

Abstract
Natural antibodies (NAb) and complement (C’) are important regulators of immune system activation. We have shown previously that the galactosyl‐α1,3‐galactosyl (Galα1,3Gal) xenoantigen and the similar ABO histo‐blood group antigens are transferred onto virus from the producer cell, resulting in sensitisation of the virus to the respective NAb in a C’‐dependent manner. Here we show that measles virus (Mv) that expresses Galα1,3Gal termini can drive the proliferation of human T cells in the presence of serum and autologous DC, whereas without such targets, measles, as expected, suppress T cell reactivity. The use of affinity‐purified NAb to Galα1,3Gal and rabbit C’ demonstrated the components in human serum responsible for this effect. Proteasome inhibition and blocking of antigen presentation showed that the increased T cell proliferation was mediated by MHC class I cross‐presentation of immune complexes. These results lend further support to the idea that polymorphic carbohydrates of the Galα1,3Gal/ABO type serve as important targets for NAb and C’ and that their expression on virus has influenced their evolution by contributing to protection against viral transmission within as well as between species. The adjuvance effect of this recognition, acting as a bridge between the natural innate and adaptive immune systems, also has important implications for vaccine development.
Keywords: Dendritic cells, Histo‐blood group, Measles, Natural antibodies, T cells
Abbreviations:
- α1,3GT:
α1,3galactosyltransferase
- C’:
complement
- Galα1,3Gal:
galactosyl‐α1,3‐galactosyl
- Mv:
measles virus
- Mv‐G1–3G:
Mv with Galα1,3Gal terminal glycosylation
- NAb:
natural antibodies
Introduction
Natural antibodies (NAb) and complement (C’) contribute to an important but not well‐defined block of both bacterial and viral spread to vital organs preceding the development of an adaptive and specific immune response 1, 2. NAb, assisted by C’, serve as endogenous adjuvants for the generation of protective CD8+ T cells after vaccination against leishmaniasis 3. Studies in mice that lack expression of secreted IgM have shown that IgM NAb are necessary components of an anti‐viral response 4. Furthermore, such NAb recognition contributes not only to direct clearance of virus but also to the stimulation of immune responses requiring macrophages and/or follicular dendritic cells (FDC) 5. DC play a major role in the initiation of a specific immune response by presenting antigens to and activating naive T cells, and many viruses have, as a consequence, evolved strategies for interfering with their functions 6. MHC class I cross‐presentation of viral antigens by DC, either by uptake of virus‐infected cells or viral immune complexes, can serve as a means by which the host cell can circumvent the viral interference 7. In addition, some viruses do not infect DC and have to be presented by this pathway for efficient T cell activation 8. In the case of measles virus (Mv), a profound immunosuppressive effect on T cells appears to be at least partially mediated through their targeting of DC 9, 10. The immunosuppression induced by measles virus is responsible for subsequent bacterial infections, making measles a world‐wide problem, particularly in developing countries.
A large proportion of NAb are directed against the terminal carbohydrate structure galactosyl‐α1,3‐galactosyl (Galα1,3Gal) as well as against the closely related ABO histo‐blood group family. This nature of the targets for many NAb might be explained by the presence of surface carbohydrate structures readily available for engagement by Ab on many bacteria and parasites 11. The nature of the target for NAb on viruses is much less clear. Since a virus must inherit its glycosylation pattern from the producer cell, questions arise as to how there could possibly be targets for NAb on viral envelopes. The production of specific NAb in only some individuals of a species (e.g. anti‐ABO) or in only some species (e.g. anti‐Galα1,3Gal) correlates with the absence of the respective carbohydrate epitope. Antibodies against the Galα1,3Gal epitope are produced by splenic B cells in both primates and mice that have been engineered to lack expression of the Galα1,3Gal structure 12, 13. It is not clear why such polymorphic carbohydrate/NAb systems have evolved. Classically, bacteria have been thought to be chiefly responsible 14. We, as well as others, have shown previously that human cells can be a target for neutralisation mediated by NAb and C’ by transfecting pig cDNA 15 encoding α1,3galactosyl‐transferase (α1,3GT), which synthesizes the Galα1,3Gal epitope 16, 17. This mimics the situation in xenotransplantation in which recognition of the Galα1,3Gal epitope leads to hyperacute rejection of pig tissue in the presence of human serum 18, 19, and it has subsequently been suggested as a means to induce rejection of tumour cells following de novo expression of the Galα1,3Gal epitope 20, 21. More recently, this phenomenon was shown to be due to the induction of specific immunity against tumour antigens 22, 23. In addition, C‐type retrovirus produced from α1,3GT‐transfected cells became sensitised to human serum containing specific NAb and C’ 16, 17. We have also shown that in addition to the Galα1,3Gal epitope, the similar ABO epitopes can appear on and sensitise measles to human non‐immune serum containing the relevant NAb and C’ 24. More recently we found that this is also true for HIV virus 25. These findings suggest that polymorphic carbohydrates and the NAb against them may have evolved partly in response to the appearance of such carbohydrates on enveloped virus being transmitted from one individual to another or between species. Further support for this contention was provided in a mathematical modeling study, which showed that the frequencies of ABO histo‐blood groups in human populations can be explained by their direct interactions with pathogens (e.g. bacteria) in combination with their appearance on viruses serving as targets for ABO NAb 26.
In this study we examine whether C’‐assisted specific NAb recognition of measles virus leads to increased T cell reactivity in the presence of DC. If such an effect on T cells is seen, this would point out a potentially important function for polymorphic terminal carbohydrates on viruses beyond the neutralising effect previously observed 16, 17, 24, 25, 27. Our data show that this is the case and indicate that this mechanism could potentially be utilised to increase cytotoxic T cell responses in viral vaccinations without the need for other adjuvants.
Results
Expression of Galα1,3Gal terminal glycosylation on measles
Carbohydrate termini on HT1080 cells were modified by stable transfection with cDNA encoding α1,3GT, denoted HT‐1,3GT, or with the empty plasmid as a control (HT1080–0) 16. Different measles preparations with (Mv‐G1–3G) or without (Mv) Galα1,3Gal terminal glycosylation were produced by infecting HT‐1,3GT or HT1080–0 cells. The amount of Galα1,3Gal terminal glycosylation was assessed by a capture‐ELISA assay (Fig. 1) 24. A terminal galactose‐specific lectin, BSI‐B4, was used to show that measles produced from the Galα1,3Gal‐expressing HT‐1,3GT cells acquired the terminal glycosylation, whereas those expressed in the HT1080–0 did not. These preparations were then used to determine the effect of terminal Galα1,3Gal glycosylation on human T cell responses in the presence of autologous DC and either autologous serum or affinity‐purified NAb against Galα1,3Gal terminal carbohydrates and rabbit C’.
Figure 1.
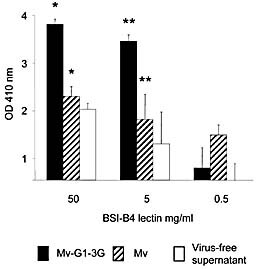
Capture ELISA showing expression of the Galα1,3Gal antigen on measles virus. Microwell plates were coated with an anti‐measles hemagglutinin mAb. Virus‐containing supernatants from cells transfected with an α1,3GT cDNA clone were diluted with respective supernatant from uninfected cells and added at an identical 5 × 105 PFU/mL. Subsequently, an HRP‐labelled Galα1,3Gal‐specific lectin from B. simplicifolia (BSI‐B4) was added at different concentrations (50, 5 and 0.5 μg/mL), after which the wells were incubated with peroxidase substrate and the absorbance analyzed on a plate reader. The results, expressed as arbitrary units, are normalised to the background value of the virus‐free supernatant incubated with 0.5 μg/mL BSI‐B4 and are shown as the mean ± SEM of duplicate samples. Significant differences were found between Mv‐G1–3G and Mv at both 50 and 5 μg/mL BSI‐B4 staining (*p<0.02 and **p<0.05, respectively).
Suppression of T cell proliferation by measles virus is reversed by serum and C’
In a series of experiments, 2.5 × 104 DC were incubated together with 2.5 × 103 live viral particles as determined by a plaque assay on Vero cells, as previously described 24. These amounts of DC and viral particles were found to be optimal from several experiments using different amounts and conditions (data not shown). A representative experiment performed in triplicate using these conditions is shown (Fig. 2a). T cell and DC exposure to measles in the absence of human serum resulted in complete obliteration of T cell proliferation, indicating viral suppression of the T cell response, as previously documented 28–30. Both viruses, i.e. displaying Galα1,3Gal or not, exhibited a similar effect on T cell proliferation (p<0.001). In the presence of autologous serum deprived of C’, exposure to either virus (Mv‐G1–3G or Mv) resulted in low proliferation (p<0.006 and p<0.01, respectively). When rabbit C’ was added to heat‐inactivated serum, more pronounced T cell proliferation was seen; moreover, proliferation was significantly higher when Mv‐G1–3G virus (with the NAb epitope) was used as compared to Mv (without the epitope) (p<0.03).
Figure 2.
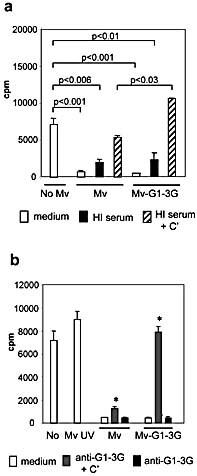
Galα1,3Gal on the virus reverses measles‐induced suppression of T cell proliferation. Measles virus produced from HT1080–0 (Mv) or HT‐1,3GT (Mv‐G1–3G) cells were exposed to a source of NAb and C’ in triplicate series, and DC‐mediated T cell proliferation was measured using [3H]‐thymidine incorporation (cpm). (a) Autologous heat‐inactivated (HI) human serum with or without added C’ was used. Significant differences between T cell proliferation are indicated by the respective p values. The results are representative of several individual experiments. (b) Measles virus, UV‐irradiated or not, was exposed to Galα1,3Gal‐specific NAb with or without rabbit C’ (*p<0.001, T cell proliferation Mv versus Mv‐G1–3G, in the presence of purified NAb and C’). The results are representative of two individual experiments.
T cell proliferation in the presence of measles together with NAb and C'
To ascertain whether NAb and C’ alone could result in the triggering of T cell proliferation, we used T cells and DC from the same donor and affinity‐purified Ab against Galα1,3Gal together with rabbit C’ instead of serum. As shown in Fig. 2b, measles without Ab and C’ again resulted in a drastic suppression as compared to the background proliferation without virus. This level of suppression was also seen when Mv (without NAb epitopes) was used in the presence of Galα1,3Gal‐specific NAb and C’. However, when Mv‐G1–3G (with the NAb epitope) was used, proliferation was restored to background levels (p<0.001). The use of UV‐irradiated measles, however, yielded normal or even above background levels of proliferation even in the absence of NAb and C’, indicating that the virus needed to replicate in the DC in order to suppress T cell proliferation. We hypothesized that the high background proliferation was due to the presence of ‘xenoantigens’ in the fetal calf serum (FCS) included in the media. We next assessed whether Galα1,3Gal‐specific NAb and C’ and their target on measles could increase T cell proliferation on their own, in the absence of background proliferation caused by FCS‐associated antigens.
T cell proliferation in the presence of measles, serum and reduced FCS concentration
When a similar T cell proliferation experiment was performed in only 3% FCS, as a means of reducing the amount of xenogeneic background antigens in the cultures, and using autologous serum as the source of NAb and C’, ‘no measles’ virus incubation as well as UV‐irradiated Mv (without Galα1,3Gal) resulted, as expected, in reduced autologous T cell proliferation (Fig. 3a), as compared to the previous experiment using 10% FCS in combination with purified NAb and C’ (Fig. 2b). However, when UV‐irradiated Mv‐G1–3G virus was used with intact human serum, drastically increased C’‐dependent proliferation resulted (p<0.001).
Figure 3.
T cell proliferation following exposure of autologous DC differentiated in 3% FCS to differentially glycosylated measles viruses. (a) UV‐irradiated non‐infectious measles virus produced from HT1080 (Mv) or HT‐1,3GT (Mv‐G1–3G) cells were exposed to fresh human serum with or without (heat‐inactivated, HI) intact C’ in triplicate series. (b) Infectious measles viruses (Mv or Mv‐G1–3G) were exposed to Galα1,3Gal‐specific NAb, with or without C’, in triplicate series. (c) UV‐irradiated non‐infectious measles (Mv or Mv‐G1–3G) were exposed to Galα1,3Gal‐specific NAb, with or without C’, in triplicate series. In addition, conditions with medium alone were used in all three experiments. DC were subsequently added, and autologous T cell proliferation was measured using [3H]‐thymidine incorporation. *Significant differences in T cell proliferation were observed between Mv and Mv‐G1–3G with intact fresh human serum (p<0.001), with affinity‐purified NAb and C’ using infectious virus (p<0.001) and with affinity‐purified NAb and C’ using UV‐irradiated non‐infectious virus (p<0.05).
T cell proliferation when using NAb, C’ and reduced amounts of FCS
In order to determine that it is the Galα1,3Gal moieties in combination with specific NAb and C’ that result in increased T cell proliferation in this autologous setting with low background proliferation, we next repeated the experiments using affinity‐purified Ab against Galα1,3Gal together with rabbit C’ instead of serum in the presence of only 3% FCS. It is well known that measles readily infects DC, upon which it efficiently inhibits T cell activation 28–30; after UV irradiation, however, measles virus does not induce inhibition 28. We thus conducted these experiments with both live infectious measles and UV‐irradiated measles shown to lack any infectious particles as detected by a highly sensitive Vero cell plaque assay (data not shown) 24 to investigate whether infection of the DC (or not) has an effect on the degree of NAb‐ and C’‐dependent T cell activation. As shown in Fig. 3b and 3c, this resulted in significantly higher T cell proliferation only in the presence of NAb against Galα1,3Gal and C’ and only when using the Mv‐G1–3G virus with NAb targets (p<0.001 and p<0.05). This effect appeared to be largely independent of whether the measles was live and thus infectious or UV‐irradiated and therefore non‐infectious, thus indicating that opsonisation and uptake of virus‐NAb‐C’ complexes rather than infecting particles resulted in T cell activation. Furthermore, the results when using UV‐irradiated non‐infectious measles with both serum and affinity‐purified NAb and C’ in the low background setting indicated that it is very unlikely that the T cell proliferation was simply due to a neutralising effect of the NAb and C’.
Cells with Galα1,3Gal can substitute for virus in inducing T cell proliferation
In order to further address the formal possibility that the T cell activation could be due mainly to neutralisation by NAb and C’ and to compare the measles results with another source of antigens, we next investigated whether UV‐irradiated producer cells of the measles virus are able to substitute for the virus in driving T cell proliferation. The results show that the allogeneic HT‐1,3GT producer cells of Mv‐G1–3G virus were able to substitute for the virus and appeared to provide antigenic stimulation driving T cell proliferation via the DC cultures (Fig. 4a). However, while Mv‐G1–3G virus‐stimulated proliferation was again strictly C’‐dependent, the HT‐1,3GT‐driven proliferation appeared to be less so. When the Mv producer cells HT1080–0 were used, no T cell proliferation resulted, again mirroring the results using Mv produced from HT1080–0 (Fig. 4b). Proliferation using DC and medium only or no T cells resulted in levels at or below the levels seen in Fig. 3b (data not shown).
Figure 4.
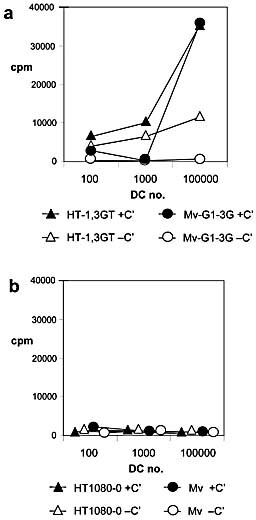
T cell proliferation following exposure of differing amounts of autologous DC to differentially glycosylated cell lines or measles with autologous human serum. (a) Irradiated HT‐1,3GT cells or Mv‐G1–3G produced from these cells were exposed to human serum with (solid symbols) or without (heat‐inactivated, open symbols) intact C’. (b) Irradiated HT1080–0 cells or Mv produced from these cells were exposed to human serum with (solid symbols) or without (heat‐inactivated, open symbols) intact C’. In all experiments DC and T cells were subsequently added, and autologous T cell proliferation was measured using [3H]‐thymidine incorporation.
DC in the co‐culture do not display increased general endocytosis or activation
The increase in T cell proliferation may be related to an altered ability of DC to endocytose in general and/or to become activated. We therefore tested whether or not DC incubated with different measles viruses and/or mAb and/or C’ were able to maintain similar levels of endocytosis. We used the fluid phase markers FITC‐dextran and rhodamine‐BSA in combination with flow cytometric analysis. The FITC‐dextran was internalised and accumulated in a time‐dependent manner in DC (Fig. 5). Similar results were observed for rhodamine‐BSA uptake (data not shown). The mean value of fluorescence was then compared for the different conditions and the different viruses after 20 min of endocytosis; no differences in uptake were observed for the FITC‐dextran (Fig. 5). Very similar results were obtained when using Rhodamine‐BSA to study uptake under the different conditions (data not shown).
Figure 5.
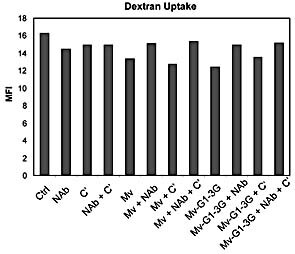
Fluid‐phase endocytosis by DC was assessed by FITC‐dextran internalisation as quantified by FACS analysis. During FITC‐dextran internalisation, DC were incubated with or without control measles virus (Mv) or measles virus expressing Galα1,3Gal epitopes (Mv‐G1–3G) in the presence or absence of the purified NAb against Galα1,3Gal epitopes and/or C’. The relative uptake was determined after 20 min incubation. Values on the y‐axis indicate the mean fluorescence intensity (MFI).
We then tested whether or not the incubation of DC with measles viruses under the different conditions modulated the membrane expression of markers potentially involved in the activation of T cells. We observed that incubation of DC with the different viruses and under the different conditions was associated with strong activation of DC in all cases. However, no differences in the levels of CD83 and CD86 were observed between Mv and Mv‐G1–3G under the different conditions (Fig. 6). Moreover, the expression of CD46, the measles virus receptor at the cell surface of DC, was similar following incubation of DC with measles viruses having the Galα1,3Gal epitope or not and in the presence or absence of NAb and/or C’ (data not shown). In addition, no differences were found when comparing the expression levels of MHC class I or class II on the DC (data not shown).
Figure 6.
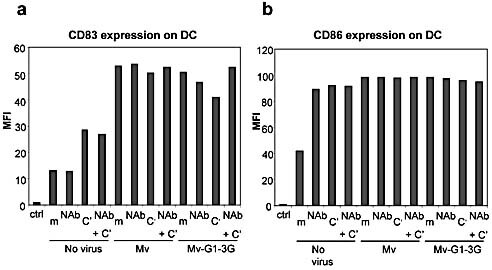
Expression of CD83 (a) or CD86 (b) on the cell surface of DC. The DC were incubated with or without control measles virus (Mv) or measles virus expressing Galα1,3Gal epitopes (Mv‐G1–3G) and in the presence, or absence (m) of purified baboon antibodies against Galα1,3Gal epitopes (NAb) and/or C’, and cell surface expression of the respective co‐stimulation molecule was determined by a FACS analysis using specific mAb. Values on the y‐axis indicate the mean fluorescence intensity (MFI).
T cell proliferation is the result of MHC class I cross‐presentation
In order to examine whether the NAb/C’‐mediated T cell proliferation was due to antigen presentation and to assess to what extent such antigen presentation is mediated through the MHC class I pathway, we used lactacystin, a specific inhibitor for this pathway (e.g. after receptor‐mediated uptake of virus) 31. Lactacystin treatment drastically reduced the purified NAb‐ and C’‐mediated proliferation down to background levels, whereas anti‐Galα1,3Gal and C’ again resulted in significantly higher T cell proliferation with Mv‐G1–3G virus as compared to with Mv (Fig. 7a). Since these data strongly indicate that mainly MHC class I presentation of viral antigens is responsible for the T cell proliferation, we next confirmed this finding by showing that a functionally blocking MHC class I mAb (W6/32) 32 was able to also reduce the T cell proliferation to background levels (Fig. 7a). In addition, to exclude the possibility that lactacystin was having a general toxic effect on the cells, thereby causing reduced proliferation of T cells, we confirmed that the same concentration of lactacystin did not significantly inhibit proliferating T cells in an allogeneic mixed lymphocyte reaction (Fig. 7a) or in the presence of anti‐CD3 and IL‐2 (Fig. 7b). Since it has been shown that lactacystin may inhibit NF‐kappa B activity as well as cause apoptosis of some cells 33, we also confirmed that the degree of MHC class I expression on the DC was not significantly affected by lactacystin incubation (Fig. 7c) and that lactacystin did not induce a significant degree of apoptosis in either proliferating T cells or activated mature DC (Fig. 7d).
Figure 7.
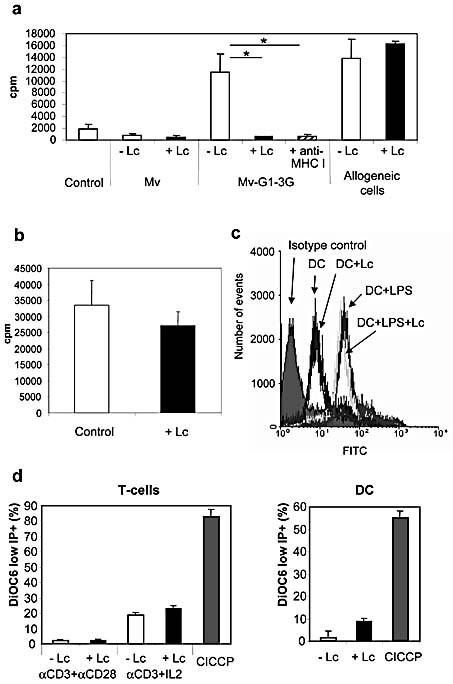
T cell proliferation is dependent on MHC class I presentation. (a) Measles without (Mv) or with (Mv‐G1–3G) Galα1,3Gal were exposed to purified NAb with added C’ in triplicate series. DC were subsequently added, and autologous T cell proliferation was measured using [3H]‐thymidine incorporation in the presence or absence of lactacystin (Lc) or a blocking anti‐MHC class I mAb. When present, the lactacystin and mAb were able to significantly inhibit proliferation (both *p<0.004). (b) Proliferating T cell cultures were treated with the same concentration of lactacystin as in (a) and did not significantly differ from the control culture. (c) Non‐activated as well as activated DC were shown to have similar MHC class I expression levels with and without lactacystin. (d) No significant increase in apoptotic activity could be seen in either two types of proliferating T cells or in activated DC following lactacystin treatment. CICCP represents control treatment to induce maximal apoptosis.
Mainly CD8+ T cells expand as a result of the Galα1,3Gal recognition
We next determined the phenotype of the expanded population of T cells following incubation with Mv‐G1–3G. In the presence of Mv‐G1–3G plus NAb and C’, the percentage of CD8+ T cells in a co‐culture with DC and T cells was increased by 54% as compared to a co‐culture with Mv (without the Galα1,3Gal epitope), representing an absolute increase in CD8+ T cells. No increase in the proportion of CD4+ T cells was seen under the same conditions (Fig. 8a), and no significant T cell death was observed. We also confirmed that a vast majority of the CD8+ T cells following proliferation were TCRαβ+, thus representing classical T cells; no change was seen after addition of lactacystin (Fig. 8b).
Figure 8.
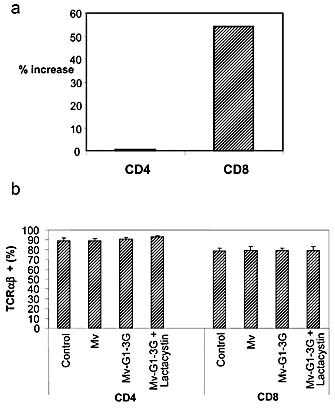
T cell numbers and phenotype after proliferation. (a) Increase in T cell numbers after incubation with Mv‐G1–3G versus Mv. Differences are indicated as the percentage of increased T cell numbers after incubation with Mv‐G1–3G as compared to with Mv in the presence of Galα1,3Gal‐specific NAb and C’. (b) The percentage of TCRαβ+ cells among total CD4+ and CD8+ cells after each treatment condition is indicated.
Discussion
Cells as well as viruses display specific terminal carbohydrate epitopes that serve as targets for NAb. In the presence of C’, NAb are able to destroy cells expressing their carbohydrate epitopes and neutralise viruses carrying such epitopes 16. In vivo, this kind of virus neutralisation can be envisaged to occur in serum or certain other biological fluids (e.g. mucosa) associated with epithelia directly or indirectly exposed to the environment. We addressed the possibility that recognition of specific polymorphic carbohydrates (e.g. Galα1,3Gal) on viruses by NAb, with or without the assistance of C’, might lead not only to neutralisation of the virus but also to increased activation of T cells. Our results indicate that expression of Galα1,3Gal histo‐blood group‐like epitopes for NAb on viruses (i.e. measles) leads to increased T cell proliferation in the presence of human serum and autologous DC. This proliferation appears to be entirely dependent on the presence of intact C’. We then proceeded to show that affinity‐purified NAb against Galα1,3Gal together with C’ results in the same effect, indicating that these are the components in human serum responsible for driving the T cell proliferation. Since the donors were immune against measles, we conclude that the NAb‐mediated proliferation must exceed any specific anti‐measles Ab‐mediated effects. The fact that UV‐irradiated non‐infectious measles virus was able to stimulate the same degree of NAb‐assisted proliferation when using both human serum and affinity‐purified anti‐Galα1,3Gal and C’ showed that neither the DC nor the T cells needed to be infected by measles virus for the proliferation to occur. One alternative explanation for our measles data would be that the normal suppressive effect of measles is overcome simply by a reduction in the number of measles virus particles available for infection of the DC. We feel that this alternative explanation can be ruled out as the main contributing mechanism, however, since: (i) The measles‐driven Galα1,3Gal‐dependent proliferation remains significantly above background levels when the FCS concentration is lowered to 3%, in combination with decreased T cell proliferation when DC numbers were titrated down stepwise to zero (Fig. 4 and data not shown); (ii) The α1,3GT‐transfected cells alone are able to support similar proliferation, whereas the same cell line without Galα1,3Gal epitopes on the cell surface is not (Fig. 4), a mechanism which has been shown to be the explanation for why the Galα1,3Gal epitope has been successfully used to initiate an anti‐tumour cell response in vitro as well as in vivo 20–23. Since this represents an allogeneic situation, the antigens that are presented to the responding T cells directly or indirectly could be either major (i.e. MHC class I or II molecules) or other minor antigens. The fact that they appeared to be efficiently presented to the T cells only in the presence of NAb and their carbohydrate targets on the cells would indicate that the cells needed to be lysed and/or cell fragments containing antigens needed to form immune complexes, thus essentially mimicking the situation with the measles virus; (iii) We showed that lactacystin, an inhibitor of proteasome‐dependent MHC class I antigen presentation, inhibits the proliferation whilst not causing inhibition of an allogeneic mixed lymphocyte reaction, NF‐kappa B inhibition or apoptosis. This observation warrants further detailed investigation to elucidate the exact function of the pathway in this system, e.g. by the use of measles‐specific T cells. However, the fact that mainly classical CD8+ TCRαβ+ T cells proliferated after NAb, C’ and Galα1,3Gal target interaction, together with to the fact that both lactacystin treatment and anti‐MHC mAb blocking could efficiently inhibit the proliferation and the finding that non‐infectious virus could be used, clearly showed that the T cell proliferation is the result of MHC class I antigen presentation of measles antigens by way of the cross‐presentation pathway. This pathway has previously been shown to be responsible for virus‐specific antibody‐mediated enhancement of viral antigen presentation 31.
Morbilliviruses (i.e. viruses related to measles and distemper) are well known to cross species barriers, often with devastating consequences. NAb recognition of such viruses (as well as potentially other emerging viral types, e.g. SARS and avian influenza) may therefore be an important contributor to protecting against cross‐species transmission. It is also very likely that the same effect on T cells would result if the Galα1,3Gal were replaced by the very similar ABO histo‐blood group antigens and the NAb directed against them, even if this remains to be demonstrated.
It has been suggested that recognition by NAb and C’ can lead to rapid T‐independent increases in B cell responses. Such B cell triggering, the mechanisms of which are far from clear, is believed to be partly mediated by macrophages and follicular dendritic cells (FDC) in the marginal zone of lymph nodes 5 following the binding of immune complexes to C’ receptors on their respective cell surfaces. It is thus possible that NAb of the anti‐Galα1,3Gal histo‐blood group‐like type recognizing virus could lead to increased T‐independent B cell responses using the same mechanism. However, it is even less clear what happens to Ab‐ and C’‐opsonised immune complexes, e.g. virus, when they encounter Fc receptors and/or C’ receptors on peripheral tissue DC of various types. One possibility is that such virus‐DC complexes are transported to the lymph nodes. Our current observations indicate that such a virus‐containing complex can also be taken up and presented by monocyte‐derived DC to T cells, leading to T cell activation and proliferation, although we have not directly addressed here what Fc receptors and/or C’ receptors may be involved in such uptake.
We recently showed that the combination of differential use of host receptors carrying ABO antigens by pathogens (e.g. bacteria) and viral transmission of ABO antigens from one host to another could explain the presence and population frequencies of these carbohydrate structures and the NAb against them 26. A recent study of the transmission of the SARS virus to care workers from a single SARS patient in Hong Kong very interestingly showed a highly significant protective effect in patients with the O histo‐blood group type 34, thus suggesting the possibility that NAb against viral ABO antigens can have a protective effect. Our present study further supports the role of terminal carbohydrate structures and the Ab directed against them in the first line of defence against viral infections and adds another dimension to previous results showing neutralisation by NAb mediated by Galα1,3Gal and ABO antigens on virus 24 by suggesting that an adaptive T cell‐mediated immune response is promoted. This is very much in keeping with a recently published study showing that a model antigen, BSA, is as immunogenic when attached to the Galα1,3Gal antigen as BSA with a standard adjuvant when injected into α1,3GT knockout mice producing antibodies against Galα1,3Gal 35. In addition, another recent publication showed that the in vitro synthesis of Galα1,3Gal epitopes on the gp120 envelope protein leads to a drastic increase in the immunizing capacity against HIV virus in both the B cell as well as, importantly, the T cell compartment in vivo in Galα1,3Gal knockout mice 36.
Most viruses, except e.g. arthropod‐borne viruses, infect a new host by way of mucosal entry, such as lung or gut. Apart from mononuclear cells present in lumenal secretions, DC are known to patrol the sub‐ and intra‐epithelial spaces along various mucosal membranes 37. There are several possible ways by which a virus can adhere to, infect or be taken up into such DC without having to undergo a round of replication in the new host; they could be transcytosed through M‐cells lining e.g. gut epithelium 38, be transported across epithelial cells 39 or even access DC directly in the lumen, since DC have been shown to penetrate tight junctions between the epithelial cells in order to ‘sample the environment’ outside the epithelial barrier 40. IgA as well as IgM, both rich sources of NAb of this type, are present in great numbers at these sites 41. We now show that such uptake and processing of virus that has not yet replicated in the new host and therefore still carries the carbohydrate epitopes determined by the previous host is possible, assisted by NAb and C’ and can be independent of infection of the DC. Such recognition could result in an important ‘head start’ for the adaptive immune response, as assisted by an NAb repertoire determined by factors other than previous viral exposure. In addition, it would ensure that virus types that do not normally infect DC are quickly handled by DC, for example through the recently described Toll‐like receptor (TLR) 3 and 7 pathways for recognition of viral RNA 42–44, and would enhance e.g. IL‐4, IL‐6 and/or IFN‐α/β production, in particular in a newly infected naive individual. Measles recognition via this pathway could include the wild‐type measles‐specific activation of TLR2 signalling 45. This type of ‘natural recognition’ of viruses constitutes a parallel example to the recently described natural adjuvance effect of NAb and C’ recognition of leishmaniasis 3. In our case, we also define a target for the NAb and C’ as a terminal carbohydrate of the histo‐blood group‐like type.
Viral vaccine formulations are often far from optimal and in some cases simply fail (e.g. HIV). One possible avenue for improvement has involved work with viral subunit vaccines. For such uses, as well as in other vaccine situations, an improved CD8+ T cell‐mediated cytotoxic response would be crucial. It has been shown that cytotoxic responses can be drastically enhanced by the cross‐presentation pathway for MHC class I/CD8 46, including responses against viruses in vivo 47. In an influenza virus model, it was shown that intact C’ is of crucial importance for CD4+ as well as CD8+ T cell activation 48. The recent in vivo demonstration that Galα1,3Gal epitopes conjugated to BSA or gp120 from HIV can result in a very impressive adjuvance effect, with resulting levels of antibody and cytokine as well as cytotoxic responses as good as or even better than in the presence of Freund's complete adjuvant alone 35, 36, in combination with the involvement of NAb, C’ and MHC class I presentation in the effect on T cell proliferation seen here with live or UV‐inactivated measles carrying antigenic carbohydrates, shows that further studies into the effect such recognition may have on vaccination are warranted. It would be particularly interesting to see what effect this glycosylation would have in the context of different subunit or split viral vaccines, where a great need for safe but efficient adjuvant alternatives exists.
Materials and methods
Cells and virus production
Human HT1080 cells were previously transfected with porcine cDNA for α1,3GT, HT‐1,3GT; or a mock construct, HT1080–0 16. The Loss strain of measles 49 was kindly provided by Dr. D. Brown at the Health Protection Agency, Colindale, UK. This measles strain was passaged through the human HT1080 cells and subsequently used to infect either α1,3GT cDNA‐transfected HT‐1,3GT or mock‐transfected HT1080–0 cells, from which supernatants containing virus (referred to as Mv‐G1–3G or Mv, respectively) were harvested upon maximal cytopathic effect, as previously described 24. Cell debris was cleared from the viral supernatants by centrifugation, titred on monkey Vero cells as plaque forming units (PFU)/mL and aliquots stored at –80°. Subsequent to thawing, aliquots of the virus were exposed to 15 min UV irradiation (253 nm) on ice. Nunc Maxisorp 96‐well plates were coated overnight at 4°C with 1 μg anti‐measles hemagglutinin mAb (clone ZD6, Biodesign Intl., Kennebunk, ME). The wells were washed using 0.1% Tween 20 in PBS, blocked with a PBS solution containing 0.1% Tween 20 and 1% bovine serum albumin (DB) for 1 h at 25°C and again washed. Viral supernatants (Mv‐G‐1–3G or Mv), diluted with respective uninfected cell supernatants, were added at identical 5 × 105 PFU/mL and incubated at 37°C for 30 min. As controls, virus‐free supernatants from the different uninfected cells were always included. New washes were followed by the addition of an HRP‐labelled Galα1,3Gal‐specific lectin from Bandeiraea simplicifolia, BSI‐B4 (Sigma), at three different dilutions in DB and incubated at 37°C for 1 h. The plate was subsequently incubated with freshly made OPD peroxidase substrate (FastTM; Sigma) for 30 min, and absorbance was analyzed at 450 nm on a Dynatech MRX plate reader.
Culture of human monocyte‐derived DC and isolation of T cells
Human monocytes from normal adult peripheral blood were prepared using LPS‐free lymphoprep (Nycomed Pharma, Norway) and used for further differentiation into DC, as previously described 50. Briefly, the resulting cells were resuspended in RPMI medium, plated in endotoxin‐free culture plates and incubated for 2 h to allow monocyte adhesion. Subsequently, adherent cells were incubated for 5 days at 37°C/5% CO2 in RPMI medium containing 104 U/mL GM‐CSF, 104 U/mL IL‐4 and 10% FCS, allowing differentiation into immature DC. After 5 days non‐adherent cells were removed, washed and immunodepleted using anti‐CD3 and anti‐CD19 immunobeads (Dynal AS, Norway). Autologous T cells were isolated simultaneously from the non‐adherent fraction of peripheral blood cells following centrifugation and washes; after immunodepletion with anti‐HLA‐DR, anti‐CD19 and anti‐CD14 immunobeads (Dynal AS, Norway), the cells were stored at –80°. Local Ethical Committee permission was received for these studies, and informed consent from the participating laboratory worker volunteers was obtained.
T cell proliferation assay
Immature DC (2.5 × 104/well unless otherwise stated) were γ‐irradiated and incubated for 2 h with 2.5 × 103 Mv‐G1–3G or Mv particles, UV‐irradiated or not, in the presence of 20% human autologous immune serum with or without heat‐inactivation of C’ for 30 min at 56°C. Alternatively, 10 µg/mL affinity‐purified baboon anti‐Galα1,3Gal, a gift from BioTransplant Inc. (Charlestown, MA) and 1% rabbit C’ serum (Sigma) was used instead of human serum. In one experiment virus was substituted for by 103 irradiated cells of the different virus‐producing cell lines HT1080–0 or HG‐1,3GT, and in another experiment 2.5 × 104 allogeneic DC were used as stimulators. Subsequently, 2.5 × 104 T cells were added in an equal volume, resulting in a 1:2 dilution of the previously added components; in some experiments lactacystin (10 µM; Sigma) or 50 ug/mL of the blocking anti‐MHC class I mAb W6/32 (a kind gift from P. Kourilsky, Paris) 32 was added to selected cultures. Cells were incubated at 37°C for 4 days in 3% FCS. [3H]‐thymidine (1 µCi) was added to cultures, which were then left for an additional 24 h. On day 5, cultures were harvested and assayed using a Wallac Trilux Microbeta scintillation counter. In another proliferation protocol, T cells were obtained from non‐adherent PBMC by negative selection using anti‐CD20, anti‐CD56 and anti‐CD14 mAb conjugated to magnetic beads (Miltenyi Biotec, Auburn, CA). The cells were then stimulated for 4 days using 0.25 µg anti‐CD3 mAb (OKT3; Ortho Biotech, Bridgewater, NJ) and 100 IU/mL IL‐2 (Chiron, Suresnes, France) in RPMI/10% FCS, with or without the addition of 10 µM lactacystin.
Fluid phase marker endocytosis
DC were incubated for 2 days with or without control measles virus (Mv) or measles virus expressing the Galα1,3Gal epitope (Mv‐G1–3G), in the presence or absence of purified baboon antibodies against the Galα1,3Gal epitope and rabbit C’ serum (Sigma). Cells were then incubated with either FITC‐dextran or rhodamine‐BSA (Sigma), 5 mg/mL in RPMI, at 37°C. After the indicated times, the cells were washed three times with PBS at 4°C. The amount of FITC accumulated in DC was quantitatively determined using a Beckman‐Coulter Epics XL flow cytometer.
Expression of membrane markers on DC and T cells
DC were incubated for 2 days with or without control measles viruses (Mv) or measles viruses expressing the Galα1,3Gal epitope (Mv‐G1–3G), in the presence or absence of purified baboon antibodies against the Galα1,3Gal epitope and rabbit C’ serum (Sigma). In one experiment DC were incubated with 100 ng/mL LPS (Sigma) and/or 10 μg lactacystin. Cells were then washed twice with PBS at 4°C and incubated with mAb against human MHC class I (Diaclone, France), CD46, CD80, CD86 (all Santa Cruz Biotechnology, CA), CD4 (Diaclone) or CD8 (Diaclone) directly conjugated with FITC or PE for 45 min at 4°C. Cells were then washed three times with PBS at 4°C and directly analyzed using a Beckman‐Coulter Epics XL flow cytometer or a FACScan (Becton Dickinson).
Apoptosis assay
Purified T cells were stimulated with anti‐CD3 mAb (0.25 μg/mL OKT3; Ortho Biotech) and IL‐2 (100 IU/mL) or with OKT3 and anti‐CD28 (1 µg/mL; eBioscience, San Diego, CA) for 4 days and incubated in the presence or absence of 10 µM lactacystin for 2 days. Living cells were determined by FACS analysis using propidium iodine (PI) added 10 min before analysis at 4°C to evaluate the integrity of the cellular membrane and DiOC6 (a marker of the integrity of the mitochondrial transmembrane potential, ΔΨm). Dying cells were PI+ and/or DiOC6low. As an apoptosis control for DiOC6 staining, cells were incubated for 10 min with 5 μM carbonyl cyanide m‐chlorophenyl hydrazone (CICCP; Sigma), which induced a complete ΔΨm loss. Similarly, mature DC were activated with LPS for 2 days and were treated or not with lactacystin. The viability of the cells was determined as for lymphocytes, and the percentage of dying cells (PI+ and/or DiOC6low) was assessed.
Statistical analysis
Statistical analyses were performed using a paired Student's t‐test for in vitro experiments. A difference was considered as significant when p<0.05.
Acknowledgements
This work was supported by a summer studentship (to E. B.) from the Child Health Research Appeal Trust at the UCL Institute of Child Health. Part of the work was funded by a project grant from the Wellcome Trust (to K. G.). We thank Dr. Siobhan Burns for help with DC culture methods, Prof. Christine Kinnon and Dr. Inger Gjertsson for critical reading of the manuscript and BioTransplant Inc. for the gift of affinity‐purified antibodies.
Footnotes
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
WILEY‐VCH
References
- 1. Ochsenbein, A. F., Fehr, T., Lutz, C., Suter, M., Brombacher, F., Hengartner, H. and Zinkernagel, R. M., Control of early viral and bacterial distribution and disease by natural antibodies. Science 1999. 286: 2156–2159. [DOI] [PubMed] [Google Scholar]
- 2. Carroll, M. C., The complement system in regulation of adaptive immunity. Nat. Immunol. 2004. 5: 981–986. [DOI] [PubMed] [Google Scholar]
- 3. Stager, S., Alexander, J., Kirby, A. C., Botto, M., Rooijen, N. V., Smith, D. F., Brombacher, F. and Kaye, P. M., Natural antibodies and complement are endogenous adjuvants for vaccine‐induced CD8+ T cell responses. Nat. Med. 2003. 9: 1287–1292. [DOI] [PubMed] [Google Scholar]
- 4. Baumgarth, N., Herman, O. C., Jager, G. C., Brown, L. E., Herzenberg, L. A. and Chen, J., B‐1 and B‐2 cell‐derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 2000. 192: 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ochsenbein, A. F. and Zinkernagel, R. M., Natural antibodies and complement link innate and acquired immunity. Immunol. Today 2000. 21: 624–630. [DOI] [PubMed] [Google Scholar]
- 6. Kaiserlian, D. and Dubois, B., Dendritic cells and viral immunity: friends or foes? Semin. Immunol. 2001. 13: 303–310. [DOI] [PubMed] [Google Scholar]
- 7. Larsson, M., Beignon, A. S. and Bhardwaj, N., DC‐virus interplay: a double edged sword. Semin. Immunol. 2004. 16: 147–161. [DOI] [PubMed] [Google Scholar]
- 8. Larsson, M., Fonteneau, J. F., Somersan, S., Sanders, C., Bickham, K., Thomas, E. K., Mahnke, K., and Bhardwaj, N., Efficiency of cross presentation of vaccinia virus‐derived antigens by human dendritic cells. Eur. J. Immunol. 2001. 31: 3432–3442. [DOI] [PubMed] [Google Scholar]
- 9. Servet‐Delprat, C., Vidalain, P. O., Valentin, H. and Rabourdin‐Combe, C., Measles virus and dendritic cell functions: how specific response cohabits with immunosuppression. Curr. Top. Microbiol. Immunol. 2003. 276: 103–123. [DOI] [PubMed] [Google Scholar]
- 10. Schneider‐Schaulies, S., Klagge, I. M. and ter Meulen, V., Dendritic cells and measles virus infection. Curr. Top. Microbiol. Immunol. 2003. 276: 77–101. [DOI] [PubMed] [Google Scholar]
- 11. Moxon, E. R., Rainey, P. B., Nowak, M. A. and Lenski, R. E., Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 1994. 4: 24–33. [DOI] [PubMed] [Google Scholar]
- 12. Xu, Y., Yang, Y. G., Ohdan, H., Ryan, D., Harper, D., Wu, C., Kruger‐Grey, H. S. et al., Characterization of anti‐Gal antibody‐producing cells of baboons and humans. Transplantation 2006. 81: 940–948. [DOI] [PubMed] [Google Scholar]
- 13. Ohdan, H., Swenson, K. G., Kruger‐Gray, H. S., Yang, Y. G., Xu, Y., Thall, A. D. and Sykes, M., Mac‐1‐negative B‐1b phenotype of natural antibody‐producing cells, including those responding to Gal alpha 1,3Gal epitopes in alpha 1,3‐ galactosyltransferase‐deficient mice. J. Immunol. 2000. 165: 5518–5529. [DOI] [PubMed] [Google Scholar]
- 14. Muschel, L. H., Blood groups, disease, and selection. Bacteriol. Rev. 1966. 30: 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strahan, K. M., Gu, F., Preece, A. F., Gustavsson, I., Andersson, L. and Gustafsson, K., cDNA sequence and chromosome localization of pig alpha 1,3galactosyltransferase. Immunogenetics 1995. 41: 101–105. [DOI] [PubMed] [Google Scholar]
- 16. Takeuchi, Y., Porter, C. D., Strahan, K. M., Preece, A. F., Gustafsson, K., Cosset, F.‐L., Weiss, R. A. and Collins, M. K. L., Sensitisation of cells and retroviruses to human serum by alpha(1–3)galactosyltransferase. Nature 1996. 379: 85–88. [DOI] [PubMed] [Google Scholar]
- 17. Rother, R. P., Fodor, W. L., Springhorn, J. P., Birks, C. W., Setter, E., Sandrin, M. S., Squinto, S. P. and Rollins, S. A., A novel mechanism of retrovirus inactivation in human serum mediated by anti‐alpha‐galactosyl natural antibody. J. Exp. Med. 1995. 182: 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandrin, M. S. and McKenzie, I. F. C., Gala(1,3)Gal, the major xenoantigen(s) recognised in pigs by human natural antibodies. Immunol. Rev. 1994. 141: 169–190. [DOI] [PubMed] [Google Scholar]
- 19. Gustafsson, K., Strahan, K. and Preece, A., α1,3Galactosyltransferase: a target for in vivo genetic manipulation in xenotransplantation. Immunol. Rev. 1994. 141: 59–70. [DOI] [PubMed] [Google Scholar]
- 20. Galili, U. and LaTemple, D. C., Natural anti‐Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol. Today 1997. 18: 281–285. [DOI] [PubMed] [Google Scholar]
- 21. Jäger, U., Takeuchi, Y. and Porter, C. D., Induction of complement attack on human cells by Gal(alpha1,3)Gal xenoantigen expression as a gene therapy approach to cancer. Gene Therapy 1999. 6: 1073–1083. [DOI] [PubMed] [Google Scholar]
- 22. Rossi, G. R., Mautino, M. R., Unfer, R. C., Seregina, T. M., Vahanian, N. and Link, C. J., Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha(1,3)‐galactosyl epitopes. Cancer Res. 2005. 65: 10555–10561. [DOI] [PubMed] [Google Scholar]
- 23. Rossi, G. R., Unfer, R. C., Seregina, T. and Link, C. J., Complete protection against melanoma in absence of autoimmune depigmentation after rejection of melanoma cells expressing alpha(1,3)galactosyl epitopes. Cancer Immunol. Immunother. 2005. 54: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Preece, A. F., Strahan, K. M., Devitt, J., Yamamoto, F. and Gustafsson, K., Expression of ABO or related antigenic carbohydrates on viral envelopes leads to neutralization in the presence of serum containing specific natural antibodies and complement. Blood 2002. 99: 2477–2482. [DOI] [PubMed] [Google Scholar]
- 25. Neil, S. J., McKnight, A., Gustafsson, K. and Weiss, R. A., HIV‐1 incorporates ABO histo‐blood group antigens that sensitise virions to complement‐mediated inactivation. Blood 2005. 105: 4693–4699. [DOI] [PubMed] [Google Scholar]
- 26. Seymour, R. M., Allan, M. J., Pomiankowski, A. and Gustafsson, K., Evolution of the human ABO polymorphism by two complementary selective pressures. Proc. Biol. Sci. 2004. 271: 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeuchi, Y., Cosset, F.‐L. C., Lachmann, P. J., Okada, H., Weiss, R. A. and Collins, M. K. L., Type C retrovirus inactivation by human complement is determined by both the viral genome and the producer cell. J. Virol. 1994. 68: 8001–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grosjean, I., Caux, C., Bella, C., Berger, I., Wild, F., Banchereau, J. and Kaiserlian, D., Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J. Exp. Med. 1997. 186: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fugier‐Vivier, I., Servet‐Delprat, C., Rivailler, P., Rissoan, M. C., Liu, Y. J. and Rabourdin‐Combe, C., Measles virus suppresses cell‐mediated immunity by interfering with the survival and functions of dendritic and T cells. J. Exp. Med. 1997. 186: 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schnorr, J. J., Xanthakos, S., Keikavoussi, P., Kampgen, E., ter Meulen, V. and Schneider‐Schaulies, S., Induction of maturation of human blood dendritic cell precursors by measles virus is associated with immunosuppression. Proc. Natl. Acad. Sci. USA 1997. 94: 5326–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Villinger, F., Mayne, A. E., Bostik, P., Mori, K., Jensen, P. E., Ahmed, R. and Ansari, A. A., Evidence for antibody‐mediated enhancement of simian immunodeficiency virus (SIV) Gag antigen processing and cross presentation in SIV‐infected rhesus macaques. J. Virol. 2003. 77: 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barnstable, C. J., Bodmer, W. F., Brown, G., Galfre, G., Milstein, C., Williams, A. F. and Ziegler, A., Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens‐new tools for genetic analysis. Cell 1978. 14: 9–20. [DOI] [PubMed] [Google Scholar]
- 33. Bellas, R. E., FitzGerald, M. J., Fausto, N. and Sonenshein, G. E., Inhibition of NF‐kappa B activity induces apoptosis in murine hepatocytes. Am. J. Pathol. 1997. 151: 891–896. [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng, Y., Cheng, G., Chui, C. H., Lau, F. Y., Chan, P. K., Ng, M. H., Sung, J. J. and Wong, R. S., ABO blood group and susceptibility to severe acute respiratory syndrome. JAMA 2005. 293: 1450–1451. [DOI] [PubMed] [Google Scholar]
- 35. Benatuil, L., Kaye, J., Rich, R. F., Fishman, J. A., Green, W. R. and Iacomini, J., The influence of natural antibody specificity on antigen immunogenicity. Eur. J. Immunol. 2005. 35: 2638–2647. [DOI] [PubMed] [Google Scholar]
- 36. Abdel‐Motal, U., Wang, S., Lu, S., Wigglesworth, K. and Galili, U., Increased immunogenicity of human immunodeficiency virus gp120 engineered to express Galalpha1–3Galbeta1–4GlcNAc‐R epitopes. J. Virol. 2006. 80: 6943–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaiserlian, D. and Etchart, N., Entry sites for oral vaccines and drugs: A role for M cells, enterocytes and dendritic cells? Semin. Immunol. 1999. 11: 217–224. [DOI] [PubMed] [Google Scholar]
- 38. Wolf, J. L., Rubin, D. H., Finberg, R., Kauffman, R. S., Sharpe, A. H., Trier, J. S. and Fields, B. N., Intestinal M cells: a pathway for entry of reovirus into the host. Science 1981. 212: 471–472. [DOI] [PubMed] [Google Scholar]
- 39. Bomsel, M., Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat. Med. 1997. 3: 42–47. [DOI] [PubMed] [Google Scholar]
- 40. Rescigno, M., Urbano, M., Valzasina, B., Francolini, M., Rotta, G., Bonasio, R., Granucci, F. et al., Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001. 2: 361–367. [DOI] [PubMed] [Google Scholar]
- 41. Neutra, M. R. and Kozlowski, P. A., Mucosal vaccines: the promise and the challenge. Nat. Rev. Immunol. 2006. 6: 148–158. [DOI] [PubMed] [Google Scholar]
- 42. Diebold, S. S., Montoya, M., Unger, H., Alexopoulou, L., Roy, P., Haswell, L. E., Al Shamkhani, A. et al., Viral infection switches non‐plasmacytoid dendritic cells into high interferon producers. Nature 2003. 424: 324–328. [DOI] [PubMed] [Google Scholar]
- 43. Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. and Reis e Sousa, C., Innate antiviral responses by means of TLR7‐mediated recognition of single‐stranded RNA. Science 2004. 303: 1529–1531. [DOI] [PubMed] [Google Scholar]
- 44. Lund, J. M., Alexopoulou, L., Sato, A., Karow, M., Adams, N. C., Gale, N. W., Iwasaki, A. and Flavell, R. A., Recognition of single‐stranded RNA viruses by Toll‐like receptor 7. Proc. Natl. Acad. Sci. USA 2004. 101: 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bieback, K., Lien, E., Klagge, I. M., Avota, E., Schneider‐Schaulies, J., Duprex, W. P., Wagner, H. et al., Hemagglutinin protein of wild‐type measles virus activates toll‐like receptor 2 signaling. J. Virol. 2002. 76: 8729–8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moron, G., Dadaglio, G. and Leclerc, C., New tools for antigen delivery to the MHC class I pathway. Trends Immunol. 2004. 25: 92–97. [DOI] [PubMed] [Google Scholar]
- 47. Villinger, F., Mayne, A. E., Bostik, P., Mori, K., Jensen, P. E., Ahmed, R. and Ansari, A. A., Evidence for antibody‐mediated enhancement of simian immunodeficiency virus (SIV) Gag antigen processing and cross presentation in SIV‐infected rhesus macaques. J. Virol. 2003. 77: 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kopf, M., Abel, B., Gallimore, A., Carroll, M. and Bachmann, M. F., Complement component C3 promotes T cell priming and lung migration to control acute influenza virus infection. Nat. Med. 2002. 8: 373–378. [DOI] [PubMed] [Google Scholar]
- 49. Sinitsyna, O. A., Khudaverdyan, O. E., Steinberg, L. L., Nagieva, F. G., Lotte, V. D., Dorofeeva, L. V., Rozina, E. E. and Boriskin, Y., Further‐attenuated measles vaccine: virus passages affect viral surface protein expression, immunogenicity and histopathology pattern in vivo. Res. Virol. 1990. 141: 517–531. [DOI] [PubMed] [Google Scholar]
- 50. Burns, S., Thrasher, A. J., Blundell, M. P., Machesky, L. and Jones, G. E., Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood 2001. 98: 1142–1149. [DOI] [PubMed] [Google Scholar]