

Abstract
Background
Elacestrant is an oral selective estrogen receptor (ER) degrader. This phase 1b open-label, non-randomized study (RAD1901-106) was initiated to determine the effect of elacestrant on the availability of ER in lesions from postmenopausal women with ER+ advanced breast cancer (ABC) using 16α-18F-fluoro-17β-estradiol positron emission tomography with low-dose computed tomography (FES-PET/CT).
Methods
Eligible patients were postmenopausal women with ER+, HER2− ABC; tumor progression after ≥ 6 months of 1–3 lines of endocrine treatment for ABC; and measurable or evaluable disease. Two 8-patient cohorts were enrolled: one treated with 400 mg elacestrant once daily (QD) and one treated with 200 mg elacestrant QD with dose escalation to 400 mg QD after 14 days. Elacestrant was dosed continuously until progressive disease, toxicity, or withdrawal. FES-PET/CT was performed pre-dose at baseline and 4 h post-dose on day 14. The primary endpoint was the percentage difference in FES uptake in tumor lesions (maximum 20) after 14 days of treatment compared to baseline. Overall response was investigator-assessed by Response Evaluation Criteria in Solid Tumors [RECIST] version 1.1.
Results
Patients (n = 16; median age, 53.5 years) had ABC with a median 2.5 prior lines of endocrine therapy. Median reduction in tumor FES uptake from baseline to day 14 was 89.1% (Q1, Q3: 75.1%, 94.1%) and was similar in both cohorts (89.1% [Q1, Q3: 67.4%, 94.2%], 200/400 mg and 88.7% [Q1, Q3: 79.5%, 94.1%], 400 mg). Residual ER availability (> 25% persistence in FES uptake) on day 14 was observed in 3 patients receiving 200/400 mg (3/78, 37.5%) and 1 patient receiving 400 mg (1/8, 12.5%). The overall response rate (ORR) was 11.1% (1 partial response), and clinical benefit rate (CBR) was 30.8%. Median percentage change in FES uptake did not correlate with ORR or CBR. Adverse events occurring in > 20% of the patients were nausea (68.8%), fatigue (50.0%), dyspepsia (43.8%), vomiting (37.5%), and decreased appetite, dysphagia, and hot flush (31.3% each). Most events were grade 2 in severity.
Conclusion
Elacestrant 200 mg and 400 mg QD greatly reduced ER availability measured by FES-PET/CT. In a heavily pretreated population, elacestrant was associated with antitumor activity.
Trial registration
ClinicalTrials.gov, NCT02650817. Registered on 08 January 2016
Keywords: Advanced breast cancer, Elacestrant, Endocrine therapy, Estrogen receptor, 16α-18F-fluoro-17β-estradiol (FES), Hormonal therapy, Metastatic breast cancer, Positron emission tomography (PET), RAD1901
Background
Hormone receptor-positive breast cancer accounts for approximately 70 to 80% of breast cancers [1, 2] for which endocrine treatment in both the adjuvant and advanced settings is recommended [3–5]. Available endocrine therapies decrease estrogen production (aromatase inhibitors), antagonize the estrogen receptor (ER) (selective estrogen receptor modulators [SERMs], e.g., tamoxifen), or degrade the ER (selective estrogen receptor degraders [SERDs], e.g., fulvestrant). Fulvestrant is the only approved SERD for the treatment of postmenopausal women with hormone receptor-positive advanced breast cancer. Fulvestrant has demonstrated valuable clinical benefit and prolonged progression-free survival (PFS) compared to anastrozole [6]. However, fulvestrant is limited by its pharmacokinetic (PK) properties and intramuscular route of administration, underscoring the need for novel ER antagonists that are efficacious and provide a more favorable PK profile [7, 8].
16α-18F-fluoro-17β-estradiol positron emission tomography (FES-PET) has been used in a variety of preclinical and clinical studies to detect ER expression in breast cancer. FES-PET is a non-invasive imaging modality that can be used to assess ER status of a tumor, potentially replacing tumor biopsy [9]. It can visualize ER occupation with SERMs and ER downregulation with SERDs due to reduced uptake of the FES tracer and has the potential to predict response based on the degree of downregulation [10–13]. A reduction of ≥ 75% was reported to be associated with a longer PFS in patients receiving fulvestrant compared to patients with a lower reduction in FES uptake (11.7 months vs 3.3 months, respectively; P < 0.05) [14].
Elacestrant is an investigational, nonsteroidal, oral SERD. In ER+ breast cancer cell lines, elacestrant showed dose-dependent ER degradation and inhibited estradiol-dependent induction of ER target gene transcription and cell proliferation [7, 8, 15]. In patient-derived xenograft models of heavily pretreated patients and in the ER+ MCF-7 breast cancer cell line xenograft model, elacestrant inhibited estradiol-activated tumor growth [7, 8, 15]. Elacestrant has also demonstrated antitumor activity in breast cancer models harboring mutations in estrogen receptor alpha gene (ESR1) known to confer resistance (e.g., Y537S, D538G) and those resistant to cyclin-dependent kinase 4,6 (CDK4/6) inhibitors [7, 16, 17].
In the clinical setting, elacestrant 400 mg daily has demonstrated an objective response rate (ORR) of 19.4% and median PFS of 4.5 months in a phase 1 trial (RAD1901-005) of heavily pretreated postmenopausal women with ER+/human epidermal growth factor receptor (HER)2− advanced/metastatic breast cancer (ABC/mBC) [18].
While the RAD1901-005 phase 1 trial was ongoing, the current phase 1b study (RAD1901-106) was initiated to determine the effect of elacestrant treatment on the availability of ER in lesions from patients with ABC using FES-PET with low-dose computed tomography (FES-PET/CT) imaging as a measure of ER downregulation. Additionally, the trial was designed to assess the safety, preliminary efficacy, correlation of ER availability with response, and PK of elacestrant in postmenopausal women with ER+ ABC.
Methods
RAD1901-106 (NCT02650817) was a phase 1b, open-label, non-randomized, multicenter, international study conducted at 5 centers (3 in the Netherlands and 2 in Belgium) between February 2016 and August 2018. The study protocol and relevant supporting information were approved by the institutional review board at each participating site or by a national central review board. The trial was performed in accordance with ethical principles consistent with the Declaration of Helsinki and International Council of Harmonisation/Good Clinical Practice and applicable regulatory requirements. Each trial participant provided written informed consent.
The primary endpoint was the percentage difference in FES uptake in tumor lesions (up to a maximum of 20 lesions) after 14 days of treatment with elacestrant compared to baseline. Secondary endpoints were correlation of changes in FES uptake after elacestrant treatment to clinical responses measured by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 [19], preliminary antitumor effects of elacestrant, safety and tolerability of elacestrant, and elacestrant PK. An exploratory endpoint was correlation of tumor response with ESR1 mutations detected in circulating tumor DNA (ctDNA).
Patients
Eligible patients were women ≥ 18 years of age who were postmenopausal (defined as > 56 years old with amenorrhea for > 12 months, < 56 years old with amenorrhea for > 12 months plus serum estradiol < 20 pg/mL and follicle-stimulating hormone > 40 mIU/mL, or prior bilateral ovariectomy). Patients had histologically proven ER+ (defined as ≥ 1% staining by immunohistochemistry [20]), HER2− ABC (either inoperable primary breast cancer or mBC); tumor progression after ≥ 6 months of 1–3 lines of systemic endocrine treatment for mBC; measurable disease according to RECIST v1.1 or evaluable disease; Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0–2; life expectancy > 3 months; and adequate organ function. Prior CDK4/6 inhibitor therapy was allowed.
Exclusion criteria included liver-only metastases (due to high physiological liver uptake of FES, liver metastases are not evaluable by FES-PET/CT imaging); untreated or symptomatic central nervous system metastases; and prior treatment with tamoxifen or fulvestrant therapy < 42 days before first FES-PET/CT scan (since these agents may influence ER occupancy), other anticancer endocrine therapy < 14 days before first elacestrant dose, or chemotherapy < 28 days before first elacestrant dose. Full eligibility criteria are listed in an Additional file.
Study procedures
The study consisted of a screening period of up to 21 days prior to the first dose of elacestrant (with the exception of tumor imaging tests, which were performed within 28 days of the first elacestrant dose); an open-label treatment period; and a follow-up period during which patients were followed for adverse events (AEs) and concomitant medication use for 30 days after the final dose of elacestrant or until resolution/stabilization of all AEs to grade ≤ 2.
A total of 8 patients were initially enrolled and treated with 400 mg elacestrant capsule once daily (QD)—the recommended phase 2 dose determined in study RAD1901-005. Following a protocol amendment, a second cohort of 8 patients was enrolled and treated with 200 mg elacestrant capsule QD for 14 days to assess target engagement of a lower dose that might be utilized when the 400-mg dose could not be tolerated alone or if elacestrant was given in combination with another agent. After 14 days, the dose was escalated to 400 mg QD. Elacestrant was dosed continuously with 28-day treatment cycles until progressive disease [PD], unacceptable toxicity, or patient/investigator decision to withdraw. Patients were instructed to take elacestrant at approximately the same time each day, approximately 30 min after a light meal; to fast for ≥ 1 h after dosing; and to remain upright for ≥ 2 h after taking elacestrant. After the first 14 days of treatment, dosing delays of ≤ 7 days were permitted. During the study, no hormonal medications, anticancer therapy, palliative radiotherapy, or strong CYP3A inducers or inhibitors were permitted.
Assessments
At baseline, day 14 and day 28 of each cycle, and at end of treatment, patients underwent physical examination, ECOG PS assessment, and hematology and chemistry laboratory evaluations. Patients also underwent 12-lead electrocardiogram and coagulation evaluations at these time points, except day 14. Transvaginal ultrasound was performed at screening and end of treatment.
FES-PET/CT imaging
Imaging with FES-PET/CT was performed pre-dose at baseline and approximately 4 h post-dose (Tmax) on day 14 of cycle 1. Patients were given a single bolus injection of ~ 200 MBq FES prior to whole-body PET/CT imaging. Scan routines were conducted according to the European protocol for standardization of 18F whole-body PET studies, and quantification was performed using European Association of Nuclear Medicine (EANM) Research Ltd. (EARL) reconstruction according to the European standard for multicenter trials, as described in an Additional file [21, 22]. The percentage difference in background-corrected FES uptake in tumor lesions (up to an arbitrary maximum of 20 lesions) after 14 days of treatment with elacestrant compared to baseline was calculated. A relative decrease of < 75% in the median (background-corrected) tumor FES uptake and an absolute tumor lesion standardized uptake value (SUV)max ≥ 1.5 was defined as incomplete reduction in ER availability.
ESR1 mutation analysis
Blood samples were collected for circulating tumor DNA (ctDNA) analysis at screening, day 28 of cycles 1–3, and at the end of treatment. The OncoBEAM™ assay (Sysmex Inostics, Baltimore, MD) was used to determine ESR1 mutational status in ctDNA. This is a digital droplet PCR (ddPCR) assay that detects 12 mutations in the ligand binding domain of ESR1 (E380Q, S463P, V524E, P535H, L536H/P/Q/R, Y537C/N/S, D538G) [23].
Pharmacokinetic analysis
Blood samples were collected for PK analysis at baseline (pre-dose and 4 h post-dose), pre-dose on day 14 of cycle 1, and day 28 of cycles 1–3. Elacestrant plasma concentrations were determined using a validated ultra-performance liquid chromatography with tandem mass spectrometric detection method with a linear range of 0.05 to 100 ng/mL and a lower limit of quantification of 0.05 ng/mL (PRA Health Sciences, Assen, The Netherlands).
Tumor assessments
Tumor assessments were performed at screening, every 2 cycles, and end of treatment. Response was evaluated by the investigator using RECIST v1.1. Clinical benefit rates (CBR) at 16 weeks and 24 weeks were defined as the proportion of patients who had confirmed CR or PR any time during the study, or stable disease (SD) that lasted at least 16 and 24 weeks, respectively.
Safety assessments
Adverse events were collected throughout the study up to 30 days following the final elacestrant dose. The Medical Dictionary for Regulatory Activities version 17.1 was used for coding AEs, and the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 was used to grade AE severity.
Statistical methods
The study planned to enroll a total of 16 patients (8 per cohort). Percentage change in median FES uptake between baseline and day 14 was summarized as median and interquartile range due to slightly skewed data for each dose cohort and the overall population.
The intention-to-treat (ITT) population included all patients who received at least 1 full or partial dose of elacestrant. Best overall response was calculated for the response evaluable population, which included all patients who had measurable disease at baseline and at least 1 post-baseline RECIST assessment on any lesions (target or nontarget) and/or had a new lesion. Best overall response was summarized by dose cohort and reported as percentage with the 2-sided 95% exact confidence interval (CI) using the Clopper-Pearson method. The CBR was calculated for the clinical benefit evaluable population, which included all patients who had measurable and/or evaluable disease at baseline and at least 1 RECIST assessment post-baseline on any lesion (target or nontarget) and/or had a new lesion. Clinical benefit rate was summarized by dose cohort along with the 95% CI. Correlation between the median percentage change in FES uptake and best overall response was evaluated using the Spearman’s rank correlation coefficient. Kaplan-Meier methods were used to analyze PFS. Elacestrant plasma concentrations were summarized over time for each dose cohort. Adverse events and changes in vital signs, laboratory tests, and ECGs were summarized descriptively.
Results
A total of 16 patients were enrolled and all patients received at least 1 dose of elacestrant and had paired baseline and day 14 FES-PET imaging. All patients have discontinued treatment, 12 (75.0%) due to radiographic or clinical disease progression, 3 (18.8%) due to an adverse event, and 1 (6.3%) for a protocol violation (treatment interruption > 7 days) (Additional file Fig. S1). Baseline characteristics were similar between the 2 dose cohorts (Table 1). The median age was 53.5 (range 43–84) years. All patients had ABC with a median number of prior lines of endocrine therapy of 2.5; no patients had prior CDK4/6 inhibitor therapy. Nine patients (56.3%) had ESR1 mutation detected by ctDNA at baseline (D538G, n = 6; Y537S, n = 5; Y537C, n = 2; Y537N, n = 2; E380Q, n = 1; L536P, n = 1).
Table 1.
Demographics and baseline characteristics
| Characteristic | Elacestrant dose cohort | ||
|---|---|---|---|
| 200/400 mg (N = 8) | 400 mg (N = 8) | Overall (N = 16) | |
| Median age (range), years | 57.0 (49, 74) | 53.0 (43, 84) | 53.5 (43, 84) |
| Female, n (%) | 8 (100) | 8 (100) | 16 (100) |
| ECOG performance status, n (%) | |||
| 0 | 3 (37.5) | 3 (37.5) | 6 (37.5) |
| 1 | 5 (62.5) | 4 (50.0) | 9 (56.3) |
| 2 | 0 | 1 (12.5) | 1 (6.3) |
| Ductal carcinoma, n (%) | 8 (100) | 8 (100) | 16 (100) |
| Median time since breast cancer diagnosis (Q1, Q3), yearsa | 12.5 (5.8, 16.7) | 6.0 (4.2, 11.0) | 8.5 (5.2, 13.7) |
| Stage IV, n (%) | 8 (100) | 8 (100) | 16 (100) |
| Visceral diseaseb, n (%) | 5 (62.5) | 5 (62.5) | 10 (62.5) |
| Bone-only disease, n (%) | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| ESR1 mutationc, n (%) | 4 (50.0) | 5 (62.5) | 9 (56.3) |
| Median number of lines of prior anticancer therapy (Q1, Q3), n | |||
| Total, for ABC | 3.0 (1.5, 3.0) | 3.0 (2.0, 3.0) | 3.0 (2.0, 3.0) |
| Endocrine therapy, for ABC | 3.0 (1.5, 3.0) | 2.0 (2.0, 3.0) | 2.5 (2.0, 3.0) |
| Chemotherapy, for ABC | 0 (0, 1.0) | 0.5 (0, 1.0) | 0 (0, 1.0) |
| Adjuvant endocrine therapy, n (%) | 7 (87.5) | 6 (75.0) | 13 (81.3) |
| Prior fulvestrant, n (%) | 4 (50.0) | 2 (25.0) | 6 (37.5) |
| Prior mTOR inhibitor, n (%) | 3 (37.5) | 3 (37.5) | 6 (37.5) |
| Prior CDK4/6 inhibitor, n (%) | 0 | 0 | 0 |
CDK4/6, cyclin-dependent kinase 4,6; ECOG, Eastern Cooperative Oncology Group; mTOR, mammalian target of rapamycin
aMedian time since breast cancer diagnosis regardless of stage
bIncludes the liver, lung, and pleura
cMutations detected include D538G (n = 6), Y537S (n = 5), Y537C (n = 2), Y537N (n = 2), E380Q (n = 1), and L536P (n = 1)
FES tumor uptake
Percentage change from baseline in tumor FES uptake was assessed in all 16 patients. The median reduction in tumor FES uptake from baseline to day 14 was 89.1% (Q1, Q3: 75.1%, 94.1%; Table 2). Median reduction in FES uptake was similar in both dose cohorts (89.1% [Q1, Q3: 67.6%, 94.2%] in the 200/400 mg cohort and 88.7% [Q1, Q3: 79.5%, 94.1%] in the 400 mg cohort). The reduction in FES uptake was independent of baseline ctDNA ESR1 mutation status (median reduction in FES uptake 90.2% [Q1, Q3: 80.2%, 96.4%] in patients with ESR1 mutation and 86.3% [Q1, Q3: 71.3%, 91.2%] in patients without ESR1 mutation). A representative pre- and post-elacestrant FES-PET scan in a patient with extensive metastases is shown in Fig. 1. Residual ER availability (defined as > 25% persistence in FES uptake) was observed in 1 patient in the 400-mg elacestrant cohort (1/8, 12.5%) and 3 patients in the 200/400-mg elacestrant cohort (3/8, 37.5%; Fig. 2). Among 9 patients with baseline ESR1 mutation, 8 had at least 1 post-baseline ESR1 result. Among these 8 patients, 5 had reduction in mutation allele frequency and 3 did not. There was no difference between percent change in FES uptake between the 2 groups (P > 0.05 calculated using the Wilcoxon rank sum test).
Table 2.
Percentage change in FES uptake from baseline to day 14 for the two different dose cohorts
| Elacestrant dose cohort | |||
|---|---|---|---|
| Parameter | 200/400 mg (N = 8) | 400 mg (N = 8) | Overall (N = 16) |
| Number evaluated, n (%) | 8 (100)a | 8 (100) | 16 (100) |
| Mean (SD), % | − 82.6 (15.5) | − 86.0 (10.2) | − 84.3 (12.8) |
| Median (Q1, Q3), % | − 89.1 (− 94.2, − 67.6) | − 88.7 (− 94.1, − 79.5) | − 89.1 (− 94.1, − 75.1) |
ITT intention to treat, FES 16α-18F-fluoro-17β-estradiol
aOne patient who had < − 100% change (i.e., > 100% reduction) was included as − 100%
Fig. 1.
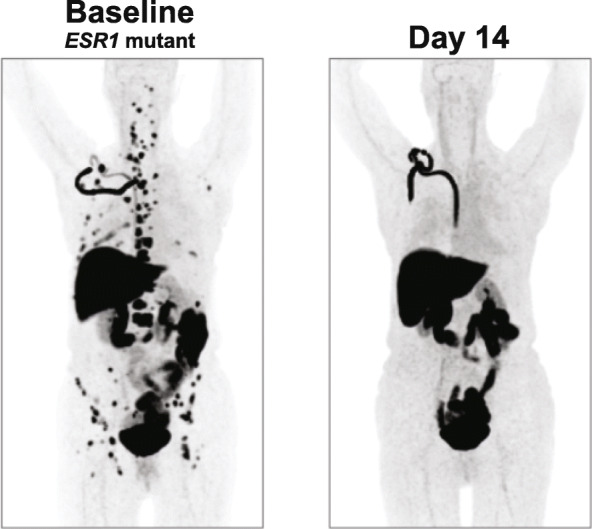
Reduction in FES uptake after elacestrant treatment in a patient with ESR1 mutation. Images depict FES-PET scan in the patient at baseline (left) and 14 days after receiving elacestrant 400 mg daily (right), at which time a 96.6% reduction in FES uptake was observed. Physiologic FES uptake/excretion is observed in the liver, intestines, bladder, and port-a-cath infusion line; pathologic uptake is observed in bone lesions and lymph nodes
Fig. 2.

Percentage change from baseline to day 14 in median FES uptake for individual patients in the intention-to-treat population. One patient who had < − 100% change (i.e., > 100% reduction) was included as −100%. Response per RECIST (PR, SD, PD, NE) for each patient and patients with CBR at 24 weeks (†) are noted. FES, 16α-18F-fluoro-17β-estradiol
Antitumor efficacy
The ORR was 11.1% (Table 3); of 9 patients evaluable for overall response, 1 partial response was observed (in a patient without an ESR1 mutation); the time to response was 7.9 weeks and the duration of response was 22 weeks. The CBR was 30.8%; 4 out of 13 patients evaluable for CBR had stable disease at 24 weeks. Of these 4 patients, 1 had an ESR1 mutation. No significant correlation was found between the median percentage change in FES uptake and best overall response (Spearman’s rank correlation coefficient − 0.13; P > 0.7). The patient who experienced a partial response had a 78.9% reduction in FES uptake. In the ITT population, median PFS was 5.3 months (95% CI, 1.7, 17.9).
Table 3.
Efficacy endpoints in evaluable populations
| Parameter | Elacestrant dose cohort | ||
|---|---|---|---|
| 200/400 mg | 400 mg | Overall | |
| Response | N = 4 | N = 5 | N = 9 |
| ORR, % | 0 | 20.0 | 11.1 |
| Partial response, n (%) | 0 | 1 (20.0) | 1 (11.1) |
| Duration of response, weeks | – | 22 | 22 |
| Time to response, weeks | – | 7.9 | 7.9 |
| Stable disease, n (%) | 2 (50.0) | 3 (60.0) | 5 (55.6) |
| Progressive disease, n (%) | 2 (50.0) | 1 (20.0) | 3 (33.3) |
| CBR | N = 6 | N = 7 | N = 13 |
| 16 weeks, n (%) | 3 (50.0) | 4 (57.1) | 7 (53.8) |
| 24 weeks, n (%) | 1 (16.7) | 3 (42.9) | 4 (30.8) |
| Median PFS (95% CI), months | 3.6 (0.7, 17.9) | 6.9 (0.7, NA) | 5.3 (1.7 17.9) |
CBR clinical benefit rate, ORR objective response rate, PFS progression-free survival
Pharmacokinetics of elacestrant
Pharmacokinetics were evaluated in all 16 patients. Over the first 2 weeks of treatment, the geometric mean elacestrant plasma concentrations were approximately twice as high for patients receiving 400 mg QD as those for patients receiving 200 mg QD (59.8 ng/mL vs 27.4 ng/mL at cycle 1, day 1, hour 4 post-dose, respectively, and 70.1 ng/mL versus 20.1 ng/mL at cycle 1, day 14, pre-dose, respectively; Fig. 3). Elacestrant plasma concentrations at additional timepoints are summarized in Additional file Table S1.
Fig. 3.

Geometric mean elacestrant plasma concentrations over time (N = 16). Vertical bars indicate standard error of the mean
Safety
The median duration of elacestrant treatment was 4.3 months (range, 0.4–23.2). Patients in the 400-mg cohort remained on treatment longer than the 200/400-mg cohort (median 5.2 months [range, 0.7–23.2] vs 1.8 [range, 0.4–18.1] months, respectively). One patient receiving the initial dose of 200 mg did not dose escalate to 400 mg per protocol due to a serious AE of grade 3 esophagitis that was assessed as possibly related to elacestrant by the investigator and that resulted in treatment discontinuation. Two additional patients discontinued treatment due to AEs: grade 2 nausea, fatigue, and anorexia in 1 patient, and grade 2 cough and grade 1 upper extremity peripheral edema in 1 patient. Among all patients, 4 (25.0%) had dose delay and 2 (12.5%) had dose reduction due to a treatment-emergent AE (TEAE). All patients experienced at least 1 TEAE (Table 4). The most common TEAEs occurring in > 20% of patients were nausea (n = 11, 68.8%), fatigue (n = 8, 50.0%), dyspepsia (n = 7, 43.8%), vomiting (n = 6, 37.5%), decreased appetite, dysphagia, and hot flush (n = 5, 31.3% each; Table 4). The majority of patients (n = 10, 62.5%) had grade 2 TEAEs. Five grade 3 TEAEs occurred in 4 patients; no grade 4 or 5 events occurred (Table 5). Adverse events assessed as related to elacestrant by the investigator occurred in 15 patients (93.8%), with gastrointestinal disorders being the most commonly reported. No consistent trends were observed in clinically significant changes in laboratory parameters, vital signs, or electrocardiogram parameters.
Table 4.
Adverse events occuring in ≥ 10% of the ITT population
| Adverse event (AE), n (%) | Elacestrant dose cohort | ||
|---|---|---|---|
| 200/400 mg (N = 8) | 400 mg (N = 8) | Overall (N = 16) | |
| At least 1 AE | 8 (100) | 8 (100) | 16 (100) |
| Nausea | 5 (62.5) | 6 (75.0) | 11 (68.8) |
| Fatigue | 4 (50.0) | 4 (50.0) | 8 (50.0) |
| Dyspepsia | 2 (25.0) | 5 (62.5) | 7 (43.8) |
| Vomiting | 5 (62.5) | 1 (12.5) | 6 (37.5) |
| Decreased appetite | 4 (50.0) | 1 (12.5) | 5 (31.3) |
| Dysphagia | 1 (12.5) | 4 (50.0) | 5 (31.3) |
| Hot flush | 2 (25.0) | 3 (37.5) | 5 (31.3) |
| Hypertension | 0 | 3 (37.5) | 3 (18.8) |
| Arthralgia | 1 (12.5) | 2 (25.0) | 3 (18.8) |
| Dizziness | 2 (25.0) | 1 (12.5) | 3 (18.8) |
| Dyspnea | 0 | 3 (37.5) | 3 (18.8) |
| Abdominal pain upper | 2 (25.0) | 1 (12.5) | 3 (18.8) |
| Back pain | 0 | 2 (25.0) | 2 (12.5) |
| Diarrhea | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Esophageal pain | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Neck pain | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Pain in extremity | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Cough | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Nail discoloration | 1 (12.5) | 1 (12.5) | 2 (12.5) |
| Anemia | 0 | 2 (25.0) | 2 (12.5) |
ITT intention to treat
Table 5.
Grade 3 adverse events, serious adverse events, and adverse events leading to discontinuation
| Adverse event (AE), n (%) | Elacestrant dose cohort | ||
|---|---|---|---|
| 200/400 mg (N = 8) | 400 mg (N = 8) | Overall (N = 16) | |
| Grade 3 AEsa | 1 (12.5) | 3 (37.5) | 4 (25.0) |
| Anemia | 0 | 1 (12.5) | 1 (6.3) |
| Circulatory collapse | 0 | 1 (12.5) | 1 (6.3) |
| Esophagitis | 1 (12.5) | 0 | 1 (6.3) |
| Cystitis | 0 | 1 (12.5) | 1 (6.3) |
| Pulmonary embolism | 0 | 1 (12.5) | 1 (6.3) |
| Serious AEs | 1 (12.5) | 2 (25.0) | 3 (18.8) |
| Esophagitis | 1 (12.5) | 0 | 1 (6.3) |
| Amnesia | 0 | 1 (12.5)b | 1 (6.3) |
| Circulatory collapse | 0 | 1 (12.5)b | 1 (6.3) |
| Presyncope | 0 | 1 (12.5)b | 1 (6.3) |
| Pulmonary embolism | 0 | 1 (12.5)c | 1 (6.3) |
| Dyspnea | 0 | 1 (12.5)c | 1 (6.3) |
| AEs leading to elacestrant discontinuation | 3 (37.5) | 0 | 3 (18.8) |
| Esophagitis | 1 (12.5) | 0 | 1 (6.3) |
| Nausea | 1 (12.5)d,e | 0 | 1 (6.3) |
| Fatigue | 1 (12.5)d,e | 0 | 1 (6.3) |
| Anorexia | 1 (12.5)d,e | 0 | 1 (6.3) |
| Cough | 1 (12.5)d,f | 0 | 1 (6.3) |
| Upper extremity peripheral edema | 1 (12.5)d,f | 0 | 1 (6.3) |
aThere were no grade 4 AEs
bThese events occurred in the same patient
cThese events occurred in the same patient
dThese events occurred during treatment with elacestrant 400 mg
eGrade 2 nausea, fatigue, and anorexia occurred in the same patient
fGrade 2 cough and grade 1 upper extremity peripheral edema occurred in the same patient
Discussion
In the current study, the investigational oral SERD elacestrant decreased ER availability for binding 17β-estradiol to tumor metastases (assessed by reduction in FES uptake quantified using FES-PET/CT imaging) by a median of 89%, with 12 of 16 patients demonstrating a reduction of ≥ 75%. A reduction of ≥ 75% is suggested to be clinically relevant since this cut-off was found to be associated with a longer PFS in patients receiving fulvestrant compared to patients with a lower reduction in FES uptake [14].
In our phase 1 study, the proportion of patients with a ≥ 75% reduction in ER availability was 62.5% in the cohort receiving a dose of 200 mg elacestrant QD and 88% in the cohort receiving the recommended phase 2 dose (RP2D) of 400 mg QD. These proportions appear to be similar or higher than the 63% reported for treatment with fulvestrant [14], suggesting an equivalent or better reduction in ER availability by elacestrant at both the 200 mg and 400 mg doses. While the proportion of patients with a ≥ 75% reduction in ER availability with the elacestrant 200 mg dose at day 14 was lower than the 400 mg RP2D dose, this lower reduction in FES uptake should not lead to the conclusion that elacestrant 200 mg is not efficacious. It should also be noted that the reduction in FES uptake for 200 mg elacestrant was similar to what was reported for fulvestrant 500 mg at day 28 [14]; therefore, these data suggest that elacestrant 200 mg may be a potential option for patients who require a dose reduction from the 400 mg RP2D due to toxicity.
Patients enrolled in the current study were heavily pretreated (median 3 prior lines of therapy in all settings, median 2.5 lines of endocrine therapy in the advanced setting, 38% prior fulvestrant); however, none had prior CDK4/6 inhibitor exposure. Moreover, 56% of the patients had a detectable ESR1 mutation by ctDNA at baseline, as is expected with extensive prior endocrine therapy [24, 25]. Despite these unfavorable prognostic characteristics, elacestrant demonstrated antitumor activity with an ORR of 11% and a median duration of response of 22 weeks. A 24-week CBR was observed in 31% of patients and the median PFS was 5.3 months. There was no correlation between reduction in ER availability to treatment response; however, the sample size evaluable for response in our study was too small to draw any conclusions (n = 9 evaluable for tumor overall response with 1 partial response). Other investigators have observed a correlation between degree of ER blockade on FES-PET and clinical response to tamoxifen and clinical benefit with fulvestrant [26, 27]. Dehdashti et al. reported a mean SUV decrease of 2.7 in responders versus 0.8 in nonresponders (P = 0.04) in a study of 11 women with newly diagnosed mBC [26]. Mortimer et al. reported a mean SUV decrease of 2.5 in responders versus 0.5 in nonresponders (P = 0.0003), which corresponded to a mean percentage decrease in SUV of 54.8% in responders versus 19.4% in nonresponders (P = 0.0003), in 40 women with locally advanced, chest-wall recurrent, or mBC [27]. It should be noted that neither study utilized RECIST criteria to define response or PET image reconstruction according to EANM/EARL guidelines. EARL/EANM accreditation is recommended for proper FES-PET implementation to avoid erroneous results [21, 22].
FES-PET scanning has not been routinely available at clinical sites and has generally been reserved for investigational purposes at select academic centers. As more data become available from ongoing trials validating the safety and predictive accuracy of FES-PET imaging, it could replace or supplement tumor biopsy. FES-PET allows the level and heterogeneity of ER expression across the full burden of disease to be evaluated to help guide treatment selection and characterize distinct subsets of patients [9]. The noninvasiveness of FES-PET could also facilitate serial imaging to monitor effect of ER-targeted therapies on treatment; ineffective agents could be switched earlier if reduction in ER availability is not observed. The first FES-PET imaging agent specifically indicated for use in patients with recurrent or metastatic breast cancer was approved by the FDA in May 2020 [28]. Availability of this agent in the US may lead to more widespread use for diagnostic purposes.
Strengths of this study are that ER target engagement was assessed with robust FES-PET/CT imaging, which allows whole-body visualization and quantification of ER availability on ER-expressing metastases and has shown predictive value for response on fulvestrant [12, 14]. The imaging was performed at an EARL/EANM-accredited academic center that routinely uses this technology. This study also assessed antitumor activity using the stringent RECIST criteria to measure response. The sample size was relatively modest for a phase 1 FES-PET/CT imaging study (only 8 patients were included per cohort), which limited the study’s capability to evaluate the correlation between reduction in ER availability and treatment response.
Conclusions
This study demonstrated that elacestrant greatly reduces ER availability, as measured by FES-PET/CT imaging, at doses of 200 mg and 400 mg QD, to a similar, if not better, extent than that reported for fulvestrant. In a heavily pretreated population, including 56% of patients with ESR1 mutation, elacestrant was associated with antitumor activity. The safety profile of elacestrant was acceptable, consisting predominately of low-grade upper gastrointestinal toxicity. The risk-benefit profile supports further development of elacestrant in ER+/HER2− ABC. This study supports the use of elacestrant 400 mg as the recommended dose for future studies. A randomized phase 3 study comparing elacestrant 400 mg to standard-of-care endocrine monotherapy (EMERALD: NCT03778931) in postmenopausal women and men with ER+/HER2− ABC is currently ongoing.
Supplementary information
Additional file 1: Table S1. Elacestrant plasma concentrations over time. Figure S1. Patient disposition.
Acknowledgements
We thank Geraldine Gebhart, MD, from the Institut Jules Bordet for performing the FES-PET and CT scans and Phillips Gilmore Oncology Communications Inc., for professional assistance with manuscript preparation, which was funded by Radius Health, Inc.
Abbreviations
- ABC
Advanced or metastatic breast cancer
- AE
Adverse event
- ctDNA
Circulating tumor DNA
- CBR
Clinical benefit rate
- CDK4/6
Cyclin-dependent kinase 4/6
- CR
Complete response
- CI
Confidence interval
- CYP
Cytochrome P450
- ECOG
Eastern Cooperative Oncology Group
- ER
Estrogen receptor
- ESR1
Estrogen receptor 1 (estrogen receptor gene alpha)
- FES
16α-18F-fluoro-17β-estradiol
- HR
Hormone receptor
- HER2
Human epidermal growth factor receptor 2
- ITT
Intention to treat
- mBC
Metastatic breast cancer
- PK
Pharmacokinetic
- ORR
Objective response rate
- QD
Once daily
- PR
Partial response
- PET
Positron emission tomography
- PFS
Progression-free survival
- RECIST
Response Evaluation Criteria in Solid Tumors
- SERD
Selective estrogen degraders
- SD
Stable disease
- SUV
Standardized uptake value
- TEAE
Treatment-emergent adverse event
Authors’ contributions
Radius Health conceived and designed the trial in collaboration with clinical and academic investigators (EdV and AWJMG) and developed the statistical plan for the trial. Data acquisition was performed by Radius Health. Analysis and interpretation of data were performed by AWJMG, YW, MGC, and RGB. Administrative, technical, and material support were provided by YW, MGC, and RGB. Study supervision was provided by individual investigators (AJ, EdV, CWMdHvO, PN, CMV, AWJMG, PA). This manuscript was principally written by AJ with assistance from MGC and was critically reviewed, edited, and approved by all authors.
Funding
This work was supported by Radius Health, Inc. on behalf of its wholly owned subsidiary, Radius Pharmaceuticals, Inc.
Availability of data and materials
Data that underlie the results reported in a published article may be requested for further research 6 months after FDA or EMA approval or 18 months after trial completion (whichever is latest). Radius will review requests individually to determine whether (i) the requests are legitimate and relevant and meet sound scientific research principles and (ii) are within the scope of the participants’ informed consent. Prior to making data available, requestors will be required to agree in writing to certain obligations, including without limitation, compliance with applicable privacy and other laws and regulations. Proposals should be directed to info@radiuspharm.com.
Ethics approval and consent to participate
The study protocol and relevant supporting information were approved by the institutional review board at each participating site or by a national central review board. The trial was performed in accordance with ethical principles consistent with the Declaration of Helsinki and International Council of Harmonisation/Good Clinical Practice and applicable regulatory requirements. Each trial participant provided written informed consent.
Consent for publication
Not applicable.
Competing interests
AJ: No competing interest.
EdV: Institutional financial support for advisory role from Daiichi Sankyo, Merck, NSABP, Pfizer, Sanofi, and Synthon and institutional financial support for clinical trials or contracted research from Amgen, AstraZeneca, Bayer, Chugai Pharma, CytomX Therapeutics, G1 Therapeutics, Genentech, Nordic Nanovector, Radius Health, Regeneron, Roche, Servier, and Synthon.
CWMdHvO: Institutional financial support for clinical trials or contracted research from BMS, Boeringher Ingelheim, Crystal Therapeutics, CytomX Therapeutics, G1 Therapeutics, Pfizer, Takeda, and Synthon.
PN: No competing interest.
CMV: No competing interest.
AWJMG: No competing interest.
PA: Honoraria/advisory fees from Novartis, Roche, Macrogenics, G1 Therapeutics, Servier, Synthon, Amcure, Amgen, and Boehringer Ingleheim and travel grants from MSD, Pfizer, and Roche.
YW, RGB, and MGC are employees and stockholders of Radius Health, Inc.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13058-020-01333-3.
References
- 1.Huang HJ, Neven P, Drijkoningen M, Paridaens R, Wildiers H, Van Limbergen E, et al. Association between tumour characteristics and HER-2/neu by immunohistochemistry in 1362 women with primary operable breast cancer. J Clin Pathol. 2005;58:611–16. [DOI] [PMC free article] [PubMed]
- 2.Malmgren JA, Mayer M, Atwood MK, Kaplan HG. Differential presentation and survival of de novo and recurrent metastatic breast cancer over time: 1990–2010. Breast Cancer Res Treat. 2018;167:579–90. [DOI] [PMC free article] [PubMed]
- 3.Cardoso F, Senkus E, Costa A, Papadopoulos E, Aapro M, André F, et al. 4th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 4) Ann Oncol. 2018;29:1634–1657. doi: 10.1093/annonc/mdy192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rugo HS, Rumble RB, Macrae E, Barton DL, Connolly HK, Dickler MN, et al. Endocrine therapy for hormone receptor-positive metastatic breast cancer: American Society of Clinical Oncology guideline. J Clin Oncol. 2016;34:3069–103. [DOI] [PubMed]
- 5.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: breast cancer. Version 2.2019. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 14 Aug 2019.
- 6.Robertson JFR, Bondarenko IM, Trishkina E, et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet. 2016;388:2997–3005. [DOI] [PubMed]
- 7.Bihani T, Patel HK, Arlt H, Tao N, Jiang H, Brown JL, et al. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin Cancer Res. 2017;23:4793–804. [DOI] [PubMed]
- 8.Wardell SE, Nelson ER, Chao CA, Alley HM, McDonnell DP. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer. 2015;22:713–24. [DOI] [PMC free article] [PubMed]
- 9.Chae SY, Ahn SH, Kim SB, et al. Diagnostic accuracy and safety of 16α-[18F]fluoro-17β-oestradiol PET-CT for the assessment of oestrogen receptor status in recurrent or metastatic lesions in patients with breast cancer: a prospective cohort study. Lancet Oncol. 2019;20(4):546–55. [DOI] [PubMed]
- 10.Venema CM, Apollonio G, Hospers GA, et al. Recommendations and Technical Aspects of 16α-[18F]Fluoro-17β-Estradiol PET to Image the Estrogen Receptor In Vivo: The Groningen Experience. Clin Nucl Med. 2016;41(11):844–51. [DOI] [PubMed]
- 11.Wang Y, Ayres KL, Goldman DA, et al. 18F-Fluoroestradiol PET/CT Measurement of Estrogen Receptor Suppression during a Phase I Trial of the Novel Estrogen Receptor-Targeted Therapeutic GDC-0810: Using an Imaging Biomarker to Guide Drug Dosage in Subsequent Trials [published correction appears in Clin Cancer Res. 2019 Feb 15;25(4):1435]. Clin Cancer Res. 2017;23(12):3053–60. [DOI] [PMC free article] [PubMed]
- 12.van Kruchten M, de Vries EG, Brown M, de Vries EFJ, Glaudemans AWJM, Dierckx RAJO, et al. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol. 2013;14:e465–475. [DOI] [PubMed]
- 13.van Kruchten M, Glaudemans AWJM, de Vries EFJ, Schröder CP, de Vries EGE, Hospers GAP. Positron emission tomography of tumour [18F]fluoroestradiol uptake in patients with acquired hormone-resistant metastatic breast cancer prior to oestradiol therapy. Eur J Nucl Med Mol Imaging. 2015;42:1674–81. [DOI] [PMC free article] [PubMed]
- 14.van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov. 2015;5:72–81. [DOI] [PubMed]
- 15.Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26:948–56. [DOI] [PMC free article] [PubMed]
- 16.Patel HK, Tao N, Lee KM, et al. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. 2019;21(1):146. doi: 10.1186/s13058-019-1230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel H, Tao N, Arlt H, Bihani T. Anti-tumor activity of elacestrant (RAD1901) in models harboring ESR1 mutations resistant to standard of care therapies. Cancer Res 2019b;79 (4 Supplement) P6-20-08.
- 18.Kaklamani V, Bardia A, Wilks S, et al. Final analysis of phase 1 study of elacestrant (RAD1901), a novel selective estrogen receptor degrader (SERD), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. Abstract PD7-07. Presented at: San Antonio: San Antonio Breast Cancer Symposium; 2019.
- 19.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed]
- 20.Hammond MEH, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–3543. [DOI] [PMC free article] [PubMed]
- 21.Boellaard R, Delgado-Bolton R, Oyen WJ, Giammarile F, Tatsch K, Eschner W, et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: version 2.0. Eur J Nucl Med Mol Imaging. 2015;42:328–54. [DOI] [PMC free article] [PubMed]
- 22.European Association of Nuclear Medicine (EANM) Research Ltd (EARL). FDG-PET/CT accreditation. http://earl.eanm.org/cms/website.php?id=/en/projects/fdg_pet_ct_accreditation.htm. Accessed 7 Oct 2019.
- 23.Sysmex Inostics, Inc. OncoBEAM mutation testing. Sysmex-inostics.com. https://www.sysmex-inostics.com/oncobeam-mutation-testing. Accessed 16 June 2020.
- 24.Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–15. [DOI] [PMC free article] [PubMed]
- 25.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67. [DOI] [PMC free article] [PubMed]
- 26.Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ, Siegel BA. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur J Nucl Med 1999;26:51–56. [DOI] [PubMed]
- 27.Mortimer JE, Dehdashti F, Siegel BA, Trinkaus K, Katzenellenbogen JA, Welch MJ. Metabolic flare: indicator of hormone responsiveness in advanced breast cancer. J Clin Oncol 2001;19:2797–2803. [DOI] [PubMed]
- 28.Cerianna [package insert] New York: Zionexe USA; 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Elacestrant plasma concentrations over time. Figure S1. Patient disposition.
Data Availability Statement
Data that underlie the results reported in a published article may be requested for further research 6 months after FDA or EMA approval or 18 months after trial completion (whichever is latest). Radius will review requests individually to determine whether (i) the requests are legitimate and relevant and meet sound scientific research principles and (ii) are within the scope of the participants’ informed consent. Prior to making data available, requestors will be required to agree in writing to certain obligations, including without limitation, compliance with applicable privacy and other laws and regulations. Proposals should be directed to info@radiuspharm.com.