

Abstract
Background
Tuberous sclerosis complex (TSC) is an autosomal-dominant hereditary disease characterized by hamartomas of multiple organ systems, including the brain, skin, heart, kidney and lung. Genetically, TSC is caused by pathogenic variants in the TSC1 or TSC2 gene.
Case presentation
We reported a sporadic case of a 32-year-old Han Chinese male diagnosed with TSC, whose spouse had a history of two spontaneous miscarriages and an induced abortion of a 30-week fetus identified with cardiac rhabdomyoma by ultrasound. A novel heterozygous missense variant in the TSC2 gene (Exon35:c.4511 T > C:p.L1504P) was identified in the male patient and the aborted fetus by next-generation sequencing, but not in his wife or both his parents. According to the ACMG/AMP criteria, this variant was classified as a “likely pathogenic” variant.
Conclusion
The novel TSC2:c.4511 T > C variant identified was highly likely associated with TSC and could potentially lead to adverse reproductive outcomes. IVF-ET and pre-implantation genetic diagnosis for TSC are recommended for this patient in the future to prevent fetal TSC.
Keywords: Tuberous sclerosis complex, TSC1, TSC2, Novel variant, Next generation sequencing, Preimplantation genetic testing
Background
Tuberous sclerosis complex (TSC) is an autosomal dominant hereditary disease that is characterized by hamartomas of multiple organ systems, including the brain, skin, heart, kidney and lung. Symptoms of the central nervous system include epilepsy, learning disabilities, behavioural problems, and autism. Additionally, 70–80% of TSC patients develop kidney manifestations, usually angiomyolipoma and renal cysts. Genetically, TSC is caused by pathogenic variants in the TSC1 or TSC2 gene [1], accounting for approximately 10–30% and nearly 70% of all cases respectively [2]. Variants in TSC2 cause more severe symptoms [3]. In this study, we identified a novel de novo variant in TSC2 in a male TSC patient and his aborted 30-week fetus with cardiac rhabdomyoma by ultrasonography, a typical symptom of fetal TSC.
Case presentation
The patient (proband) was a 32-year-old male, who was admitted to our reproductive centre with a referral for “recurrent spontaneous miscarriage of his spouse”. He had a history of head injury at the age of 6 years old, accompanied by brief loss of consciousness, but no skull fracture or intracranial haemorrhage occurred. One year after, fibrous nodules gradually appeared on the tip of his nose, nasolabial groove, cheek and mandible. He started to experience recurring epilepsy more than 10 attacks a year, which manifested as loss of consciousness, spastic convulsions of the limbs, eyes-rolling, occasional foaming at the mouth and biting of the tongue and lip, no loss of bowel or bladder control. The frequency of epilepsy attacks gradually decreased after the patient was 14 years old. At the age of 18, the patient underwent laser treatment for facial nodules so the facial nodules are currently atypical. The growth, development and intelligence of the paternal sample was basically unaffected by the disease. The patient denied any histories of other chronic diseases, smoking or drinking. His parents were healthy without any family history.
Physical examination of the patient revealed diffuse, dark brown, tough, bean-sized papules on the face, shark skin-like patches on his back and waist, and patchy hypopigmentation on his limbs and trunk (Fig. 1). Laboratory biochemical tests (blood, urine, stool, liver function, kidney function, electrolyte, coagulation function, blood glucose, and thyroid function) were within the normal range. The result of his G-banding chromosomal analysis was 46XY 22 ps + (Fig. 2a). No abnormality was found in his semen volume, sperm concentration, liquefaction time, motility, and morphology. The clinical diagnosis was TSC and genetic testing was recommended to confirm the clinical diagnosis.
Fig. 1.

Photographs of the paternal sample showing (a) facial angiofibromas and (b) hypopigmented macules on the back
Fig. 2.

Chromosomal analysis results for (a) the paternal sample and (b) the maternal sample
The male patient’s spouse was healthy. The result of her G-banding karyotype analysis was 46, XX (Fig. 2b). She was married to the patient at the age of 28 and experienced three pregnancies. The first two were miscarriages with arrested intrauterine fetal development at the first trimester of unidentified causes. No genetic analysis were performed on these fetuses. The third pregnancy resulted in induced abortion at 30 weeks into gestation due to “abnormal echogenic nodules in the fetal thoracic cavity and fetal oedema, indicative of cardiac rhabdomyoma, pericardial effusion and ascites” as diagnosed by three-dimensional ultrasonography (Fig. 3). The couple refused fetal autopsy, but the fetal tissue sample was collected.
Fig. 3.

The results of fetal ultrasound showed (a, b) cardiac rhabdomyosarcoma, (c) pericardial effusion, and (d) ascites
Peripheral whole blood was collected from the male patient with his informed consent, from which the genomic DNA was extracted and subjected to next-generation sequencing (NGS) by Beijing CIC Clinical Laboratory (Beijing, China) as previously described [4]. The sequencing data were analyzed for point mutations, copy number variations, and chimeric genes in TSC1 and TSC2, the two candidate pathogenic genes of TSC. The average sequencing depth for target genes was 199.65 × .
NGS analysis identified a single-base heterozygous variant of NM_000548.3:exon35:c.4511 T > C:p.L1504P in the paternal sample’s TSC2 gene (Chr16:2134969, CDS34 gene subregion) (Table 1). We verified this variant in the DNA extracted from the peripheral blood samples of the patient’s parents by Sanger sequencing. The result showed that neither of the male patient’s parents carried this TSC2 variant (Fig. 4).
Table 1.
Summary of the novel TSC2 variant
| Gene | TSC2 | |
|---|---|---|
| Inheritance | de novo | |
| Location | Exon35 | |
| Clinical significance | Likely pathogenic [PS2, PM2, PM5, PP3] | |
| In silico analysis | Polyphen2 | Probably damaging (score:0.999 for sensitivity 0.14 and specificity 0.95) |
| Mutation Taster | Disease causing (prob: 0.999); | |
| Amino acid sequence changed; | ||
| Known disease mutation at this position (HGMD CM091108); | ||
| Protein features (might be) affected; | ||
| Splice site changes | ||
| PROVEAN | Deleterious (score: −5.807) | |
| Population frequency | 1000 Genomes | Absence |
| ExAC | Absence | |
Fig. 4.

Heterozygous TSC2:c.4511 T > C variant (arrow) was identified in the paternal sample (proband) but not in his parents
Sequencing results were analyzed and compared online with the Geneious Prime software (Geneious Biologics). The TSC2:c.4511 T > C:p.L1504P variant has not been described previously. However, another substitution at the same nucleotide (TSC2:c.4511 T > A:p.L1504H) has been reported from the HGMD database (http://www.hgmd.cf.ac.uk/ac) to be confirmed as pathogenic (HGMD CM091108). The frequency of this identified variant among the population was analyzed using the 1000 Genomes (http://browser.1000genomes.org/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), and Exome Variant Server (http://evs.gs.washington.edu/EVS) databases. This variant was absent from all databases. PROVEAN (http://provean.jcvi.org/index.php), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/) were used to analyze the effect of this TSC2 gene variation on protein structure and function, which all predicted the TSC2:p.L1504P variant to be deleterious or disease causing with high confidence scores. (Table 1).
Then we isolated genomic DNA from the peripheral blood of the patient (paternal sample), his wife (maternal sample), and the tissue sample of the aborted 30-week fetus, and analyzed the specific SNP sites at and near TSC2 gene (NM_000548.3 chr16:2097472–2,138,713 forward transcript) using multiplex PCR combined NGS by Beijing Garbo Medical Laboratory Co. LTD (Beijing, China). The sequencing depths were 236×, 212×, and 305× respectively for the paternal sample, maternal sample and aborted fetus. The same heterozygous variant TSC2:c.4511 T > C was detected in the aborted fetus, whereas the maternal sample did not harbour this variant. The family pedigree is shown in Fig. 5.
Fig. 5.
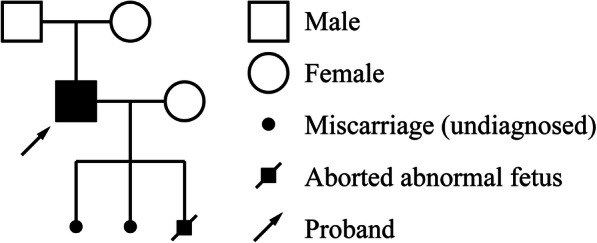
Family pedigree of the case
Discussion and conclusions
In this case, we identified a previous undescribed de novo heterozygous TSC2:c.4511 T > C:p.L1504P variant in a TSC patient whose wife had multiple miscarriages and in one aborted abnormal fetus by NGS.
TSC is an autosomal dominant neurocutaneous syndrome with an estimated incidence of 1/6000 to 1/10000 [5]. Sporadic TSC accounts for approximately 2/3 of all cases and family inheritance for approximately 1/3, and there is no difference in prevalence between men and women. The contribution of TSC1 and TSC2 variants is similar in inherited genetic cases of TSC, while TSC2 mutations are 4–5 times more common in sporadic TSC cases. The main types of variant in TSC1 are point mutations (nonsense, missense, splicing site, frameshift), and deletion/insertion of small fragments [6, 7]. Most of the variants in TSC2 are missense and nonsense mutations, less frequently small/large fragment deletions, and splice site mutations, which are often accompanied by genetic recombination [8, 9]. According to the LOVD reported mutation database, the ratio of the frequencies of TSC2 variants vs. TSC1 variants was approximately 7:3.
Different TSC1 and TSC2 variants lead to varied clinical phenotypes. Therefore, an increasing number of studies have focused on the correlation between phenotype and genotype in TSC patients [10, 11]. Curatolo et al. found that the clinical symptoms caused by TSC2 variants were more serious than those by TSC1 variants, the former typically included mental retardation, epilepsy, and facial angiofibroma [12]. In this case, the patient presented severe epileptic seizures and facial angiofibroma, which matches the typical manifestation of TSC2 mutation. As no other variants was identified in the patient’s TSC1 or TSC2 gene, it is highly likely that this novel variant caused this sporadic TSC case in the patient.
According to the ACMG/AMP criteria for classifying pathogenic variants [13], this variant was qualified for the following criteria: PS2 (de novo in a patient with the disease and no family history), PM2 (absent from controls in Exome Sequencing Project, 1000 Genomes or ExAC), PM5 (novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before), PP3 (multiple lines of computational evidence support a deleterious effect on the gene or gene product). Although the aborted fetus had been identified with cardiac rhabdomyoma typical of fetal TSC [14], as the patient refused fetal autopsy, we did not have strong evidence to make a definite diagnosis of TSC in the fetus. Therefore this variant was not qualified for the criterion PP1 (co-segregation with disease in multiple affected family members in a gene definitively known to cause the disease). Taken together, this new TSC2 variant was considered as a “likely pathogenic” variant.
TSC could adversely affect fetal health. Fetal and maternal TSC can be complicated by preeclampsia, intrauterine growth retardation, preterm labour, pre-term premature rupture of membrane, oligohydramnios, polyhydramnios, hydrops, abruption, haemorrhage from rupture renal tumor, renal failure and fetal demise [15]. There has been no study on the rate of spontaneous miscarriage of fetus from paternal TSC. As no genetic tests had been carried out in the patient’s previous two miscarriages, it was unknown whether the spontaneous miscarriages resulted from fetal TSC.
Considering the potential pathogenicity of this variant, in vitro fertilization and embryo transplantation (IVF-ET) and pre-implantation genetic diagnosis (PGD) for TSC are recommended for this patient in the future to prevent fetal TSC. Future pregnancy should also require close monitoring by fetal ultrasound, echocardiography, and MRI imaging.
In conclusion, we identified a sporadic novel variant in TSC2 gene in association with the TSC phenotype. This will further expand the TSC mutation database and improve our understanding of the molecular pathogenesis of TSC. TSC, as a genetic disease, can present clinical manifestations at an early age or even prenatally, which deteriorate as the patient ages. There is no effective treatment for TSC at present. With PGD, it is possible for families with TSC to bear healthy offspring.
Acknowledgements
We acknowledge Dr. Jiahui Wang for providing critical revision and language editing for the manuscript. We thank the family who agreed to participate in this study.
Abbreviations
- TSC
Tuberous sclerosis complex
- NGS
Next-generation sequencing
- PGD
Preimplantation genetic diagnosis
- IVF-ET
In vitro fertilization and embryo transfer
Authors’ contributions
SZH and HCB performed physical examinations and wrote the draft. SZH also collected the clinical data and wrote the manuscript. NL provided guidance on genetics and interpreted the G-banding karyotype analysis and PGT results. XW performed the experiments and acquired data. JL designed the study, performed data analyses and contributed to manuscript revisions. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Availability of data and materials
The reference sequence for validation of the c.4511 T > C:p.L1504P variant in the TSC2 gene was acquired from the NCBI Nucleotide database by using accession number NM_000548.3. The raw sequencing data of NGS are available in the NCBI’s Sequence Read Archive (SRA) with accession number PRJNA659398 (SRR12538044, SRR12538043, SRR12538042, and SRR12538041). The datasets generated and/or analysed during the current study are available in HGMD database (http://www.hgmd.cf.ac.uk/ac), 1000 Genomes database (http://browser.1000genomes.org/), Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), Exome Variant Server database (http://evs.gs.washington.edu/EVS), PROVEAN (http://provean.jcvi.org/index.php), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/).
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Yantai Yuhuangding Hospital and conducted in accordance with approved guidelines. Written informed consent to participate was obtained from the proband, the proband’s wife and the proband’s parents before collecting blood samples and fetal DNA samples.
Consent for publication
We confirm that all participants in the case report gave their consent for their medical data to be published. A copy of the written consent is available for review by the editor of this journal on request.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shunzhi He and Na Lv contributed equally to this work.
Contributor Information
Shunzhi He, Email: tophha@126.com.
Na Lv, Email: 1048181326@qq.com.
Hongchu Bao, Email: bhch1005@126.com.
Xiong Wang, Email: wx83905@163.com.
Jing Li, Email: acoco2019@163.com.
References
- 1.Hoogeveen-Westerveld M, Wentink M, van den Heuvel D, Mozaffari M, Ekong R, Povey S, et al. Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with tuberous sclerosis complex. Hum Mutat. 2011;32(4):424–435. doi: 10.1002/humu.21451. [DOI] [PubMed] [Google Scholar]
- 2.Habib SL. Tuberous sclerosis complex and DNA repair. Adv Exp Med Biol. 2010;685:84–94. doi: 10.1007/978-1-4419-6448-9_8. [DOI] [PubMed] [Google Scholar]
- 3.Van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet. 2012;20(5):510–515. doi: 10.1038/ejhg.2011.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Jin H, Han F, Cui Y, Chen J, Yang C, et al. Homozygous DNAH1 frameshift mutation causes multiple morphological anomalies of the sperm flagella in Chinese. Clin Genet. 2017;91(2):313–321. doi: 10.1111/cge.12857. [DOI] [PubMed] [Google Scholar]
- 5.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 6.Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13(6):731–741. doi: 10.1038/sj.ejhg.5201402. [DOI] [PubMed] [Google Scholar]
- 7.Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121(3–4):389–400. doi: 10.1007/s00439-006-0308-9. [DOI] [PubMed] [Google Scholar]
- 8.Hung CC, Su YN, Chien SC, Liou HH, Chen CC, Chen PC, et al. Molecular and clinical analyses of 84 patients with tuberous sclerosis complex. BMC Med Genet. 2006;7:72. doi: 10.1186/1471-2350-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langkau N, Martin N, Brandt R, Zugge K, Quast S, Wiegele G, et al. TSC1 and TSC2 mutations in tuberous sclerosis, the associated phenotypes and a model to explain observed TSC1/ TSC2 frequency ratios. Eur J Pediatr. 2002;161(7):393–402. doi: 10.1007/s00431-001-0903-7. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Xiong S, Wu L, Chopra M, Hu X, Wu B. A novel TSC2 missense variant associated with a variable phenotype of tuberous sclerosis complex: case report of a Chinese family. BMC Med Genet. 2018;19(1):90. doi: 10.1186/s12881-018-0611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang G, Shi ZN, Meng Y, Shi XY, Pang LY, Ma SF, et al. Phenotypic and genotypic characterization of Chinese children diagnosed with tuberous sclerosis complex. Clin Genet. 2017;91(5):764–768. doi: 10.1111/cge.12920. [DOI] [PubMed] [Google Scholar]
- 12.Curatolo P, Moavero R, Roberto D, Graziola F. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22(4):259–273. doi: 10.1016/j.spen.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dragoumi P, O'Callaghan F, Zafeiriou DI. Diagnosis of tuberous sclerosis complex in the fetus. Eur J Paediatr Neurol. 2018;22(6):1027–1034. doi: 10.1016/j.ejpn.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Sharma N, Sharma S, Thiek JL, Ahanthem SS, Kalita A, Lynser D. Maternal and fetal tuberous sclerosis: do we know enough as an obstetrician? J Reprod Infertil. 2017;18(2):257–260. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The reference sequence for validation of the c.4511 T > C:p.L1504P variant in the TSC2 gene was acquired from the NCBI Nucleotide database by using accession number NM_000548.3. The raw sequencing data of NGS are available in the NCBI’s Sequence Read Archive (SRA) with accession number PRJNA659398 (SRR12538044, SRR12538043, SRR12538042, and SRR12538041). The datasets generated and/or analysed during the current study are available in HGMD database (http://www.hgmd.cf.ac.uk/ac), 1000 Genomes database (http://browser.1000genomes.org/), Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/), Exome Variant Server database (http://evs.gs.washington.edu/EVS), PROVEAN (http://provean.jcvi.org/index.php), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org/).