

Abstract
Purpose
To determine, using an aptamer-based technology in patients with intermediate age-related macular degeneration (AMD), (1) if there is a difference in plasma levels of 4979 proteins in patients with and without reticular pseudodrusen (RPD), and (2) if plasma levels of proteins are related to time to conversion to advanced AMD.
Methods
Patients with intermediate AMD and RPD were identified from an AMD registry. Relative concentrations of each protein were log (base 2) transformed and compared between patients with and without RPD using linear regression. A Cox proportional hazards survival model was fit to each aptamer to quantify associations with time to conversion. A pathway analysis was conducted in converters versus non-converters using the Reactome database.
Results
Of the 109 intermediate AMD patients, 39 had bilateral RPD (36%). Two proteins, TCL1A and CNDP1, were lower in patients in the intermediate AMD group with RPD. Twenty-one patients converted to advanced AMD with a median time to conversion of 25.2 months (range, 2.3–48.5 months) and median follow-up time in non-converters of 26.4 months (range, 0.03–49.7 months). Several proteins (lysozyme C, TFF3, RNAS6, and SAP3) distinguished patients who converted from those who did not convert to advanced AMD. The top conversion pathways included tumor necrosis factors bind their physiological receptors, digestion and absorption, signaling by activin, and signaling by TGF-β family members.
Conclusions
We identified a protein signature related to RPD, as well as to conversion to advanced AMD. The pathway analysis suggests that dysfunction of critical systemic pathways may have links to conversion to advanced AMD.
Translational Relevance
Biomarkers identified in plasma likely reflect systemic alterations in protein expression in patients with intermediate AMD.
Keywords: aptamer-based proteomics, intermediate age-related macular degeneration, reticular pseudodrusen
Introduction
Age-related macular degeneration (AMD) is a leading cause of vision loss among elderly individuals.1 This debilitating disease impacts as many as 11 million people in the United States.2 The advanced stages of the disease have two forms: neovascular AMD (NV) and geographic atrophy (GA).3,4 Treatment options are limited to individuals with the neovascular form of the disease; however, these interventions only serve to attenuate progress of the disease and do not cure the disease. AMD has many adverse sequelae, including the loss of the ability to drive5 and to read.6 Moreover, there are economic costs7 and a significant burden for the caregiver of the AMD patient.8 For this reason, AMD is emerging as a critical public health concern among older populations.9
The destructive pathologic process related to AMD is primarily located in the macula.3 A deposition of extracellular material called drusen begins to appear early in the natural history of AMD. Based on histologic analyses, drusen deposits are located between the basal lamina of the retinal pigment epithelium (RPE) and Bruch's membrane. Originally described in 1990,10 other deposits referred to as reticular pseudodrusen (RPD) have been described in patients with AMD. Unlike drusen, these deposits are located in the subretinal space internal to the RPE and are often referred to as subretinal drusenoid deposits. These deposits share many properties with drusen but differ in lipid composition.11
Detection of RPD has improved with advances in imaging modalities.11 This has led to an increased interest among researchers to understand the clinical significance of these subretinal lesions.12 Relationships have been described for the presence of RPD and increasing age, female sex, and genetic variants.11,13–18 Pathologic changes in the choroid11 and hypothesized dysregulation of cholesterol homeostasis19 have also been shown to be associated with RPD. Moreover, the presence of RPD has been shown to be associated with an increased risk of progression to advanced AMD, specifically GA.20
The focus of research from our group has been to investigate systemic biomarkers related to AMD. Using our AMD registry and biorepository,21 we have recently reported several proteins linked with the presence of advanced AMD22,23 and intermediate AMD.24 In the present study, we build on our biomarker research and focus on a cohort of patients with the intermediate phenotype of AMD. To our knowledge, we are presenting the first large-scale biomarker discovery study to utilize a multiplexed aptamer-based proteomic technology in intermediate AMD. We addressed two research questions in this hypothesis-generating study: (1) to determine if there is a difference in systemic levels of a panel of 4979 proteins in patients with and without RPD, and (2) to determine if systemic levels of proteins are related to time to conversion to advanced forms of AMD.
Materials and Methods
Overview of the Colorado AMD Registry
We conducted this cohort study by using records and samples from an AMD research registry and repository (described in detail elsewhere21–24) developed by the Department of Ophthalmology at the University of Colorado School of Medicine. For this study, we focused on patients in the registry with the intermediate form of AMD.25 For our first research question, protein signatures were compared in intermediate AMD patients with bilateral RPD and without RPD. To address our second research question, protein signatures were compared in patients who progressed to the advanced forms of AMD (converters) and non-converters. All patients received care at the UCHealth Sue Anschutz-Rodgers Eye Center. The registry was approved by the Colorado Multiple Institutional Review Board. The research adhered to the tenets of the Declaration of Helsinki. Informed written consent was obtained from all patients in the registry after explanation of the nature and possible consequences of the research. Enrollment in the registry is ongoing.
All patients consented to (1) a review of the medical history, (2) collection of an ethylenediaminetetraacetic acid (EDTA) plasma sample, and (3) review and disease phenotype classification of image data, including a color fundus photograph, fundus autofluorescence (FAF), near-infrared fundus reflectance (NIR), and spectral domain–optical coherence tomography (SD-OCT). Ocular exclusion criteria for patients were panretinal photocoagulation or anti-vascular endothelial growth factor injections for diabetic retinopathy, branch and central retinal vein occlusion, any active ocular inflammatory disease, or a severe decrease in visual acuity secondary to a preexisting severe retinal disease other than AMD.
Image Review
Images, described above, were performed upon enrollment into the study and were reviewed by two vitreo–retinal specialists (M.T.M., F.S.S.). For this study, we examined only the images from the enrollment visit. The image review was focused on an examination of the anatomic macula, which includes the entire area between the retinal vascular arcades. The images were categorized into early, intermediate, or advanced AMD using the classification described by Ferris et al.25 Discrepancies were resolved by a third vitreo–retinal specialist (N.M.). The presence or absence of RPD was also determined. Our definition of the presence of RPD was RPD observed on FAF and/or NIR imaging and confirmed on SD-OCT.
Study Specific Inclusion and Exclusion Criteria
To address our research questions and to maintain a homogeneous cohort, we reviewed the records of non-Hispanic White patients who were recruited into the registry between July 2014 and December 2017. From this dataset, 118 patients with intermediate AMD (advanced and early AMD were removed from the analytic dataset) were identified. A second image review was conducted by N.M. and A.G.P. on this study group to specifically (1) confirm no misclassification of the presence or absence of RPD, and (2) to apply exclusions specific to this study (other retina co-morbidities, n = 5; unilateral RPD, n = 4). Following this review, 109 cases with intermediate AMD and bilateral presence or absence of RPD remained in the final analytic dataset. For the time to conversion analysis we used the last appointment with a retinal specialist as the censoring time. Other risk factors in this dataset included age, sex, family history of AMD, body mass index (BMI), and select co-morbidities.
Collection and Processing of the Plasma Sample
All samples were taken at the time of the multimodal imaging. The EDTA tube was spun at 3000 revolutions per minute in a cooled (4°C) centrifuge for 10 minutes to isolate plasma. The average time from phlebotomy to spin was low at 2.6 minutes ± 1.7 SD (range, 0–10 minutes). The aliquots of plasma were immediately stored in a –80°C freezer.
SomaScan Assay
The SomaScan assay (SomaLogic, Boulder, CO) is described elsewhere.26–28 In brief, a plasma sample in each well of a 96-well plate was incubated with a mixture of the 4979 SOMAmer reagents (SomaLogic). Two sequential bead-based immobilization and washing steps, coupled with kinetic challenge with polyanionic competitors, eliminated unbound or nonspecifically bound proteins and SOMAmer reagents, leaving only protein target-bound SOMAmer reagents. These remaining SOMAmer reagents were isolated, and each reagent was quantified simultaneously on a custom hybridization array (Agilent, Santa Clara, CA). The amount of each SOMAmer reagent measured was quantitatively proportional to the protein concentration in the original sample, as described elsewhere.26–28
For the current version of the SomaScan assay, the coefficients of variation (CVs) for all analytes, including intra- and inter-assay variation, were measured in plasma using triplicate technical replicates for three different clinical samples across 15 independent runs. The median CV for all analyte measurements in plasma was 5.0%, and 90% of analytes had a CV < 12.0%, obtained by averaging CV results over the three clinical samples. In an independent study, the SomaScan assay with plasma samples has recently demonstrated a high degree of stability and reproducibility.29
Statistical Analysis
Descriptive statistics included percentages, means, and standard deviations. We examined the relationship of categorical risk factors using the χ2 test or Fisher's exact test. Differences in continuous variables were compared using two-sample t-tests. Odds ratios and corresponding 95% confidence intervals were estimated using univariate logistic regressions. Relative concentrations for each of 4979 aptamer targets were log (base 2) transformed and compared between subjects with and without RPD using linear regression. P values were adjusted for multiple comparisons using the false discovery rate (FDR) as described by Benjamini and Hochberg.30 Ten subjects with no record of a follow-up visit (and therefore no follow-up image) with a retinal specialist were removed from the conversion analysis. For this analysis, we fit a Cox proportional hazards survival model to each aptamer to quantify the associations with time to conversion. We then conducted a pathway analysis on this group. The pathway analysis detects coordinated changes in levels of proteins in the same pathway and is useful for providing mechanistic insights by evaluating the combined association of groups of proteins that function in the same pathway.
Pathway analyses were conducted using pathways downloaded from Reactome31–33 and a functional class scoring approach, appropriate for platforms where proteins are selected a priori,34 using the P values as the protein-level statistics for the 4979 aptamer targets that were measured.35 This functional class scoring approach differs from an enrichment analysis in that it does not specifically test whether the pathways are enriched with a larger than expected number of significant proteins and therefore also does not require a cut-off to be applied to each protein. The underlying inference from the functional class scoring approach is testing whether the pathway contains at least one measured protein that significantly differed between groups or whether a subset of proteins in the pathway has coordinated differences. Pathways were ranked based on the unadjusted P values calculated using a permutation approach that appropriately accounts for the correlation among proteins, permuting group labels using 1000 permutations.36
Results
We show in Table 1 the differences in demographic variables and select co-morbidities between patients with bilateral RPD, n = 39 (36%), and without RPD, n = 70 (64%). Higher frequencies of a family history of AMD and treated hypertension were found in patients with RPD compared with patients without RPD. In Figure 1, we demonstrate differences in proteins in patients with and without RPD, where a difference of 1 on a log (base 2) scale corresponds to a fold change of 2. As seen, TCL1A and CNDP1 were the top RPD-related proteins and were lower in intermediate patients with bilateral RPD compared with intermediate patients with no RPD. Neither of these proteins was significant following multiple-comparisons testing. Most of the remaining proteins were clustered in the bottom part of the plot. More detailed information on the top two proteins is shown in Table 2, including the UniProt number, full name, and other names for the protein. The ranking of all proteins is shown in the Supplementary Table S1.
Table 1.
Characteristics of Those With and Without RPD in Intermediate AMD
| Intermediate AMD (n = 109) | ||||
|---|---|---|---|---|
| RPD (n = 39) | No RPD (n = 70) | Odds Ratio (95% CI) | P* | |
| Sex, female, n (%) | 25 (64) | 49 (70) | 0.8 (0.3–1.8) | 0.53 |
| Family history of AMD, n (%) | ||||
| None | 14 (36) | 40 (57) | 1 | |
| Yes | 18 (46) | 18 (26) | 2.9 (1.2–7.0) | |
| Uncertain | 7 (18) | 12 (17) | 1.7 (0.5–5.1) | 0.07 |
| Age, mean (SD) | 78 (7.1) | 76 (6.7) | 1.1 (0.9–1.1) | 0.09 |
| BMI | 37 | 67 | ||
| Mean (SD) | 26.2 (4.6) | 26.4 (5.1) | 1.0 (0.9 – 1.1) | 0.78 |
| Smoking, n (%) | ||||
| Never | 16 (41) | 37 (53) | 1 | |
| Current | 1 (3) | 1 (1) | 2.3 (0.1 – 39) | |
| Former | 22 (56) | 32 (46) | 1.6 (0.7 – 3.5) | 0.40† |
| History of, n (%) | ||||
| Type 2 diabetes | 5 (13) | 10 (14) | 0.9 (0.3–2.8) | 0.83 |
| Treated hypertension | 29 (74) | 36 (51) | 2.7 (1.2–6.5) | 0.02 |
| Kidney disease | 8 (21) | 7 (10) | 2.3 (0.8–7.0) | 0.13 |
| Stroke | 0 (0) | 6 (9) | — | 0.09† |
| Peripheral vascular disease | 5 (13) | 9 (13) | 1.0 (0.3–3.2) | 0.99 |
| Atrial fibrillation | 2 (5) | 5 (7) | 0.7 (0.1–3.8) | 0.99† |
| Cardiac disease | 17 (44) | 27 (39) | 1.2 (0.6–2.7) | 0.61 |
P values obtained from χ2 tests for categorical variables and t-tests for continuous variables unless noted otherwise.
P values obtained from Fisher's exact test.
Figure 1.
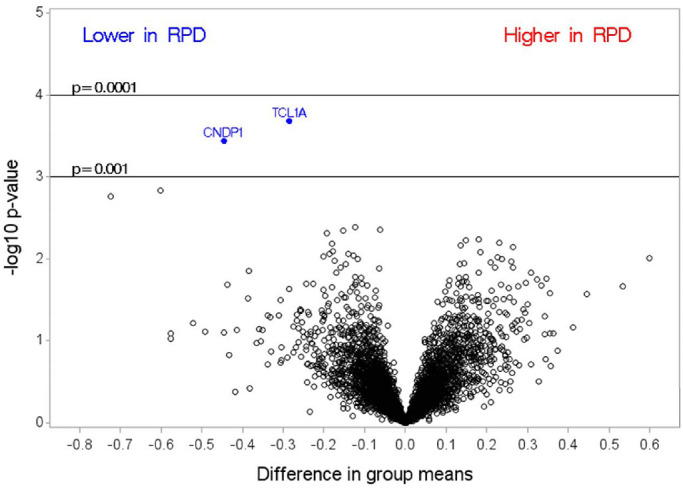
Differences between group means in intermediate AMD patients with and without RPD, where a difference of 1 on a log (base 2) scale corresponds to a fold change of 2.
Table 2.
Top-Ranked Aptamers Comparing RPD Versus No RPD
| UniProt Number | Target | Recommended and Alternative Names | Estimate (SE) | Raw P | fdr P |
|---|---|---|---|---|---|
| P56279 | TCL1A | T-cell leukemia/lymphoma protein 1A; oncogene TCL-1; protein p14 TCL1 | –0.28 (0.07) | <0.001 | 0.90 |
| Q96KN2 | CNDP1 | β-Ala-His dipeptidase; CNDP dipeptidase 1; carnosine dipeptidase 1; glutamate carboxypeptidase-like protein 2; serum carnosinase | –0.45 (0.12) | <0.001 | 0.90 |
The characteristics of patients who did and did not progress to advanced AMD are shown in Table 3. There were 21 converters who converted to advanced AMD (7 GA, 13 NV, and one who converted to both NV and GA) with a median time to conversion of 25.2 months (range, 2.3–48.5 months) and median follow-up time in non-converters of 26.4 months (range, 5.5–49.7 months). The association between the presence of RPD and time to conversion is shown in Figure 2. The log-rank test suggests that there may be a trend for a shorter time to conversion in patients who have RPD compared to those who do not have RPD (P < 0.07).
Table 3.
Characteristics of Patients Who Did and Did Not Convert to AMD
| Intermediate AMD (n = 99) | ||||
|---|---|---|---|---|
| Non-Converters (n = 78) | Converters (n = 21) | Odds Ratio (95% CI) | P* | |
| RPD, n (%) | 24 (31) | 10 (48) | 2.0 (0.8–5.5) | 0.15 |
| Sex, female, n (%) | 54 (69) | 13 (62) | 0.7 (0.3–2.0) | 0.53 |
| Family history of AMD, n (%) | ||||
| None | 39 (50) | 11 (52) | 1 | |
| Yes | 23 (29) | 8 (38) | 1.2 (0.4–3.5) | |
| Uncertain | 16 (21) | 2 (10) | 0.4 (0.1–2.2) | 0.51† |
| Age (y), mean (SD) | 76 (6.9) | 80 (6.1) | 1.10 (1.02–1.19) | 0.02 |
| BMI | 76 | 21 | ||
| Mean (SD) | 26.2 (5.2) | 26.5 (4.2) | 1.0 (0.9–1.1) | 0.84 |
| Smoking, n (%) | ||||
| Never | 38 (49) | 11 (52) | 1 | |
| Current | 2 (3) | 0 | — | |
| Former | 38 (49) | 10 (48) | 0.9 (0.3–2.4) | 0.99† |
| History of, n (%) | ||||
| Type 2 diabetes | 12 (15) | 3 (14) | 0.9 (0.2–3.6) | 0.99† |
| Treated hypertension | 44 (56) | 15 (71) | 1.9 (0.7–5.5) | 0.21 |
| Kidney disease | 10 (13) | 2 (10) | 0.7 (0.1–3.6) | 0.99† |
| Stroke | 4 (5) | 2 (10) | 1.9 (0.3–11.4) | 0.60† |
| Peripheral vascular disease | 10 (13) | 4 (19) | 1.6 (0.4–5.7) | 0.49† |
| Atrial fibrillation | 3 (4) | 3 (14) | 4.2 (0.8–22.4) | 0.11† |
| Cardiac disease | 31 (40) | 9 (43) | 1.1 (0.4–3.0) | 0.80 |
P values obtained from χ2 tests for categorical variables and t-tests for continuous variables.
P values obtained from Fisher's exact test.
Figure 2.
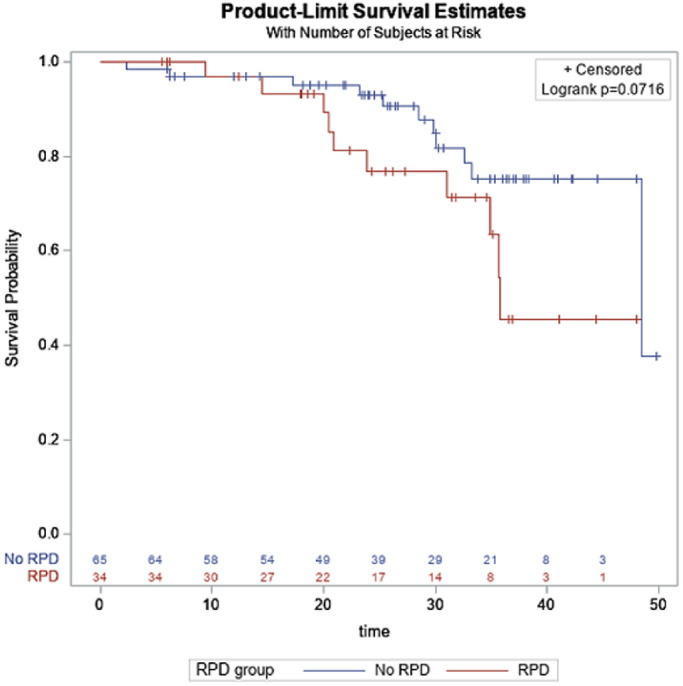
Association between the presence of reticular pseudodrusen and time to conversion.
Several proteins were related to conversion to advanced AMD (Fig. 3). In Table 4, the top-ranked proteins from the plot are presented in more detail, including the UniProt number, full name, and other names for the protein. As shown, the top four proteins—lysozyme, TFF3, RNAS6, and SAP3—had highly significant unadjusted associations with the time to conversion (hazard ratios = 5.6, 3.0, 3.9, and 10.1, respectively). The differences in these protein levels were mainly driven by neovascular AMD, and for all, except RNAS6, levels were also elevated in those who converted to GA (Fig. 4). None of the conversion proteins was significant following multiple-comparisons testing. The ranking of all proteins is shown in Supplementary Table S2.
Figure 3.
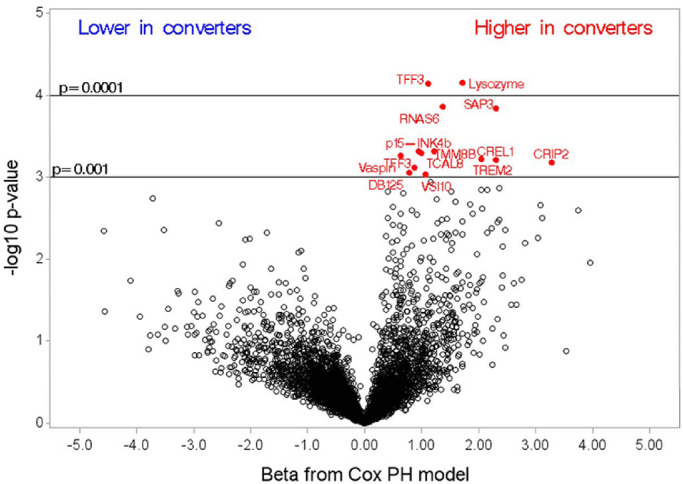
Parameter estimates from the Cox proportional-hazards model assessing association with time to conversion.
Table 4.
Top-Ranked Aptamers Associated With Time to Conversion
| UniProt Number | Target | Recommended and Alternative Names | Estimate (SE) | Hazard Ratio | Raw P | fdr P |
|---|---|---|---|---|---|---|
| P61626 | Lysozyme C | — | 1.73 (0.43) | 5.62 | <0.001 | 0.18 |
| Q07654 | TFF3 | Trefoil factor 3; intestinal trefoil factor; polypeptide P1.B | 1.11 (0.28) | 3.04 | <0.001 | 0.18 |
| Q93091 | RNAS6 | Ribonuclease K6 | 1.37 (0.36) | 3.94 | <0.001 | 0.18 |
| P17900 | SAP3 | Ganglioside GM2 activator; cerebroside sulfate activator protein; GM2-AP; sphingolipid activator protein 3 | 2.31 (0.61) | 10.08 | <0.001 | 0.18 |
Figure 4.

Boxplots displaying the distribution of the protein aptamer levels for the top conversion-related proteins by AMD type. The box extends to the 25th and 75th percentiles, the line and diamond correspond to the median and mean values, respectively. The whiskers show values within 1.5 times the interquartile range. Individual points are overlaid on top to show the raw values. The one subject who converted to both GA and NV is included in the NV group for this plot.
The high-signal proteins noted in the time-to-conversion analysis were of interest. The top four pathways with at least 10 of the aptamers included are displayed in Table 5. The rankings of all of the pathways can be found in Supplementary Table S3.
Table 5.
Pathway Analysis: Top Four Converter Pathways
| Rank* | Pathway Name | k | Raw P | Adjusted P |
|---|---|---|---|---|
| 1 | TNFs bind their physiological receptors | 29 | 0.013 | 0.62 |
| 2 | Digestion and absorption | 14 | 0.013 | 0.92 |
| 3 | Signaling by activin | 12 | 0.016 | 0.94 |
| 4 | Signaling by TGF-β family members | 58 | 0.024 | 0.58 |
Table includes pathways with a minimum of 10 proteins and are sorted by raw P value.
Discussion
We embarked on this study to profile the plasma proteome in a highly multiplexed manner to discover potential markers of intermediate AMD with RPD and to identify subjects with a higher likelihood for progression from intermediate to advanced AMD. We hypothesized that there are predisposing systemic proteins and pathways for the local disease related to AMD that could be detected in the circulation. The first key finding was that, apart from two proteins (TCL1A and CNDP1), no difference was found in the proteins measured in patients with and without RPD. The second key finding was that, when we examined time to conversion from intermediate to advanced AMD, we found higher levels of several proteins in converters versus non-converters. Although the differences were not significant following multiple-comparisons testing, the magnitude of the relationship37 of these proteins with conversion was meaningful. The final finding was that the key pathways linked with conversion had connections with inflammation, immune/tissue homeostasis, and digestion and absorption.
For this study, we used an aptamer-based multiplexed proteomic assay (SomaScan assay) currently capable of measuring 4979 proteins. The assay utilizes a collection of DNA-based aptamers with base modifications that resemble side chains of amino acids overrepresented in interaction surfaces of protein–protein complexes (e.g., antibody–protein antigen interactions). These base modifications have expanded the range of proteins for which aptamers with high affinity and specificity and slow dissociation kinetics can be selected.26 These binding properties are intimately tied to the exquisite shape complementary with which aptamers engage distinct epitopes on protein surfaces, in a manner that is highly similar to that of antibodies.38 The highly specific nature of protein measurements obtained by the SomaScan assay, as well as the reproducibility and stability of the measurements, have been reported in several recent studies.29,39,40
The protein TCL1A was one of two proteins significantly associated with the presence of RPD. TCL1A is part of a family of oncoproteins that enhance cell transformation and are important in tumor progression.41 The second protein was CNDP1, an enzyme with antioxidant, anti-inflammatory, and anti-aggregate characteristics that degrades carnosine and is found in muscle and brain tissue. An inherited deficiency of carnosinase is related to the neurometabolic disorder carnosinemia. This disorder is clinically characterized by mental deterioration, spastic paraplegia, and retinitis pigmentosa.42,43 Altered levels of carnosinase have also been described in other neurological and systemic disorders.44,45
The most highly ranked protein tied to conversion to advanced AMD was lysozyme C, which has an important role in innate immunity. Lysozymes have a bacteriolytic function and have strong ties to the macrophage/monocyte system. As reviewed by Ragland and Criss,46 lysozymes are an integral part of immune system. Originally described in 1922 by Alexander Fleming as a substance in the nasal mucus that could kill bacteria, lysozymes are now recognized to be found in many body fluids and to influence resolution of inflammation especially on mucosal surfaces. Importantly, mutations of this antimicrobial protein are reported to have links with systemic hereditary amyloidosis.47,48 The second ranked protein was TFF3, a member of the trefoil factor (TFF) family. These proteins are secreted with mucins and have an important role in wound healing, mucosal protection and repair, and inhibition of apoptosis. Dysregulated expression of these proteins is also associated with several tumors and other disorders.49,50 It is noteworthy that TFF peptides have been found to be expressed in the human lacrimal sac and nasolacrimal duct51 and to be proliferative and pro-apoptotic in the murine retina.52 These peptides also support corneal wound healing.53
Another top-ranked protein was RNA6. This protein is derived from epithelial cells and is an antimicrobial peptide against Gram-positive and Gram-negative bacteria.54 It is of interest that our top-ranked proteins linked with progression to advanced AMD were all antimicrobial proteins. This finding is aligned with recent hypotheses that the microbiota may have a role in diseases of the retina.55–57 The last of the highly ranked proteins found in the patients who converted to advanced AMD was SAP3 (ganglioside GM2 activator), which stimulates the breakdown of ganglioside GM2 and glycolipid GA2 by β-hexosaminidase A.33 A genetic deficiency of either the β-hexosaminidase A or the GM2 activator can result in the neurological disorder and cherry-red foveal spots in the retina.58,59
With regard to the pathway analysis, we found several pathways highly ranked in their relationship with conversion to advanced AMD. The most highly ranked pathway was the tumor necrosis factors (TNF) pathway. First isolated in 1984, this superfamily of TNF proteins, which exert their physiological effects by binding to their receptors on cell surfaces, consists of cytokines, mainly derived from macrophages and monocytes. As reviewed by Aggarwal,60 although TNFs regulate normal functions such as immune responses, hematopoiesis, and morphogenesis, they also have a role in tumorigenesis, transplant rejection, septic shock, viral replication, bone resorption, rheumatoid arthritis, and diabetes. In the latter situations, these cytokines have links with cellular proliferation, survival, differentiation, or apoptosis. Indeed, anti-TNF therapy is approved for several inflammatory diseases.60
Signaling by transforming growth factor beta (TGF-β) family members was another pathway ranked high in our pathway analysis. The TGF-β superfamily of cytokines regulate cellular functions during development and are important in maintaining tissue homeostasis. Activin, also represented in our pathway analysis, is another member of this family of cytokines. TGF-β is a cytokine regulator that can either inhibit or stimulate cell proliferation. Dysregulation of TGF-β signaling is related to the pathogenesis of several connective tissue diseases.61 There are also reports of an emerging role of TGF-β in AMD and that TGF-β signaling pathways may be a target for treatment in, specifically, the neovascular form of advanced AMD.62 Indeed, a TGF-β inhibitor has been shown to have a role in decreasing the development of choroidal neovascular (CNV) lesions in a rat model of CNV induction.63 This family of proteins also has been found to be downregulated in the aqueous of patients with neovascular AMD.64 TGF-β also has a critical role in immune homeostasis, as dysregulation of this signaling pathway is related to inflammatory diseases and the development of tumors.65,66 Another top-ranked but smaller pathway was digestion and absorption, which refers to digestion of fats, carbohydrates, and proteins in the intestine and movement of the products of digestion out of the lumen of the intestine.33 Collectively, the top-ranked pathways described in this study suggest that dysfunction of critical systemic pathways may have links to conversion to the advanced forms of AMD.
The main limitation of the study was sample size, which may have impacted the results of our first research question. Apart from two proteins, the remainder of the proteins examined were not significantly related to the outcome, RPD. Sample size also limited our ability to stratify by conversion to either neovascular AMD or GA, which may have a different underlying pathogenesis. Another limitation was that, when adjusted for multiple comparisons, all of the unadjusted significant findings became non-significant. Because this was a small, hypothesis-generating study, we view the application of the FDR to be less important.67 Our a priori interest was focused more on the ranking of the proteins (not impacted by the FDR) than on finding a cut-off based on statistical significance. We believe that with this approach we found some interesting high-signal protein signatures, especially in our group of patients who progressed to advanced AMD. Moreover, the risk estimates37,67 associated with the protein signatures in the conversion analysis were impressive. Reporting of these results is an important first step in our research. Recruitment into our AMD registry is ongoing. With a larger sample size, we hope to validate our results in a future intermediate AMD cohort.
Another limitation of this study was that we did not examine samples such as aqueous to determine if our systemic findings were also found in samples closer to the source of the local pathology of AMD. Indeed, the longstanding concept is that AMD is linked with local inflammatory events. This is supported by the findings of complement components, as constituents of drusen,68 markers of complement activation in the aqueous humor,69 and the complement membrane attack complex in the choriocapillaris.70,71 The results of the pathway analysis from this hypothesis-generating study suggest that markers of systemic inflammation may also be contributing to intermediate AMD progression. Apart from a small number of studies of the complement system in mainly advanced AMD,23,72–79 the significance of the presence of systemic biomarkers of inflammation in AMD and specifically in intermediate AMD has not been adequately addressed. We addressed this knowledge gap in a recent study of 17 complement factors in patients with intermediate AMD. After adjusting for multiple-comparisons testing, we found significantly altered levels of nine complement factors in the cases versus controls with no AMD.24 The results of that complement study and this proteomic study suggest that the role of systemic inflammation in intermediate AMD progression requires a more comprehensive investigation to specifically determine if select inflammatory markers may be both a target and a biomarker for potential intervention strategies to prevent transition to the devastating forms of advanced AMD. To this end, we will continue to recruit and follow this valuable cohort of patients with intermediate AMD. Moreover, moving forward we will also examine genetic variants of the complement system and their relationship with AMD progression as correlated with systemic biomarkers. It is established that polymorphisms of the central complement factor C3 and variants of CFH, CFI, and CFB, factors that affect C3 activation or degradation of its active products, are risk factors for AMD.68,80–83 As we expand this intermediate AMD cohort, we will direct our attention to the role of polymorphisms called complotypes (defined as any inherited pattern of genetic variants in complement genes that alters risk for both inflammatory disorders and infectious diseases involving the complement system84,85) in intermediate AMD. We also suggest the need for an in-depth proteomic analysis of eye bank eyes with different stages of AMD to determine if there are different patterns and profiles of proteins across the phenotypes of AMD. Strengths of our study include the careful phenotyping of the intermediate AMD cases using multimodal imaging, the meticulous collection of the plasma samples, the short time to freezer storage, and the large range of proteins studied, including many low-abundance proteins.
In summary, we found that, apart from two proteins, aptamer targets did not distinguish patients with intermediate AMD who did and did not have bilateral RPD. We found several proteins and pathways of interest in patients who converted to advanced AMD versus those who did not convert to advanced AMD. Ultimately, we would like to build a predictive model of epidemiological, environmental, genetic, and systemic factors that may predict conversion to advanced AMD. The long-term goal is to find a protein or a pathway that could be a target for intervention early in the course of AMD.
Supplementary Material
Acknowledgments
Supported by a Macula Society Research Grant supported by Regeneron; by a Challenge Grant to the Department of Ophthalmology from Research to Prevent Blindness; and by the Frederic C. Hamilton Macular Degeneration Center.
This work was presented at the 43rd Annual Macula Society Meeting, Rancho Bernardo, San Diego, CA, February 19–22, 2020.
Disclosure: A.M. Lynch, None; B.D. Wagner, None; A.G. Palestine, None; N. Janjic, SomaLogic (E, I); J.L. Patnaik, None; M.T. Mathias, None; F.S. Siringo, None; N. Mandava, SomaLogic (C)
References
- 1. Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012; 379: 1728–1738. [DOI] [PubMed] [Google Scholar]
- 2. Bright Focus Foundation. Understanding your diagnosis: quick facts about age-related macular degeneration (AMD). Available at: https://www.brightfocus.org/macular/information. Accessed August 24, 2020.
- 3. Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003; 48: 257–293. [DOI] [PubMed] [Google Scholar]
- 4. Holz FG, Strauss EC, Schmitz-Valckenberg S, van Lookeren Campagne M. Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology. 2014; 121: 1079–1091. [DOI] [PubMed] [Google Scholar]
- 5. Patnaik JL, Pecen PE, Hanson K, et al.. Driving and visual acuity in patients with age-related macular degeneration. Ophthalmol Retina. 2019; 3: 336–342. [DOI] [PubMed] [Google Scholar]
- 6. Taylor DJ, Jones L, Binns AM, Crabb DP. ‘You've got dry macular degeneration, end of story’: a qualitative study into the experience of living with non-neovascular age-related macular degeneration. Eye (Lond). 2020; 34: 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pezzullo L, Streatfeild J, Simkiss P, Shickle D. The economic impact of sight loss and blindness in the UK adult population. BMC Health Serv Res. 2018; 18: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gohil R, Crosby-Nwaobi R, Forbes A, Burton B, Hykin P, Sivaprasad S. Caregiver burden in patients receiving ranibizumab therapy for neovascular age related macular degeneration. PLoS One. 2015; 10: e0129361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bressler NM, Bressler SB, Congdon NG, et al.. Potential public health impact of Age-Related Eye Disease Study results: AREDS report no. 11. Arch Ophthalmol. 2003; 121: 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mimoun G, Soubrane G, Coscas G. [Macular drusen]. J Fr Ophtalmol. 1990; 13: 511–530. [PubMed] [Google Scholar]
- 11. Spaide RF, Ooto S, Curcio CA. Subretinal drusenoid deposits AKA pseudodrusen. Surv Ophthalmol. 2018; 63: 782–815. [DOI] [PubMed] [Google Scholar]
- 12. Hogg RE. Reticular pseudodrusen in age-related macular degeneration. Optom Vis Sci. 2014; 91: 854–859. [DOI] [PubMed] [Google Scholar]
- 13. Wilde C, Poostchi A, Mehta RL, et al.. Prevalence of reticular pseudodrusen in an elderly UK Caucasian population-The Bridlington Eye Assessment Project (BEAP): a cross-sectional study (2002-2006). Eye (Lond). 2018; 32: 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boddu S, Lee MD, Marsiglia M, et al.. Risk factors associated with reticular pseudodrusen versus large soft drusen. Am J Ophthalmol. 2014; 157: 985–993.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Finger RP, Chong E, McGuinness MB, et al.. Reticular pseudodrusen and their association with age-related macular degeneration: the Melbourne Collaborative Cohort Study. Ophthalmology. 2016; 123: 599–608. [DOI] [PubMed] [Google Scholar]
- 16. Buitendijk GH, Hooghart AJ, Brussee C, et al.. Epidemiology of reticular pseudodrusen in age-related macular degeneration: the Rotterdam Study. Invest Ophthalmol Vis Sci. 2016; 57: 5593–5601. [DOI] [PubMed] [Google Scholar]
- 17. Wu Z, Ayton LN, Luu CD, Baird PN, Guymer RH. Reticular pseudodrusen in intermediate age-related macular degeneration: prevalence, detection, clinical, environmental, and genetic associations. Invest Ophthalmol Vis Sci. 2016; 57: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 18. Smith RT, Merriam JE, Sohrab MA, et al.. Complement factor H 402H variant and reticular macular disease. Arch Ophthalmol. 2011; 129: 1061–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Curcio CA, Messinger JD, Sloan KR, McGwin G, Medeiros NE, Spaide RF. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: morphology, prevalence, topography, and biogenesis model. Retina. 2013; 33: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Domalpally A, Agron E, Pak JW, et al.. Prevalence, risk, and genetic association of reticular pseudodrusen in age-related macular degeneration: Age-Related Eye Disease Study 2 Report 21. Ophthalmology. 2019; 126: 1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lynch AM, Patnaik JL, Cathcart JN, et al.. Colorado age-related macular degeneration registry: design and clinical risk factors of the cohort. Retina. 2019; 39: 656–663. [DOI] [PubMed] [Google Scholar]
- 22. Lynch AM, Wagner BD, Weiss SJ, et al.. Proteomic profiles in advanced age-related macular degeneration using an aptamer-based proteomic technology. Transl Vis Sci Technol. 2019; 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lynch AM, Mandava N, Patnaik JL, et al.. Systemic activation of the complement system in patients with advanced age-related macular degeneration. Eur J Ophthalmol. 2019: 1120672119857896. [DOI] [PubMed] [Google Scholar]
- 24. Lynch AM, Palestine AG, Wagner BD, et al.. Complement factors and reticular pseudodrusen in intermediate age-related macular degeneration staged by multimodal imaging. BMJ Open Ophthalmol. 2020; 5: e000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferris FL 3rd, Wilkinson CP, Bird A, et al.. Clinical classification of age-related macular degeneration. Ophthalmology. 2013; 120: 844–851. [DOI] [PubMed] [Google Scholar]
- 26. Rohloff JC, Gelinas AD, Jarvis TC, et al.. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids. 2014; 3: e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lynch AM, Wagner BD, Deterding RR, et al.. The relationship of circulating proteins in early pregnancy with preterm birth. Am J Obstet Gynecol. 2016; 214: 517.e1–517.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gold L, Ayers D, Bertino J, et al.. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010; 5: e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim CH, Tworoger SS, Stampfer MJ, et al.. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci Rep. 2018; 8: 8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamini Y, Drai D, Elmer G, Kafkafi EN, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001; 125: 279–284. [DOI] [PubMed] [Google Scholar]
- 31. Fabregat A, Sidiropoulos K, Garapati P, et al.. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2016; 44: D481–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milacic M, Haw R, Rothfels K, et al.. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers (Basel). 2012; 4: 1180–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reactome. Available at: http://www.reactome.org. Accessed August 24, 2020.
- 34. Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012; 8: e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poole W, Gibbs DL, Shmulevich I, Bernard B, Knijnenburg TA. Combining dependent P-values with an empirical adaptation of Brown's method. Bioinformatics. 2016; 32: i430–i436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Freedman D, Lane D. A nonstochastic interpretation of reported significance levels. J Bus Econ Stat. 1983; 1: 292–298. [Google Scholar]
- 37. Ranganathan P, Pramesh CS, Buyse M. Common pitfalls in statistical analysis: the perils of multiple testing. Perspect Clin Res. 2016; 7: 106–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gelinas AD, Davies DR, Janjic N. Embracing proteins: structural themes in aptamer-protein complexes. Curr Opin Struct Biol. 2016; 36: 122–132. [DOI] [PubMed] [Google Scholar]
- 39. Sun BB, Maranville JC, Peters JE, et al.. Genomic atlas of the human plasma proteome. Nature. 2018; 558: 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Candia J, Cheung F, Kotliarov Y, et al.. Assessment of variability in the SOMAscan assay. Sci Rep. 2017; 7: 14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teitell MA. The TCL1 family of oncoproteins: co-activators of transformation. Nat Rev Cancer. 2005; 5: 640–648. [DOI] [PubMed] [Google Scholar]
- 42. Gjessing LR, Lunde HA, Morkrid L, et al.. Inborn errors of carnosine and homocarnosine metabolism. J Neural Transm Suppl. 1990; 29: 91–106. [DOI] [PubMed] [Google Scholar]
- 43. Murphey WH, Lindmark DG, Patchen LI, Housler ME, Harrod EK, Mosovich HL. Serum carnosinase deficiency concomitant with mental retardation. Pediatr Res. 1973; 7: 601–606. [DOI] [PubMed] [Google Scholar]
- 44. Caruso G, Caraci F, Jolivet RB. Pivotal role of carnosine in the modulation of brain cells activity: multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog Neurobiol. 2019; 175: 35–53. [DOI] [PubMed] [Google Scholar]
- 45. Bhosale SD, Moulder R, Venalainen MS, et al.. Serum proteomic profiling to identify biomarkers of premature carotid atherosclerosis. Sci Rep. 2018; 8: 9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ragland SA, Criss AK. From bacterial killing to immune modulation: recent insights into the functions of lysozyme. PLoS Pathog. 2017; 13: e1006512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pepys MB, Hawkins PN, Booth DR, et al.. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993; 362: 553–557. [DOI] [PubMed] [Google Scholar]
- 48. Swaminathan R, Ravi VK, Kumar S, Kumar MVS, Chandra N. Lysozyme: a model protein for amyloid research. Adv Protein Chem Struct Biol. 2011; 84: 63–111. [DOI] [PubMed] [Google Scholar]
- 49. Busch M, Dunker N. Trefoil factor family peptides–friends or foes? Biomol Concepts. 2015; 6: 343–359. [DOI] [PubMed] [Google Scholar]
- 50. Braga EN, Hoffmann W, Brierley SM, Muttenthaler M. Trefoil factor family: unresolved questions and clinical perspectives. Trends Biochem Sci. 2019; 44: 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paulsen FP, Hinz M, Schaudig U, Thale AB, Hoffmann W. TFF peptides in the human efferent tear ducts. Invest Ophthalmol Vis Sci. 2002; 43: 3359–3364. [PubMed] [Google Scholar]
- 52. Paunel-Gorgulu AN, Franke AG, Paulsen FP, Dunker N. Trefoil factor family peptide 2 acts pro-proliferative and pro-apoptotic in the murine retina. Histochem Cell Biol. 2011; 135: 461–473. [DOI] [PubMed] [Google Scholar]
- 53. Schulze U, Hampel U, Sel S, et al.. Trefoil factor family peptide 3 (TFF3) is upregulated under experimental conditions similar to dry eye disease and supports corneal wound healing effects in vitro. Invest Ophthalmol Vis Sci. 2014; 55: 3037–3042. [DOI] [PubMed] [Google Scholar]
- 54. Becknell B, Eichler TE, Beceiro S, et al.. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int. 2015; 87: 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rowan S, Taylor A. The role of microbiota in retinal disease. Adv Exp Med Biol. 2018; 1074: 429–435. [DOI] [PubMed] [Google Scholar]
- 56. Zinkernagel MS, Zysset-Burri DC, Keller IB, et al.. Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci Rep. 2017; 7: 40826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rullo J, Far PM, Quinn M, et al.. Local oral and nasal microbiome diversity in age-related macular degeneration. Scientific reports. 2020; 10: 3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schroder M, Schnabel D, Suzuki K, Sandhoff K. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 1991; 290: 1–3. [DOI] [PubMed] [Google Scholar]
- 59. Brownstein S, Carpenter S, Polomeno RC, Little JM. Sandhoff's disease (GM2 gangliosidosis type 2). Histopathology and ultrastructure of the eye. Arch Ophthalmol. 1980; 98: 1089–1097. [DOI] [PubMed] [Google Scholar]
- 60. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003; 3: 745–756. [DOI] [PubMed] [Google Scholar]
- 61. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016; 8: a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang K, Li H, Sun R, et al.. Emerging roles of transforming growth factor β signaling in wet age-related macular degeneration. Acta Biochim Biophys Sin (Shanghai). 2019; 51: 1–8. [DOI] [PubMed] [Google Scholar]
- 63. Zarranz-Ventura J, Fernandez-Robredo P, Recalde S, et al.. Transforming growth factor-β inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS One. 2013; 8: e65434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tosi GM, Neri G, Caldi E, et al.. TGF-β concentrations and activity are down-regulated in the aqueous humor of patients with neovascular age-related macular degeneration. Sci Rep. 2018; 8: 8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Batlle E, Massague J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019; 50: 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen YG, Wang Q, Lin SL, Chang CD, Chuang J, Ying S-Y. Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Exp Biol Med (Maywood). 2006; 231: 534–544. [DOI] [PubMed] [Google Scholar]
- 67. Vasilopoulos T, Morey TE, Dhatariya K, Rice MJ. Limitations of significance testing in clinical research: a review of multiple comparison corrections and effect size calculations with correlated measures. Anesth Analg. 2016; 122: 825–830. [DOI] [PubMed] [Google Scholar]
- 68. Hageman GS, Anderson DH, Johnson LV, et al.. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005; 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schick T, Steinhauer M, Aslanidis A, et al.. Local complement activation in aqueous humor in patients with age-related macular degeneration. Eye (Lond). 2017; 31: 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Whitmore SS, Sohn EH, Chirco KR, et al.. Complement activation and choriocapillaris loss in early AMD: implications for pathophysiology and therapy. Prog Retin Eye Res. 2015; 45: 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mullins RF, Schoo DP, Sohn EH, et al.. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014; 184: 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009; 50: 5818–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lechner J, Chen M, Hogg RE, et al.. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: complement activation in AMD. Immun Ageing. 2016; 13: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sivaprasad S, Adewoyin T, Bailey TA, et al.. Estimation of systemic complement C3 activity in age-related macular degeneration. Arch Ophthalmol. 2007; 125: 515–519. [DOI] [PubMed] [Google Scholar]
- 75. Scholl HP, Charbel I P, Walier MJ, et al.. Systemic complement activation in age-related macular degeneration. PLoS One. 2008; 3: e2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Machalinska A, Dziedziejko V, Mozolewska-Piotrowska K, Karczewicz D, Wiszniewska B, Machaliński B. Elevated plasma levels of C3a complement compound in the exudative form of age-related macular degeneration. Ophthalmic Res. 2009; 42: 54–59. [DOI] [PubMed] [Google Scholar]
- 77. Lores-Motta L, Paun CC, Corominas J, et al.. Genome-wide association study reveals variants in CFH and CFHR4 associated with systemic complement activation: implications in age-related macular degeneration. Ophthalmology. 2018; 125: 1064–1074. [DOI] [PubMed] [Google Scholar]
- 78. Ristau T, Paun C, Ersoy L, et al.. Impact of the common genetic associations of age-related macular degeneration upon systemic complement component C3d levels. PLoS One. 2014; 9: e93459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Smailhodzic D, Klaver CC, Klevering BJ, et al.. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology. 2012; 119: 339–346. [DOI] [PubMed] [Google Scholar]
- 80. Fritsche LG, Igl W, Bailey JN, et al.. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016; 48: 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Klein RJ, Zeiss C, Chew EY, et al.. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012; 217: 1034–1046. [DOI] [PubMed] [Google Scholar]
- 83. Seddon JM, Rosner B. Validated prediction models for macular degeneration progression and predictors of visual acuity loss identify high-risk individuals. Am J Ophthalmol. 2019; 198: 223–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012; 33: 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heurich M, Martinez-Barricarte R, Francis NJ, et al.. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. 2011; 108: 8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.