

Abstract
A 24-year-old woman with a medical history of chronic lower extremity oedema, abdominal pain, diarrhoea and recurrent pulmonary infections presented with sepsis from right lower extremity cellulitis. Blood cultures grew Morganella morganii. Laboratory evaluation revealed lymphopaenia, hypogammaglobulinaemia, a low CD4+ T-cell count and nutritional deficiencies resulting from protein-losing enteropathy (PLE). CT showed small bowel wall thickening in the jejunum and ileum. Primary intestinal lymphangiectasia (PIL) was the likely diagnosis that explained her PLE and immunodeficiencies. Video capsule endoscopy is an important diagnostic tool for distal small bowel pathology and confirmed patchy areas of lymphangiectasia of the jejunum and ileum. Secondary causes of lymphangiectasia were ruled out. Clinically significant immunodeficiency from PIL has not been frequently documented, and this case adds to the literature of rare infections associated with PIL. Treatment with intravenous antibiotics resolved her septicaemia, while dietary modifications improved her oedema, abdominal pain and diarrhoea.
Keywords: endoscopy, malabsorption, general practice / family medicine, infectious diseases
Background
Protein-losing enteropathy (PLE) is a rare cause of oedema but should be considered when evaluation fails to yield evidence of cardiac disease, proteinuria, malnutrition or liver disease. Since the first description of PLE in 1947, it has been associated with over 60 conditions, mostly of the gastrointestinal (GI) tract.1 Disease categories that result in PLE include erosive mucosal diseases (ie, Crohn’s disease), diseases without mucosal erosion (ie, coeliac disease) and gastric PLE (ie, Menetrier’s disease), and disorders with increased lymphatic pressure (ie, primary intestinal lymphangiectasia (PIL)).1 PIL is a rare disease primarily of childhood but has been diagnosed across all age groups.2 3 The loss of lymph from the small intestine results in lymphopaenia and hypogammaglobulinaemia, resulting in subsequent immunodeficiency.2 While oedema, abdominal pain and cellulitis are common diagnoses in an acute care setting, combining these brought forth a rare unifying diagnosis. We present a case of PIL in a young woman presenting to the hospital with sepsis from a lower extremity cellulitis and M. morganii bacteraemia. Her underlying PLE and immunodeficiency increased her susceptibility to an otherwise uncommon infection. Our case adds to the limited documentation of clinically significant infections occurring in patients with PIL. We also discuss the clinical features, pathophysiology and diagnosis of PIL, with an emphasis on the diagnostic evaluation of distal small bowel involvement that is not amenable to biopsy by conventional endoscopy.
Case presentation
A 24-year-old woman with a recent evaluation for PLE presented to the emergency department for 2 days of right lower extremity pain, swelling, erythema and fever. She denied trauma, animal bites or injection drug use into the right lower extremity. Her medical history was notable for chronic diarrhoea and abdominal pain, lower extremity oedema since the age of 16, history of recurrent respiratory infections and a stroke at the age of 21. She had no history of diabetes and renal, cardiac or liver disease. She had no prior surgeries. Her father had a stroke in his early 50s. She drank alcohol occasionally but denied tobacco or illicit drug use. There was no recent travel. On review of systems, she endorsed vomiting but denied paroxysmal nocturnal dyspnoea, orthopnea, dyspnoea on exertion, haematemesis or haematochezia.
On presentation to the emergency department, her temperature was 37.6°C, with a heart rate of 134 beats/min, a respiratory rate of 22 breaths/min, blood pressure of 122/79 mm Hg and oxygen saturation was 98% on room air. She appeared uncomfortable but was in no acute distress. Skin exam revealed a swollen, erythematous, indurated right lower extremity without wounds. Bilateral lower extremities had 3+ pitting oedema; however, the right lower extremity was comparatively larger. Jugular venous pressure was normal. The cardiovascular exam revealed normal heart sounds without murmurs, rubs or gallops. Lungs were clear to auscultation. The abdomen was mildly distended with diffuse tenderness to palpation but no rebound or guarding. There were no stigmata of chronic liver disease. Neurological exam revealed a left sixth cranial nerve palsy and 4+/5 strength in the left upper extremity that was chronic from her prior stroke. There was no lymphadenopathy. Examination of the joints was normal.
Laboratory evaluation revealed low potassium of 3 mmol/L; the remainder of serum chemistries were normal. Renal function was normal with a serum creatinine of 0.82 mg/dL. Complete blood count with differential revealed a white blood cell count of 2.6 k/μL, haemoglobin of 136 g/L, platelet count of 101 k/μL and a low absolute lymphocyte count of 0.66 k/μL (1.20–3.70 k/μL). Urinalysis was negative for proteinuria or haematuria. Albumin was 1.7 g/dL; total protein was 3.9 g/dL; the remainder of the hepatic panel was normal. She was started on broad-spectrum antibiotics and admitted to the hospital for management of sepsis due to her lower extremity soft tissue infection. Two days later, her blood cultures returned positive for M. morganii.
The prior evaluation for suspected PLE revealed an elevated stool alpha-1 antitrypsin (A1AT)>113 mg/dL; this exceeded the reference measuring range, rendering the lab unable to calculate the stool A1AT clearance. Serum A1AT was 162 mg/dL. Her prior evaluation for chronic diarrhoea included a thyroid-stimulating hormone, coeliac panel, faecal calprotectin, stool PCR, and stool ova and parasite testing, which were unrevealing. Evaluation for her oedema included a right upper quadrant ultrasound, which showed no evidence of cirrhosis, and a normal transthoracic echocardiogram. A CT of the abdomen and pelvis with contrast (figure 1) revealed ascites, a patent inferior vena cava, and diffuse small bowel wall thickening without evidence of obstruction, adenopathy or masses.
Figure 1.
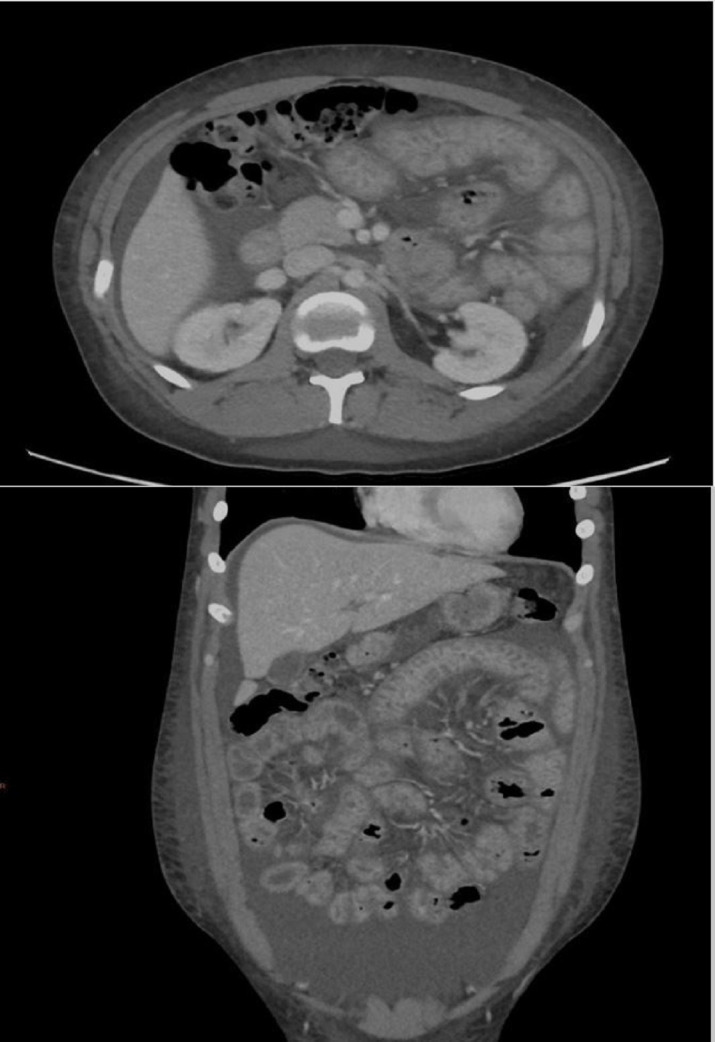
CT of the abdomen and pelvis. (A) Axial view showing small bowel wall thickening and oedema. Surrounding ascitic fluid is present; inferior vena cava and intra-abdominal vasculature is patent. (B) Coronal view showing small bowel wall thickening. The liver contour is normal. Bowel loops are non-dilated without evidence of obstruction. Ascitic fluid, again demonstrated with body wall anasarca, is also present.
Investigations
Our patient, with oedema, chronic diarrhoea and a reported history of recurrent respiratory infections, was now presenting with M. morganii bacteraemia, cellulitis and marked bilateral pitting oedema. Her evaluation for common causes of oedema had returned normal. The elevated stool A1AT was highly suggestive of PLE despite it being a random value. After her infection resolved on intravenous antibiotics, the evaluation centred on finding an aetiology of PLE that also explained her chronic GI symptoms, lymphopaenia and recurrent infections.
Further testing of her immunological status included a negative HIV test. Immunoglobulin testing revealed an IgA level of 67 mg/dL (reference range: 67–288 mg/dL), an IgG level of 143 mg/dL (reference range: 505–1365 mg/dL)and an IgM level of 55 mg/dL (reference range: 67–354 mg/dL). A CD4+ T-cell count was 261/μL (reference range: 496–1647/μL). Antinuclear antibodies were undetectable.
Push enteroscopy and colonoscopy with biopsies were performed to evaluate the aetiology of PLE and did not reveal evidence of Crohn’s disease, coeliac disease, Menetrier’s disease, nor did they reveal pathological lesions typical of lymphangiectasia. However, the abnormalities on CT scan were located more distally in the small bowel. Therefore, video capsule endoscopy was performed, which revealed localised, patchy regions of lymphangiectasia in the jejunum and ileum (figure 2).
Figure 2.

Video capsule endoscopy image of the jejunum showing lymphangiectasia. Lacteals of the villi are prominent with a ‘snowflake’ appearance.
Differential diagnosis
In this patient with pitting oedema, lymphopaenia, hypogammaglobulinaemia, a low CD4+ T-cell count, and evidence of intestinal lymphangiectasia on capsule endoscopy, we considered PIL versus secondary causes of lymphangiectasia. Other aetiologies of PLE caused by increased lymphatic pressure are presented in box 1. The echocardiogram did not reveal cardiac causes that would obstruct intestinal venous outflow. CT of the abdomen revealed patent abdominal vasculature and no lymphadenopathy or masses to suggest neoplastic disease. Push enteroscopy and colonoscopy did not show evidence of Crohn’s disease or coeliac disease. There were no other clinical features to suggest connective tissue disease or sarcoidosis, and ANA testing was negative. She had no history of prolonged fevers or articular symptoms to suggest disease due to Tropheryma whipplei and duodenal biopsy specimens were negative for T. whipplei and tuberculosis. Syndromes associated with PIL include von Recklinghausen, Turner (XO), Noonan, Klippel-Trenaunay and Hennekam syndrome.2 However, these are associated with intellectual disability, facial and skeletal dysmorphic features, and seizures, which were not present in our patient. A thorough evaluation ruled out secondary causes of lymphangiectasia. Therefore, PIL was the unifying diagnosis that explained the patient’s clinical and laboratory abnormalities.
Box 1. Conditions associated with protein-losing enteropathy from increased lymphatic pressure.
A. Obstruction A: obstruction to venous outflow
Budd-Chiari syndrome.17
Cardiac causes: constrictive pericarditis, congestive heart failure, tricuspid regurgitation.17
Superior vena cava thrombosis.17
Portal hypertensive gastropathy.17
B. Neoplastic
Intestinal lymphoma.17
Post-transplant lymphoproliferative disease.17
Neoplastic disease of mesenteric lymphatics.17
C. Connective tissue and autoimmune diseases
D. Infectious
E. Primary or congenital
Primary intestinal lymphangiectasia.17
Treatment
The initial M. morganii bacteraemia and cellulitis completely resolved after a 14-day course of intravenous levofloxacin. After resolution of the initial infection, management focused on her oedema and new diagnosis of PIL. Her management of PIL included a high-protein, low-fat diet supplemented with medium-chain triglycerides.4 5 The low-fat diet leads to a reduction in lymphatic pressure, while medium-chain triglycerides bypass the intestinal lymphatics to help maintain optimal nutritional status. The reduction in lymphatic pressure halts the ongoing leakage of lymphatic protein and lipids into the intestinal lumen. Over time, this results in normalisation of protein levels and a reduction in peripheral oedema. For patients who fail to respond to dietary modification, alternative treatments include octreotide, tranexamic acid, antiplasmin and surgical resection of localised intestinal lymphangiectasia.4 5 The evaluation for nutritional deficiencies revealed a low vitamin D, vitamin B12 and ferritin and she was started on a multivitamin and vitamin D.
Outcome and follow-up
Since starting the modified diet, her diarrhoea, ascites and abdominal pain resolved at a 6-month follow-up. Her oedema improved over several months but did not fully resolve. She had no recurrent sinopulmonary or skin and soft tissue infections. She was evaluated by neurology to see if her prior stroke was related to her diagnosis of PIL as there have been case reports of a hypercoagulable state caused by antithrombin III loss in these patients.6 However, at the time of evaluation, she was already adhering to a modified diet and had normal antithrombin III, protein C and S levels. Long-term management will require ongoing assessment of nutritional status and screening for premature osteoporosis. She will also need monitoring for signs and symptoms concerning for lymphoma, as this occurs in up to 5% of patients diagnosed with PIL.5
Discussion
The diagnosis of PLE is based on elevated stool A1AT levels. Both random stool A1AT7 and 24-hours stool A1AT clearance8 have been studied. A 24-hour stool collection with calculation of the A1AT clearance is preferred as random samples may misclassify patients with abnormal serum A1AT levels or faecal weight. Stool A1AT levels are used because A1AT is not degraded by luminal proteases, and its molecular weight is similar to albumin, making it an ideal surrogate for albumin losses into the GI tract.8
PIL is diagnosed by the endoscopic identification of lymphangiectasia, followed by the histopathological findings on biopsy confirming the presence of dilated mucosal and submucosal lymphatic vessels.2 5 In our patient, the distal and patchy involvement localised to the jejunum and ileum precluded biopsy during push enteroscopy. Video capsule endoscopy and double-balloon enteroscopy can aid in diagnosis when patients have distal disease.9 Alternatively, a high-fat meal challenge before endoscopy can improve the diagnostic yield.10 Findings on capsule endoscopy include oedematous mucosa with whitish, swollen villi.9 An alternate strategy would have been surgical resection and biopsy, as this is both a diagnostic and treatment modality.11 However, surgical biopsy or treatment carried increased risks in the setting of her immunodeficiency and recent infection. Therefore, once endoscopic evidence of intestinal lymphangiectasia was discovered, ruling out secondary aetiologies of lymphangiectasia was crucial prior to diagnosing PIL.
The clinical manifestations of PIL are due to either congenital malformation or obstruction of the intestinal lymphatics.4 Obstruction of the intestinal lymphatic channels results in the loss of protein and lymphatic fluid into the intestinal lumen, which exceeds the rate of synthesis resulting in the clinical manifestations of oedema, hypoalbuminaemia, hypogammaglobulinaemia, nutritional deficiencies and lymphopaenia.4 The loss of lymphocytes and CD4+ T cells results in impaired cell-mediated immunity and increased risk of infection. Pleural effusion, pericardial effusion and ascites may occur as well.2 Chronic diarrhoea is the main GI symptom; however, severe small bowel oedema resulting in mechanical obstruction has been reported.2 Nutritional deficiencies associated with PIL include a loss of fat-soluble vitamins and possible development of osteomalacia and osteoporosis.4 Secondary causes (box 1) have similar pathophysiology in that venous congestion, malignant infiltration or inflammatory mucosal swelling all result in the obstruction to intestinal lymphatic flow.
Both genetic mutations and altered regulatory molecules involved in lymphangiogenesis are hypothesised to play a role in the pathogenesis of intestinal lymphangiectasia.9 Genetic syndromes for which intestinal lymphangiectasia is present include lymphoedema-distichiasis (LD) syndrome and Hennekam syndrome. FOXC2 gene mutations are present in the LD syndrome, which is associated with other cardiac defects and distichiasis (double set of eyelashes).12 Hennekam syndrome is associated with FAT4 mutations as well as genetic variants of the CCBE1 and ADAMTS3 genes.13 These disorders are associated with other anomalies, including cardiac defects, skeletal anomalies and dysmorphic facies that were not present in our patient. Altered expressions of VEGFR-3 and LYVE-1 proteins are suggested to have a role in cases of PIL previously assumed to be idiopathic.14 A 4q25 deletion has also been identified in one case of PIL.9 These genetic and molecular variants provide potential explanations and direction for future study in patients with PIL who lack other clinical features suggestive of a known hereditary syndrome associated with lymphangiectasia.
Our patient suffered significant immunological abnormalities classically associated with PIL. The resultant lymphopaenia and low CD4+ T-cell count are caused by the loss of lymphocyte-rich fluid into the GI tract.15 M. morganii is a Gram-negative bacillus involved in skin and soft tissue infections, urinary tract infections and hepatobiliary infections. These infections mostly occur in older patients in hospital-acquired settings rather than in young, community-dwelling adults.16 The hypogammaglobulinaemia, impaired cell-mediated immunity and impaired host defences made her susceptible to this severe infection from cellulitis and explained her history of recurrent sinopulmonary infections. Despite these known immunological abnormalities, reports of significant infectious complications of PIL are limited to a few case reports, including cryptococcal meningitis and group G streptococcal empyema.2 Our case adds to the growing literature of atypical infections in patients with PIL. A low-fat, high-medium-chain triglyceride diet is designed to reduce lymphatic pressure and the loss of protein and lymphocytes into the intestinal lumen. Six months after starting this diet, our patient had a significant reduction in oedema and had not suffered a recurrent infection.
Learning points.
Primary intestinal lymphangiectasia (PIL) is a disorder caused by intestinal lymphatic outflow obstruction that can occur at any age.
Clinical manifestations of PIL include oedema, diarrhoea, serous effusions, nutritional deficiencies, lymphopaenia, hypogammaglobulinaemia and low CD4+ T-cell count.
Diagnosis of PIL is based on the endoscopic findings of lymphangiectasia, which include whitish, swollen villi with a ‘snowflake’ appearance followed by histological findings of dilated mucosal and submucosal lymphatic vessels.
Capsule endoscopy is an important diagnostic tool when evaluating suspected PIL that cannot be visualised or biopsied on standard endoscopy.
Atypical infections and sepsis are rare but important causes of morbidity in patients with PIL.
Acknowledgments
We acknowledge the patient for providing consent for the writing and publication of this article.
Footnotes
Contributors: All three authors cared for the patient and contributed to the manuscript. JMC and SN conducted a literature review and manuscript write-up. AET performed the endoscopy, provided clinical images and figure legends, and helped revise the manuscript critically for important intellectual content. All authors gave their final approval of the version to be published.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Levitt DG, Levitt MD. Protein losing enteropathy: comprehensive review of the mechanistic association with clinical and subclinical disease states. Clin Exp Gastroenterol 2017;10:147–68. 10.2147/CEG.S136803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vignes S, Bellanger J. [Primary intestinal lymphangiectasia (Waldmann's disease)]. Rev Med Interne 2018;39:580–5. 10.1016/j.revmed.2017.07.009 [DOI] [PubMed] [Google Scholar]
- 3.Huber X, Degen L, Muenst S, et al. Primary intestinal lymphangiectasia in an elderly female patient: a case report on a rare cause of secondary immunodeficiency. Medicine 2017;96:e7729. 10.1097/MD.0000000000007729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ingle SB, Hinge Ingle CR. Primary intestinal lymphangiectasia: minireview. World J Clin Cases 2014;2:528–33. 10.12998/wjcc.v2.i10.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman HJ, Nimmo M. Intestinal lymphangiectasia in adults. World J Gastrointest Oncol 2011;3:19–23. 10.4251/wjgo.v3.i2.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amtage F, Marouf W, Hetzel A, et al. Acquired prothrombotic state due to protein-losing enteropathy as a rare cause for ischemic stroke? Eur J Neurol 2007;14:e7–8. 10.1111/j.1468-1331.2006.01656.x [DOI] [PubMed] [Google Scholar]
- 7.Thomas DW, Sinatra FR, Merritt RJ. Random fecal alpha-1-antitrypsin concentration in children with gastrointestinal disease. Gastroenterology 1981;80:776–82. 10.1016/0016-5085(81)90140-2 [DOI] [PubMed] [Google Scholar]
- 8.Strygler B, Nicar MJ, Santangelo WC, et al. Alpha 1-antitrypsin excretion in stool in normal subjects and in patients with gastrointestinal disorders. Gastroenterology 1990;99:1380–7. 10.1016/0016-5085(90)91165-3 [DOI] [PubMed] [Google Scholar]
- 9.Oh TG, Chung JW, Kim HM, et al. Primary intestinal lymphangiectasia diagnosed by capsule endoscopy and double balloon enteroscopy. World J Gastrointest Endosc 2011;3:235–40. 10.4253/wjge.v3.i11.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J, Kong M-S. Primary intestinal lymphangiectasia diagnosed by endoscopy following the intake of a high-fat meal. Eur J Pediatr 2008;167:237–9. 10.1007/s00431-007-0445-8 [DOI] [PubMed] [Google Scholar]
- 11.Huber T, Paschold M, Eckardt AJ, et al. Surgical therapy of primary intestinal lymphangiectasia in adults. J Surg Case Rep 2015;2015:rjv081. 10.1093/jscr/rjv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yildirim-Toruner C, Subramanian K, El Manjra L, et al. A novel frameshift mutation of FOXC2 gene in a family with hereditary lymphedema-distichiasis syndrome associated with renal disease and diabetes mellitus. Am J Med Genet A 2004;131:281–6. 10.1002/ajmg.a.30390 [DOI] [PubMed] [Google Scholar]
- 13.Ivanovski I, Akbaroghli S, Pollazzon M, et al. Van Maldergem syndrome and Hennekam syndrome: further delineation of allelic phenotypes. Am J Med Genet A 2018;176:1166–74. 10.1002/ajmg.a.38652 [DOI] [PubMed] [Google Scholar]
- 14.Hokari R, Kitagawa N, Watanabe C, et al. Changes in regulatory molecules for lymphangiogenesis in intestinal lymphangiectasia with enteric protein loss. J Gastroenterol Hepatol 2008;23:e88–95. 10.1111/j.1440-1746.2007.05225.x [DOI] [PubMed] [Google Scholar]
- 15.Fuss IJ, Strober W, Cuccherini BA, et al. Intestinal lymphangiectasia, a disease characterized by selective loss of naive CD45RA+ lymphocytes into the gastrointestinal tract. Eur J Immunol 1998;28:4275–85. [DOI] [PubMed] [Google Scholar]
- 16.Erlanger D, Assous MV, Wiener-Well Y, et al. Clinical manifestations, risk factors and prognosis of patients with Morganella morganii sepsis. J Microbiol Immunol Infect 2019;52:443–8. 10.1016/j.jmii.2017.08.010 [DOI] [PubMed] [Google Scholar]
- 17.Feldman F, Brandt F, Mark F, et al. Sleisenger and Fordtran’s gastrointestinal and liver disease : pathophysiology/diagnosis/management. Tenth edition, 2016. [Google Scholar]