomes. Attempts to block metabolically vulnerable positions adjacent to the basic nitrogen by alkyl substitution typically gave rise to higher hERG inhibition, and compounds of this class were reported to have other off-target activities.24,28
In contrast, the individual isomers of 14, compounds 17–20, while showing broadly similar use-dependent block of NaV1.3, possessed differential effects in their other properties (Table 2). Syn-isomers 17 and 20 showed a trend toward lower blockade of hERG while, interestingly, there was a clear diastereomeric preference for the 2-(S)-stereochemistry alpha to the carbonyl (19, 20) giving the lowest rat microsomal clearance. Of these analogs 2-(S)-5-(R) isomer 20 had the most attractive overall profile exhibiting a tonic selectivity of 31, and an analog program was initiated around 20 while maintaining clogP values between 2.5 and 3.5. In addition to measuring rat clearance and hERG inhibition, in a number of cases, the potential for brain exposure was assessed by measuring the fraction unbound in rat plasma and brain tissue (Table 3).
Table 2. Isomers of Phenyl Prolineamide 14.

Table 3. Analogs of Phenyl Prolinamide 20.

Changing the ring from a pyrrolidine to a piperidine (21, 22 absolute stereochemistry unassigned) led to a significant increase in NaV1.3 tonic block, but reduced tonic selectivity (21) or increased hERG inhibition (22). Thiomorpholine 23, in contrast, showed poorer use-dependent block. Analogs of 20 in which the fluorine atom was removed (25), or moved to a meta-substituted (24) or para-position (26), maintained good use-dependent NaV block but with a trend for increased hERG inhibition. More generally, increased hERG inhibition was observed for the more lipophilic compounds, but an attempt to reduce lipophilicity by introducing a cyano group as a terminal benzyl substituent had little impact at any positional isomers (27–29) although the ortho example 29 possessed the best overall profile. Substitution on the benzylic methylene increased hERG affinity in compound 30, but fluoro analog 31 maintained a good state-dependent profile, although tonic block was marginal. Alkylation of the pyrrolidine nitrogen (32) markedly reduced state-dependent activity.
Substitution in the 3-position of the central aromatic ring was also examined in order to modulate hERG inhibition; however, both methoxy analog 34 and cyano analog 35 gave reduced use-dependent block relative to the unsubstituted 20. A central ring pyridyl analog 36 was also detrimental to use-dependent NaV blockade. Replacement of the terminal benzyl ether with a phenyl ether was tolerated in analog 37 and a range of substituted analogs (38–40) was prepared; however, the unsubstituted ether 37 had the best overall profile. Finally, repositioning the terminal aromatic-containing group meta led to reduced sodium channel block, for both phenyl ether 41 and benzyl ether 42.
Compound 31 was not progressed as some tonic block was believed to be important to modulate overall physiological channel tone. Four compounds with high brain tissue binding, meeting the criteria of tonic block greater than 4, tonic selectivity greater than 10-fold, rat microsomal clearance less than 2.0 mL/min/g, and hERG pIC50 less than 5.0, were progressed into rat in vivo exposure studies, to determine clearance and bioavailability and measure tissue distribution in in vitro studies (Table 4).
Table 4. Rat Clearance, Tissue Penetration, and Oral Bioavailability.
| Entry | Blood Clb (mL/min/kg) | Brain:Blood ratioa | % F(po) |
|---|---|---|---|
| 20 | 39 | 6.8 | 43 |
| 26 | 91 | 3.5 | 31 |
| 29 | 37 | 0.8 | 39 |
| 37 | 40 | 4.8 | 25 |
Total by equilibrium dialysis.
Three compounds showed oral bioavailability greater than 30% and were analyzed for brain penetrance. The more polar analog 29 showed a brain:blood ratio of less than 1 but 2-fluorobenzyloxy- and 4-fluorobenzyloxy analogs 20 and 26 achieved high brain penetration. Isomer 26 was highly cleared in vivo, but compound 20 had good oral bioavailability and moderate clearance. This compound was therefore selected for further studies
The interaction of 20 with binding sites on 130 receptors, ion channels, transporters, and enzymes was examined in a radioligand binding cross screening panel. The pan-sodium channel inhibition profile of this compound is reported separately.25 The most significant off-target interaction was inhibition of MAO-B. This activity was confirmed in human liver microsomes where 20 inhibited MAO-B enzyme activity with a pIC50 of 8.4. In contrast 20 had no measurable inhibitory effect on MAO-A enzyme activity (pKi < 5).
Synthesis
Compounds in these studies were prepared by a variety of general routes typified by A–E shown in Scheme 1. Where nonstereoselective syntheses were performed, compounds were separated by chiral reverse phase HPLC. The absolute stereochemistry of compound 20 was confirmed by single crystal X-ray crystallography.26
Scheme 1. Synthetic Routes to Benzyl Glycinamides.

Typical reagents and conditions: (a) R1-benzyl-bromide, K2CO3, MeCN; (b) NaH3CN, MeOH, AcOH, substituted glycinamide; (c) LAH, THF; (d) DIPEA, THF, TfO–CH(R3)CO2Et; (e) NH3, MeOH; (f) NH2OMe, EtOH then LiBH4, ZrCl4, THF; (g) Boc2O, THF; (h) TBTU, HMDS, DIPEA, DMF; (i) TFA, DCM.
Benzyl glycinamides were synthesized by adapting a number of published methodologies (Scheme 1). Two main strategies were employed. Reductive amination of an appropriate aldehyde A1 or ketone A2 with a glycine amide27 or prolinamide24 (Route A) was used for 1–3, 11, and 16. Alternatively (Route B) alkylation of a benzylic amine such as B1(28) with an appropriate acetic ester bearing a leaving group alpha to the carbonyl, followed by amidation of the ester provided a route to 4–8. The right-hand side benzyl ether could be introduced by alkylation of the corresponding hydroxyaromatic, undertaken either prior to introducing the glycine component or with a suitably protected glycine in place (Route C). Thus, amines of structure C1 (for 12 and 13) could be prepared from parent heterocycles by ether formation then reduction. Likewise compounds of generic structure D1, in which the glycine nitrogen is in a ring fused to the central aromatic group (e.g., 9, 10), are accessible by alkylation and amidation of corresponding heterocycles (Route D).29 Compounds in which the benzylglycinamide nitrogen was also linked to the benzylic position by a pyrrolidine ring (e.g., 15) were accessible (Route E) by reductive cyclizations onto carbonyl precursors.30
For certain analogs, enantiomeric mixtures were separated by reverse phase chiral chromatography; however, an enantioselective route to the isomers of compound 14 (compounds 20 and its analogs) was developed, illustrated in Scheme 2.31 Chiral Boc-protected (S)-pyroglutamate 43 was treated with the organolithium reagent formed from 4-benzyloxy-1-bromobenzene 44 at low temperature to furnish the ketone adduct 45. Deprotection under acidic conditions formed the cyclic 2H-pyrrolidine 46. Stereoselective reduction of the double bond over a platinum catalyst installed predominantly the 2(S)-stereogenic center in 47. Hydrolysis and reprotection with Boc gave acid 49 which could be amidated to give 50 and the benzyl group removed by a second hydrogenation, this time over palladium, to give a versatile phenolic intermediate 51. Alkylation of this material to 52 followed by removal of the Boc protection afforded carboxamide 20 as the crystalline hydrochloride salt. The benzylic epimer 19 could be prepared by taking the minor isomer from the imine reduction (48) through the later stages of this sequence. The corresponding pair of diastereomers 17 and 18 was accessed by using the (R) enantiomer of 43 in the procedure.
Scheme 2. Enantioselective Synthesis of Compound 20.

Reagents and conditions: (a) nBuLi, THF, −78 °C; (b) 43 −40 °C; (c) TFA, DCM; (d) H2, Pt/C, MeOH; (e) (i) LiOH aq THF; (f) Boc2O, THF; (g) TBTU, DIPEA, DMF, HMDS; (h) H2 Pd/C MeOH; (i) (2-F)-benzyl bromide, K2CO3, MeCN; (j) AcCl, MeOH, EtOAc.
Using the methodology of Scheme 2, the different aromatic right-hand groups of 21–30 could be introduced by alkylating intermediate 51 with the corresponding substituted benzyl bromide. Analogs 21–23 could also be prepared using this route by employing piperidine or thiomorpholine analogs of Boc-pyroglutamic acid. Analogs 31 and 34–36 could be prepared by using a corresponding bromoaromatic in place of 4-benzyloxybromobenzene 44 in the lithiation step. Reductive alkylation of 20 with formaldehyde gave 32. Amidation of ester 47 with ammonia gave 42 and with methylamine gave compound 33.
A more efficient and scalable route to 20 was subsequently developed (Scheme 3) in which a Grignard formed from 4-(2-fluorobenzyoxy)-4-bromobenzene 53 was reacted with pyroglutamate 43. Hydrolysis of the resulting Boc-protected ketone 54 gave an imine 55 which could be stereoselectively hydrogenated to ester 56. Aminolysis of this ester and precipitation of the resulting amide as a hydrochloride salt furnished 20. Analogs of 20 containing phenyl ethers (37–41) can be accessed by using an appropriate bromophenyl ether in place of benzyl ether 53. The details and yields for Scheme 3 and the further development of efficient routes to 20 are described elsewhere.32
Scheme 3. Scalable Route to 20.

Reagents and conditions: (a) Mg, I2, THF, −60 °C, BrCH2CH2Br; (b) 43, THF; (c) NH4Cl/brine/IPA; (d) TFA, DCM (e) KHCO3, H2O, n-heptane; (f) H2, 5%Pt/C, EtOAc/TFA; (g) NaHCO3, H2O, MeOH; (h) NH3 in MeOH, (i) toluene, n-heptane; (j) HCl, EtOAc.
Pharmacology
Compound 20 was evaluated in the chronic constriction injury (CCI) model of neuropathic pain by oral administration at 0.5 or 5 mg/kg, BID. It significantly reversed mechanical allodynia after 6 and 8 days of dosing (Figure 3) starting during day 22. Mean free plasma concentrations of 20 achieved on Day 8 at 0.5 h postdose were 1.9 ng/mL and 23.4 ng/mL at 0.5 and 5 mg/kg, respectively. In the same study, gabapentin (30 mg/kg orally BID) was dosed as a positive control and significantly inhibited CCI induced mechanical allodynia on days 1, 6, and 8 of dosing.
Figure 3.
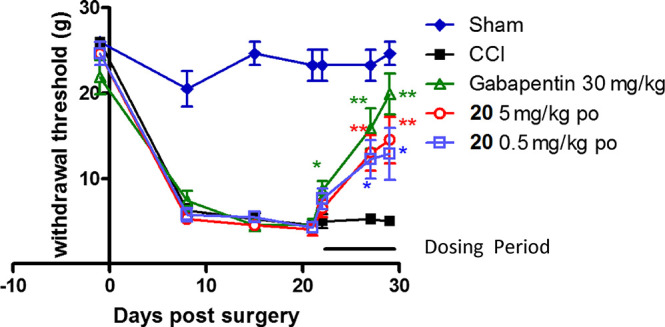
Compound 20 CCI induced static allodynia (0.5 h post dose).
Compound 20 was also evaluated in the Complete Freund’s Adjuvant (CFA) in vivo model of inflammatory pain. After single oral doses, it produced a dose-related reversal of the established CFA-induced hypersensitivity to pain, as measured by weight bearing. The ED50 for this effect was 0.91 mg/kg. A dose of 1 mg/kg po was associated with a mean free plasma concentration of 7.8 ng/mL at 1 h postdose. A second experiment was conducted in the CFA-induced knee joint model of subchronic inflammation. At an oral dose of 1 mg/kg BID, 20 showed a reversal of hypersensitivity that was equipotent to the effect produced by the nonsteroidal anti-inflammatory drug celecoxib over the 5 day dosing period.
In order to assess the centrally mediated side-effects such as sedation or ataxia, 20 was tested in a model of spontaneous locomotor activity (sLMA) of male rats. It was administered orally at doses of 60, 100, or 300 mg/kg, 30 min before the test session. No significant reduction in sLMA was observed with a dose of 60 mg/kg. The mean plasma concentration of 20 60 min postdose was 13.1 μg/mL. Higher doses of 100 or 300 mg/kg of 20 significantly reduced locomotor activity, a typical CNS side effect associated with NaV channel blockers. We concluded that 20 has a substantial therapeutic index (TI) between plasma exposures associated with central side effects and exposures required to demonstrate efficacy in the in vivo pain models.
The pharmacokinetics of 20 following a single intravenous and an oral dose were investigated in the rat, dog, marmoset, and monkey. A summary of the derived pharmacokinetic data is provided in Table 5. Blood clearance of 20 was moderate in rat and dog (46% and 39% of liver blood flow, respectively) and low (11% of liver blood flow) in monkey. The volume of distribution at steady state (Vss) was in excess of total body water in all species, suggesting moderate to extensive tissue distribution and resulting in half-lives of 0.8, 2.2, and 5 h in rat, dog, and monkey, respectively.
Table 5. Exposure of Compound 20 across Species (n = 3).
| Species | Route | Dose(mg/kg) | Tmax (h) | Cmax(ng/mL) | t1/2 (h) | Vss(L/kg) | CLb(mL/min/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|
| Rat | IV | 0.5 | 0.8 | 2 | 39 | |||
| Oral | 3 | 0.5 | 226 | 43 | ||||
| Dog | IV | 0.5 | 2.2 | 2 | 12 | |||
| Oral | 3 | 0.75 | 705 | 57 | ||||
| Monkey | IV | 0.5 | 5 | 2 | 5 | |||
| Oral | 3 | 2 | 730 | 42 |
The physicochemical properties of 20 lie within CNS drug-like space (MW 314.4 Da; cLogP 1.6 and pKa 7.6). These properties confer excellent aqueous solubility (solubility of HCl salt: water 16.3 mg/mL; FeSSIF 6.9 mg/mL) and high membrane permeability (Papps = 386 nm/s at pH 7.4) in the MDCK cell assay. 20 has been assigned BCS Class I (high solubility, high permeability).
Compound 20 has a low potential for clinically relevant drug–drug interactions. Against a panel of CYP450 enzymes, it showed low levels of inhibition (pIC50: 1A2, 13 μM; 2A6, >100 μM; 2B6, 31 μM; 2C8, >100 μM; 2C9, >100 μM; 2C19, 79 μM; 2D6, >100 μM; 3A4, >100 μM vs atorvastatin, mizazolam, and nifedipine). Furthermore in the same panel of CYP450 enzymes it showed no time/mechanism dependent inhibition. Up to concentrations of 100 μM, 20 is not an inhibitor of P-glycoprotein and is not an inhibitor of OATP1B1, BCRP, OATP1B3, OAT1, or OCT2.
After successful first-time-in-human enabling studies (genetic toxicology, safety pharmacology, and 28-day repeat dose toxicology in rat and dog) 20 was progressed into clinical Phase I studies in healthy volunteers. A single ascending dose study demonstrated that 20 had a good tolerability profile and was well absorbed with a dose related increase in exposure (Figure 4).33 In a repeat dose study, steady state blood levels were achieved rapidly and 20 exhibited a 12 h half-life consistent with twice-daily dosing.34
Figure 4.

Plasma concentration–time profile for 20.
Conclusion
In summary, we report the discovery of 20, now vixotrigine,35 a novel use-dependent voltage-gated sodium channel blocker with MAO-B activity. Vixotrigine has broad spectrum in vivo antihyperalgesic efficacy. It has successfully completed ICH mandated toxicology, genotoxicity, and safety pharmacology studies and, subsequently, a suite of Phase I human volunteer studies. Vixotrigine is well tolerated in humans at predicted pharmacologically active exposures and shows rapid absorption, high oral bioavailability, and a half-life of 12 h in humans sufficient to support BID or TID dosing in the clinic. A Phase II study has been conducted showing efficacy in Trigeminal Neuralgia and is reported elsewhere.36
Glossary
Abbreviations
- FU
fraction unbound
- MAO
monoamine oxidase
- ADME
absorption, distribution, metabolism, and elimination
- CCI
chronic constriction injury
- CFA
Complete Freund’s Adjuvant
- BID
twice a day
- sLMA
spontaneous locomotor activity
- ICH
International Council for Harmonisation
The authors declare the following competing financial interest(s): D.W. holds stock in Biogen Inc. and was an employee of Convergence Pharmaceuticals Ltd. and Biogen Inc. at the time of the conduct of this work. His involvement ended after leaving Biogen in 2017, except in the creation of this manuscript.
Author Status
§ C.L. and G.A. were project leaders on this work at GlaxoSmithKline prior to the acquisition of this project by Convergence Pharmaceuticals Ltd.
References
- Mantegazza M.; Curia G.; Biagini G.; Ragsdale D. S.; Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- Zuliani V.; Rivara M.; Fantini M.; Costantino G. Sodium channel blockers for neuropathic pain. Expert Opin. Ther. Pat. 2010, 20, 755–779. 10.1517/13543771003774118. [DOI] [PubMed] [Google Scholar]
- Witty D. R.; MacPherson D. T.; Giblin G. M. P.. Sodium channel Blockers for the Treatment of Pain. In Comprehensive Medicinal Chemistry III; Chackamannil S., Rotella D., Ward S. E., Eds; Elsevier, 2017; pp 131–175. [Google Scholar]
- Curia G.; Biagini G.; Perucca E.; Avoli M. Lacosamide: a new approach to target voltage-gated sodium currents in epileptic disorders. CNS Drugs 2009, 23, 555–568. 10.2165/00023210-200923070-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk B.; Czuczwar S. J. Efficacy, safety, and potential of extended-release lamotrigine in the treatment of epileptic patients. Neuropsychiatr. Dis. Treat. 2010, 6, 145–150. 10.2147/NDT.S6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahesh Kumar S.; Suresh I.; Satish Kumar C. H.; Swaroop Kumar S. L. V. V. S. N. K.; Vijey Aanandhi M. Role of carbamazepine in trigeminal neuralgia (TN). Journal of Advances in Drug Research 2011, 1, 68–81. [Google Scholar]
- Catterall W. A.; Goldin A. L.; Waxman S. G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Savio-Galimberti E.; Gollob M. H.; Darbar D. Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front. Pharmacol. 2012, 3, 124. 10.3389/fphar.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino A.; Maertens C.; Ohno K.; Shen X.-M.; Fukuda T.; Harper T. C.; Cannon S. C.; Engel A. G. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7377–7382. 10.1073/pnas.1230273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.-T.; Po-Yuan Huang P.-Y.; Chen-Tung Yen C.-T.; Chih-Cheng Chen C.-C.; Lee M.-J. A Novel SCN9A Mutation Responsible for Primary Erythromelalgia and Is Resistant to the Treatment of Sodium Channel Blockers. PLoS One 2013, 8, e55212. 10.1371/journal.pone.0055212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J. M. G.; Han C.; Merkies I. S. J.; Macala L. J.; Lauria G.; Gerrits M. M. G; Dib-Hajj S. D.; Faber C. G.; Waxman S. G. Small nerve fibres, small hands and small feet: a new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV1.7 mutation. Brain 2012, 135 (Pt 2), 345–358. 10.1093/brain/awr349. [DOI] [PubMed] [Google Scholar]
- Zakrzewska J. M. Trigeminal neuralgia. Br. Med. J. 2014, 348, 474. 10.1136/bmj.g474. [DOI] [PubMed] [Google Scholar]
- https://www.aans.org/Patients/Neurosurgical-Conditions-and-Treatments/Trigeminal-Neuralgia.
- Bu H.-Z.; Kang P.; Deese A. J.; Zhao P.; Poo W. F. Human in vitro glutathionyl and protein adducts of carbamazepine-10,11-epoxide, a stable and pharmacologically active metabolite of carbamazepine. Drug Metab. Dispos. 2005, 33, 1920–1924. 10.1124/dmd.105.006866. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj A.; Priestly T.. Voltage Gated Sodium Channels. In Ion Channels from structure to function; Kew J., Davies C., Eds.; Oxford University Press: Oxford, 2010; pp 131–171. [Google Scholar]
- For a description of channelopathies associated with NaV1.5 see:; Remme C. A. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. 10.1113/jphysiol.2013.256461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitges M.; Chiu L. M.; Guarneros A.; Nekrassov V. Effects of carbamazepine, phenytoin, lamotrigine, oxcarbazepine, topiramate and vinpocetine on Na+ channel-mediated release of [3H]glutamate in hippocampal nerve endings. Neuropharmacology 2007, 52, 598–605. 10.1016/j.neuropharm.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Johannesen L.; Vicente J.; Gray R.; Galeotti L.; Loring Z.; Garnett Ce; Florian J.; Ugander M.; Stockbridge N.; Strauss D. Improving the Assessment of Heart Toxicity for All New Drugs Through Translational Regulatory Science. Clin. Pharmacol. Ther. 2014, 95, 501–508. 10.1038/clpt.2013.238. [DOI] [PubMed] [Google Scholar]
- Lorberg B.; Youssef N. A.; Bhagwagar Z. Lamotrigine-associated rash: to rechallenge or not to rechallenge?. Int. J. Neuropsychopharmacol. 2009, 12, 257–265. 10.1017/S1461145708009504. [DOI] [PubMed] [Google Scholar]
- IonWorks Assay: HEK293 cells expressing the NaV1.3 ion channel were grown in DMEM-F12 + 10% FBS culture media at 37 °C. At a confluency of 50–70% cells were dissociated from culture flasks and triturated to ensure unicellular cell suspension. Currents were recorded at room temperature (21–23°C) using the IonWorksHT planar array electrophysiology technology (Molecular Devices Corp.). After cell addition a seal test was performed prior to antibiotic (amphotericin) circulation to achieve intracellular access. The intracellular solution contained the following (in mM): K-gluconate 100,KCI 40 mM, MgCI2 3.2, EGTA 3, HEPES 5, adjusted to pH 7.3. The external solution contained the following (in mM): CaCI2 0.9, KCI 2.7, KH2PO4 1.1, MgCI2 0.5, NaCI 136.9, NaHPO4 8. Trains of 10 20 ms-long pulses delivered at a frequency of 10 Hz, from the holding potential of −90 mV to 0 mV, were applied every 30 s. Test compounds were applied for 3–3.5 min. Tonic block was assessed by measuring the effect of test compounds on the peak current elicited by the first pulse in each train (i.e. a stimulation frequency of 0.033 Hz, and the equation: % Tonic block = (1 – (Drug1/Ctl1)) × 100. Data were fit using a logistic function in order to determine the IC50 and expressed as the negative log of the IC50 concentration (plC50). Use-dependent (UD) block was calculated using the following equation: % UD block = (1 – ((Drug10/drug1)/(CtH 0/Ctl1))) × 100. Data presented here are the negative log of the concentration that caused a 15% inhibition (UD15) of the sodium current (pUD15).
- Pevarello P.; Bonsignori A.; Dostert P.; Heidempergher F.; Pinciroli V.; Colombo M.; McArthur R. A.; Salvati P.; Post C.; Fariello; Ruggero G.; Varasi M. Synthesis and Anticonvulsant Activity of a New Class of 2-[(Arylalkyl)amino]alkanamide Derivatives. J. Med. Chem. 1998, 41, 579–590. 10.1021/jm970599m. [DOI] [PubMed] [Google Scholar]
- Jarvis M. F.; Honore P.; Shieh C.-C.; Chapman M.; Joshi S.; Zhang X.-F.; Kort M.; Carroll W.; Marron B.; Atkinson R.; Thomas J.; Liu D.; Krambis M. Y.; McGaraughty S.; Chu K.; Roeloffs R.; Zhong C.; Mikusa J. P.; Hernandez G.; Gauvin D.; Wade C.; Zhu C.; Pai M.; Scanio M.; Shi L.; Drizin I.; Gregg R.; Matulenko M.; Hakeem A.; Gross M.; Johnson M.; Marsh K.; Wagoner P. K.; Sullivan J. P.; Faltynek C. R.; Krafte D. S. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 8520–8525. 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M. R.; Kirschmann G.; Laedermann C. J.; Abriel H.; Decosterd I. Rufinamide Attenuates Mechanical Allodynia in a Model of Neuropathic Pain in the Mouse and Stabilizes Voltage-gated Sodium Channel Inactivated State. Anesthesiology 2013, 118, 160–172. 10.1097/ALN.0b013e318278cade. [DOI] [PubMed] [Google Scholar]
- Caccia C.; La Porta E.; Maestroni S.; Melloni P.; Sabido D. C.; Salvati P.. Prolinamide derivatives as sodium and/or calcium channel blockers or selective MAO-B inhibitors. EP1524265A1.
- Hinkley C.; Hajos M.; Kuryshev Y.; Sers A.; Barre A.; Buisson B.; Naik H.. Characterisation of Vixotrigine, a Broad Spectrum Voltage-Gated Sodium Channel Blocker. Mol. Pharmacol. Submitted for publication 2020. [DOI] [PubMed] [Google Scholar]
- Gopinathan N.; Irdam E.; Kiesman W.; Kwok A.; Lin Y.; Osei-Yeboah F.; Peterson M.; Tran K.; Vasudevan K.. Crystalline forms of 5-(4-{[(2-fluorophenyl)methyl]oxy}phenyl)prolinamide hydrochloride. PCT Int. Appl. WO 2018213686, 22 November 2018.
- Baxter E. W.; Reitz A. B.. Reductive aminations of carbonyl compounds with borohydride and borane reducing agents. Organic Reactions; Hoboken: New York, 2002, 59, 1–714. 10.1002/0471264180.or059.01 [DOI] [Google Scholar]
- Cesura A.; Rodriguez Sarmiento R. M.; Scalone M.; Thomas A. W.; Wyler R.. Preparation of isoquinolines as monoamine oxidase B inhibitors useful against Alzheimer’s disease and senile dementia. PCT Int. Appl. WO 2003091219, 6 November 2003.
- Manini P.; D’Ischia M.; Prota G. A Novel Octahydropyridobenzothiazepine Metabolite in Human Urine: Biomimetic Formation from the Melanogen 5-S-Cysteinyldopa and Formaldehyde via a Peculiar Sulfur-Controlled Double Pictet-Spengler. J. Org. Chem. 2000, 65, 4269–4273. 10.1021/jo991969c. [DOI] [PubMed] [Google Scholar]
- Snoeck H. J. M.Fused imidazole derivatives for improving oral bioavailability of pharmaceutical agents. PCT Int. Appl. WO 9913871, 25 March 1999.
- Alvaro G.; Andreotti D.; Bergauer M.; Giovannini R.; Marasco A.. Prolinamide derivatives as sodium channel modulators. PCT Int. Appl. WO 2007042250, April 19, 2007.
- Giblin G. M. P.; Heseltine A.; Kiesman W.; MacPherson D. T.; Ramsden J.; Williams M. T.; Witty D. R.. Org. Process Res. Dev. 2020, manuscript in preparation. [Google Scholar]
- Naik H.; Steiner D.; Versavel M.; Palmer J.; Fong R. Safety, Tolerability, and Pharmacokinetics of Single Doses of the Nav1.7 Selective Sodium channel Blocker Vixotrigine in Healthy Volunteers. American Society for Clinical Pharmacology and Therapeutics 119th Annual Meeting 2018.
- Naik H.; Steiner D.; Versavel M.; Palmer J.; Fong R. Safety, Tolerability, and Pharmacokinetics of Repeat Doses of the Nav1.7 Selective Sodium channel Blocker Vixotrigine in Healthy Volunteers.. American Society for Clinical Pharmacology and Therapeutics 119th Annual Meeting 2018.
- Also reported as CNV1014802, BIIB074.
- Zakrzewska J. M.; Palmer J.; Morisset V.; Giblin G. M.; Obermann M.; Ettlin D. A.; Cruccu G.; Bendtsen L.; Estacion M.; Derjean D.; Waxman S. G.; Layton G.; Gunn K.; Tate S. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017, 16, 291–300. 10.1016/S1474-4422(17)30005-4. [DOI] [PubMed] [Google Scholar]