

Abstract
The behavioral responses exerted by spinal administration of the opioid-neurotensin hybrid peptide, PK23, were studied in adult male rats. The antinociceptive effect upon exposure to a thermal stimulus, as well as tolerance development, was assessed in an acute pain model. The PK23 chimera at a dose of 10 nmol/rat produced a potent pain-relieving effect, especially after its intrathecal administration. Compared with intrathecal morphine, this novel compound was found to possess a favourable side effect profile characterized by a reduced scratch reflex, delayed development of analgesic tolerance or an absence of motor impairments when given in the same manner, though some animals died following barrel rotation as a result of its i.c.v. administration (in particular at doses higher than 10 nmol/rat). Nonetheless, these results suggest the potential use of hybrid compounds encompassing both opioid and neurotensin structural fragments in pain management. This highlights the enormous potential of synthetic neurotensin analogues as promising future analgesics.
Key words: Analgesia, Receptor binding, Hybrid compound, Tolerance, Side effects
Graphical abstract
PK23 is a bifunctional compound consisting of both opioid and neurotensin fragments fused into one molecule. It mediates dose- and time-dependent antinociceptive responses when administered either centrally or peripherally, likely due to its interaction with μ opioid and neurotensin type 1 (NTS1) receptors.

1. Introduction
Opioid-based hybrid structures have been proposed as a putative valuable alternative to the use of strong opioids administered alone or in combination with other drugs. The development of such novel compounds for pain management is directed by the objective of reducing possible adverse side effects as well as to minimize the risk for drug interactions. Indeed, prolonged use of opioids that are considered the most potent and effective analgesics1 to date is commonly accompanied by burdensome side effects (e.g., constipation, vomiting, nausea, tolerance, physical dependence, itch, motor impairment, and an increased risk of respiratory depression2, 3, 4, 5, 6, 7). Therefore, much effort is dedicated to the discovery of innovative drugs that would retain their opioid activity, but were less likely to cause severe adverse effects that require medical intervention.
In our recent work, we presented several hybrid compounds that encompassed two pharmacologically distinct pharmacophores confined in one molecular scaffold, which resulted in potent drug-like molecules with dual activity8,9. These two chimeras (i.e., AA3052 and [Ile9]PK20) were identified as promising pain killers, devoid of several opioid-induced side effects8. This illustrates how administration of two active entities joined chemically into one molecule may significantly improve physicochemical and pharmacokinetic properties of individual molecules. This enables the drug to reach its specific target without modifying other properly functioning processes in the body10,11. Additionally, apart from an improved biological activity, such drugs could also prove useful for the treatment of other comorbidities12, 13, 14.
In the present study, we focused on a novel hybrid that combines opioid and neurotensin (NT)-like fragments. Of note, neurotensin was previously documented for its opioid-independent role in pain modulation15, 16, 17. Also, by inhibiting dopamine release18, NT may prove efficient in modulating the rewarding effect of opioids. Herein presented is an opioid-derived hybrid peptide codenamed PK23, the third compound of the series designed by our group that encompasses an NT pharmacophore. This structure, however, differs from both PK20 or [Ile9]PK20 by i) the d-amino acid residue located in the second position from the N-terminal, as well as ii) the presence of unnatural amino acids in the first and ninth positions. This bivalent drug lacks both the dimethyl tyrosine and tert-leucine present in PK2012 that could result either in a lower μ-opioid receptor binding affinity or reduced plasma stability. Additionally, incorporation of polar amino acid residues in the chain, particularly alanine, may result in lower blood–brain barrier (BBB) permeability19. We herein investigated how these modifications impact on the biological activity of this new hybrid compound. Apart from preliminary in vitro studies, we evaluated the antinociceptive properties of PK23 upon various delivery routes and examined some possible undesirable side effects arising from its repeated administration.
2. Materials and methods
2.1. Drugs and chemicals
The opioid–NT hybrid peptide—PK23 (Tyr-d-Ala-Phe-Phe-Lys-Lys-Pro-Phe-Ile-Leu-OH) was designed and prepared at the Department of Neuropeptides of the Mossakowski Medical Research Centre PAS (Warsaw, Poland) by manual solid-phase peptide synthesis using N-fluorenylmethoxycarbonyl (Fmoc) chemistry. Morphine was obtained from Polfa (Warsaw, Poland) and similarly to the chimera tested, was dissolved in saline. NT as well as naltrexone, an opioid receptor antagonist together with SR 48692 and NTRC 824, NTS1 and NTS2 receptor selective antagonists, respectively, were purchased from Tocris Bioscience (Germany). The anaesthetic drugs ketamine and xylazine were purchased from Biowet (Pulawy, Poland) while isoflurane was obtained from Baxter (Cracow, Poland).
Guanosine-5-[γ-35S]-triphosphate (1000 Ci/mmol, [35S]GTPγS) was purchased from Hartmann Analytic (Braunschweig, Germany), whereas [3H]DAMGO was purchased from PerkinElmer (Waltham, MA, USA). EDTA, bovine serum albumin (BSA), polyethylenimine (PEI), tris(hydroxymethyl)amino-methane (Tris), guanosine 5′-diphosphate (GDP), unlabeled GTPγS, and peptidase inhibitors—bestatin, bacitracin and phosphoramidon were purchased from Sigma–Aldrich (St. Louis, MO, USA).
2.2. Animals
Adult male Wistar rats (225–250 g body weight) were provided by the Animal facility of the Medical University of Warsaw. The animals were housed under standard laboratory conditions (at ambient temperature of 22 ± 2 °C, 12–12 h light–dark cycle). Soon after weaning, animals were housed separately in plastic cages with soft bedding, food and water available at all times, except during the course of the experiments.
The experimental protocols for animal usage were approved by the II Local Commission for the Care and Use of Laboratory Animals for Experimental Procedures in Warsaw (Permit numbers: 27/2015 and 167/2016) and adhered to guidelines published in the European directive 2010/63/EU on the protection of animals used for scientific purposes.
2.3. Ligand-stimulated functional [35S]GTPγS assay
2.3.1. Cell culture and membrane preparation
2.3.1.1. Opioid receptors
Forebrain membrane fractions were prepared according to the method previously described by Benyhe et al20. Briefly, rats were decapitated and the whole brain (without cerebellum) was quickly removed and homogenized in 30 volumes (mL/g wet mass) of 50 mmol/L Tris-HCl buffer (pH 7.4) with a Teflon-glass homogenizer. The same procedure was used for spinal cord specimens. The homogenates were then centrifuged at 18,000 rpm (Microfuge 20, Beckman Coulter Life Science, Poland) for 20 min at 4 °C. The resulting pellets were resuspended in a fresh 50 mmol/L Tris-HCl buffer (30 volume/weight) and then incubated for 30 min at 37 °C. Subsequently, the centrifugation step was repeated. The final pellets were resuspended in 5 volumes of 50 mmol/L Tris-HCl (pH 7.4) buffer containing 0.32 mol/L sucrose and stored at −80 °C until future use. For binding assays, membranes were thawed and then resuspended in 50 mmol/L Tris-HCl (pH 7.4) buffer and centrifuged at 18,000 rpm for 20 min at 4 °C in order to remove sucrose. The resulting pellets were suspended in ice-cold TEM buffer [50 mmol/L Tris-HCl (pH 7.2), 2 mmol/L EDTA, 1 mmol/L EGTA, 5 mmol/L MgCl2] to obtain a protein concentration of approx. 40 μg/mL per assay.
2.3.1.2. NTS1 receptors
Transfected CHO cells expressing the rat NTS121 were grown in MEM alpha medium supplemented with 10% foetal calf serum, 85 μg/mL streptomycin and 85 U/mL penicillin. Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2–95% air.
2.3.2. GTPγS binding assays for μ-opioids receptors
Membrane fractions from rat brain and spinal cord (∼10 μg of protein/tube) were incubated at 30 °C for 60 min in Tris-EGTA buffer (50 mmol/L Tris-HCl, 1 mmol/L EGTA, 3 mmol/L MgCl2, 100 mmol/L NaCl, pH 7.4) containing [35S]GTPγS (0.05 nmol/L) and increasing concentrations (10−9 to 10−5 mol/L) of the compounds tested in the presence of 30 μmol/L GDP in a final volume of 1 mL. Non-specific binding was determined in the presence of 10 μmol/L unlabeled GTPγS and subtracted from total binding to determine specific binding. The reaction was initiated by addition of [35S]GTPγS and terminated by filtrating the samples through Whatman GF/B glass fiber filters. Filters were washed three times with ice-cold 50 mmol/L Tris-HCl buffer (pH 7.4) using the Brandel M24R Cell Harvester, then dried. Bound radioactivity was detected in an UltimaGold™ scintillation cocktail (Packard, Meriden, CT, USA). Agonist-induced receptor-mediated G protein stimulation was expressed as percentage over the specific [35S]GTPγS binding observed in the absence of ligands (basal activity). [35S]GTPγS binding assays were performed in duplicate and all experiments were carried out three times independently.
2.3.3. [35S]GTPγS binding assays for CHO cells stably expressing the NTS1 receptors
The specific binding of [35S]GTPγS was measured in cell homogenates as previously described22. Briefly, cell homogenates (25–50 μg) were resuspended in binding buffer (50 mmol/L Tris-HCl (pH 7.4) containing 5 mmol/L MgCl2, 1 μmol/L 1–10 phenanthroline, 0.1% BSA, 1 μmol/L GDP, 150 mmol/L NaCl and 1 mmol/L dithiothreitol). The homogenate was incubated for 40 min in the presence of 0.05 nmol/L [35S]GTPγS at 37 °C. Non-specific binding of [35S]GTPγS was measured in the presence of 0.1 mmol/L Gpp(NH)p. Incubation was terminated by addition of 3 mL ice-cold binding buffer and the suspension was immediately filtered through GF/B glass fibre filters and washed twice with the same buffer using a 24-channel Brandel Harvester (SEMAT, St Albans, Hertfordshire, UK). Radioactivity trapped on the filter was estimated by scintillation counting. Results are expressed as mentioned above (see Section 2.3.2).
2.4. Surgical procedures performed for further determination of drug-mediated analgesia
2.4.1. Intracerebroventricular catheterization
For the intracerebroventricular (central, i.c.v.) drug administration, rats were implanted with cannulas according to the previously published protocol9. Briefly, rats were deeply anesthetized with a mixture of ketamine (87 mg/kg, i.p.) and xylazine (13 mg/kg, i.p.). A longitudinal skin incision was made along the midline to expose the skull. Polyethylene cannulas (PE 10, Intramedic™ Polyethylene Tubing, Clay–Adams Company, New York, USA; ID = 0.28 mm, OD = 0.61 mm, and 5 cm in length) were inserted into the right lateral ventricle. The cannula was secured by dental cement (Ketac™ Cem radiopaque, 3M ESPE, Germany). After surgery, animals were placed in separate cages and the experiment began on the fifth day after surgery. Animals showing signs of motor impairment were excluded from the experiment and immediately euthanized.
2.4.2. Intrathecal catheterization
Intrathecal (spinal, i.t.) administration of test drugs was performed using a method previously described by Yaksh and Rudy23. Rats were anaesthetized with a mixture of ketamine/xylaxine as detailed here above and placed in a stereotaxic head holder. A silicone catheter (PE 10, Intramedic™ Polyethylene Tubing, Clay–Adams Company, New York, USA; ID = 0.28 mm, OD = 0.61 mm, and 13 cm in length) was inserted into the intrathecal space through the atlanto-occipital membrane, positioning the tip at the L1 spinal level. Dental cement was used to fix the guide cannula to the skull. After surgery, animals with no obvious motor deficits were allowed to recover for 4 days before pharmacological experiments. However, during this time, rats were habituated to the experimenter in order to minimize stress. Animals showing paralysis or other signs of motor impairment after recovering from anaesthesia were immediately euthanized.
After each cannulation (i.c.v. or i.t.) the cannula/catheter placement was verified by an injection of 5% methylene blue and further checking for distribution within either the cerebroventricular or spinal subarachnoid space, respectively.
2.5. Surgical procedures performed for further determination of drug-induced hypotension and respiratory depression
Rats were i.t. catheterized as described in Section 2.4.2. For measurements of cardiovascular and respiratory parameters, animals were anesthetized with an intraperitoneal injection of 600 mg/kg urethane and 120 mg/kg alpha-chloralose (Sigma–Aldrich, Poznan, Poland). The depth of anaesthesia was assessed by monitoring any changes in blood pressure to a noxious pinch to the tail. Next, animals were placed in a supine position on a heating pad so that rectal temperature was kept between 37 and 38 °C. Tracheotomy was performed in the neck. A catheter was inserted into the femoral artery. Cardiovascular and respiratory responses to i.t. PK23 (10 nmol/rat) injection were examined right after the end of surgical procedures.
2.6. Drug administration and treatment
2.6.1. Acute pain model
The PK23 hybrid peptide was dissolved in sterile 0.9% NaCl and brought to room temperature. For single i.t. and i.c.v. injections, four various dosages of the peptide (0.5, 2, 5 and 10 nmol/rat) were administered through the catheter at a maximal volume of 10 and 3 μL, respectively. Noteworthy, the dose range used in the current experiment, in particular for i.t. administration, is similar to those used in our previous studies8,12, so the impact of amino acids replacement on drug biological activity could be observed. For i.p. administration, two doses of 1.8 and 2.7 μmol/rat were used. Control animals received saline. In order to determine the involvement of the opioidergic and NTergic systems in analgesia, animals were pre-treated with an opioid receptor antagonist, naltrexone (NTX), an NTS1 antagonist SR 48692, or an NTS2 antagonist, NTRC 82424. Each antagonist, except for NTX, was dissolved in saline with 5% DMSO supplemented with one drop of Tween 80 (Sigma–Aldrich) as vehicle, and injected i.t. at 10 μg/rat, 10 min before PK23 administration. Morphine (10 nmol/rat, i.t.; dissolved in sterile 0.9% NaCl) was used as a reference analgesic.
Rats (n = 5 per group) were intravenously injected with PK23 (1, 2.5 and 5 mg/kg m.c.). Briefly, the rat's tail was immersed in hot-water (42 °C) for 40–50 s in order to stimulate dilation of the tail veins. The tail was dried and the test chimera was injected. Nocifensive responses were measured individually in each animal in the tail-flick test at selected time intervals for 2 h. Subsequently, in order to preliminary determine in vivo BBB permeability, brains were removed for further HPLC studies (HPLC conditions described in Section 2.9).
2.6.2. Tolerance and cross-tolerance study
In order to evaluate tolerance to analgesic effects of PK23 and morphine, escalating doses of these drugs (0.5, 2, 5 and 10 nmol/rat) were administered intrathecally (i.t.) for 8 consecutive days. Measurements of nociceptive response to thermal stimuli were conducted daily at 30 min post-injection. On the 1st and 4th day (or 3rd day in case of morphine-treated group), the dose–response curves were generated and the ED50 values were calculated to determine the potency of tested drugs.
A state of tolerance was indicated by a progressive decrease in analgesic effect and an increase in ED50 values due to rightward shift in the acute dose–response curve. For morphine treated group, dose–response curve was generated on the third day of the experiment due to dramatic decline of analgesic potency in further days (antinociceptive effect was less than 50% MPE at the highest dose). Additionally, the range of tested doses in tolerance study was limited by the development of hyperalgesia resulting in lack of dose-dependent effect in morphine treated group.
The dose of 10 nmol/rat of tested drugs was selected to clearly present the changes in antinociceptive effect including hyperalgesia over the 8-day period of administration. Additionally, cross-tolerance between both drugs was investigated on the 9th day with a single injection of morphine (10 nmol/rat, i.t.) to the repeated PK23 group or a single administration of PK23 (10 nmol/rat, i.t.) to the morphine-treated group. The pain response was measured on the same day 30 min post-administration, and presented as MPE (%).
2.6.3. Cardiovascular and respiratory effects
To study any adverse effects of the hybrid on respiration, heart rate and blood pressure anesthetized rats were injected i.t. with a single antinociceptive dose of PK23 (10 nmol/rat), and NaCl at interval of at least 40 min. The effects of PK23 and saline were registered for 30 min.
2.7. Behavioral experiments
2.7.1. Tail-flick test
The nociceptive threshold was measured in the tail-flick apparatus (Ugo Basil 37360, Italy). Briefly, the tail was exposed to a powerful light beam and the tail-flick response latency was recorded. A 15 s cut-off time was chosen to prevent tissue damage. Measurements were taken in triplicate at baseline and 15, 30, 60, 90 and 120 min post-injection (acute pain model) or every 24 h after drug administration (subchronic administration).
2.7.2. Evaluation of motor impairment
We adapted a previous protocol described for this model9. Briefly, animals were trained to walk on the Rotarod apparatus (model 47700; Ugo Basile S.R.L., Italy) at an accelerating speed of 2–20 rpm (model 47700; Ugo Basile S.R.L., Italy) per 5 min for a maximal cut-off time of 300 s, according to the method described by Chu et al.25 Baseline values were recorded for each rat before i.t. administration of the drug tested or vehicle. Next, animals were randomly divided into the three following groups: i) group one received the PK23 chimera (10 nmol/rat), ii) group two received morphine (10 nmol/rat) and iii) group three was administered with saline (0.9% NaCl). Rats were placed on the rotating cylinder and the latency to fall was recorded. Each rat was subjected to three trials, 5 min apart. No injuries were caused by falling from the apparatus.
2.7.3. Evaluation of scratching and grooming behaviour in rats
The number of scratching and grooming events exhibited by the animals after i.t. injection of morphine (10 nmol/rat), PK23 (10 nmol/rat) or saline were monitored. Measurements were scored by individuals who were blinded to the experimental conditions, observing only one rat at a time. Rats were placed separately in Plexiglas boxes (18 cm × 23 cm × 20 cm) and habituated for 30 min. Observation for the morphine, PK23 and control groups started 2 min after drug administration. The behavioural responses were scored in 10 min intervals within a 30 min observation period. Scratching behaviour was defined as short-duration episodes of back-and-forth scraping across the body surface with the claws of the hind paw, whereas a facial grooming event was an episode of head- or face-washing movements with the forepaws.
2.8. Determination of drug-induced cardiovascular and respiratory effects in anesthetized rats
Blood pressure and heart rate in rats challenged with i.t. PK23 (10 nmol/rat) were measured via a catheter inserted into the femoral artery and connected to a BP-2 blood pressure monitor (Columbus Instruments, Columbus, OH, USA). Tidal volume signals were recorded with a pneumotachograph head attached to the tracheal cannula connected with a Research Pneumotach System (RSS 100 HR, Hans Rudolph Inc., Kansas City, MO, USA) and a computerized recording system (KORR Medical Technologies Inc., Salt Lake City, UT, USA) for measuring respiratory frequency (f), tidal volume (VT) and minute ventilation (VE). The recordings were registered on an Omnilight 8M36 apparatus (Honeywell, Tokyo, Japan).
The values of f, VT, VE, mean arterial blood pressure (MAP) and heart rate (HR) were calculated by averaging the variables measured for five sequential respiratory cycles just before drug injection, and at chosen time points of post–challenge phase. The maximum values were chosen from the computed time points ranging from 1 to 5 min.
2.9. In vitro plasma stability study
For plasma stability studies of PK23, control rats (n = 3) were anaesthetized with isoflurane and whole blood samples were collected via cardiac puncture into violet BD Vacutainer tubes. Blood was centrifuged for 4 °C for 10 min at 18,000 rpm (Microfuge 20, Beckman Coulter Life Science, Poland) to remove the fibrin clot and plasma was carefully transferred into eppendorf tube using a pipette. Fifty μL of PK23 solution in water (1000 μg/mL) was added to 400 μL of plasma sample and incubated at 37 °C for up to 24 h (incubation times were set to 0, 1, 2, 4, 6, 8, 12 and 24 h). Prior to UHPLC analysis, PK23-enriched plasma samples (25 μL) were quenched with 200 μL of acetonitrile for protein precipitation, vortex mixed for 1 min, kept at 4 °C for 15 min and then centrifuged at 4 °C for 15 min at 17,000 rpm. The resulting supernatants (ca. 100 μL) were transfered into vials with glass inserts and analyzed by UHPLC–DAD–MS. UHPLC–DAD–MS analysis was conducted using a Dionex Ultimate 3000RS system coupled with an Amazon SL ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany). The ion trap AmazonSL mass spectrometer was equipped with an ESI interface. The eluate was introduced into the ESI interface of the mass spectrometer without splitting. The parameters for the ESI source were as follows: nebulizer pressure 40 psi; dry gas flow 9 L/min; dry temperature 145 °C; and capillary voltage 4.5 kV. The analysis was carried out using scanning from m/z 70 to 2200. Mass spectra were recorded in negative-ion mode. The separation was carried out on a Kinetex XB-C18 (150 mm × 2.1 mm × 1.7 μm, Phenomenex, Torrance, CA, USA) column maintained at 25 °C. The mobile phases were 0.1% HCOOH in water (A) and 0.1% HCOOH in acetonitrile (B), and elution was conducted with the following gradient: 0 min, 5% B; 15 min, 50% B; and 20 min, 65% B. The flow rate was 0.3 mL/min, and the sample injection volume was 5 μL. The UV–Vis spectra of the detected compounds were recorded over the 190–450 nm range. The chromatogram was recorded at 205 nm. Quantification of PK23 was performed using a MS detector. Extracted ion chromatograms (EIC) for all samples at m/z = 1272 amu were calculated and the area of the detected peak at ca.12.6 min was measured.
Calibration curves based on the amount injected (ng) versus peak area were plotted. Linear regression fit was used for the data obtained. Residual compound concentration in plasma was calculated. Next, the plot showing the logarithm of the remained fraction of PK23 vs. incubation time was generated. The linear fit model was used. The t1/2 was calculated using the slope (b) of the generate cure according to the following formula: t1/2 = 0.693/b.
2.10. Statistical analyses
Group size was determined based on power analysis. Additionally, block randomization method was used in order to allocate animals into respective treatment groups. The experimenters were all blinded to treatments. Data were analysed with the use of GraphPad Prism v.5.0 software (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Figures were generated by the same software, except for data obtained from binding studies. In this case, experimental data were analysed and graphically processed by the GraphPad Prism software (version 4.00 for Windows, GraphPad Software). In binding experiments, displacement curves were fitted by non-linear regression using the one-site competition fitting option with no ligand depletion model. Results were expressed as EC50 values ± SEM (standard error of mean).
Results from behavioral studies were expressed as a percent of the maximal possible effect (MPE, %) using Eq. (1):
| (1) |
where pdr, br and co refer to post drug response, baseline response and cut-off value, respectively. Raw data from nociceptive tests were converted to AUC, calculated by the trapezoidal rule, at specific time points. Statistical analysis of the in vivo time–course curves was conducted by two-way or one-way ANOVA followed by the Bonferroni's post-test, unless otherwise stated. Whereas corresponding AUC data were analyzed using one-way ANOVA followed by the Dunnett's post hoc test. Differences in values were considered significant at ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001.
The ED50 was defined as the drug dose that induced a 50% MPE measured by the tail-flick test. ED50 values were calculated using either the linear regression method (i.v. drug administration) or nonlinear sigmoidal dose response (i.t. drug administration).
Differences in ED50 values for tolerance study were considered significant when confience intervals did not overlap.
Cardiovascular and respiratory parameters were expressed as means ± SEM. Non-parametric U Mann–Whitney test was used for the comparisons of parameters between PK23 and saline treatments, while the Wilcoxon's signed-ranks test was used for comparisons within groups. A P < 0.05 was considered statistically significant.
3. Results
3.1. PK23 stimulation of opioid and NTS1 receptors
The effect of PK23 on the G protein activation efficacy (Emax) and potency (pEC50) was tested in [35S]GTPγS binding assays in comparison with morphine and NT (Table 1). Analyses of concentration–response curves from data obtained both in spinal cord or brain samples permitted to derive pEC50 values supportive for an interaction of PK23 with both types of receptors tested. Importantly, similarly to morphine, the potency and efficacy of this chimeric peptide at μ-opioid receptors appeared greater in brain than spinal cord homogenates.
Table 1.
Pharmacological characterization of morphine, NT and PK23 in promoting [35S]GTPγS binding (see text for details).
| Peptide | Potency pEC50±SEM (EC50, nmol/L) |
Efficacy (Emax), stimulation±SEM (%) over the basal |
||||
|---|---|---|---|---|---|---|
| Opioid receptor mediated effect |
NTS1 receptor mediated effect | Opioid receptor mediated effect |
NTS1 receptor mediated effect | |||
| Brain | Spinal cord | Brain | Spinal cord | |||
| Morphine | 8.46 ± 0.10 (3.5)25 | 6.92 ± 0.16 (120) | N/A | 146.9 ± 4.725 | 98.6 ± 5.0 | N/A |
| NT | N/A | N/A | 8.50 ± 0.20 (3.31)8 | N/A | N/A | 146.6 ± 0.038 |
| PK23 | 6.71 ± 0.51 (197) | 5.91 ± 0.25 (1222) | 6.95 ± 0.16 (113) | 110.5 ± 2.5 | 53.7 ± 7.2 | 104.4 ± 6.5 |
3.2. Dose- and time-dependent antinociceptive response induced by PK23
In the radiant heat tail-flick test, PK23 exhibited a time- and dose-dependent analgesic effect after supraspinal (i.c.v.), spinal (i.t.) and peripheral (i.v.) administration (Fig. 1). Since ED50 represents a dose required to achieve 50% of the maximal effect, there was no posibility to produce the ED50 value for i.c.v PK23. This resulted from the fact that higher doses generated serious adverse effect that did not allow further testing, while lower shown less that 50% activity; although for PK23 (i.t.) the ED50 value was 4.38 nmol/rat (i.t.). The maximal efficacy of PK23 was obtained after i.t. administration (Fig. 1B). A strong pain-relieving effect, with the MPE (%) value of 93.20 ± 2.65%, was obtained 30 min-post-injection with the highest PK23 dose tested (10 nmol/rat). The response to PK23 was transient as the post-treatment MPE (%) values reached baseline at 90 min after administration. On the contrary, the weakest activity was observed in rats receiving an i.p. administration of PK23 (Fig. 1C). Unexpectedly, whilst the lower dose of 10 mg/kg resulted in a modest analgesic effect [MPE (%) = 9.45 ± 0.81%] seen in the first 30 min post-administration, a trend for increased pain sensitivity was observed when injected at a dose of 15 mg/kg. The responses at both doses were calculated to be statistically significant at 15 min (P < 0.01), 30 min (P < 0.001) and 60 min (P < 0.001), respectively [data not shown in Fig. 1; F4,48 = 9.21 (P < 0.0001) and F1,12 = 50.50 (P < 0.0001)].
Figure 1.

Time- and dose-dependent analgesic effect induced by (A) i.c.v., (B) i.t., and (C) i.p., and (D) i.v. PK23 administration. The analgesic effect of a single bolus administration of PK23 in rats was measured with the tail-flick test. Dose–response curves derived from AUC calculated after PK23 treatment are presented as insert within each panel. Individual data were compared to the control group injected with saline. Results were analysed either with two-way ANOVA followed by Bonferroni's post-test (dose–response curves) or one-way ANOVA followed by Dunnett's post hoc test (AUC calculation). Significant differences between animals receiving PK23 and control groups were indicated as: ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001, respectively. n = 6–7 rats/group. F4,120 = 44.89 (P < 0.0001) and F4,30 = 10.26 (P < 0.0001) for i.c.v. administration; F4,120 = 279.23 (P < 0.0001) and F4,30 = 118.70 (P < 0.0001) for i.t. administration; F4,72 = 4.70 (P = 0.0020) and F2,18 = 6.34 (P = 0.0082) for i.p. injection, and F4,80 = 151.02 (P < 0.0001) and F3,20 = 76.01 (P < 0.0001) for i.v. administration.
PK23 also induced a modest antinociceptive effect in the tail-flick test following i.c.v. administration (Fig. 1A). In fact, maximal analgesia was obtained with a dose of 10 nmol/rat and reached MPE (%) values of 15.79 ± 1.41% (30 min post-administration). However, central administration of PK23 at this dose resulted in visible abnormalities of motor behavior manifested by short barrel rotation.
Intrathecal administration of 10 nmol/rat of PK23 produced a comparable analgesic effect as an equimolar dose of morphine (Fig. 2). However, these similarities in terms of response magnitude were observed only at 15 min [MPE (%) = 84.50 ± 3.84% for morphine and MPE (%) = 80.71 ± 4.11% for PK23] and 30 min post-administration [MPE (%) = 86.23 ± 4.76% for morphine and MPE (%) = 93.20 ± 2.65% for PK23]. A noticeable and significant attenuation of PK23-induced effect in comparison to morphine was noticed at later time points (P < 0.001).
Figure 2.

Comparison of the antinociceptive effect mediated by i.t. administered PK23 (10 nmol/rat) and morphine (10 nmol/rat) as the response to noxious thermal stimuli (Tail-flick test). Two-way ANOVA followed by the Bonferroni's post-test revealed significant differences between PK23 vs. morphine (∗∗∗P < 0.001), morphine vs. control ($$$P < 0.001) and PK23 vs. control (###P < 0.001); n = 7 rats/group. F4,72 = 118.49 (P < 0.0001) and F2,18 = 224.51 (P < 0.0001).
Intravenous (i.v.) PK23 administration revealed that this compound could also induce a dose-dependent antinociceptive response (ED50 = 2.33 mg/kg) as measured by the tail-flick assay. The maximal possible effect was observed for the 5 mg/kg dose (Fig. 1D) at the 5 min time point with an MPE (%) value equal to 73.55 ± 4.69%. After this time, the pain-relieving effect (practically for every dose tested) decreased dramatically.
3.3. Contribution of opioid and NT receptors to PK23-mediated spinal analgesia
Animals were pre-injected with specific competitive antagonists of opioid receptors (NTX), the NTS1 (SR48692) or NTS2 receptors (NTRC 824) in order to delineate the involvement of these molecular targets in the effect of PK23. Pharmacological blockade of the μ-opioid and NTS1 receptors was found to significantly attenuate the analgesic effect obtained with i.t. PK23 (Fig. 3). Thus, injection of NTX (26.4 nmol/rat) considerably reduced the analgesic response evoked by PK23 throughout the 120 min observation period with maximal inhibition after 15 min [from MPE (%) = 80.71 ± 4.11% for PK23 alone to MPE (%) = 12.12 ± 1.89% observed after co-administration with NTX] and 30 min [from MPE (%) = 93.20 ± 2.65% for PK23 alone to MPE (%) = 21.95 ± 2.94% observed after co-administration with NTX]. Similarly, administration of SR 48692 resulted in an inhibition of the PK23 pain-relieving effect at both 15 min [up to MPE (%) = 53.36 ± 2.99%] and 30 min [up to MPE (%) = 55.90 ± 5.71%] post-injection, respectively. In fact, two-way ANOVA revealed significant differences (P < 0.001) between time–course curves and AUC values for PK23 alone or PK23 with NTX and SR 48692, respectively (Fig. 3A and B). In contrast, NTRC 824 (17.5 nmol/rat) did not influence the analgesic response to PK23 (P > 0.05). Estimation of the AUC for NTRC 824 with PK23 indeed confirmed the absence of inhibition, suggesting the lack of involvement of NTS2 receptor.
Figure 3.

Effect of the non-selective opioid receptor antagonist NTX (26.4 nmol/rat) and the selective NTS1 and NTS2 receptor antagonists: SR 48692 (17 nmol/rat) and NTRC 824 (17.5 nmol/rat), respectively, on the analgesic effect of PK23 (10 nmol/rat). Antagonists were administered i.t. 10 min before PK23 and the response to the thermal stimulus was measured at different time points (panel A). Data from panel A were combined allowing estimate inhibition as AUC values (panel B). Results were analyzed by two-way ANOVA (panel A) or one-way ANOVA (panel B), revealing significant differences between PK23 alone and antagonist pre-treated PK23 groups. ∗∗∗P < 0.001 (PK23 vs. PK23 + NTX and vs. PK23 + SR 48692). From the panel B–Dunnett's post-test showed significances between PK23 and its combination with NTX (P < 0.001) and PK23 vs. PK23 + SR 48692 (P < 0.05); n = 7 rats/group. F4,96 = 239.84 (P < 0.0001) and F3,24 = 22.56 (P < 0.0001) for panel A.
3.4. Assessment of the occurrence of tolerance following administration of PK23
3.4.1. Tolerance development
Repeated i.t. administration of the chimeric compound PK23 for 8 consecutive days was performed in order to determine whether PK23-induced effects would decline over time as a consequence of tolerance. Although tolerance developed, likely resulting from the activity of the opioid pharmacophore moiety, the loss of PK23-induced analgesia was induced at a slower rate than for morphine administered at the same dose of 10 nmol/rat (Fig. 4B). In fact, on day 8 while the MPE (%) value calculated for PK23 was estimated at 35.73 ± 5.16%, the MPE (%) for morphine was −16.89 ± 1.52%, reflecting the development of hyperalgesia (Fig. 4B). Additionally, intrathecal PK23 chimera or morphine was reported to exert different pharmacological actions mediated by variations in receptor interactions.
Figure 4.

Dose– (A) and time–response (B) curves of PK23 and morphine following intrathecal (i.t.) administration in the tail-flick test. Panel A: the dose–response relationship was examined on day 1 for both drugs and on day 3 (for morphine) or 4 (for PK23), respectively. Compounds were administered at the dose range of 0.5–10 nmol/rat. Panel B: Drugs were intrathecally administered at a dose of 10 nmol/rat for 8 consecutive days, and the effect was measured at 30 min time-point. Data are presented as the percentage of maximum possible effect (MPE, %)±SEM. Two-way ANOVA (panel B) showed significant differences between PK23 and morphine (###P < 0.001; post-hoc), PK23 and control (∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001; post-hoc) and between morphine and control ($P < 0.05, $$P < 0.01 and $$$P < 0.001; post-hoc); n = 6–10 rats/group. Asterisk (∗) represents the ED50 value for morphine which was calculated for the effect observed in days 1 and 3. The dose of 10 nmol/rat of morphine was not able to induce antinociception equal 50% of the maximum effect. Administration of higher doses resulted in loss of dose-dependency; n = 7 rats/group. F8,144 = 171.89 (P < 0.0001) and F2,18 = 415.68 (P < 0.0001) for panel B.
Differences in tolerance development between tested drugs were supported by the shifts in ED50 (with 95% CI), based upon peak dose–response relationship values, and was shown in Table 2. Indeed, a repeated morphine treatment produced a 3.2-fold increase in the acute ED50 values in the tail-flick test reflecting a significant loss of analgesic potency. In the contrary, chronic treatment of a tested hybrid resulted only in 1.22-fold increase of the morphine ED50 values. Thus, development of tolerance to intrathecal PK23 seems to be delayed as compared with morphine.
Table 2.
ED50 values for morphine and PK23 analgesia in the rat tail-flick test after repeated intrathecal treatment.
| Group | ED50 vaule (nmol, i.t.)a | Shift | |
|---|---|---|---|
| PK23 | Day 1 | Day 4 | 1.22∗ |
| 4.38 (3.86–4.96) | 5.36 (4.67–6.17) | ||
| Morphine | Day 1 | Day 3 | 3.20 |
| 2.82 (2.33–3.41) | 9.03 (7.03–11.61) | ||
The ED50 values were calculated from dose–response curve. For morphine-treated group, dose–response curve should have been generated following 3 days treatment period due to the serious side effects (e.g., hyperalgesia) revealing in further days of administration and resulting in lack of dose-dependent effect. ∗Statistically insignificant.
Mean (CI 95%).
In the study of cross-tolerance, animals treated for 8 consecutive days with PK23 were found to retain analgesic response to single injection of morphine (i.t.) on day 9 compared with the saline-treated group. In these conditions, the magnitude of the response to morphine [MPE (%) = 33.10 ± 5.44%] was decreased compared to day 1 and was similar to the response to PK23 alone measured on day 8 [MPE (%) = 35.73 ± 5.16%] (Fig. 5A). Similarly, in an analogous study (Fig. 5B), administration of PK23 on day 9 neither stopped nor reduced/improved hyperalgesia induced by morphine (day 8, i.t.). The MPE value were −6.89 ± 1.52% vs. −14.38 ± 2.71%, respectively. These suggest the existence of cross-tolerance between PK23 and morphine.
Figure 5.

Panels A and B represent cross-tolerance of PK23 or morphine with morphine or PK23, respectively, administered on day 9. Measurements of the nociceptive response to thermal stimuli were conducted daily (30 min post-treatment), except for day 1; day 1 reflects the baseline value (measurements performed before drug administration). In panels A and B, asterisks (one-way ANOVA with Bonferroni's post-test) indicate significant differences in the effect obtained between PK23 (day 1) vs. PK23 (day 8) (∗∗∗P < 0.001; panel A), PK23 (day 1) vs. PK23+morphine (day 9) (∗∗∗P < 0.001; panel A), morphine (day 1) vs. morphine (day 8) (∗∗∗P < 0.001; panel B) and morphine (day 1) vs. morphine + PK23 (day 9) (∗∗∗P < 0.001; panel B), respectively. No statistical differences were observed in case of morphine or PK23 (day 8) vs. morphine + PK23 or PK23+morphine (day 9), respectively; n = 7 rats/group. F4,34 = 89.66 (P < 0.0001) for panel A and F4,34 = 360.0 (P < 0.0001) for panel B.
3.4.2. Locomotor impairment
As shown in Fig. 6, rats having received 10 nmol/rat of PK23 (i.t.) did not exhibit any impairment of motor activity. Furthermore, when compared with morphine, significant differences appeared during the entire testing period (*P < 0.05, **P < 0.01 and ***P < 0.001). In fact, at 30 min after administration, the latency to fall was reduced from 286.50 ± 4.39 s (for PK23) to 222.33 ± 8.44 s (for morphine, respectively; F2,15 = 10.44, P = 0.0014).
Figure 6.
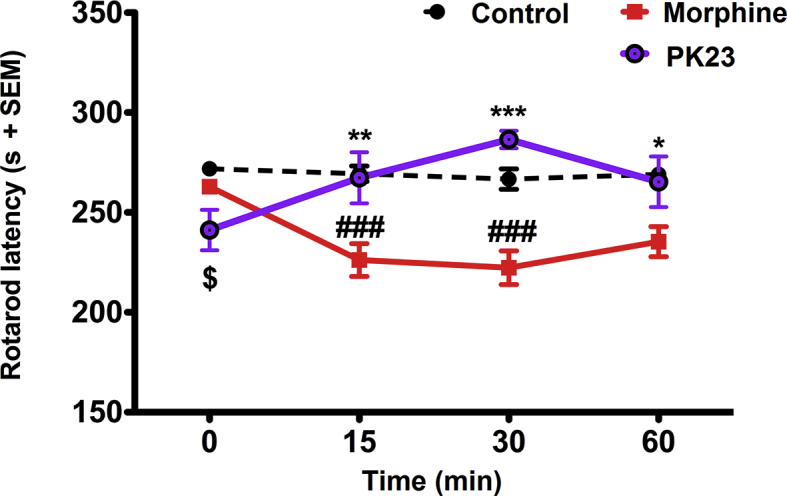
The effect of PK23 on locomotor activity in comparison to morphine. Drugs were given i.t. at a dose of 10 nmol/rat. Significant differences were shown for PK23 vs. morphine (∗P < 0.05, ∗∗P < 0.01 and ∗∗∗P < 0.001; Bonferroni's post-hoc test), PK23 vs. control ($P < 0.05; post-hoc test) and morphine vs. control (###P < 0.001; post-hoc test); n = 6 rats/group. F3,45 = 0.32 (P=0.8144) and F2,15 = 10.44 (P = 0.0014).
3.4.3. PK23 induces less scratching and grooming behaviour
The PK23 hybrid peptide was found to elicit time-dependent scratching and grooming behaviours in rats (Fig. 7A–D). However, at a dose of 10 nmol/rat i.t., the number of grooming events induced by the chimeric compound was significantly lower as compared to morphine-treated rats either on day 1 (F2,15 = 13.54, P = 0.0004) or day 9 (F2,15 = 26.79, P < 0.0001; Fig. 7A and C, respectively). Similarly, on day 9, there was a notable difference in the scratching response elicited by both drugs examined (Fig. 7D). Indeed, within the first 30 min post-injection, morphine was found to produce a strong scratch reflex in rats that weakened over time. In contrast, PK23-treated animals demonstrated a time-dependent increase in scratching behaviour (Fig. 7D). Noticeably, by the end of the experiment (20–30 min post-administration), there were no significant differences in such behaviours between animals receiving either morphine or PK23 (P > 0.05). It's worth mentioning that morphine-treated rats also exhibited additional behaviours that were merely absent in PK23-treated rats. These included wet-dog shakes and teeth chattering.
Figure 7.

Comparison of grooming (left column) and scratching (right column) responses elicited by PK23 and morphine administered i.t. at a dose of 10 nmol/rat on day 1 (panels A and B) and day 9 (panels C and D). Two-way ANOVA followed by Bonferroni's post-test indicated significant differences (∗P < 0.05; ∗∗P<0.01; ∗∗∗P<0.001) between control and drug-treated groups (morphine or PK23) and between PK23 and morphine-treated animals (#P < 0.05; ###P< 0.001); n = 6 rats/group. F2,30 = 0.70 (P = 0.5034) and F2,15 = 13.54 (P = 0.0004) for panel A; F2,30 = 0.64 (P = 0.5626) and F2,15 = 0.62 (P = 0.5529) for panel B; F2,30 = 3.69 (P = 0.0368) and F2,15 = 26.78 (P < 0.0001) for panel C and F2,30 = 0.09 (P = 0.9131) and F2,15 = 47.33 (P < 0.0001) for panel D.
3.5. Cardiovascular and respiratory effects induced by intrathecal PK23
Blood pressure and respiratory function in the PK23-treated group (10 nmol/rat, i.t) was compared with saline-treated rats. As presented in Table 3, no changes in ventilation or heart rate were reported after i.t administration of PK23. A visible, but insignificant decrease in arterial blood pressure (MAP) was noted immediately after injection that lasted for 30 s. At 1 min, the depressor response translated into an insignificant increase in MAP lasting about 5 min. MAP values returned to baseline within 10 min.
Table 3.
Cardiovascular and respiratory effects of i.t. injected PK23.
| Factor | PK23 |
NaCl |
||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | 0 min | MAX | 10 min | Baseline | 0 min | MAX | 10 min | |
| VE, (mL/min) | 121 ± 12 | 118 ± 10 | 122 ± 13 | 113 ± 14 | 120 ± 8 | 119 ± 9 | 124 ± 5 | 120 ± 7 |
| HR (beat/min) | 436 ± 24 | 439 ± 24 | 435 ± 31 | 418 ± 32 | 404 ± 33 | 403 ± 32 | 415 ± 33 | 411 ± 36 |
| MAP (mm Hg) | 83 ± 4 | 76 ± 5 | 97 ± 8 | 87 ± 7 | 83 ± 5 | 83 ± 6 | 87 ± 6 | 84 ± 7 |
VE, minute ventilation; HR, heart rate; MAP, mean arterial blood pressure; Baseline, before injection, 0 min, early phase after injection, MAX, maximum value selected between 1 and 5 min after injection; n = 5 for PK23 and n = 4 for NaCl treated rats.
3.6. Stability of PK23 in rat plasma in vitro and blood–brain barrier permeability after i.v. administration
Plasma stability of PK23 was evaluated. The compound was not fully metabolized within the total experiment time (24 h). The peak representing PK23 was observed on BPC recorded in negative ion mode throughout the experiment (Supporting Information Fig. S1). Starting from 1 h incubation the occurrence of two peaks (A and B, in Fig. S1) in BPC was observed. Compounds A and B are the metabolic products of PK23 produced by plasma protease activity. The tentative chemical structures of A and B were proposed in Supporting Information Fig. S2 (Supplementary file). Compound A is a product of leucine and isoleucine cleavage at the C-end of PK23. Compound B was proposed as PK23 after the loss of phenylalanine present at the N-end of the investigated peptide.
Apart from plasma stability studies, also brain penetration of PK23 was assessed in our studies. Indeed, after a single intravenous administration at a dose of 5 mg/kg, the test chimera was found to penetrate the BBB, which was determined by HPLC experiments using brain homogenates (Supporting Information Fig. S3B).
4. Discussion
This paper describes the biological activity of PK23, a novel compound combining an opioid- and an NT-like pharmacophores. The presented chimeric ligand was found to exert a strong antinociceptive effect, but noticeably weaker in comparison to a previously reported PK20 and [Ile9]PK20 opioid–NT hybrids8,12. Indeed, both PK20 and its analogue were shown to induce substantial pain-relief at doses approximately 500-times (0.02 nmol/rat; i.t.) and 100-times (0.1 nmol/rat; i.t.) lower than PK23 administered via the same route. However, intrathecal administration of PK20 at a dose higher than 10 nmol/rat led to sudden death of animals (data not published). Similar results were observed for our compound but administered intracerebroventricularly at 10 nmol/rat and higher. This in consequence did not allow us performing a dose−response analysis that would give a complete picture after its i.c.v. administration. Despite the relatively modest analgesic efficacy, data obtained with this new compound, however, provide essential information regarding structure modifications crucial for obtaining a potent analgesic with reduced side effects. In fact, substitution of 2′,6′-dimethyltyrosine (Dmt) for the N-terminal Tyr in opioid peptides (as in PK20 chimera) has recently been shown as a promising tool for improving opioid receptor affinity and biological activity. However, it was shown that accordingly to the “message-address” concept26,27 of opioid ligands, the lack of modification of the Tyr1 residue (as in PK23) that comprises the message component may still result in high affinity towards the opioid receptor, but with improved safety profile of the compound. Additionally, the attached modified pharmacophore of NT(8–13) may serve as an address domain which can modulate opioid peptide receptor selectivity, while maintaining specific interactions with the NT receptor. Furthermore, it was noticed that PK23 shows structural homology with the amphibian skin opioid peptide dermorphin (Tyr-d-Ala-Phe-Gly-Tyr-Pro-Ser-NH2), especially considering the first two N-terminally located amino acid residues that are also shared among all deltorphins. Moreover, this dipeptide fragment differentiates PK23 from its analogue, [Ile9]PK20. However, considering that Dmt incorporation within the N-terminal part of opioids plays an essential role in conferring high affinity to μ-opioid receptors, the role and functional diversity of various d-amino acids in position 2 of the sequence still remain controversial28,29. Thus, the functionality of the specific d-amino acid residue while preserving the subsequent amino acids requires further examination. Nevertheless, the PK23-mediated pain-reducing activity was strongly evidenced after spinal administration (Figure 1, Figure 2) suggesting that the main site of action of this opioid−NT hybrid peptide, is located at the spinal cord level. In fact, both the i.p. as well as i.v. delivery of PK23 was found to exert rather weak antinociceptive responses when compared to its central administration (Fig. 1C and D, respectively). However, although opioid-based ligands can produce analgesic effect through peripheral mechanisms, particularly in inflammatory state, one could assume their capacity to penetrate the blood–brain barrier. Indeed, brain uptake of PK23 following its single intravenous administration (5 mg/kg) to rats was shown in Fig. S3. Such a result seems to be intriguing since—according to the “Lipinski's Rule of Five”—the optimal compound's molecular mass desired for its sufficient BBB penetration should not exceed 400 Da; of note, the molecular weight of the PK23 chimera is equal 1273 Da. Nevertheless, further studies with multiple dosing may be performed in order to determine the potential of PK23 chimera to accumulate within the brain.
Apart from the analgesic effect, opioids and various opioid-based compounds are well-known for their ability to induce clinically significant side effects, in particular tolerance and dependence. The hybrid approach is aimed particularly at developing improved compounds with fewer or more discrete side effects. In 2016, we demonstrated that the responses obtained after administration of an opioid hybrid peptide or a combination of its pharmacophores are different8. In particular, the hybrid which combines two distinct activities in a single molecular entity, caused no impairments of motor function. Similar results were later obtained for PK23 (Fig. 6). Furthermore, the analgesic properties of this compound were preserved for a longer period of time as compared with morphine after subchronical administration (Fig. 4B). Importantly, morphine induced a severe loss of analgesic activity as soon as 3 days after initiating treatment. This may result from the different pharmacokinetic profiles of these two drugs. However, the mean elimination half-life of morphine ranges from 1.5 to 3.4 h30, while the half-life of PK23 reaches similar values (t1/2 at 3.13 h). However, being an analogue of PK20 (t1/2 = 31.45 h), PK23 was anticipated to show similar of extended half-life values13. We also cannot exclude the hypothesis that the biochemical pathways responsible for termination of the compound actions and desensitization of functional μ-opioid receptors differ.
The development of opioid receptor ligands associated with reduced tolerance is of major importance31. New ligands could be considered as substitutes for classical opioids or could be used in opioid rotation protocols to obtain a balance between pain control and the intensity of adverse effects.
There is evidence for the role of dopamine D2 receptors in the development of morphine tolerance as well as on some motor behaviors32,33. The possible activation of this dopamine receptor by PK23 was therefore examined in [35S]GTPγS binding assay in transfected cells31. Dopamine produced a robust response (Emax = 140.92%), while PK23 stimulated G protein activation with much lower efficacy (Emax = 117.13%) (Kleczkowska and Hermans, unpublished data). Even more convincing, while the response to dopamine was considerably reduced in the presence of domperidone, this dopamine receptor antagonist was without influence on the response to PK23. Although we did not further address the mechanism of lower side effect liability of PK23, some hypotheses explaining this phenomenon exist. Firstly, the effect observed may result from the activity of the NT-like pharmacophore, attachment of which could have resulted in the reduction of potency and selectivity of the compound to the μ-opioid receptors. Such a phenomenon reflecting changes in the aforementioned features of a hybrid compound in comparison to a mixture of its pharmacophores, was already reported by Kleczkowska et al8. Secondly, this effect may be due to some putative activity of PK23 at δ-opioid receptors. The involvement of this type of receptors in the development of opioid tolerance was confirmed in transgenic δ-opioid receptor knock-out mice34. Hence, δ-opioid antagonists were found effective in preventing morphine tolerance35. It is possible that with a putative mixed μ/δ activity, PK23 could modulate opioid receptor-mediated tolerance and dependence.
PK23-mediated antinociception and sustained pain-relieving effect after repeated administration could also be the consequence of its functional interaction with other nociceptive systems. Indeed, animals treated with doses higher than 10 nmol (i.c.v.) of PK23 exhibited behaviors such as barrel rotation, which are related neither to opioid nor NT receptors. Also, death occurred in some cases. Blockage of the β-adrenergic receptor was found to exert antinociceptive effects and thus could represent a potential target for PK2336.
Apart from the hallmark features of opioids, like tolerance and dependence liability, NT is also known to elicit side effects like anorexia nervosa37,38 or hypotension39. In our study, we did not observe reduced food intake or changes in the body temperature (data not shown) following subchronic i.t. administration of PK23. Additionally, PK23 affected neither the respiratory system nor induce hypotension. However, considering that the biological activity of PK23 resulted rather from the synergistic interactions between both of its pharmacophores, and given a strong involvement of the NTS1 receptor agonist activity, we expected this peptide to alter blood pressure. However, a single i.t. injection of PK23 in anaesthetized animals did not significantly change either blood pressure, heart rate or ventilation. What is more important, by careful dose titration and the right selection of the delivery route, we managed to avoid a prolonged hypotensive response described previously after i.t and i.v. administration of neurotensin (NT)39, 40, 41.
Several studies have reported that morphine elicits scratching and grooming behaviours in rodents, together with a series of other spontaneous signs accompanying nociceptive responses42,43. In contrast, NT suppresses both grooming and scratching behaviour induced by several endogenous peptides, including β-endorphins and bombesin44,45. Such observations are highly consistent with our data (Fig. 7). In fact, PK23 chimera was found to reduce or delay grooming and scratching behaviours, which could be explained by the presence of an NT-like structural component. Whilst morphine significantly increased the scratching reflex in rats, especially on day 9 (Fig. 7C), the effect induced by the tested peptide was almost similar to that observed for control. This however, was true only for the first 20 min post-administration as a similar response to PK23 and morphine occurred at later time points (between 20 and 30 min). Furthermore, attenuation of grooming behaviour reported for i.t. administered PK23 chimera (Fig. 7B and D) was also observed in our current study (in a substance P-induced grooming model; data not shown). These results clearly show that the C-terminal NT-like fragment is strongly involved not only in the antinociceptive activity exhibited by the chimera, but also in other of its properties.
5. Conclusions
The herein presented that the opioid–NT bivalent compound PK23 seems a promising analgesic with a favourable profile in terms of side effects, like reduced scratch reflex, delayed development of tolerance and no obvious signs of motor impairment when compared with the gold standard, morphine. However, numerous additional studies need to be performed in order to determine the possible mechanism of action of this novel chimera. Furthermore, extended characterization of the analgesic and possible side effects exerted by PK23 need to be performed to get a complete picture of the compound's activity.
Acknowledgments
Research equipment implemented with CPB infrastructure financed by the EU—the European Regional Development Fund within the Operational Program “Innovative economy” for 2007–2013. This research did not receive any specific grants from funding agencies from the public, commercial, or not-for-profit sectors.
Author contributions
Patrycja Kleczkowska and Karolina Frączek participated in research design; Karolina Frączek, Mattia Ferraiolo, Kaja Kasarello, Anna Erdei, Kamila Kulik, Agnieszka Kowalczyk, Piotr Wojciechowski, Dorota Sulejczak, Piotr Sosnowski, and Sebastian Granica conducted the experiments; Emmanuel Hermans, Magdalena Bujalska-Zadrozny, Sandor Benyhe, Katarzyna Kaczynska, Lukasz Nagraba, Artur Stolarczyk, Agnieszka Cudnoch-Jedrzejewska and Patrycja Kleczkowska contributed reagents or analytic tool; Karolina Frączek, Mattia Ferraiolo, Kaja Kasarello, Anna Erdei, Sebastian Granica, Katarzyna Kaczynska and Patrycja Kleczkowska performed data analysis; Karolina Frączek, Emmanuel Hermans, Anna Erdei, Sebastian Granica, Sandor Benyhe, Katarzyna Kaczynska and Patrycja Kleczkowska wrote or contributed to the writing of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.04.014.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.DeHaven-Hudkins D.L., Burgos L.C., Cassel J.A., Daubert J.D., DeHaven R.N., Mansson E. Loperamide (ADL 2-1294), an opioid antihyperalgesic agent with peripheral selectivity. J Pharmacol Exp Therapeut. 1999;289:494–502. [PubMed] [Google Scholar]
- 2.Harned M., Sloan P. Safety concerns with long-term opioid use. Expet Opin Drug Saf. 2016;15:955–962. doi: 10.1080/14740338.2016.1177509. [DOI] [PubMed] [Google Scholar]
- 3.Lewis A.B., Ferry D.A., Sadeghi M. Fetal cardiovascular and breathing movement responses to endogenous opiates. Biol Neonate. 1986;50:278–287. doi: 10.1159/000242610. [DOI] [PubMed] [Google Scholar]
- 4.Walsh T.D. Prevention of opioid side effects. J Pain Symptom Manag. 1990;5:362–367. doi: 10.1016/0885-3924(90)90031-e. [DOI] [PubMed] [Google Scholar]
- 5.Webster L.R., Camilleri M., Finn A. Opioid-induced constipation: rationale for the role of norbuprenorphine in buprenorphine-treated individuals. Subst Abuse Rehabil. 2016;7:81–86. doi: 10.2147/SAR.S100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitman C.B., Reid M.W., Arnold C., Patel H., Ursos L., Sa’adon R. Balancing opioid-induced gastrointestinal side effects with pain management: insights from the online community. J Opioid Manage. 2015;11:383–391. doi: 10.5055/jom.2015.0288. [DOI] [PubMed] [Google Scholar]
- 7.Yuill M.B., Zee M.L., Marcus D., Morgan D.J. Tolerance to the antinociceptive and hypothermic effects of morphine is mediated by multiple isoforms of c-Jun N-terminal kinase. Neuroreport. 2016;27:392–396. doi: 10.1097/WNR.0000000000000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleczkowska P., Hermans E., Kosson P., Kowalczyk A., Lesniak A., Pawlik K. Antinociceptive effect induced by a combination of opioid and neurotensin moieties vs. their hybrid peptide [Ile9]PK20 in an acute pain treatment in rodents. Brain Res. 2016;1648:172–180. doi: 10.1016/j.brainres.2016.07.039. [DOI] [PubMed] [Google Scholar]
- 9.Kowalczyk A., Kleczkowska P., Rękawek M., Kulik K., Lesniak A., Erdei A. Biological evaluation and molecular docking studies of AA3052, a compound containing a μ-selective opioid peptide agonist DALDA and d-Phe-Phe-d-Phe-Leu-Leu-NH2, a substance P analogue. Eur J Pharmaceut Sci. 2016;93:11–20. doi: 10.1016/j.ejps.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Dietis N., Guerrini R., Calo G., Salvadori S., Rowbotham D.J., Lambert D.G. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effects profile. Br J Anaesth. 2009;103:38–49. doi: 10.1093/bja/aep129. [DOI] [PubMed] [Google Scholar]
- 11.Morphy R., Rankovic Z. Designed multiple ligands: an emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 12.Kleczkowska P., Kosson P., Ballet S., van den Eynde I., Tsuda Y., Tourwe D. PK20, a new opioid–neurotensin hybrid peptide that exhibits central and peripheral antinociceptive effects. Mol Pain. 2010;6:86. doi: 10.1186/1744-8069-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleczkowska P., Kawalec M., Bujalska-Zadrozny M., Filip M., Zablocka B., Lipkowski A.W. Effects of the hybridization of opioid and neurotensin pharmacophores on cell survival in rat organotypic hippocampal slice cultures. Neurotox Res. 2015;28:352–360. doi: 10.1007/s12640-015-9553-9. [DOI] [PubMed] [Google Scholar]
- 14.Kaczyńska K., Kogut E., Zając D., Jampolska M., Andrzejewski K., Sulejczak D. Neurotensin-based hybrid peptide's anti-inflammatory activity in murine model of a contact sensitivity response. Eur J Pharmaceut Sci. 2016;93:84–89. doi: 10.1016/j.ejps.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Behbehani M.M., Pert A. A mechanism for the analgesic effect of neurotensin as revealed by behavioural and electrophysiological techniques. Brain Res. 1984;324:35–42. doi: 10.1016/0006-8993(84)90619-x. [DOI] [PubMed] [Google Scholar]
- 16.Clineschmidt B.V., McGuffin J.C., Bunting P.B. Neurotensin: antinocisponsive action in rodents. Eur J Pharmacol. 1979;54:129–139. doi: 10.1016/0014-2999(79)90415-1. [DOI] [PubMed] [Google Scholar]
- 17.Clineschmidt B.V., Martin G.E., Veber D.F. Antinocisponsive effects of neurotensin and neurotensin-related peptides. Ann NY Acad Sci. 1982;400:283–306. doi: 10.1111/j.1749-6632.1982.tb31576.x. [DOI] [PubMed] [Google Scholar]
- 18.Binder E.B., Kinkead B., Owens M.J., Nemeroff C.B. Neurotensin and dopamine interactions. Pharmacol Rev. 2001;53:453–486. [PubMed] [Google Scholar]
- 19.Chakrabarti A.C. Permeability of membranes to amino acids and modified amino acids: mechanisms involved in translocation. Amino Acids. 1994;6:213–229. [PubMed] [Google Scholar]
- 20.Benyhe S., Farkas J., Tóth G., Wollemann M. Met5-enkephalin-Arg6-Phe7, an endogenous neuropeptide, binds to multiple opioid and nonopioid sites in rat brain. J Neurosci Res. 1997;48:249–258. doi: 10.1002/(sici)1097-4547(19970501)48:3<249::aid-jnr7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 21.Hermans E., Geurts M., Maloteaux J.M. Agonist and antagonist modulation of [35S]GTP γS binding in transfected CHO ells expressing the neurotensin receptor. Br J Pharmacol. 1997;121:1817–1823. doi: 10.1038/sj.bjp.0701334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermans E., Maloteaux J.M., Octave J.N. Phospholipase C activation by neurotensin and neuromedin N in Chinese hamster ovary cells expressing the rat neurotensin receptor. Mol Brain Res. 1992;15:332–338. doi: 10.1016/0169-328x(92)90126-v. [DOI] [PubMed] [Google Scholar]
- 23.Yaksh T.L., Rudy T.A. Analgesia mediated by direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- 24.Thomas J.B., Giddings A.M., Wiethe R.W., Olepu S., Warner K.R., Sarret P. Identification of N-[(5-{[(4-methylphenyl)sulfonyl]amino}-3-(trifluoroacetyl)-1H-indol-1-yl)acetyl]-l-leucine (NTRC-824), a neurotensin-like nonpeptide compound selective for the neurotensin receptor type 2. J Med Chem. 2014;57:7472–7477. doi: 10.1021/jm500857r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu L.C., Tsaur M.L., Lin C.S., Hung Y.C., Wang T.Y., Chen C.C. Chronic intrathecal infusion of gabapentin prevents nerve ligation-induced pain in rats. Br J Anaesth. 2011;106:699–705. doi: 10.1093/bja/aer063. [DOI] [PubMed] [Google Scholar]
- 26.Portoghese P.S., Sultana M., Takemori A.E. Design of eptidomimetic delta opioid receptor antagonists using the message-address concept. J Med Chem. 1990;33:1714–1720. doi: 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- 27.Metzger T.G., Paterlini M.G., Portoghese P.S., Ferguson D.M. Application of the message-address concept to the docking of naltrexone and selective naltrexone-derived opioid antagonists into opioid receptor models. Neurochem Res. 1996;21:1287–1294. doi: 10.1007/BF02532369. [DOI] [PubMed] [Google Scholar]
- 28.Bryant S.D., Jinsmaa Y., Salvadori S., Okada Y., Lazarus L.H. Dmt and opioid peptides: a potent alliance. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- 29.Dooley C.T., Chung N.N., Wilkes B.C., Schiller P.W., Bidlack J.M., Pasternak G.W. An all d-amino acid opioid peptide with central analgesic activity from a combinatorial library. Science. 1994;266:2019–2022. doi: 10.1126/science.7801131. [DOI] [PubMed] [Google Scholar]
- 30.Koyyalagunta D. Opioid analgesics. In: Waldman S.D., editor. Pain management. Elsevier Saunders; Philadelphia: 2007. pp. 939–964. [Google Scholar]
- 31.Koener B., Focant M.C., Bosier B., Maloteaux J.M., Hermans E. Increasing the density of the D2L receptor and manipulating the receptor environment are required to evidence the partial agonist properties of aripiprazole. Prog Neuro-Psychopharmacol Biol Psychiatry. 2012;36:60–70. doi: 10.1016/j.pnpbp.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Dai W.L., Xiong F., Yan B., Cao Z.Y., Liu W.T., Liu J.H. Blockade of neuronal dopamine D2 receptor attenuates morphine tolerance in mice spinal cord. Sci Rep. 2016;6:38746. doi: 10.1038/srep38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viaro R., Calcagno M., Marti M., Borrelli E., Morari M. Pharmacological and genetic evidence for pre- and postsynaptic D2 receptor involvement in motor responses to nociceptin/orphanin FQ receptor ligands. Neuropharmacology. 2013;72:126–138. doi: 10.1016/j.neuropharm.2013.04.046. [DOI] [PubMed] [Google Scholar]
- 34.Zhu Y., King M.A., Schuller A.G., Nitsch J.F., Reidl M., Elde R.P. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 35.Abdelhamid E.E., Sultane M., Portoghese P.S., Takemor A.E. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J Pharmacol Exp Therapeut. 1991;258:299–303. [PubMed] [Google Scholar]
- 36.Harris G.C., Aston-Jones G. Beta-adrenergic antagonists attenuate somatic and aversive signs of opiate withdrawal. Neuropsychopharmacology. 1993;9:303–311. doi: 10.1038/npp.1993.66. [DOI] [PubMed] [Google Scholar]
- 37.Cooke J.H., Patterson M., Patel S.R., Smith K.L., Ghatei M.A., Bloom S.R. Peripheral and central administration of xenin and neurotensin suppress food intake in rodents. Obesity. 2009;17:1135–1143. doi: 10.1038/oby.2008.652. [DOI] [PubMed] [Google Scholar]
- 38.Stanley B.G., Hoebel B.G., Leibowitz S.F. Neurotensin: effects of hypothalamic and intravenous injections on eating and drinking in rats. Peptides. 1983;4:493–500. doi: 10.1016/0196-9781(83)90054-2. [DOI] [PubMed] [Google Scholar]
- 39.Zogovic B., Pilowsky P.M. Intrathecal neurotensin is hypotensive, sympathioinhibitory and enhances the baroreflex in anaesthetized rats. Br J Pharmacol. 2012;166:378–389. doi: 10.1111/j.1476-5381.2011.01760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaczynska K., Szereda-Przestaszewska M. Cardio-respiratory effects of systemic neurotensin injection are mediated through activation of neurotensin NTS1 receptors. Eur J Pharmacol. 2012;69:245–250. doi: 10.1016/j.ejphar.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 41.Oishi M., Inagaki C., Fujiwara M., Takaori S., Yajima H., Akazawa Y. Possible mechanisms of the triphasic effects of neurotensin on the rat blood pressure. Jap J Pharmacol. 1981;31:1043–1049. doi: 10.1254/jjp.31.1043. [DOI] [PubMed] [Google Scholar]
- 42.Sakurada T., Wako K., Sakurada C., Manome Y., Tan-No K., Sakurada S. Spinally-mediated behavioural responses evoked by intrathecal high-dose morphine: possible involvement of substance P in the mouse spinal cord. Brain Res. 1996;724:213–221. doi: 10.1016/0006-8993(96)00319-8. [DOI] [PubMed] [Google Scholar]
- 43.Sakurada T., Komatsu T., Sakurada S. Mechanisms of nociception evoked by intrathecal high-dose morphine. Neurotoxicology. 2005;26:801–809. doi: 10.1016/j.neuro.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 44.van Wimersma Greidanus T.B., Maigret C., Ten Haaf J.A., Spruijt B.M., Colbern D.L. The influence of neurotensin, naloxone, and haloperidol on elements of excessive grooming behavior induced by ACTH. Behav Neural Biol. 1986;46:137–144. doi: 10.1016/s0163-1047(86)90612-6. [DOI] [PubMed] [Google Scholar]
- 45.van Wimersma Greidanus T.B., van de Brug F., de Bruijckere L.M., Pabst P.H., Ruesink R.W., Hulshof R.L. Comparison of bombesin-, ACTH-, and beta-endorphin-induced grooming. Antagonism by haloperidol, naloxone, and neurotensin. Ann NY Acad Sci. 1988;525:219–227. doi: 10.1111/j.1749-6632.1988.tb38607.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.