

Abstract
Angiokinases, such as vascular endothelial-, fibroblast- and platelet-derived growth factor receptors (VEGFRs, FGFRs and PDGFRs) play crucial roles in tumor angiogenesis. Anti-angiogenesis therapy using multi-angiokinase inhibitor has achieved great success in recent years. In this study, we presented the design, synthesis, target identification, molecular mechanism, pharmacodynamics (PD) and pharmacokinetics (PK) research of a novel triple-angiokinase inhibitor WXFL-152. WXFL-152, identified from a series of 4-oxyquinoline derivatives based on a structure–activity relationship study, inhibited the proliferation of vascular endothelial cells (ECs) and pericytes by blocking the angiokinase signals VEGF/VEGFR2, FGF/FGFRs and PDGF/PDGFRβ simultaneously in vitro. Significant anticancer effects of WXFL-152 were confirmed in multiple preclinical tumor xenograft models, including a patient-derived tumor xenograft (PDX) model. Pharmacokinetic studies of WXFL-152 demonstrated high favourable bioavailability with single-dose and continuous multi-dose by oral administration in rats and beagles. In conclusion, WXFL-152, which is currently in phase Ib clinical trials, is a novel and effective triple-angiokinase inhibitor with clear PD and PK in tumor therapy.
Key words: Drug synthesis, Tumor, Anti-angiogenesis therapy, Multi-angiokinase inhibitor, Pharmacokinetic
Abbreviations: ATCC, American Type Culture Collection; AUC, area under the plasma concentration–time curve; CE, collision energy; CL, systemic clearance; Cmax, maximum plasma concentration; EC, vascular endothelial cell; ECM, endothelial cell medium; ERKs, extracellular signal-regulated kinases; FGF, fibroblast growth factor; FGFRs, fibroblast growth factor receptors; HBVPs, human brain vascular pericytes; HUVECs, human umbilical vein endothelial cells; IC50, half maximal inhibitory concentration; IHC, immunohistochemistry; i.v., intravenous injection; LC–MS, liquid chromatography mass spectrometry; LLOQ, lower limit of quantification; MRM, multiple reaction monitoring; MsOH, methane sulfonic acid; NMR, nuclear magnetic resonance; PD, pharmacodynamics; PDB, protein data bank; PDGF, platelet-derived growth factor; PDGFRs, platelet-derived growth factor receptors; PDX, patient-derived tumor xenograft; PK, pharmacokinetics; PM, pericyte medium; p.o., per os; QC, quality control; RE, values and relative error; RSD, relative standard deviation; RTKs, receptor tyrosine kinases; TGI, tumor growth inhibition rate; TLC, thin-layer chromatography; Tmax, time the maximum concentration occurred; ULOQ, up limit of quantitation; Vdss, volume of distribution at steady state; VEGF, vascular endothelial growth factor; VEGFRs, vascular endothelial growth factor receptors
Graphical abstract
WXFL-152 is a novel and effective triple-angiokinase inhibitor with clear pharmacodynamics and pharmacokinetics in tumour therapy. Recently, WXFL-152 as a novel clinical candidate is being tested in phase Ib clinical trials. Further clinical trials will focus on the treatment of solid tumors and pulmonary fibrosis.

1. Introduction
Vascular endothelial-, fibroblast- and platelet-derived growth factor receptors (VEGFRs, FGFRs and PDGFRs) are a class of receptor tyrosine kinases (RTKs), which contain extracellular domain of ligand binding sites, hydrophobic helicity of single transmembrane, and intracellular kinases domains1,2. Angiokinases, such as the intracellular kinases domains of VEGFRs, FGFRs and PDGFRs, which mainly promote angiogenesis, are proven to be clinical targets in cancer therapy3, 4, 5, 6. When the growth factors, such as VEGF, FGF and PDGF, bind the correspondingly extracellular domains of the RTKs, the angiokinase signals will be activated and deliver growth signals intracellularly by transferring a phosphate group of ATP to the tyrosine residue. Overactivated angiokinase signals contributed to the angiogenesis, tumorigenesis, metastasis and progression7,8.
Tumor angiogenesis is a complicated process that involves multiple angiokinase signals and angiogenesis-related cells (vascular endothelial cell, pericytes and smooth muscle cells)9. VEGF/VEGFR2 signals promote tumor angiogenesis by stimulating the growth of adjacent vascular endothelial cells (ECs) to form new vascular sprouts in tumor tissue10,11. The PDGF-BB produced by ECs promotes the pericytes (PDGFR-positive) to enwrap and stabilize the tumor vasculature, and protect the new vascular sprouts by PDGF/PDGFR signalling12. Abnormal FGF/FGFRs not only promote tumor development by directly driving cancer cells and endothelial cells proliferation, but also support tumor angiogenesis by synergizing with the VEGF and PDGF signalling pathways13, 14, 15. Fibroblast growth factor 2, also known as bFGF, is an important pro-angiogenic factor in tumor angiogenesis16,17. bFGF and VEGF synergistically increased vascular density and permeability18. The simultaneous overexpression of bFGF and PDGF-BB in mouse fibrosarcoma results in the formation of high density primary vascular plexus19. Additionally, bFGF can stimulate the expression of PDGFR-α and -β in activated endothelial cells, thus stimulating the tumor angiogenesis by promoting PDGF/PDGFRβ signalling pathway20. This synergistic effect of multiple angiokinase signals in tumor angiogenesis may limit the anti-tumor effect of single-target therapy21. Some drugs targeting VEGF or VEGFR2 have been approved for cancer therapy, such as bevacizumab (monoclonal antibody targets VEGF) and apatinib (VEGFR2 inhibitor)22. However, limited clinical benefits have been observed during anti-angiogenic therapy for cancer by blocking VEGF/VEGFR2 signals alone23. The reasons are that alternative mechanism of tumor adaptation to VEGF/VEGFRs inhibition includes the induction of compensatory pro-angiogenic growth signals, such as FGF/FGFRs, and the protection effect of pericytes to ECs drived by the PDGF/PDGFRs signal24,25. Anti-angiogenesis therapy targeting multiple specific angiokinases, such as nintedanib (VEGFRs, FGFRs and PDGFRs inhibitor), provided great clinical benefits for cancer therapy26, 27, 28.
In our previous study, we obtained the quinoline-thiourea derivative WXFL-255 from a series of 4-oxyquinoline derivatives as a potent angiokinase inhibitor (targeting VEGFRs, FGFRs and PDGFRs)29. To improve its selectivity for VEGFR2, FGFR1 and PDGFRβ, further optimization on its four substituents (R1‒R4) was performed. A novel and priority candidate WXFL-152 was identified based on the structure–activity relationship (SAR) study. The aim of the study was to clarify the design, synthesis, target identification, molecular mechanism, pharmacodynamics (PD) and pharmacokinetics (PK) of WXFL-152.
2. Results and discussions
2.1. Chemistry and SAR study
The compound WXFL-255, 4-(3-chloro-4-(3-cyclopropylthioureido)phenoxy)-7-methoxyquinoline-6-carboxamide, is a potential triple-angiokinase inhibitor (IC50 values are 9.4 (VEGFR2), 188.0 (FGFR1), 143.0 (PDGFRβ), 90.2 (HUVEC) nmol/L, respectively) obtained in our previous study as shown in Fig. 1A and Scheme 129. In this study, WXFL-255, as the lead compound, was optimized in its four major groups (R1–R4) based on the consideration of lipophilicity, lipid water distribution coefficient under physiological conditions, apparent volume of distribution and the affinity toward the targets, as shown in Fig. 1B.
Figure 1.

Typical optimization routes of the clinical candidate compound D4 (WXFL-152). (A) WXFL-255 as the lead compound was obtained from a series of 4-oxyquinoline derivatives. (B) WXFL-152 was identified based on the four major groups optimization of the lead compound (R1–R4). (C) and (D) Molecular docking simulation of WXFL-152 with VEGFR2 and FGFR1.
Scheme 1.

Synthesis of the lead compound WXFL-25529. Reagents and conditions: (a) 1A, 2,2-dimethyl-1,3-dioxane-4,6-dione, trimethyl orthoformate, 90 °C, 1 h; (b) 1B, diphenyl ether, 220 °C, 1 h, methyl tert-butyl ether, 140 °C, 12 h; (c) 1C, NaOH, water and methanol, 30 °C, 2 h; (d) 1D, N,N-dimethylformamide, 1,2-dichloroethane, thionyl chloride, 110 °C, 2 h; (e) 1E, dichloromethane, aqueous ammonia, 25 °C, 0.5 h; (f) 1F, 3-chloro-4-nitro-phenol, 140 °C, 12 h; (g) 1G, Fe, HOAc, 80 °C, 16 h; (h) 1H, 1,1′-thiocarbonyldi-2(1H)-pyridone, dioxane, 15 °C, 1 h, refluxing, 12 h; (i) lead compound WXFL-255, cyclic propylamine, 25 °C, 16 h.
Partially typical compounds (A1‒A10) with R1 substituted groups, including various amide, ester, heterocycle and cyan, were showed in Table 1, and their synthetic routes as shown in Scheme 2. Based on a kinase inhibition assay for VEGFR2, FGFR1 and PDGFRβ, amide-substituted in R1 of the quinoline ring was determined to be superior to the other alternatives. Especially the N,N-disubstituted amide derivatives significantly decreased the inhibitory activity of the three kinases. Interestingly, amide-substituted compounds A1 (IC50 = 67.6 nmol/L), A5 (IC50 = 101.5 nmol/L) and ester-substituted compound A2 (IC50 = 72.5 nmol/L) even surpassed the lead compound (IC50 = 143.0 nmol/L) in the inhibition of PDGFRβ. However, the compounds showed no superiority in the inhibition of VEGFR2 and FGFR1. The ester-substituted compound A2 was effective at inhibiting VEGFR2 and PDGFRβ, but failed to inhibit FGFR1 (IC50 values are 28.1 (VEGFR2), 2394.9 (FGFR1) and 72.5 (PDGFRβ) nmol/L, respectively).
Table 1.
VEGFR2/FGFR1/PDGFRβ inhibition of the typical compounds A1‒A10 obtained by R1 group optimization.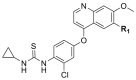
| Compd. | Structure or R1 | IC50 (nmol/L) |
||
|---|---|---|---|---|
| VEGFR2 | FGFR1 | PDGFRβ | ||
| Lead compd. | 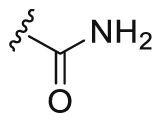 |
9.4 | 188.0 | 143.0 |
| A1 |  |
20.1 | 293.7 | 67.6 |
| A2 |  |
28.1 | 2394.9 | 72.5 |
| A3 | 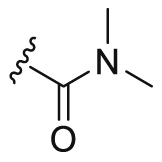 |
503.4 | 8130.1 | 414.3 |
| A4 | 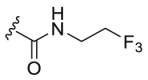 |
79.5 | 4436.8 | 316.1 |
| A5 |  |
156.4 | 8262.5 | 101.5 |
| A6 | 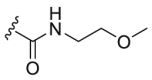 |
119.9 | 4235.1 | 240.6 |
| A7 |  |
116.3 | 6804.1 | 210.2 |
| A8 | 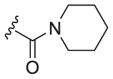 |
1668.4 | >10,000 | 1050.6 |
| A9 | 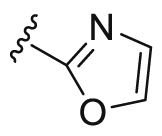 |
101.3 | 2644.5 | 135.6 |
| A10 | 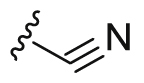 |
172.4 | 1903.3 | 504.0 |
Scheme 2.

Synthesis of A route (compounds A1‒A10). Reagents and conditions: (A)-(a) thionyl chloride, trimethyl orthoformate, 90 °C, 1 h; (b) 3-chloro-4-nitro-phenol, chlorobenzene, 140 °C, 15 h; (c) Fe, NH4Cl, 90 °C, 2 h; (d) 1,1′-thiocarbonyldi-2(1H)-pyridone, 25 °C, 0.5 h, 120 °C, 12 h; (e) cyclopropylamine, 25 °C, 2 h; (f) LiOH, 25 °C, 2 h; (g) amine, 25 °C, 15 h; (B)-(a) dimethyl 1,2-vinylene carbonate, 190 °C, 0.5 h; (b) Fe, NH4Cl, 80 °C, 1 h; (c) 1,1′-thiocarbonyldi-2(1H)-pyridone, dioxane, 65 °C, 1 h; (d) cyclic propylamine, 25 °C, 1 h; (C)-(a) LiOH, THF, 20 °C, 4 h; (b) NH2Cl, HATU, DIPEA, DCM, 20 °C, 12 h; (c) thionyl chloride, 80 °C, 3 h; (d) Fe, NH4Cl, 90 °C, 2 h; (e) 1,1′-thiocarbonyldi-2(1H)-pyridone, 10 °C, 10 min, 110 °C, 2 h, cyclic propylamine, 20 °C, 2 h.
Alkyl-, alkoxy-, and heterocycle-substituted groups in R2 were synthesized and tested for kinase inhibition. The typical compounds B1–B3 were shown in Table 2 and the synthetic routes as shown in Scheme 3. The results indicated that bulky group decreased the inhibition of VEGFR2, FGFR1 and PDGFRβ significantly. However, extending the straight chain in R2 increased the inhibition of PDGFRβ, such as the compound B2. Unfortunately, the activity against VEGFR2 and FGFR1 of compound B2 was significantly decreased.
Table 2.
VEGFR2/FGFR1/PDGFRβ inhibition of the typical compounds B1–B3 obtained by R2 group optimization.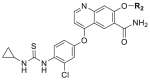
| Compd. | Structure or R2 | IC50 (nmol/L) |
||
|---|---|---|---|---|
| VEGFR2 | FGFR1 | PDGFRβ | ||
| Lead compd. | CH3 | 9.4 | 188.0 | 143.0 |
| B1 | 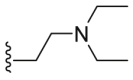 |
335.2 | 9032.0 | 388.9 |
| B2 | 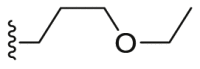 |
26.7 | 216.4 | 56.9 |
| B3 | 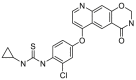 |
226.8 | 777.1 | 409.8 |
Scheme 3.

Synthesis of B route (compounds B1–B3). Reagents and conditions: (A)-(a) BBr3, 25 °C, 16 h; (b) KCO4, bromide, DMF, 80 °C, 16 h; (c) Zn, AcOH, 80 °C, 16 h; (d) 1,1′-thiocarbonyldi-2(1H)-pyridone, 80 °C, 16 h; (e) cyclopropylamine, 20 °C, 16 h; (B)-(a) trioxymethylene, H2SO4, 70 °C, 2.5 h; (b) Raney Ni, MeOH, 25 °C, 0.5 h; (c) 1,1′-thiocarbonyldi-2(1H)-pyridone, 70 °C, 1 h, 25 °C, cyclopropylamine, 1 h.
A series of novel candidates C1–C41 with amino-substituted groups including –NH2, –NH–R′ (R′ were alkyl, benzene series, heterocycle and cyclane) and a nitrogen heterocyclic ring in the R3 position were designed, synthesized and tested as shown in Table 3 and Scheme 4. Since the R3 group was located in the hydrophobic pockets of the VEGFR2, FGFR1 and PDGFRβ, the different size of the three kinase cavities determined the selection of the R3 group. The kinase inhibition results showed that cyclopropane amine-substituted in R3 demonstrated the best inhibition of VEGFR2 (IC50 = 9.4 nmol/L), and dimethylamino-substituted compound C2 in R3 had the best inhibitory effect on FGFR1 (IC50 = 63.0 nmol/L). The 2-thioxoimidazolidin-4-one-substituent compound C41 in the thiourea-structure exhibited the best inhibition of PDGFRβ (IC50 = 12.2 nmol/L) but poor inhibition of FGFR1.
Table 3.
VEGFR2/FGFR1/PDGFRβ inhibition of the typical compounds C1–C41 obtained by R3 group optimization.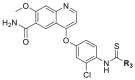
| Compd. | Structure or R3 | IC50 (nmol/L) |
||
|---|---|---|---|---|
| VEGFR2 | FGFR1 | PDGFRβ | ||
| Lead compd. | 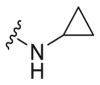 |
9.4 | 188.0 | 143.0 |
| C1 | 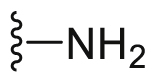 |
32.7 | 436.4 | 77.2 |
| C2 | 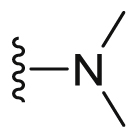 |
78.6 | 63.0 | 311.1 |
| C3 | 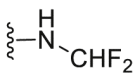 |
10.0 | 129.9 | 84.3 |
| C4 | 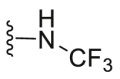 |
22.6 | 378.4 | 139.9 |
| C5 | 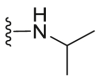 |
30.1 | 645.7 | 78.5 |
| C6 | 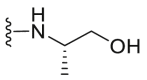 |
110.3 | 634.7 | 89.1 |
| C7 | 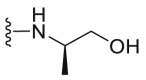 |
55.1 | 415.9 | 80.1 |
| C8 | 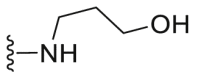 |
59.7 | 1216.3 | 87.3 |
| C9 | 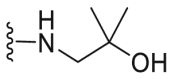 |
244.7 | 4358.0 | 696.7 |
| C10 | 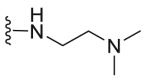 |
2353.5 | 6757.7 | 62.8 |
| C11 | 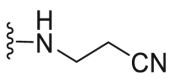 |
208.5 | 1934.4 | 254.6 |
| C12 | 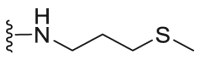 |
52.2 | 765.7 | 19.7 |
| C13 | 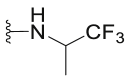 |
216.9 | 1583.1 | 508.6 |
| C14 | 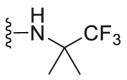 |
234.4 | 1420.8 | 28.1 |
| C15 | 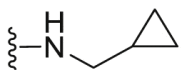 |
16.6 | 406.2 | 31.1 |
| C16 | 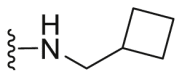 |
20.5 | 1103.5 | 161.4 |
| C17 | 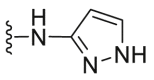 |
72.7 | 1831.4 | 347.8 |
| C18 | 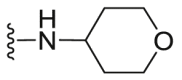 |
339.0 | 5073.1 | 178.4 |
| C19 | 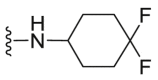 |
1649.1 | 7616.3 | 167.3 |
| C20 | 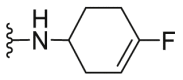 |
1103.5 | 7478.2 | 399.5 |
| C21 |  |
107.5 | 2671.4 | 54.2 |
| C22 | 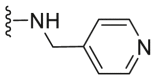 |
195.8 | 1729.0 | 62.8 |
| C23 | 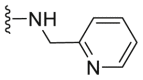 |
372.6 | 2748.4 | 73.3 |
| C24 | 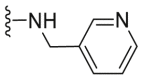 |
127.6 | 2933.7 | 37.0 |
| C25 |  |
74.1 | 6100.4 | 193.7 |
| C26 |  |
104.6 | 906.7 | 24.7 |
| C27 | 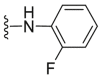 |
138.8 | 2798.5 | 37.3 |
| C28 | 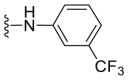 |
117.6 | 3541.8 | 563.7 |
| C29 | 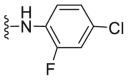 |
307.5 | 9154.3 | 2040.9 |
| C30 | 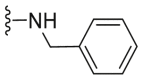 |
675.8 | 4898.2 | 361.0 |
| C31 | 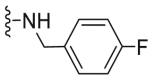 |
206.8 | 3608.5 | 85.9 |
| C32 | 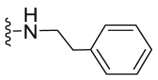 |
255.3 | 3377.2 | 186.4 |
| C33 | 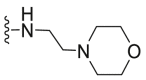 |
397.9 | 3754.0 | 41.7 |
| C34 | 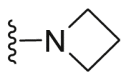 |
63.6 | 70.4 | 63.1 |
| C35 | 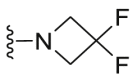 |
36.1 | 135.8 | 184.2 |
| C36 | 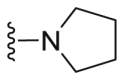 |
25.8 | 226.8 | 867.9 |
| C37 |  |
79.6 | 475.7 | 974.5 |
| C38 | 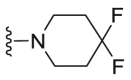 |
194.0 | 457.3 | 323.7 |
| C39 | 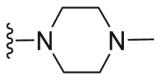 |
472.2 | >10,000 | 221.9 |
| C40 | 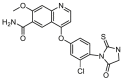 |
68.1 | 1258.7 | 156.5 |
| C41 | 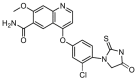 |
51.7 | 1565.8 | 12.2 |
Scheme 4.

Synthesis of C route (compounds C1–C43). Reagents and conditions: amines react with 1H to form thiourea under the similar reaction conditions of the last step of lead compound WXFL-255, see Supporting Information.
Compounds D1‒D11, which contained substitutes on the benzene and benzoheterocycle in the R4 position, were obtained and tested as shown in Table 4 and Scheme 5. The R4 substituted phenyl group was inserted into another hydrophobic pocket near the hinge region of the kinases, so the aryl heterocycles substitution of the nonpolar phenyl weakened the binding force to the pocket, and decreased the inhibition of VEGFR2, FGFR1 and PDGFRβ, such as D7‒D10. On the contrary, appropriate substituents connected to the phenyl could increase the van de Waals of the compounds with the kinase hydrophobic pockets, and improve the activity against the kinases. The compounds with the benzene ring substituted by two groups in the R4 position tended to inhibit VEGFR2, FGFR1 and PDGFRβ simultaneously. Interestingly, the compound D4 (WXFL-152) containing the 1-chloro-2-fluorobenzene group in R4 was identified as the priority candidate for further testing in vitro and in vivo based on the cytotoxicity on HUVECs induced by VEGFA-165 (Table 5).
Table 4.
VEGFR2/FGFR1/PDGFRβ inhibition of the typical compounds D1‒D11 obtained by R4 group optimization.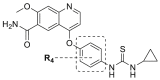
| Compd. | Structure or R4 | IC50 (nmol/L) |
||
|---|---|---|---|---|
| VEGFR2 | FGFR1 | PDGFRβ | ||
| Lead compd. |  |
9.4 | 188.0 | 143.0 |
| D1 | 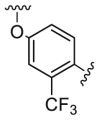 |
16.2 | 242.4 | 406.1 |
| D2 | 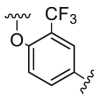 |
2043.5 | 4923.5 | 956.3 |
| D3 | 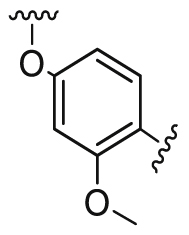 |
231.0 | 3144.0 | 54.0 |
| D4/WXFL-152 | 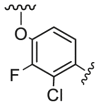 |
7.0 | 69.0 | 31.0 |
| D5 | 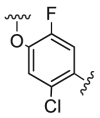 |
25.2 | 713.0 | 68.8 |
| D6 | 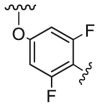 |
64.0 | 541.0 | 162.0 |
| D7 | 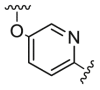 |
8803.8 | >10,000 | 3807.4 |
| D8 | 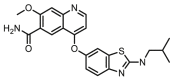 |
4802.4 | 3683.8 | 175.8 |
| D9 | 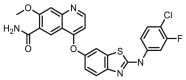 |
>10,000 | >10,000 | >10,000 |
| D10 | 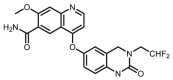 |
375.9 | 6042.3 | 3183.4 |
| D11 | 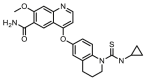 |
75.7 | 3704.6 | 857.4 |
Scheme 5.

Synthesis of D route (compounds D1‒D11). Reagents and conditions: (A) the synthesis methods are similar to the synthesis of lead compound (from 1E to lead compound); (B)-(a) HBr, reflux, 4 h; (b) compound 1E, 100 °C, 0.5 h; (c) aldehyde, STAB, 100 °C, 12 h; (C)-(a) Renny Ni/H2, 20 °C, 1 h; (b) CDI, DMF, 70 °C, 16 h; (c) BBr3, DMF, 78 °C, 0.5 h; (d) compound 1E, Cs2CO3, 100 °C, 1.5 h; (D)-(a) PtO2, MeOH, 45 °C, 24 h; (b) BBr3, CHCl2, 15 °C, 2 h; (c) compound 1E, Cs2CO3, NMP, 120 °C, 2 h; (d) cyclopropylthiocyanate, THF, 25 °C, 12 h.
Table 5.
Cytotoxicity of optimized compounds on HUVECs (induced by 20 ng/mL VEGFA-165), HBVPs (induced by 20 ng/mL PDGF-BB) and inhibition of optimized compounds on VEGFR2/FGFR1/PDGFRβ.
| Compd. | IC50 |
||||
|---|---|---|---|---|---|
| VEGFR2 (nmol/L) | FGFR1 (nmol/L) | PDGFRβ (nmol/L) | HUVECs (nmol/L) | HBVPs (μmol/L) | |
| Lead compd. | 9.4 | 188.0 | 143.0 | 90.2 | / |
| D4/WXFL-152 | 7.0 | 69.0 | 31.0 | 71.0 | 1.94 |
| A1 | 20.1 | 293.7 | 67.6 | 228.8 | / |
| B2 | 26.7 | 216.4 | 56.9 | 145.9 | / |
| C2 | 78.6 | 63 | 311.1 | >10,000.0 | / |
| C3 | 10.0 | 129.9 | 84.3 | 31.0 | / |
| C4 | 22.6 | 378.4 | 139.9 | 30.9 | / |
| C16 | 20.5 | 1103.50 | 161.4 | 20.7 | / |
| C25 | 74.1 | 6100.40 | 193.7 | 84.1 | / |
| Nintedanib | 8.8 | 93.9 | 35.4 | 41.0 | 1.67 |
The method for synthesis of WXFL-152 was demonstrated in Scheme 6. The Cl of quinoline was replaced by a phenoxy to obtain compound b. WXFL-152 was obtained by the condensation of compound b and amine with 1,1′-thiocarbonyldi-2(1H)-pyridone. Then, WXFL-152 reacted with methane sulfonic acid (MsOH) to yield WXFL-152 mesylate as described in our previous patent30. The detailed synthesis methods, including the reagents and conditions of the other compounds mentioned in this study, were described in our previous patent29. The mesylate form of WXFL-152 was used for further biological tests, including PD and PK.
Scheme 6.

The synthesis route of WXFL-152 and its mesylate.
2.2. Molecular modelling of WXFL-152
Molecular docking simulations of WXFL-152 with VEGFR2 and FGFR1 were performed using AutoDock Vina. The co-crystal structure of VEGFR2 complexed with sorafenib (protein data bank, PDB ID: 4ASD) was used as a template. As shown in Fig. 1C, the quinoline ring and the thiourea of WXFL-152 bound to Cys919 (the hinge region amino acid) and Glu885 on VEGFR2 in the form of hydrogen bonds, thus preventing ATP from binding to the ATP binding site of VEGFR2. In the co-crystal structure molecular modelling of FGFR1 with ponatinib (PDB ID: 4V04), the quinoline ring and the thiourea group of WXFL-152 bound to Ala564 and Glu531 on FGFR1 via forming hydrogen bonds to prevent ATP from binding to the ATP binding site of FGFR1 (Fig. 1D).
2.3. Kinase inhibition assay of WXFL-152
Invitrogen Z′-LYTE® Kinase detection technology was used to analyse the optimized compounds and profile the kinase inhibition of WXFL-152. As shown in Table 6, the target kinases of WXFL-152 were VEGFR types 1, 2 and 3 (IC50s: 30.9, 7.0 and 8.5 nmol/L, respectively); PDGFRα and PDGFRβ (IC50s: 135.0 and 31.0 nmol/L); and FGFR types 1, 2, 3 and 4 (IC50s: 69.0, 15.5, 132.0 and 131.0 nmol/L, respectively). WXFL-152 also inhibited MAP4K4, MINK, TNIK, AUR2, ABL and LCK, with IC50 values of 79.0, 55.0, 118.0, 262.0, 520.0 and 252.0 nmol/L, respectively. WXFL-152 showed almost no inhibitory activity against 16 other selected human protein kinases. These data demonstrated that WXFL-152 is a potent triple inhibitor targeting VEGFRs, PDGFRs and FGFRs. In addition, we tested and compared the kinases inhibition activity of WXFL-152 with that of nintedanib. Angiokinases, such as VEGFRs, FGFRs and PDGFRs, are not only a pro-angiogenic factors, but also a pro-fibrotic factors31. Nintedanib, which targets VEGFRs, FGFRs and PDGFRs simultaneously, has achieved great clinical success in tumor therapy and idiopathic pulmonary fibrosis (IPF). The lead compound WXFL-255 has the similar targets with nintedanib. Thus, nintedanib was chosen as the control compound in our study. The results showed that WXFL-152 and nintedanib demonstrated similar inhibition effect on VEGFR2 (i.e., IC50 value of WXFL-152 vs. nintedanib: 7.0 ± 2.0 vs. 8.8 ± 3.6 nmol/L), FGFR1 (IC50 value of WXFL-152 vs. nintedanib: 69.0 ± 30.0 vs. 93.9 ± 37.0 nmol/L) and PDGFRβ (IC50 value of WXFL-152 vs. nintedanib: 31.0 ± 7.0 vs. 35.4 ± 14.0 nmol/L).
Table 6.
In vitro kinase inhibition profile of WXFL-152.
| Kinase | IC50 (nmol/L) | Kinase | IC50 (nmol/L) |
|---|---|---|---|
| VEGFR1 | 30.9 | EphA2 | >3333 |
| VEGFR2 | 7.0 | GSK3β | >10,000 |
| VEGRR3 | 8.5 | MAP4K4 | 79.0 |
| FGFR1 | 69.0 | JAK2 | >10,000 |
| FGFR2 | 15.5 | JNK1 | >10,000 |
| FGFR3 | 132.0 | LCK | 252.0 |
| FGFR4 | 131.0 | MAPKAPK2 | >10,000 |
| PDGFRα | 135.0 | MINK | 55.0 |
| PDGFRβ | 31.0 | MST4 | >10,000 |
| ABL | 520.0 | p38α | >3333 |
| AUR2 | 262.0 | PDK1 | >10,000 |
| CAMK2α | >10,000 | PKA | >10,000 |
| CDK2 | >10,000 | SRC | 2648.0 |
| CHK1 | >10,000 | TAOK2 | >10,000 |
| CK1α | >10,000 | TNIK | 118.0 |
| DYRK3 | >10,000 | / | / |
2.4. WXFL-152 inhibited HUVECs proliferation, tube formation, migration and invasion
VEGF/VEGFR2 is a key signal to induce the proliferation, migration, and tube formation of endothelial cell in tumor angiogenesis. To analyse the proliferation inhibition of WXFL-152 in vitro, VEGF-induced HUVECs proliferation treated with WXFL-152 was evaluated using a CellTiter-Glo luminescent cell viability assay. The results indicated that WXFL-152 significantly inhibited the proliferation of HUVECs (IC50 71.0 ± 5.0 nmol/L) induced by 20 ng/mL hVEGF-165. Tube formation of ECs is important for angiogenesis. WXFL-152 significantly suppressed the tube formation of HUVECs induced by 50 ng/mL hVEGF-165 at doses of 0.5 and 5.0 μmol/L (WXFL-152 treated groups vs. hVEGF-165 control group, P < 0.01). The inhibition effects at 2, 4 and 6 h were shown in Fig. 2A (representative images of tube formation of HUVECs) and B (the numbers of HUVECs tubes in the different groups and times). Wound healing assays were performed to investigate the effects of WXFL-152 on cell migration. The migration of HUVECs was appreciably inhibited by treatment with 5.0 μmol/L WXFL-152 compared with the hVEGF-165 treated control group (5.0 μmol/L, P < 0.05; 50.0 μmol/L, P < 0.001). Representative images of wound closure were shown in Fig. 2C, and the wound closure percentages in the different groups were shown in Fig. 2E. A transwell invasion assay was used to evaluate the invasion ability of HUVECs driven by 50 ng/mL hVEGF-165 under exposure to WXFL-152. The results showed that WXFL-152 significantly inhibited the invasion of HUVECs in a dose-dependent manner compared with the hVEGF-165 treated control group (0.5 μmol/L, P < 0.01; 5.0 and 50.0 μmol/L, P < 0.001). Typical images were shown in Fig. 2D, and the invasive cell number percentages were shown in Fig. 2F. In conclusion, these results confirmed that WXFL-152 was an effective bioactive inhibitor that blocked VEGF/VEGFR2 signals in cellular level.
Figure 2.

WXFL-152 inhibited HUVECs' tube formation, migration and invasion. (A) Representative images of tube formation of HUVECs in different groups. (B) The numbers of HUVECs tubes in different groups. (C) Representative images of wound closure. (D) Representative images of invasive cells in the transwell invasion assay. (E) The wound closure percentages of different groups in the wound healing assay. (F) The invasive cell number percentages of different groups in the transwell invasion assay (∗P < 0.05, ∗∗∗P < 0.01, ∗∗∗P < 0.001). Data are represented as mean ± SEM (n = 3).
2.5. WXFL-152 inhibited HBVPs proliferation
In tumor angiogenesis, ECs-secreted PDGF-BB promotes the pericytes proliferation to protect the new vascular sprouts by PDGF/PDGFR signal. To analyse the proliferation inhibition of WXFL-152 in pericytes, cell model of PDGF-BB-induced HBVPs (PDGFR-positive) proliferation and cell counting kit-8 (CCK-8) assay were performed. The results indicated that WXFL-152 significantly inhibited the proliferation of HBVPs (IC50 value of WXFL-152 vs. nintedanib: 1.94 ± 0.14 vs. 1.67 ± 0.11 μmol/L) induced by 20 ng/mL hPDGF-BB as shown in Table 5. These results indicated that WXFL-152 could not only inhibit ECs proliferation induced by hVEGF-165, but also inhibit pericytes proliferation induced by PDGF-BB in tumor angiogenesis.
2.6. WXFL-152 inhibited the VEGF/VEGFR2/ERK, FGF/FGFRs/ERK and PDGF/PDGFRβ/ERK signalling pathway
Tumor angiogenesis stimulated by multiple angiokinase signals such as VEGF/VEGFRs, FGF/FGFRs and PDGF/PDGFRs in ECs and pericytes plays a crucial role in tumorigenesis, metastasis and progression. Blocking these signals in ECs and pericytes can inhibit tumor angiogenesis and suppress tumor growth effectively32. The kinase inhibition assays showed that WXFL-152 significantly inhibited human VEGFR2, FGFRs and PDGFRβ. To reveal the further molecular mechanism of WXFL-152, Western blot assays were used to evaluate the effects on VEGF/VEGFR2, FGF/FGFRs and PDGF-BB/PDGFRβ and their downstream signalling. As shown in Fig. 3A and B, 5.0 μmol/L WXFL-152 significantly suppressed the phosphorylation of VEGFR2 and ERK induced by 50 ng/mL hVEGF 165 in HUVECs (P < 0.01). The PDGFRβ phosphorylation leads to the activation of various downstream signalling substrates, and plays important roles in cell proliferation (pericytes, vascular smooth muscle cells and fibroblasts) and blood vessel formation. As shown in Fig. 3C and D, the phosphorylation of PDGFRβ and ERK induced by 50 ng/mL PDGF-BB in HBVPs were significantly blocked by 0.5 and 5.0 μmol/L WXFL-152, respectively. The phosphorylation of FGFR1‒4 and ERK induced by 50 ng/mL bFGF in HUVECs was significantly inhibited by 5.0 μmol/L WXFL-152, respectively (P < 0.01, as shown in Fig. 3E and F). The above-mentioned results suggested that the anti-angiogenic effect of WXFL-152 inhibited the proliferation, migration, invasion, and tube formation of endothelial cells and pericytes respectively by targeting VEGFR2 and FGFRs on the endothelial cell membrane and PDGFRβ on the pericytes, as shown in Fig. 3G.
Figure 3.

WXFL-152 inhibited the VEGFR2, FGFRs and PDGFRβ/ERK signalling pathways. (A) Western blot images of VEGFR2/ERK proteins and their phosphorylation induced by 50 ng/mL VEGF-165 in HUVECs. (B) Statistical analysis of (A). (C) PDGFRβ and ERK1/2 protein and their phosphorylation induced by 50 ng/mL PDGF-BB in HBVPs. (D) Statistical analysis of (C). (E) Western blot images of FGFRs/ERK proteins and their phosphorylation forms induced by 50 ng/mL bFGF in HUVECs. (F) Statistical analysis of (E). (G) The diagram displayed the anti-angiogenesis mechanism of WXFL-152. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. Data are represented as mean ± SEM (n = 3).
2.7. WXFL-152 inhibited tumor-induced angiogenesis in a transgenic zebrafish model
To visually display the anti-tumor mechanism and effectiveness of WXFL-152 in vivo, a xenograft tumor model in the Tg (flk1:eGFP; Gata1:dsRed) double transgenic zebrafish model was established as shown in Fig. 4A. In this model, the volume and neovascularization in microtumors could be observed by high-resolution fluorescence microscopeusing time-lapse imaging strategy. Red fluorescence-labelled CT26 colon adenocarcinoma cells were microinjected into zebrafish embryos. After 7 days, endothelial cells of zebrafish in the control group had filled the inner space of the xenografted tumor and established a primary vascular network as the tumor grew. Moreover, few tumors exhibiting angiogenesis and small tumors were observed in the group treated with 0.5 and 2.0 μmol/L WXFL-152. Typical images of tumor cells (red) and tumor angiogenesis (green) in transgenic zebrafishes were shown in Fig. 4B, and the numbers of tumor vessels were counted as shown in Fig. 4C. These results not only confirmed the anti-tumor effect of WXFL-152 but also visually demonstrated neovascularization during tumor growth in vivo.
Figure 4.

WXFL-152 inhibited tumor growth in subcutaneous xenograft models of transgenic zebrafish and mice. (A) The CT-26 cell line xenograft tumor model in the Tg (flk1: eGFP; Gata1: dsRed) double transgenic zebrafish model. (B) Typical images of tumor cells (red) and tumor angiogenesis (green) in transgenic zebrafishes captured by confocal. (C) The numbers of tumor vessels in each group, ∗P < 0.05. Data are represented as mean ± SEM (n = 8). (D) WXFL-152 inhibited tumor growth in A549 and NCI-H520 cell lines subcutaneous xenograft models of mice and liver cancer PDX model. Except for the nintedanib (100 mg/kg) treatment group, the animals in the other treatment groups did not show obvious weight loss throughout the entire treatment period in the A549 lung cancer xenograft model. In the NCI-H520 lung cancer xenograft model, compared with the control group, the animals in the treatment groups showed a decrease in body weight. In the patient-derived tumor (LI-03-0020) xenograft model, there was no significant decrease in animal weight. (E) Typical H&E staining images of the vital organs (heart, liver, spleen, lung and kidney, × 400) of mice treated with WXFL-152.
2.8. WXFL-152 significantly inhibited tumors in the subcutaneous xenograft models and patient-derived tumor xenograft model
To further evaluate the anti-tumor efficacy of WXFL-152 in vivo, two non-small cell lung cancer xenograft models and a liver cancer PDX model were established in Balb/c nude mice. After 21 days of treatment with WXFL-152, the tumors were significantly inhibited by all the dose treatment groups, as shown in Fig. 4D and Table 7. The anti-tumor effects were evaluated by the relative tumor growth rate T/C (%) and tumor growth inhibition rate TGI (%) as described in the Experimental Section. In the A549 lung cancer xenograft model, 50 and 100 mg/kg WXFL-152 and 100 mg/kg nintedanib demonstrated similar significant suppression of cancer growth (T/C, 29%, 27% and 26%; TGI, 78%, 80% and 80%, P < 0.0001, respectively). In the H520 xenograft model, the T/C values were 58%, 36% and 45%, and the TGI values were 51%, 72% and 62% for the 50 and 100 mg/kg WXFL-152 and 100 mg/kg nintedanib treatment groups, respectively. WXFL-152 had better effects than nintedanib at the same dose of 100 mg/kg. In the liver cancer PDX model, 200 mg/kg WXFL-152 and 60 mg/kg sorafenib demonstrated similar therapeutic efficacies compared with the control group (T/C 35%, TGI 76%, P = 0.0097 for WXFL-152; T/C 31%, TGI 80%, P = 0.0182 for sorafenib). No obvious body weight loss observed throughout the entire treatment period except for the weight loss attributed to nintedanib (100 mg/kg) treatment in the A549 lung cancer xenograft model, suggesting that the administration of WXFL-152 was well tolerated. In addition, the vital organs (heart, liver, spleen, lung and kidney) of mice treated with WXFL-152 (100 and 200 mg/kg, p.o., QD × 3 weeks) were collected and further analysed by H&E staining. No significant pathologic changes were observed in these organs of the nude mice (Fig. 4E), suggesting that there were no obvious toxicity with continuous treatment with WXFL-152.
Table 7.
Tumor volumes, T/C and TGI after treatment of WXFL-152 in subcutaneous xenograft models of mice (mean ± SEM).
| Group | Mouse number (n) | Tumor volume at Day 21 (mm3) | T/C (%) | TGI (%) | P value |
|---|---|---|---|---|---|
| A549 Cell line model | |||||
| Control | 9 | 1489 ± 138 | / | / | / |
| WXFL-152 (50 mg/kg) | 9 | 430 ± 42 | 29 | 78 | <0.0001 |
| WXFL-152 (100 mg/kg) | 8 | 401 ± 38 | 27 | 80 | <0.0001 |
| Nintedanib (100 mg/kg) | 9 | 391 ± 43 | 26 | 80 | <0.0001 |
| NCI-H520 Cell line model | |||||
| Control | 6 | 1261 ± 186 | / | / | / |
| WXFL-152 (50 mg/kg) | 6 | 704 ± 107 | 58 | 51 | 0.0415 |
| WXFL-152 (100 mg/kg) | 6 | 478 ± 85 | 36 | 72 | 0.0014 |
| Nintedanib (100 mg/kg) | 6 | 585 ± 101 | 45 | 62 | 0.0057 |
| Liver cancer PDX model | |||||
| Control | 8 | 1026 ± 199 | / | / | / |
| WXFL-152 (200 mg/kg) | 6 | 334 ± 69 | 35 | 76 | 0.0097 |
| Sorafenib (60 mg/kg) | 6 | 304 ± 73 | 31 | 80 | 0.0182 |
2.9. WXFL-152 decreased the tumor micro-vessel density in the subcutaneous xenograft models analysed by immunohistochemistry
CD31 is one of the standard markers for detecting vascular endothelial cells in vivo. To confirm the anti-angiogenesis mechanism of WXFL-152, anti-CD31 immunohistochemical analysis of tumors from mice in the subcutaneous xenograft and PDX models was performed. Compared with those in controls, the micro-vessel density of tumors from the WXFL-152 treated groups was significantly decreased. The areas of CD31 (mean ± SEM, n = 3) were 3.38 ± 0.49, 1.05 ± 0.33, 2.20 ± 0.29 and 1.27 ± 0.48 in the control, 100 mg/kg nintedanib, 50 and 100 mg/kg WXFL-152 groups in the A549 models, respectively (Fig. 5A and B), and were 4.96 ± 0.62, 1.99 ± 0.04, 1.96 ± 0.05, 1.83 ± 0.26 in the control, 100 mg/kg nintedanib, 50 and 100 mg/kg WXFL-152 groups in the NCI-H520 model (Fig. 5C and D), respectively. In the liver PDX model, the areas of CD31 in the control and 200 mg/kg WXFL-152 groups were 0.30 ± 0.10 and 0.19 ± 0.06, respectively (Fig. 5E and F). These results indicated the significant anti-angiogenic activity of WXFL-152 even in vivo (P < 0.05), consistent with results that WXFL-152 inhibited HUVECs proliferation in vitro and tumor angiogenesis in a transgenic zebrafish model.
Figure 5.

WXFL-152 decreased the tumor micro-vessel density in the subcutaneous xenograft models analysed by immunohistochemistry. (A) The typical CD31 staining images of tumor tissues from mice of A549 subcutaneous xenograft model ( × 100). (B) The staining areas statistics of CD31 in A549 subcutaneous xenograft model, ∗P < 0.05. (C) The CD31 immunohistochemical analysis of tumors from mice in the NCI-H520 subcutaneous xenograft ( × 100). (D) The staining areas statistics of CD31 in NCI-H520 subcutaneous xenograft model, ∗P < 0.05. (E) and (F) The micro-vessel density analysis of tumors from mice in the liver PDX model ( × 100, ∗P < 0.05). Data are represented as mean ± SEM (n = 3).
2.10. Pharmacokinetic study of WXFL-152 in rats and beagles given by oral or intravenous administration with a single dose or multi-doses
To profile pharmacokinetic characteristics of WXFL-152, including absorption, distribution, metabolism and excretion, systematic pharmacokinetic studies were performed. LC–MS/MS was used to analyse the concentration of WXFL-152 in the plasma of rats and beagles. The optimized quasi-molecular and fragment ions were selected for qualitative analysis of WXFL-152 (m/z, parent ion/product ion: 461.0/404.1, collision energy (CE): 33 V) and IS (m/z, parent ion/product ion: 393.0/373.10, CE: 18 V), respectively. Multiple reaction monitoring (MRM) mode was used to monitor both quasi-molecular/fragment ions of WXFL-152 and IS for the WXFL-152 concentration quantified in rat and beagle plasma. The protein precipitation method using acetonitrile was chosen as the extraction method of WXFL-152 and IS. As shown in Supporting Information Fig. S72‒S79, no significant interference from rat and beagle plasma was observed at the retention times of WXFL-152 and IS. The retention times of WXFL-152 and IS were 1.09 and 1.49 min in rat plasma and 0.97 and 1.43 min in beagle plasma, respectively (Figs. S75 and S79). The calibration curve was linear over the concentration range of 2.0–2000 ng/mL for WXFL-152. The regression Eqs. (1), (2) obtained by least-squares regression were
| (1) |
(R2 = 0.9942, Supporting Information Fig. S80) for WXFL-152 in rat plasma and
| (2) |
(R2 = 0.9872, Supporting Information Fig. S81) in beagle plasma (Y represents the peak-area ratio of an analyte to IS, and X represents the plasma concentration of the analyte). The lower limit of quantification (LLOQ) was 2.0 ng/mL for WXFL-152 in both rat and beagle plasma. The precision and accuracy of LLOQ were acceptable, with relative standard deviation (RSD) values and relative error (RE) values within 20% for both analytes. The accuracies and precisions intra- and inter-day were within ±15%. In rat plasma analysis, the precision and accuracy were 2.7%–10.5% and 97.0%–108.6% intra-day and 4.3%–13.7% and 92.5%–103.8% inter-day. In the beagle plasma analysis, the precision and accuracy were 2.9%–9.9% and 101.7%–108.1% intra-day and 5.4%–9.3% and 102.0%–110.6% inter-day, all within the acceptable limits. The results indicated that the present method was reliable and reproducible for the simultaneous quantitative analysis of WXFL-152 in rat and beagle plasma samples.
The recoveries (CV) of WXFL-152 were 81.9%–109.1% and 87.6%–94.1% at concentrations of 2.0, 800 and 1600 nmol/L (n = 3) from rat and beagle plasma, respectively. The IS-normalized matrix factors for WXFL-152 at concentrations of 4.0 and 2000 nmol/L were 90.9 ± 12.1% and 92.1 ± 9.2% in rat plasma, and 94.7 ± 9.6% and 92.4 ± 3.8% in beagle plasma, respectively (mean ± CV%, n = 6). These results suggested that there was no matrix effect under the tested conditions. The stability of the rat and beagle analytes was investigated at three concentrations under a variety of storage and process conditions. The data showed that the stability of quality control (QC) samples was acceptable when stored at room temperature for 24 h, placed in an auto-sampler (4 °C) for 58 h, and submitted to freeze-thaw cycles (9, 15 and 33 days, at −80 °C to room temperature, deviation within 15%). The dilution integrity was confirmed for rat and beagle QC samples that exceeded the up limit of quantitation (ULOQ). The results showed that the intra- and inter-day precision and accuracy of diluted samples were within the acceptable range.
The plasma concentration–time curves of WXFL-152 in rat and beagle plasma were examined by non-compartmental analysis. The drug–time curve of single-dose WXFL-152 (rat, 3 mg/kg; beagle, 2 mg/kg) in rats and beagles (male and female, n = 3, respectively) administered by intravenous injection (i.v.) showed that WXFL-152 had similar pharmacokinetic characteristics in male and female rats or beagles after 24 h (Fig. 6A and B). Fig. 6C and D showed the drug–time curve of single-dose WXFL-152 (rat, 15, 50 and 150 mg/kg; beagle, 10, 30 and 100 mg/kg) in rats and beagles (male and female, n = 3, respectively) administered p.o. (per os) in 24 h. Drug–time curves of multi-dose WXFL-152 (rat, 50 mg/kg/day; beagle, 30 mg/kg/day, for 7 days) in rats and beagles (male and female, n = 3, respectively) administered p.o. in 24 h were shown in Fig. 6E and F. To analyse the PK of continuous multi-dose WXFL-152 in the tumor-bearing mice, lung cancer-bearing mice (A549 and H520) treated with WXFL-152 (50 and 100 mg/kg/day, for 21 days) were given another dose of the same volume of WXFL-152 by p.o. The drug–time curves were shown in Fig. 6G and H. The PK parameters for rats and beagles were summarized in Table 8, Table 9. The t1/2 values of WXFL-152 in rats and beagles varied with the dose and administration. The bioavailability (%) of WXFL-152 in rats decreased with increasing dose. The values were 42.0%, 18.7% and 13.0% when the doses were 15, 50 and 150 mg/kg in rats, respectively. This trend was not observed in beagles. The bioavailabilities (%) were 76.9%, 65.4% and 78.0% when the doses were 10, 30 and 100 mg/kg, respectively.
Figure 6.

Pharmacokinetic study of WXFL-152 in mice, rats and beagles. (A) and (B) The plasma concentration in rat and beagle were measured at 24 h with intravenous injection of WXFL-152 (rat, 3 mg/kg; beagle, 2 mg/kg) by i.v. The plasma concentration–time curve of WXFL-152 in rats (male, n = 3; female, n = 3) was shown in (A), and that for beagles (male, n = 3; female, n = 3) was shown in (B). (C) and (D) The 24 h drug–time curves of single-dose WXFL-152 (rat, 15, 50 and 150 mg/kg; beagle, 10, 30 and 100 mg/kg) in rats and beagles (male and female, n = 3, respectively) administered p.o. (E) and (F) The 24 h drug–time curves of continuous multi-dose WXFL-152 (rat, 50 mg/kg/day; beagle, 30 mg/kg/day, for 7 days) in rats and beagles (male and female, n = 3, respectively) administered p.o. (G) and (H) The 24 h drug–time curves of continuous multi-dose WXFL-152 (50 and 100 mg/kg/day, for 21 days) in tumor-bearing mice (A549 and H520) administered p.o.
Table 8.
The pharmacokinetic parameters of WXFL-152 in rat plasma after single or multi-doses (n = 6, 3 male and 3 female, mean ± SD).
| Parameter | 3 mg/kg (single dose, i.v.) | 15 mg/kg (single dose, p.o.) | 50 mg/kg (single dose, p.o.) | 50 mg/kg/day (multi-doses, p.o., 7th day) | 150 mg/kg (single dose, p.o.) |
|---|---|---|---|---|---|
| Cmax (μmol/L) | 40.4 ± 3.0 | 26.0 ± 13.3 | 32.3 ± 11.3 | 46.5 ± 13.9 | 48.7 ± 9.5 |
| Tmax (h) | / | 0.58 ± 0.20 | 0.54 ± 0.25 | 1.42 ± 0.67 | 1.58 ± 2.26 |
| t1/2 (h) | 1.18 ± 0.18 | 2.21 ± 0.39 | 2.00 ± 0.55 | 2.17 ± 0.55 | 2.54 ± 0.80 |
| Vdss (L/kg) | 0.26 ± 0.05 | / | / | / | / |
| CL (mL/min/kg) | 3.24 ± 0.81 | / | / | / | / |
| AUC0–last (μmol/L·h) | 35.0 ± 8.2 | 74.6 ± 36.3 | 110.0 ± 84.9 | 160.0 ± 60.3 | 232.0 ± 158.0 |
| AUC0–inf (μmol/L·h) | 35.2 ± 8.3 | 77.7 ± 39.6 | 110.0 ± 84.9 | 151.0 ± 63.5 | 233.0 ± 158.0 |
| Bioavailability (%) | / | 42.0 | 18.7 | / | 13.0 |
Cmax, maximum plasma concentration; Tmax, time the maximum concentration occurred; Vdss, volume of distribution at steady state; CL, systemic clearance; AUC, area under the plasma concentration–time curve.
Table 9.
The pharmacokinetic parameters of WXFL-152 in beagle plasma after single or multi-doses (n = 6, 3 male and 3 female, mean ± SD).
| Parameter | 2 mg/kg (single dose, i.v.) | 10 mg/kg (single dose, p.o.) | 30 mg/kg (single dose, p.o.) | 30 mg/kg/day (multi-doses, p.o., 7th day) | 100 mg/kg (single dose, p.o.) |
|---|---|---|---|---|---|
| Cmax (μmol/L) | 4.36 ± 0.96 | 6.28 ± 6.57 | 16.20 ± 16.50 | 6.97 ± 5.26 | 47.80 ± 20.80 |
| Tmax (h) | / | 1.25 ± 0.612 | 0.917 ± 0.585 | 1.08 ± 0.736 | 2.33 ± 0.816 |
| t1/2 (h) | 4.91 ± 2.18 | 5.77 ± 2.17 | 5.25 ± 1.03 | 12.30 ± 12.60 | 2.99 ± 0.70 |
| Vdss (L/kg) | 1.45 ± 0.33 | / | / | / | / |
| CL (mL/min/kg) | 14.5 ± 2.74 | / | / | / | / |
| AUC0–last (μmol/L·h) | 5.1 ± 0.9 | 19.6 ± 20.2 | 50.0 ± 58.8 | 23.7 ± 16.1 | 199.0 ± 90.5 |
| AUC0–inf (μmol/L·h) | 5.13 ± 0.9 | 20.2 ± 20.2 | 51.8 ± 62.3 | 37.9 ± 31.9 | 230.0 ± 51.5 |
| Bioavailability (%) | / | 76.9 | 65.4 | / | 78.0 |
Cmax, maximum plasma concentration; Tmax, time the maximum concentration occurred; Vdss, volume of distribution at steady state; CL, systemic clearance; AUC, area under the plasma concentration–time curve.
3. Conclusions
Angiokinase signals such as VEGF/VEGFRs, FGF/FGFRs and PDGF/PDGFRs promote the proliferation and cellular migration of ECs and pericytes, leading to angiogenesis in cancer. Multi-angiokinase inhibitors targeting VEGFRs, FGFRs and PDGFRs, such as lenvatinib and nintedanib, have achieved highly favourable clinical benefits in recent years.
WXFL-152, which targets VEGFRs, FGFRs and PDGFRs, was identified from a novel series of 4-oxyquinoline derivatives based on a SAR study in this work. Molecular docking simulations revealed that the quinoline ring and the thiourea group were the main important groups in the binding site of its targets. The kinase inhibition assay showed the high affinity of WXFL-152 for VEGFRs, FGFRs and PDGFRs. Inhibition of endothelial cells and peripheral cells proliferation simultaneously can achieve a better anti-angiogenesis effect. The results of proliferation assays on HUVECs and HBVPs indicated that WXFL-152 could inhibit the proliferation of ECs induced by hVEGF-165 and pericytes induced by PDGF-BB in tumor angiogenesis.
To further confirm the molecular mechanisms of WXFL-152, Western blot assay was used to investigate its biological function in cellular level that blocked angiogenic signalling pathways (including VEGF–VEGFRs/ERKs, FGF–FGFRs/ERKs, and PDGF–PDGFRs/ERKs) in HUVECs and HBVPs. Furthermore, the molecular mechanism of WXFL-152 in transgenic zebrafish and mice bearing tumor models was revealed by the analysis of confocal microscopy images and immunohistochemistry (IHC). In vivo, the significant anti-cancer effects of WXFL-152 were confirmed in multiple preclinical tumor xenograft models in mice with excellent tumor growth inhibition. In addition, pharmacokinetic studies of WXFL-152 demonstrated highly favourable bioavailability under single- and multi-dose continuous oral administration in rats and beagles.
In conclusion, WXFL-152 is a novel and effective triple-angiokinase inhibitor with clear PD and PK in tumor therapy. Recently, WXFL-152 as a novel clinical candidate is being tested in phase Ib clinical trials. Further clinical trials will focus on the treatment of solid tumors and pulmonary fibrosis.
4. Experimental
4.1. Chemistry
All starting chemicals and reagents were commercially available and used without further purification. Reaction progress was usually monitored by thin-layer chromatography (TLC) or LC–MS. The products were separated and purified by conventional preparative TLC and HPLC. 1H nuclear magnetic resonance (1H NMR) and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer (Bruker Company, Karlsruhe, Germany) using TMS as an internal standard and DMSO-d6 or CDCl3 as a solvent. The chemical shifts were reported in parts per million (ppm) relative to TMS. The mass spectral (MS) data were obtained on LC triple quadrupole mass spectrometer system (LC–MS/MS 8050 system, Shimadzu Corporation, Kyoto, Japan). The purity of compound was tested by reverse-phase HPLC (XBridge column, C18, 150 mm × 4.6 mm, 5 μm) using a Shimadzu 20A LC system (Shimadzu). The detailed synthesis methods, including reagents and conditions of all compounds mentioned in this study (Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5), were described in our previous patent27. All of the compounds identification were provided in the Supporting Information. The synthesis route of WXFL-255 (lead compound) and WXFL-152 were shown in Scheme 1, Scheme 6. The detailed methods were as follows.
4.1.1. 4-(3-Chloro-4-(3-cyclopropylthioureido)phenoxy)-7-methoxyquinoline-6-carboxamide (compound WXFL-255, lead compound)
The detailed synthesis methods, including reagents and conditions of WXFL-255 was described in our previous research20. Briefly, as shown in Scheme 1, methyl 4-amino-2-methoxybenzoate (1) reacted with trimethyl orthoformate and 2,2-dimethyl-1,3-dioxane-4,6-dione to yield compound 1A. Subsequent heating 1A at 220 °C triggered intramolecular cyclization to give quinolone 1B, which upon hydrolysis of methyl ester, chlorination of the ketone and aminolysis of the acyl chloride led to the desired chloroquinoline 1E. Then 3-chloro-4-nitro-phenol was heated with 1E to afford nitro compound 1F, which was reduced by iron to provide the amine 1G. The following isothiocyanate from the treatment of 1G with 1,1′-thiocarbonyldi-2(1H)-pyridone coupled with cyclic propylamine to produce the lead compound WXFL-255, yellow solid, 90 mg, Yield 15.63%. 1H NMR (400 MHz, methanol-d4) δ 9.03 (s, 1H), 8.93 (d, J = 6.78 Hz, 1H), 7.91 (br. s., 1H), 7.57–7.68 (m, 2H), 7.39 (d, J = 8.78 Hz, 1H), 7.21 (br. s., 1H), 4.22 (s, 3H), 2.73 (br. s., 1H), 0.53–1.09 (m, 4H). LC–MS (ESI) m/z [M+H]+: 443.0.
4.1.2. 4-(4-Amino-3-chloro-2-fluorophenoxy)-7-methoxyquinoline-6-carboxamide (compound b)
4-Chloro-7-methoxyquinoline-6-carboxamide compound a (710.4 g, 3.00 mol), sodium tert-butoxide (353 g, 3.67 mol) and 4-amino-3-chloro-2-fluorophenol (600 g, 3.71 mol) were dissolved in DMSO (21.3 L) at 20–35 °C. Under the protection of nitrogen, the reaction system was stirred at room temperature for 20 min and then stirred at 96–100 °C for 2 h. The reaction was detected by HPLC. The resulting mixture was cooled to room temperature. Water (28.4 L) was added, and the mixture was filtered and dried in vacuo to obtain compound b (purple-grey solid, 994 g). 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 2 H), 7.87 (br. s., 1H), 7.75 (br. s., 1H), 7.51 (br. s., 1H), 7.17 (br. s., 1H), 6.72–6.70 (d, J = 8.00 Hz, 1H), 6.50 (s, 1H), 5.83 (s, 2H), 4.01 (s, 3H). MS m/z [M+H]+: 362.0.
4.1.3. 4-(3-Chloro-4-(3-cyclopropylthioureido)-2-fluorophenoxy)-7-methoxyquinoline-6-carboxamide (WXFL-152)
Compound b (990 g, 2.49 mol) and 1,1′-thiocarbonyldi-2(1H)-pyridone (1020.5 g, 4.39 mol) were dissolved in THF (19.8 L) at 20–35 °C. Under the protection of nitrogen, the reaction system was stirred at 61–65 °C for 2 h. The reaction was detected by HPLC. The resulting mixture was cooled to room temperature. Cyclopropylamine (328.7 g, 5.76 mol) was added dropwise and stirred for 20 min, then filtered. The filtrate was collected. Water (140 L) was added, and the mixture was stirred for 30 min, filtered, and dried in vacuo to obtain WXFL-152 (light-brown solid, 664 g, Yield 57.9%). 1H NMR (400 MHz, CD3OD) δ 9.05 (s, 1H), 8.95 (d, J = 6.4 Hz, 1H), 7.86–7.72 (m, 1H), 7.66 (s, 1H), 7.55–7.51 (m, 1H), 7.22 (m, 2H), 4.22 (s, 3H), 2.76 (m, 1H), 0.94 (m, 2H), 0.79 (m, 2H). MS m/z [M+H]+: 461.1.
4.1.4. 4-(3-Chloro-4-(3-cyclopropylthioureido)-2-fluorophenoxy)-7-methoxyquinoline-6-carboxamide methanesulfonate (WXFL-152 methanesulfonate)
To a solution of WXFL-152 (650 g, 1.41 mol) in EtOH (18.85 L) was added MsOH (128.76 g, 1.34 mol) dropwise at 55–65 °C. The resulting mixture was cooled to room temperature and stirred for 12 h. The mixture was filtered and dried in vacuo to obtain WXFL-152 methanesulfonate (white solid, 703 g; Yield 89.6%, purity 98.93%). 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 9.07 (d, J = 6.4 Hz, 1H), 8.74 (s, 1H), 8.74–8.37 (m, 1H), 8.01 (s, 1H), 7.93 (s, 1H), 7.81–7.62 (m, 2H), 7.71 (s, 1H), 7.07–6.95 (m, 1H), 4.10 (s, 1H), 3.03–2.69 (m, 1H), 2.39 (s, 3H), 0.78–0.59 (m, 4H). 13C NMR (400 MHz, DMSO-d6) δ 182.6 (s, –C), 165.4 (s, –C), 165.0 (s, –C), 161.0 (s, –C), 151.0 (s, –C), 149.1 (d, –CH), 148.5 (s, –C), 143.7 (s, –C), 128.7 (s, –C), 124.7 (s, –CH), 121.0 (s, –CH), 113.8 (s, –C), 103.2 (s, –CH), 101.6 (s, –CH), 56.9 (s, –CH3), 39.8 (s, –CH3), 6.6 (s, –CH3). MS m/z [M+H]+: 483.1. The mesylate form of WXFL-152 was used in biological test in this study.
4.2. Cell lines and cell culture
Non-small cell lung cancer lines (A549 and NCI-H520) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL). HUVECs isolated from human umbilical veins and human brain vascular pericytes (HBVPs) isolated from human brains were obtained from ScienCell Research Laboratories, Inc. (Catalog: 8000 and 1200, CA, USA). HUVECs were grown in endothelial cell medium (ECM, Cat. #1001, ScienCell Research Laboratories, Inc.) supplemented with 5% FBS. HBVPs were grown in Pericyte Medium (PM, Cat. #1201, ScienCell Research Laboratories, Inc.) supplemented with 5% FBS. HUVECs and HBVPs at passages 3–8 were used in all studies. All cells were maintained in a humidified 5% CO2 atmosphere at 37 °C33.
4.3. Molecular docking analyses
Molecular docking analyses were performed as described in our previous study34. Briefly, the AutoDock Vina program (Scripps Research Institute, CA, USA) was used for molecular docking simulations35. WXFL-152 was prepared as a pdbqt file using Auto Dock Tools. The crystal structure of FGFR1 complexed with ponatinib (PDB ID: 4V04) and VEGFR2 complexed with sorafenib (PDB ID: 4ASD) were selected as the protein models. Then, all the water molecules and solvent molecules were removed, Gasteiger–Marsili charges were added, and non-polar hydrogens were then merged into their respective heavy atoms, as previously described36. A total of 20 docking poses were generated for WXFL-152, and the other parameters for Vina were set to their default settings. The docking results were viewed using the PyMOL program.
4.4. Kinase inhibition assay
A total of 31 human kinases (Invitrogen, CA, USA) were incubated with WXFL-152 (0.17–10,000 nmol/L, diluted with DMSO). The kinase activities were analysed by Invitrogen Z′-LYTE® Kinase detection technology according to its protocol and an Agilent Bravo Automated Liquid Handler (G5409A, CA, USA). Briefly, 2.5 μL/well WXFL-152 was added to 384-well microplate assays and centrifuged (5810R, Eppendorf, Hamburg, Germany) for 1 min (1000 rpm), and 5 μL/well kinase solutions were added and centrifuged for 1 min (1000 rpm). After incubation for 15 min at 23 °C, another 2.5 μL ATP solution was added per well and centrifuged for 1 min (1000 rpm). After incubation for 60 min at 23 °C, the reaction was stopped by the addition of 5 μL stop buffer. Envision (PerkinElmer, MA, USA) was used to analyse the readouts after incubation at 23 °C for 60 min. The IC50 values were calculated using the XLfit5 205 formula (IDBS, Guildford, UK).
4.5. Proliferation, tube formation, scratch wound and migration assays of HUVECs
The CellTiter-Glo luminescent cell viability assay (Promega, Madison, USA) was used for the proliferation activity assay following the manufacturer's instructions37. HUVECs in the logarithmic growth phase were collected and seeded in 384-well plates (1000 cells per well) with ECM basal medium containing 2% FBS. Twenty-four hours after seeding in 384-well plates, a series of WXFL-152 dilutions (final concentration from 10,000–0.038 nmol/L) with a final concentration of 20 ng/mL hVEGF-165 in ECM basal medium containing 2% FBS were added to the well by an Agilent Bravo Automated Liquid Handler (G5409A, Agilent Technologies Inc., CA, USA). Cell viability was detected after 72 h by an Envision microplate reader (PerkinElmer). The IC50 values were calculated using the XLfit5 205 formula (IDBS). Recombinant human vascular endothelial cell growth factor (hVEGF-165) was obtained from PeproTech Inc. (Suzhou, China).
HUVEC tube formation was performed as previously described38. First, 300 μL precooled Matrigel (growth factor reduced, BD Biosciences, NJ, USA) was coated on the bottom of each 48-well plate. After incubation in a humidified atmosphere of 5% CO2 at 37 °C for 1 h, HUVECs with 8 × 104 cells/well were seeded into the coated 48-well plate. Then, WXFL-152 at concentrations of 0.5, 5.0 and 50 μmol/L and/or stimulated with 50 ng/mL hVEGF-165 was added to each well after cells were seeded in triplicate. After 2, 4 and 6 h of co-culture, tube formation was visualized using an Olympus microscope (IX71, Olympus, Tokyo, Japan), and total tubes were analysed using Image-Pro Plus 7.0 software.
HUVECs were seeded in 6-well plates at a density of 4 × 105 cells/well and were maintained in a humidified atmosphere of 5% CO2 at 37 °C. After 24 h, a straight scratch was introduced into the monolayer of endothelial cells in each well. The cells were treated with WXFL-152 at concentrations of 0.5, 5.0 and 50.0 μmol/L in the ECM medium containing 20 ng/mL hVEGF-165 after being washed twice with DPBS (ScienCell Research Laboratories, Inc.). All of the straight scratches were captured by an Olympus microscope (IX71, Olympus) at 12 h. The width of the scratch was measured at two time points using Image-Pro Plus 7.0 software (Media Cybernetics, MD, USA).
For migration assays, HUVECs were seeded into the upper chamber of a transwell device (24-well plate, 2 × 104 cells/well) with the ECM medium containing 20 ng/mL VEGF-165 and 0.5, 5.0 and 50.0 μmol/L WXFL-152. Then, 600 μL ECM medium containing 20 ng/mL VEGF-165 with 10% FBS was added to the bottom of the transwell chamber. The plates were then incubated in a humidified atmosphere of 5% CO2 at 37 °C for 24 h. The cells that migrated into the pores of the inserted filter were fixed with 4% PFA solution and stained with crystal violet. The stained migrated cells were counted manually under a fluorescence microscope (IX71, Olympus).
4.6. Proliferation assay of HBVPs
To test the effect of WXFL-152 on the proliferation of HBVPs, CCK-8 (Dojindo, Kumamoto, Japan) assay was performed as described in our previous study39. HBVPs in the logarithmic growth phase were collected and seeded in 96-well plates using complete PM medium. After 24 h, the medium in 96-well plate was removed. HBVPs were subsequently treated with different concentrations of WXFL-152 (0–10 μmol/L) or nintedanib (0–10 μmol/L) with 20 ng/mL PDGF-BB in basal PM medium (1% FBS). After 48 h treatment, CCK-8 assay kit was used to detect the living cells. The IC50 values were calculated by linear regression of plots.
4.7. Western blot analysis
HUVECs and HBVPs were pre-treated with WXFL-152 (0.5, 5.0 and 50.0 μmol/L) in ECM and PM basal medium with 1% FBS for 3 h at 37 °C with 5% CO2. Following another 15 min of stimulation with 50 ng/mL VEGF-165 or 100 ng/mL bFGF for HUVECs and 50 ng/mL PDGF-BB (PeproTech Inc.), for HBVPs, cells were homogenized in cell lysis buffer containing 20 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1% Triton X-100, sodium pyrophosphate, β-glycerophosphate, EDTA, Na3VO4 and leupeptin, PMSF, and phosphatase inhibitor cocktails. The lysates were centrifuged at 13,000×g for 15 min. The protein concentration of the supernatant was measured using a BCA Protein Assay Kit (Thermo Fisher Scientific, MA, USA). Equivalent amounts of total proteins (30 μg) were loaded on 10% SDS-PAGE gel. After being transferred to PVDF membranes, the samples were incubated with primary antibodies: VEGF receptor 2 rabbit mAb (CST, MA, USA), phosphor-VEGF receptor2 (Tyr1175) rabbit mAb (CST), FGF receptor1 (9740S, CST), FGF receptor2 rabbit mAb (BD), phosphor-FGF receptor1–4 rabbit mAb (BD), PDGF receptor β rabbit mAb (CST), phosphor-PDGF receptor β (Tyr1009) rabbit mAb (CST) and mouse anti-β-actin mAb. The secondary antibodies were goat anti-rabbit IgG (HRP)-conjugated (1:10,000) or goat anti-mouse IgG (HRP)-conjugated (1:10,000).
4.8. Tumor xenograft model in zebrafish
Pharmacodynamic studies in zebrafish were performed according to our previous research37. Zebrafish experiments were approved by the Institutional Animal Care and Treatment Committee of State Key Laboratory of Biotherapy of Sichuan University (Chengdu, China). Tg (flk1: EGFP) zebrafish were bred and maintained normally (temperature, 28 °C; pH 7.2–7.4; 14 h on and 10 h off light cycle). Colon carcinoma cell line CT-26 (red fluorescence-labelled) was harvested and implanted into each zebrafish embryo through the perivitelline space using an electronically regulated air-pressure microinjector (PL1-90, Harvard Apparatus, NY, USA). WXFL-152 was added directly into the fish water at a final concentration of 2.0 and 0.5 μmol/L. DMSO was used as a vehicle control. Animals were maintained in 2 mL fish water that was changed daily. After 7 days of treatment, living zebrafish were anaesthetized with 0.003% tricaine. Digital micrographs were taken with a Zeiss Imager. Whole animal images were taken with a Zeiss Stemi 2000-C stereomicroscope with an AxioCam MRc5 digital CCD camera (Carl Zeiss Microimaging Inc., Jena, Germany). All images were taken in the same focal plane in bright-field mode and in transmitted light passing through RFP or GFP filters. To clearly image all vessels within xenograft models, 0.5–2 μm step z-stacks (512 × 512 focal planes, 50–200 μm in depth) were acquired using a 10 × or 20 × objective (Zeiss, Plan-Neofluar). Image capture and the measurement of tumor size, vessel length and vessel diameter were performed with AxioVision rel.4.8 software (Carl Zeiss Microimaging Inc.)40.
4.9. Pharmacokinetic study
To systematically reveal the pharmacokinetic characteristics of WXFL-152, a series of normalized PK methods, including HPLC–MS/MS conditions, standard solutions, calibration standards and QC sample, sample preparation, method validation, linearity of calibration curve and lower limit of quantification, were established as described in the Supporting Information. All animal experiments were approved by the Institutional Animal Care and Treatment Committee of WuXi AppTec. (Wuxi, China). Twenty-four Sprague–Dawley rats (12 male and 12 female, 180–220 g) were obtained from the Laboratory Animal Center of Shanghai Institutes for Biological Sciences of the Chinese Academy of Sciences (Shanghai, China), and 24 beagles (12 male and 12 female, 8–11 kg) were obtained from Beijing Marshall Biological Resources Co., Ltd. (Beijing, China). Twelve male or female animals (rats and beagles) were randomly divided into four treatment groups (n = 3). Animals in group 1 were administered a single dose of WXFL-152 in 20% PEG400 (3 mg/kg for rat and 2 mg/kg for beagle) by i.v. animals in group 2 (15 mg/kg for rat and 10 mg/kg for beagle), and 4 (150 mg/kg for rat and 100 mg/kg for beagle) were administered a single dose of WXFL-152 in 0.5% methocel/0.2% Tween-80 p.o. The rats in group 3 (50 mg/kg/day for rat and 30 mg/kg/day for beagle) were given WXFL-152 in 0.5% methocel/0.2% Tween-80 for seven days by p.o. The animals were fasted, except for provision of water, for 12 h before the experiment. The diet was restored after 4 h of administration. Blood samples (0.2 mL for rat and 0.6 mL for beagle) were collected from the vein (tail vein for rats and saphenous vein of the forelimb for beagles) into heparinized 1.5 mL polythene tubes at continuous points in time after oral or intravenous administration of WXFL-152. The samples were immediately centrifuged at 3000×g for 15 min. The plasma obtained was stored at −80 °C until analysis. The concentration of WXFL-152 in human plasma was detected by LC–MS/MS as described in the Supporting Information. Plasma WXFL-152 concentration–time data for each group were analysed using WinNonlin™ Version 6.3 software (Pharsight, Mountain View, CA, USA).
4.10. Anti-tumor activity and pharmacokinetic study in subcutaneous xenograft models
Female Balb/c nude mice (6–8 weeks old) were purchased from the Shanghai Laboratory Animal Research Center (Shanghai, China) and were acclimated for 1 week before the experiment. Animal experiments were approved by the Institutional Animal Care and Treatment Committee of WuXi AppTec. (Wuxi, China). All pharmacodynamics experiments of WXFL-152 were independently repeated twice. To establish a subcutaneous xenograft model, mice were injected subcutaneously with A549 and NCI-H520 cells (5 × 106 cells/100 μL). One or two weeks later, when the tumor sizes reached approximately 150 (100–200) mm3, the mice were divided into five groups in the A549 cell model and four groups in the NCI-H520 cell model, with 9 mice in each group. WXFL-152 (25, 50 and 100 mg/kg, QD × 3 weeks), nintedanib (100 mg/kg) and the control (vehicle) were administered p.o. once a day in the A549 cell model experiment. WXFL-152 (50 and 100 mg/kg), nintedanib (100 mg/kg) and the control (vehicle) were administered p.o. once a day in the NIC-H520 cell model experiment. The tumor volumes were measured using electronic digital callipers every 3–4 days and were calculated as 1/2 × length × width × width. The anti-tumor effects were evaluated based on the T/C (%) and TGI (%), as Eqs. (3), (5):
| (3) |
where TRTV: mean value of mice RTV in treatment group; CRTV: mean value of mice RTV in control group;
| RTV = Vt/V0 (4) |
where V0 is the tumor volume of the mice starting treatment, Vt is the tumor volume of the mice at the end of treatment;
| (5) |
where Vti, average tumor volume of mice in treatment group, ended treatment; Vt0, average tumor volume of mice in treatment group, starting treatment; Vi, average tumor volume of mice in control group at the end of treatment; V0, average tumor volume of mice in control group starting treatment.
To further analyse the PK characteristics in the tumor-bearing mice, animals in the WXFL-152 treatment groups were given another dose after 21 days of treatment. Orbital blood samples were collected from mice (A549-or NCI-H520-bearing mouse) into heparinized 1.5 mL polythene tubes at continuous points in time. The plasma samples were obtained, stored and analysed according to the method described in the pharmacokinetics study section.
4.11. Patient-derived tumor xenograft model
A liver cancer PDX model was established in accordance with China regulations concerning patient information and consent. A fresh specimen was collected from a patient diagnosed with liver cancer (LI-03-0020) and then xenografted subcutaneously in the interscapular space of 5-week-old male Balb/c nude mice (Shanghai Laboratory Animal Research Center) named P0. After five serial passages (P5), tumor pieces measuring approximately 30 mm3 were xenografted to every Balb/c nude mouse. When the average tumor volume reached 122 mm3, the mice were divided into three groups (n = 6). WXFL-152 (200 mg/kg), sorafenib (60 mg/kg) and a control (vehicle) were administered via oral administration once a day. Tumor volume and body weight were monitored twice a week throughout the treatment.
4.12. Histopathology and immunohistochemistry
Tumor tissues obtained from the mice in the A549, NCI-H520 and PDX models were collected for immunohistological analysis. At the end of the treatment, mice were sacrificed, and tumor tissues were obtained. IHC staining was performed to detect CD31 expression. Briefly, tissue peroxidases were inactivated by treatment with 3% H2O2 for 10 min. Then, the sections were pre-treated with antibody diluent solution containing 1% BSA, followed by 20 min incubation at room temperature with primary antibodies against CD31 (1:200 dilution, BD-550274, BD). Labelling was accomplished with HRP secondary antibodies (M2U522H, Biocare, CA, USA) and a DAB kit (ZSGB-BIO, Beijing, China) and then counterstained with haematoxylin for 5 min. The CD31 areas were captured and analysed to evaluate the micro-vessel density in tumor tissue. To observe drug toxicity, vital organs, including the heart, liver, spleen, lung and kidney, were collected and analysed by H&E staining after treatment with WXFL-152. The PerkinElmer Vectra system (PerkinElmer) with Inform 2.2.0 software was used to capture and analyse the image results. IHC staining intensity and quantity were examined by three independent pathologists who did not have authorship in this study.
Acknowledgments
This work was supported by the National Major Scientific and Technological Special Project (Nos. 2018ZX09201002, 2018ZX09711001-011 and 2019ZX09201001) and the National Natural Science Foundation of China (No. 81773375). The authors thank WuXi AppTec Ltd. provided the high-quality technology services, also thank the research platform provided by Public Health and Preventive Medicine Provincial Experiment Teaching Center at Sichuan University and Food Safety Monitoring and Risk Assessment Key Laboratory of Sichuan Province.
Author contributions
Jinliang Yang, Xiaoxin Chen, Yang Zhang, and Yuqin Yao designed this research. Yuqin Yao, Zhuowei Liu, Zhengxia Chen, Peng Li, Yang Zhang, and Chengjian Zhao performed the experiments. Yuqin Yao, Zhuowei Liu, Yang Zhang, Yuxi Wang, and Peng Li collected and interpreted data. Yuqin Yao and Manyu Zhao wrote this paper. Xiaoxin Chen and Jinliang Yang revised the paper. Jinliang Yang, Xiaoxin Chen, and Chaofeng Long were responsible for fund collection.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences..
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.04.002.
Contributor Information
Xiaoxin Chen, Email: chenzhenyu2000@zspcl.com.
Jinliang Yang, Email: jlyang01@163.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Shibuya M. VEGFR and type-V RTK activation and signaling. Cold Spring Harbor Perspect Biol. 2013;5:a009092. doi: 10.1101/cshperspect.a009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemmon M.A., Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 4.Maguire M.P., Sheets K.R., McVety K., Spada A.P., Zilberstein A. A new series of PDGF receptor tyrosine kinase inhibitors: 3-substituted quinoline derivatives. J Med Chem. 1994;37:2129–2137. doi: 10.1021/jm00040a003. [DOI] [PubMed] [Google Scholar]
- 5.Roth G.J., Heckel A., Colbatzky F., Handschuh S., Kley J., Lehmann-Lintz T. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120) J Med Chem. 2009;52:4466–4480. doi: 10.1021/jm900431g. [DOI] [PubMed] [Google Scholar]
- 6.Ma W.W., Xie H., Fetterly G., Pitzonka L., Whitworth A., LeVea C. A phase Ib study of the FGFR/VEGFR inhibitor dovitinib with gemcitabine and capecitabine in advanced solid tumor and pancreatic cancer aatients. Am J Clin Oncol. 2019;42:184–189. doi: 10.1097/COC.0000000000000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ran C., Liu H., Hitoshi Y., Israel M.A. Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res. 2013;73:1831–1843. doi: 10.1158/0008-5472.CAN-12-2460. [DOI] [PubMed] [Google Scholar]
- 8.Taeger J., Moser C., Hellerbrand C., Mycielska M.E., Glockzin G., Schlitt H.J. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Canc Therapeut. 2011;10:2157–2167. doi: 10.1158/1535-7163.MCT-11-0312. [DOI] [PubMed] [Google Scholar]
- 9.Weis S.M., Cheresh D.A. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 10.Adams R.H., Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 11.de Palma M., Biziato D., Petrova T.V. Microenvironmental regulation of tumour angiogenesis. Nat Rev Canc. 2017;17:457–474. doi: 10.1038/nrc.2017.51. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Z., Wang L., Liu X., Chen C., Wang B., Wang W. Discovery of a highly selective VEGFR2 kinase inhibitor CHMFL-VEGFR2-002 as a novel anti-angiogenesis agent. Acta pharmac Sin B. 2020;10:488–497. doi: 10.1016/j.apsb.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner N., Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Canc. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 14.Presta M., Dell'Era P., Mitola S., Moroni E., Ronca R., Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Fons P., Gueguen-Dorbes G., Herault J.P., Geronimi F., Tuyaret J., Frederique D. Tumor vasculature is regulated by FGF/FGFR signaling-mediated angiogenesis and bone marrow-derived cell recruitment: this mechanism is inhibited by SSR128129E, the first allosteric antagonist of FGFRs. J Cell Physiol. 2015;230:43–51. doi: 10.1002/jcp.24656. [DOI] [PubMed] [Google Scholar]
- 16.Ronca R., Giacomini A., Rusnati M., Presta M. The potential of fibroblast growth factor/fibroblast growth factor receptor signaling as a therapeutic target in tumor angiogenesis. Expert Opin Ther Targets. 2015;19:1361–1377. doi: 10.1517/14728222.2015.1062475. [DOI] [PubMed] [Google Scholar]
- 17.Bono F., de Smet F., Herbert C., de Bock K., Georgiadou M., Fons P. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Canc Cell. 2013;23:477–488. doi: 10.1016/j.ccr.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 18.Giavazzi R., Sennino B., Coltrini D., Garofalo A., Dossi R., Ronca R. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162:1913–1926. doi: 10.1016/S0002-9440(10)64325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nissen L.J., Cao R., Hedlund E.M., Wang Z., Zhao X., Wetterskog D. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest. 2007;117:2766–2777. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Y., Cao R., Hedlund E.M.R. Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med (Berl) 2008;86:785–789. doi: 10.1007/s00109-008-0337-z. [DOI] [PubMed] [Google Scholar]
- 21.Bergers G., Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Canc. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan J.C., Wang Z.J., Lin L., Li J.L., Wang Y., Bai H. Apatinib, a novel VEGFR inhibitor plus docetaxel in advanced lung adenocarcinoma patients with wild-type EGFR: a phase I trial. Invest N Drugs. 2019;37:731–737. doi: 10.1007/s10637-019-00735-1. [DOI] [PubMed] [Google Scholar]
- 23.Frentzas S., Simoneau E., Bridgeman V.L., Vermeulen P.B., Foo S., Kostaras E. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med. 2016;22:1294–1302. doi: 10.1038/nm.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erber R., Thurnher A., Katsen A.D., Groth G., Kerger H., Hammes H.P. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18:338–340. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 25.Gordon-Weeks A.N., Lim S.Y., Yuzhalin A.E., Jones K., Markelc B., Kim K.J. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology. 2017;65:1920–1935. doi: 10.1002/hep.29088. [DOI] [PubMed] [Google Scholar]
- 26.Bergers G., Song S., Meyer-Morse N., Bergsland E., Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H.J., Yun H.J., Kim S. Lenvatinib in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372:1868. doi: 10.1056/NEJMc1503150. [DOI] [PubMed] [Google Scholar]
- 28.Wei P., Liu B., Wang R., Gao Y., Li L., Ma Y. Discovery of a series of dimethoxybenzene FGFR inhibitors with 5H-pyrrolo[2,3-b]pyrazine scaffold: structure–activity relationship, crystal structural characterization and in vivo study. Acta pharma Sin B. 2019;9:351–368. doi: 10.1016/j.apsb.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long C., Chen Z., Chen X., Zhang Y., Liu Z., Li P., inventors. Guangdong Raynovent Biotech Co., Ltd., assignee . October 13 2016. Triazinopyrrole- and quinoline-urea derivatives as tyrosine kinase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of cancer. WO2016161952A1. [Google Scholar]
- 30.Long C., Chen Z., Chen X., Zhang Y., Liu Z., Chen S., inventors. Guangdong Raynovent Biotech Co., Ltd., assignee . April 5 2018. Crystal form and salt form of quinolinecarboxamide-thiourea and preparation method for tyrosine kinase inhibitor. WO2018059534A1. [Google Scholar]
- 31.Roth G.J., Binder R., Colbatzky F., Dallinger C., Schlenker-Herceg R., Hilberg F. Nintedanib: from discovery to the clinic. J Med Chem. 2015;58:1053–1063. doi: 10.1021/jm501562a. [DOI] [PubMed] [Google Scholar]
- 32.Chen H., Cong Q., Du Z., Liao W., Zhang L., Yao Y. Sulfated fucoidan FP08S2 inhibits lung cancer cell growth in vivo by disrupting angiogenesis via targeting VEGFR2/VEGF and blocking VEGFR2/Erk/VEGF signaling. Canc Lett. 2016;382:44–52. doi: 10.1016/j.canlet.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 33.Hu M., Zhou W., Wang Y., Yao D., Ye T., Yao Y. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2,3-dioxygenase 1. Acta Pharm Sin B. 2020 doi: 10.1016/j.apsb.2020.02.010. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H.L., Liu S., Wu C.Y., Cheng L.N., Wang Y.X., Chen K. Interactions between sirtuins and fluorogenic small-molecule substrates offer insights into inhibitor design. RSC Adv. 2017;7:36214–36222. [Google Scholar]
- 35.Trott O., Olson A.J. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G.B., Huang L.Y., Li H., Ji S., Li L.L., Yang S.Y. Identification of new p300 histone acetyltransferase inhibitors from natural products by a customized virtual screening method. RSC Adv. 2016;6:61137–61140. [Google Scholar]
- 37.Bhatia S., Krieger V., Groll M., Osko J.D., Ressing N., Ahlert H. Discovery of the first-in-class dual histone deacetylase-proteasome inhibitor. J Med Chem. 2018;61:10299–10309. doi: 10.1021/acs.jmedchem.8b01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glen H., Mason S., Patel H., Macleod K., Brunton V.G. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Canc. 2011;11:309. doi: 10.1186/1471-2407-11-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei X., Zhang T., Yao Y., Zeng S., Li M., Xiang H. Efficacy of Lenvatinib, a multitargeted tyrosine kinase inhibitor, on laser-induced CNV mouse model of neovascular AMD. Exp Eye Res. 2018;168:2–11. doi: 10.1016/j.exer.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Zhao C.J., Wang X.F., Zhao Y.W., Li Z.M., Lin S., Wei Y.Q. A novel xenograft model in Zebrafish for high-resolution investigating dynamics of neovascularization in tumors. PloS One. 2011;6 doi: 10.1371/journal.pone.0021768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.