

Abstract
Background
Myhre syndrome is a genetic disorder caused by gain of function mutations in the SMAD Family Member 4 (SMAD4) gene, resulting in progressive, proliferative skin and organ fibrosis. Skin thickening and joint contractures are often the main presenting features of the disease and may be mistaken for juvenile scleroderma.
Case presentation
We report a case of a 13 year-old female presenting with widespread skin thickening and joint contractures from infancy. She was diagnosed with diffuse cutaneous systemic sclerosis, and treatment with corticosteroids and subcutaneous methotrexate recommended. There was however disease progression prompting genetic testing. This identified a rare heterozygous pathogenic variant c.1499 T > C (p.Ile500Thr) in the SMAD4 gene, suggesting a diagnosis of Myhre syndrome. Securing a molecular diagnosis in this case allowed the cessation of immunosuppression, thus reducing the burden of unnecessary and potentially harmful treatment, and allowing genetic counselling.
Conclusion
Myhre Syndrome is a rare genetic mimic of scleroderma that should be considered alongside several other monogenic diseases presenting with pathological fibrosis from early in life. We highlight this case to provide an overview of these genetic mimics of scleroderma, and highlight the molecular pathways that can lead to pathological fibrosis. This may provide clues to the pathogenesis of sporadic juvenile scleroderma, and could suggest novel therapeutic targets.
Keywords: Scleroderma, Myhre syndrome, SMAD4
Background
Myhre syndrome is a genetic disorder often presenting in infancy, caused by a gain of function mutation in the SMAD family member 4 (SMAD4) gene causing progressive, proliferative fibrosis, occurring spontaneously or following trauma, in addition to a unique set of clinical phenotypic features described below [1–4]. Clinical manifestations of Myhre syndrome include: cardiovascular involvement in up to 70% of patients (congenital heart defects, long- and short-segment stenosis of the aorta and peripheral arteries, pericardial effusion, constrictive pericarditis, restrictive cardiomyopathy, and arterial hypertension); respiratory manifestations (choanal stenosis, laryngotracheal narrowing, obstructive airway disease, or restrictive pulmonary disease); gastrointestinal symptoms (pyloric stenosis, duodenal strictures, severe constipation); hearing loss, mild to moderate development delay, dysmorphic features and skin involvement (skin sclerosis, particularly involving the hands and extensor surfaces) leading to joint contractures [1, 5–12]. Patients presenting with predominantly skin sclerosis and contractures, cardiovascular involvement may be misdiagnosed as a having systemic sclerosis (SSc) despite the presence of other atypical features for SSc such as hearing loss and developmental delay thus causing unnecessary exposure to immunosuppression. Herein, we present a case of a 13 year-old female considered as having diffuse cutaneous systemic sclerosis, who was subsequently identified to have Myhre syndrome caused by a previously well described heterozygous c.1499 T > C variant in SMAD4. We discuss the therapeutic implications of establishing a genetic diagnosis in this case and provide an overview of genetic mimics of scleroderma.
Case presentation
A 13 year-old girl of Black African decent was referred to the scleroderma services of the rheumatology department at Great Ormond Street Hospital for Children NHS Foundation Trust, London for a second opinion with history of extensive skin thickening and widespread joint contractures, which started in infancy at the age of 9 months (Fig. 1). The skin changes started in her lower limps and over the course of 2 years spread to the arms and trunk. The joint contractures were noted approximately 2 years after the initial skin changes were observed. There was history suggestive of mild Raynaud’s phenomenon, but no digital ulceration, gastrointestinal, or respiratory symptoms of note. She was born at term with no neonatal complications. She had a past medical history of: valvar and supravalvar pulmonary artery stenosis requiring serial balloon dilatation; mild developmental delay; and conductive hearing loss. Microarray-based comparative genomic hybridization was used to exclude chromosomal abnormalities that could explain her presentation and was normal. There was no history of cancer in the immediate family.
Fig. 1.
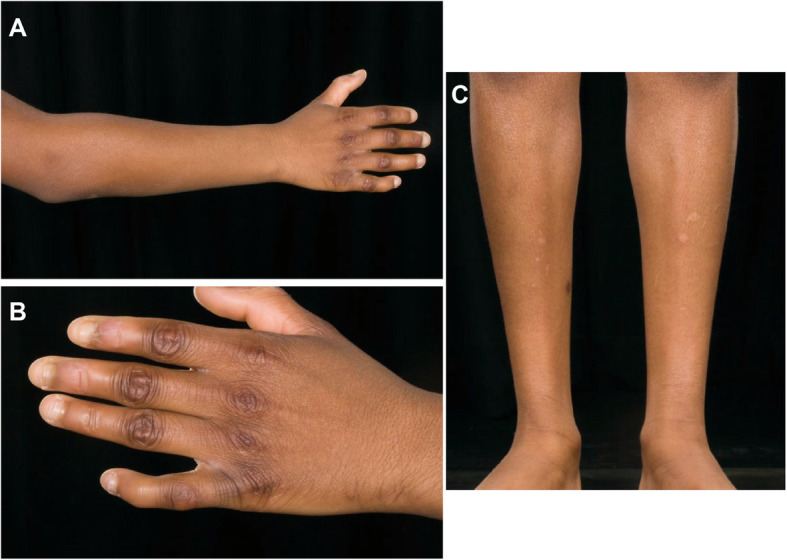
Cutaneous and skeletal manifestations of the 13 year old patient with Myhre syndrome we describe in this report. a-b Multiple joint contractures and clinodactyly. c Extensive skin thickening and muscle wasting of lower limbs in same patient
Clinical examination revealed diffusely thickened skin affecting the full length of her limbs and trunk, but sparing her face; weight 41 kg (25th centile for age), height 139 cm (2nd centile for age). She was normotensive at time of review. Delayed puberty was noted and the patient had no menarche at the age of 13 years old. There were multiple joint contractures, but no active arthritis. Cutaneous telangiectasia, fingertip ulceration and calcinosis were absent. She was also noted to have mild dysmorphic features: small eyes and ears, a broad nasal tip, a long and prominent chin, bilateral clinodactyly and mild two-three toe syndactyly. Nailfold capillaroscopy was abnormal, with evidence of dilated capillary loops, tortuosity, micro-bleeding, and widespread dropout in a pattern compatible with scleroderma-spectrum connective tissue disease (Fig. 2). Digital thermography demonstrated cold baseline cutaneous temperature of the peripheries, with some of the fingers remaining cool long after cold challenge. She tested weakly positive for antinuclear antibodies (ANA at 1:160, homogenous pattern); negative for dsDNA antibodies, rheumatoid factor and extranuclear antibodies. Complement function (alternate and classical pathways) was normal, as were levels of C3, C4, and C1q. Erythrocyte sedimentation rate and C-reactive protein were repeatedly within normal limits. Echocardiography revealed mild persistent pulmonary stenosis, a small left pulmonary artery, mild coarctation of the aorta, mild biventricular hypertrophy, but no evidence of pulmonary hypertension. Barium swallow was normal. A skeletal survey revealed advanced bone age, but no evidence of skeletal dysplasia. A skin biopsy was performed, with histology revealing hyperkeratotic epidermis, and fibrotic dermis with areas of hyalinization; adnexal structures were sparse with absence of pilosebaceous units (Fig. 3). These histological features are typically encountered in scleroderma histopathology with the exception of the hyperkeratotic epidermis, which is less often seen [11, 13, 14]. She was diagnosed with diffuse cutaneous systemic sclerosis, and treatment with oral prednisolone 2 mg/kg/day for 6 weeks, and subcutaneous methotrexate (15 mg/m2/week) started. There was deterioration in joint contractures (further loss of range of movement) and spreading of skin changes observed despite treatment. When reviewed for a second opinion at GOSH, a genetic diagnosis was suspected and genetic testing via Sanger sequencing was undertaken for some conditions that cause skin thickening, dysmorphic features and congenital heart disease. Genetic testing revealed a previously well described rare heterozygous c.1499 T > C (p.lle500Thr) class 5 variant in SMAD4 [12], suggesting a diagnosis of Myhre syndrome. Testing for variants in other relevant genes pertinent to phenotype (including PTPN11, LMNA, and MMP14) revealed no other pathogenic variants [15–26]. Parental testing confirmed this variant arose de novo in the proband. All immunosuppression was subsequently stopped, genetic counselling was provided, and the prognosis of Myhre syndrome was discussed with the patient and family.
Fig. 2.

Nailfold capillaroscopy of the 13 year old patient with Myhre syndrome we describe in this report. a-b Abnormal nailfold capillary patterns with small microbleeds, very tortuous loops and mild dilatation in a patient with Myhre syndrome suggesting an evolving microangiopathy
Fig. 3.

Skin histology of the 13 year old patient with Myhre syndrome mimicking juvenile scleroderma we describe in this report. a-b Photomicrographs of skin punch biopsy containing epidermis, dermis and superficial subcutis. There is no significant lichenoid reaction or inflammatory infiltrate (L) but the dermis shows marked replacement by hypocellular, hyalinised areas of bland collagen(R). There are no other specific features and the adnexal structures remain in this biopsy. (H&E, original magnifications Lx40 and Rx100)
Discussion
We present the case of a 13 year-old with a scleroderma-like condition, ultimately diagnosed with Myhre syndrome, a genetic disorder that may mimic juvenile scleroderma (Supplemental Table 1). Securing a molecular diagnosis in this case allowed the cessation of immunosuppression thus reducing the burden of unnecessary toxic exposure to glucocorticoids, and other ineffective immunosuppressive treatments; and facilitated genetic counselling, and prognostication. This also had implications for long term follow up as patients with Myhre syndrome require close surveillance for detection of any malignancy in view of increased risk of cancer reported in these patients [5, 12, 27]. We therefore highlight this case to raise awareness of a growing number of monogenic fibrotic disorders mimicking juvenile scleroderma which need to be considered in patients with cutaneous fibrosis beginning early in life (Table 1).
Table 1.
Monogenic disorders with a scleroderma-like phenotype. The clinical features have been summarised as described by the Online Mendelian Inheritance in Man (OMIM) [28] and Genetics Home Reference databases [29]
| Disease | Inheritance | Gene | Clinical Features |
|---|---|---|---|
| Hutchinson-Gilford Progeria | AD, AR | LMNA |
Skin: Sclerodermatous skin disease, loss of subcutaneous fat (lipodystrophy) Skeletal: Osteoporosis, joint restrictions, joint abnormalities Cardiovascular: Atherosclerosis Other: Prematurely aged appearance, postnatal onset growth retardation, hair loss (alopecia) |
| Werner syndrome | AR | WRN |
Skin: Sclerodermatous skin disease, subcutaneous calcification, ulceration Skeletal: Osteoporosis Cardiovascular: Premature arteriosclerosis Endocrine: Diabetes mellitus, hypogonadism Other: Prematurely aged appearance, short stature, alopecia, juvenile cataracts |
| Rothmund Thomson syndrome | AR | RECQL4 |
Skin: Erythematous thickened skin lesions in infancy, poikiloderma (atrophic plaques with telangiectasia), telangiectasia, atrophy, sun sensitivity Skeletal: Osteoporosis Central Nervous System: Mental retardation (rare) Endocrine: Hypogonadism Other: Prematurely aged appearance, short stature, alopecia, premature greying of hair, increased risk of malignant disease |
| Mandibular hypoplasia, deafness, progeroid features and lipodystrophy syndrome | AD | POLD1 |
Skin: Sclerodermatous skin disease, telangiectasias, atrophy, lipodystrophy Skeletal: Osteoporosis, joint contractures Endocrine: Insulin resistance, diabetes mellitus Other: Prematurely aged appearance, mandibular hypoplasia, sensorineural deafness, hepatomegaly, hepatic steatosis |
| Nestor-Guillermo Progeria Syndrome | AR | BANF1 |
Skin: Sclerodermatous skin disease (patchy) and hyperpigmentation Skeletal: Joint stiffness, joint contractures, osteoporosis, osteolysis Cardiovascular: Sinus tachycardia, prominent subcutaneous venous patterning, pulmonary hypertension Other: Prematurely aged appearance, short stature, lipoatrophy |
| Keppen-Lubinsky syndrome | AD | KCNJ6 |
Skin: Lipodystrophy, wrinkled appearance Skeletal: Joint contractures Central Nervous System: Severe mental retardation, delayed psychomotor development, hypertonia, hyperreflexia Other: Prematurely aged appearance, generalised lipodystrophy |
| Fontaine Progeroid Syndrome | AD | SLC25A24 |
Skin: Wrinkled skin, lipodystrophy, sclerodermatous skin disease Skeletal: Low bone density, delayed bone age Cardiovascular: Pulmonary artery hypertension, aortic ectasia Other: Prematurely aged appearance, short stature, intrauterine growth retardation |
| Cockayne Syndrome, Type A | AR | ERCC8 |
Skin: Cutaneous photosensitivity, scarred, pigmented, atrophy, reduced subcutaneous adipose tissue, sclerodermatous skin disease Skeletal: Flexion contractures, mild-to-moderate joint limitations Cardiovascular: Hypertension Neurological: Impaired or delayed neural development, mental retardation Other: Prematurely aged appearance, cachectic dwarfism, intrauterine growth retardation, sensorineural hearing loss, vision complications, tooth decay, hepatomegaly, splenomegaly, decreased subcutaneous adipose tissue |
| Ataxia-telangiectasia | AR | ATM |
Skin: Sclerodermatous skin disease, progeric skin changes, cutaneous telangiectasia, cafe-au-lait spots Respiratory: Bronchitis, bronchiectasis Neurological: Cerebellar ataxia, cerebellar cortical degeneration, oculomotor abnormalities, seizures, choreoathetosis, dystonia, reduced/absent deep tendon reflexes Other: Short stature |
| Myhre syndrome | AD | SMAD4 |
Skin: Sclerodermatous skin disease Skeletal: Skeletal abnormalities, joint restrictions Cardiovascular: Hypertension, congenital heart defects, aortic stenosis, aortic coarctation, pericardial fibrosis Respiratory: Laryngotracheal stenosis, respiratory failure Neurological: Mental retardation, delayed language and motor skill development, behavioural issues (autistic-like) Other: Dysmorphic facial features, short stature, hearing loss, generalised muscle hypertrophy |
| Stiff skin syndrome | AD | FBN1 |
Skin: Sclerodermatous skin disease (diffuse), lipodystrophy Skeletal: Joint restrictions, flexion contractures Other: Muscle weakness |
| Pigmentary hypertrichosis and non-autoimmune insulin-dependent diabetes mellitus (PHID) | AR | SLC29A3 |
Skin: Hyperpigmented and hypertrichotic skin lesions on lower body, sclerodermatous skin disease Skeletal: Joint contractures (elbows, fingers and toes) Abdomen: Hepatomegaly, diabetes mellitus (insulin-dependent), splenomegaly Other: Short stature, hearing loss |
| Reynolds syndrome | AD | LBR |
Skin: Sclerodermatous skin disease (tightened and shiny skin over the forearms and hands), sclerodactyly, calcinosis cutis, generalized darkening Other: Raynaud phenomenon, hepatomegaly, primary biliary cirrhosis, splenomegaly, esophageal dysfunction |
| Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome /Nakajo-Nishimura Syndrome | AR | PSMB8 |
Skin: Erythematous nodular skin lesions and plaques on the face and extremities, dry, stiff, lipodystrophy Skeletal: Joint contractures (elbow, fingers/hands, toes, feet), joint pain Muscle: Lipodystrophy, muscle weakness Other: Poor growth, hepatomegaly, splenomegaly |
| Mucolipidosis III gamma | AR | GNPTG |
Skin: Sclerodermatous skin disease Skeletal: Joint restrictions, joint stiffness, joint pain Cardiovascular: Aortic valve thickening, aortic stenosis Neurological: Mental retardation Other: Short stature |
| Hurler-Scheie syndrome / Mucopolysaccharidosis Ih/s | AR | IDUA |
Skin: Sclerodermatous skin disease Skeletal: Joint stiffness, dysostosis multiplex Cardiovascular: Thickened mitral valve leaflets, aortic valve thickening, dilated left atrium, dilated left ventricle, mild pulmonary hypertension Respiratory: Frequent respiratory infections, nasopharyngeal obstruction, tracheal stenosis Abdomen: Umbilical hernia, hepatomegaly, splenomegaly Neurological: Pachymeningitis cervicalis Other: Short stature, corneal clouding |
| Zimmermann-Laband Syndrome 1 | AD | KCNH1 |
Skin: Dry, sclerodermatous skin disease Skeletal: Scoliosis, hypoplastic distal phalanges (hands and feet), hyperextensible joints Abdomen: Hepatosplenomegaly, splenomegaly, umbilical hernia Cardiovascular: Cardiomyopathy, patent ductus arteriosus, aortic root dilatation, aortic arch dilatation Muscle: Poor muscle bulk Neurological: Hypotonia, seizures, mental retardation Other: Gingival fibromatosis, dysplastic or absent nails, hirsutism, abnormalities of the cartilage of the nose and/or ears |
| Buschke-Ollendorff syndrome | AD | LEMD3 |
Skin: Subcutaneous nontender firm nodules, subcutaneous connective tissue nevi, elastin-rich connective tissue nevi (elastoma), collagen-rich connective tissue nevi (dermatofibrosis lenticularis disseminata) Skeletal: Osteopoikilosis, joint stiffness, osteosclerosis, melorheostosis |
| Growth Retardation, Alopecia, Pseudoanodontia and Optic Atrophy (GAPO) Syndrome | AR | ANTXR1 |
Skin: Sclerodermatous skin disease, redundant, prominent scalp veins, epidermal inclusion cyst Skeletal: Delayed bone age Other: Growth retardation, alopecia, pseudoanodontia, umbilical hernia, hepatomegaly |
| Crouzon Syndrome with acanthosis nigricans | AD | FGFR3 |
Skin: Hyperpigmentation, acanthosis nigricans, melanocytic nevi, hypertrophy, sclerodermatous skin disease, redundant skin folds Skeletal: Craniosynostosis |
| Frontometaphyseal dysplasia 2 | AD | MAP 3 K7 |
Skin: Keloid formation, sclerodermatous skin disease Skeletal: Skeletal abnormalities, joint contractures Cardiovascular: Patent ductus arteriosus, bicuspid aortic valve, aortic root dilation, pulmonary valve stenosis Respiratory: Congenital stridor, subglottic stenosis, tracheal stenosis |
| Premature aging syndrome, Penttinen type | AD | PDGFRB |
Skin: Progressive cutaneous atrophy, thin translucent skin with prominent venous patterning, hypertrophic keloid-like lesions, skin retraction, sclerodermatous skin disease, lipoatrophy Skeletal: Delayed bone maturation, osteopenia, joint contractures |
| Farber Lipogranulomatosis | AR | ASAH1 |
Skin: Early-onset subcutaneous nodules, lipogranulomatosis Skeletal: Painful and progressively deformed joints, arthritis Respiratory: Laryngeal nodules Abdomen: Hepatomegaly, splenomegaly Neurological: Irritability, motor retardation, mental retardation Other: Hoarseness by laryngeal involvement |
| Amyloidosis, Primary Localised cutaneous, 3 (PLCA3) | AR | GPNMB | Skin: Amyloid disposition in the skin, hyper- and hypo-pigmented macules, mild pruritis, dry skin |
| Carney Complex, Type 1 | AD | PRKAR1A |
Skin: Cutaneous tumors, profuse pigmented skin lesions, nevi Cardiovascular: Tumors (atrial), ventricular myxoma, congestive heart failure Endocrine: Tumors, pigmented micronodular adrenal dysplasia, Cushing disease, acromegaly, thyroid follicular hyperplasia Other: Neoplasia, myxoid subcutaneous tumors, primary adrenocortical nodular hyperplasia, testicular Sertoli cell tumor (calcified), pituitary adenoma, mammary ductal fibroadenoma, schwannoma, psammomatous melanotic schwannomas, thyroid carcinoma, pheochromocytoma |
| Porphyria cutanea tarda, Porphyria, hepatoerythropoietic | AD, AR | UROD |
Skin: Sclerodermatous skin disease (diffuse), increased mechanical skin fragility after sunlight exposure (photosensitivity), vesicles, bullae and blisters on exposed areas of skin, hyperpigmentation on sun-exposed skin Abdomen: Hepatic hemosiderosis, hepatic cirrhosis, liver biopsy shows red autofluorescence and needle-like cytoplasmic inclusion bodies Other: Neoplasia, increased incidence of hepatocellular carcinoma |
| Phenylketonuria, non-PKU mild Hyperphenylalaninemia | AR | PAH |
Skin: Sclerodermatous skin disease, pale pigmentation, dry, eczema Neurological: Seizures, delayed development, mental retardation, behavioural problems and psychiatric disorders Other: Head, microcephaly, cataracts |
| Porphyria, congenital erythropoietic | AR | UROS |
Skin: Sclerodermatous skin disease, photosensitivity, blistering and scarring, hyperpigmentation, hypopigmentation Skeletal: Osteolysis, osteopenia, finger contractures Other: Short stature, conjunctivitis, corneal scarring, hypertrichosis, alopecia, porphyrin-rich gallstones, splenomegaly |
| Multicentric osteolysis, nodulosis and arthropathy (MONA) | AR | MMP2 |
Skin: Subcutaneous nodules (interphalangeal joints, knees, feet, elbows, pretibial), hyperpigmented erythematous lesions Skeletal: Osteoporosis, flexion contractures |
| Winchester syndrome | AR | MMP14 |
Skin: Sclerodermatous skin disease (patchy, dark, leathery) Skeletal: Osteopenia, osteoporosis, arthropathy, joint restrictions Cardiovascular: Heart abnormalities Other: Corneal opacity, hypertrichosis, overgrowth of the gums, coarse facial features |
| Multisystemic fibrosis-like hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP) | AD | FAM111B |
Skin: Congenital poikiloderma (face and exposed skin), telangiectatic lesions, eczema-like lesions, epidermal atrophy Respiratory: Interstitial pulmonary fibrosis Muscle: Tendon contractures, muscle weakness, myopathy Other: Congenital poikiloderma on face |
| Weill-Marchesani syndrome 1 | AR | ADAMTS10 |
Skin: Sclerodermatous skin disease Skeletal: Joint stiffness, joint restrictions Cardiovascular: Heart defects, aortic valve stenosis, pulmonary valve stenosis, ductus arteriosus, ventricular septal defect Neurological: Mild mental retardation Other: Short stature, brachydactyly, eye anomalies |
| Weill-Marchesani syndrome 4 (WMS-like syndrome) | AR | ADAMTS17 |
Skin: Sclerodermatous skin disease Skeletal: Joint stiffness Cardiovascular: Cardiac defects (uncommon) Other: Short stature, severe myopia, acute and/or chronic glaucoma, cataract |
| Frank-Ter Haar Syndrome | AR | SH3PXD2B |
Skin: Sclerodermatous skin disease (face), acne conglobata Skeletal: Osteolysis, osteopenia, osteoporosis, shortened bowed long bones, flexion deformities of fingers Other: Growth retardation, glaucoma, brachycephaly, wide fontanels, prominent forehead, hypertelorism, prominent eyes, malocclusion |
| Geleophysic dysplasia 3 | AD | LTBP3 |
Skin: Sclerodermatous skin disease Skeletal: Joint restrictions, delayed bone age Cardiovascular: Pulmonary hypertension Respiratory: Dyspnea, tracheal stenosis, respiratory failure Other: Short stature, marked brachydactyly, hepatomegaly |
| Geleophysic dysplasia 1 | AR | ADAMTSL2 |
Skin: Sclerodermatous skin disease Skeletal: Osteopenia, shortened long tubular bones, short hands and feet, joint contractures, joint restrictions, delayed bone age Cardiovascular: Progressive cardiac valvular thickening, cardiac failure, mitral stenosis, tricuspid stenosis, aortic stenosis Respiratory: Tracheal stenosis, respiratory insufficiency Neurological: Developmental delay, seizures Other: Short stature, ‘happy’ appearance with full cheeks, shortened nose, wide mouth, hepatomegaly |
| Mucolipidosis II Alpha/Beta | AR | GNPTAB |
Skin: Sclerodermatous skin disease, cavernous hemangioma Skeletal: Skeletal abnormalities, moderate joint restrictions, osteopenia Cardiovascular: Cardiomegaly, congestive heart failure, hypertrophic cardiomyopathy, cardiac murmur, aortic insufficiency Respiratory: Recurrent bronchitis, recurrent pneumonia Abdomen: Umbilical hernia, hepatomegaly Neurological: Developmental delay, severe psychomotor retardation Other: Progressive failure to thrive, Hurler-like body configuration, marked growth retardation, coarse facial features, abdominal protuberance, hoarse voice |
| Hypertrophic Osteoarthropathy, Primary, Autosomal Recessive 1 / Cranioosteoarthropathy | AR | HPGD |
Skin: Sclerodermatous skin disease, pachydermia, furrowed, oily, seborrhea, redundant, palmoplantar hyperkeratosis, eczema Skeletal: Digital clubbing, osteoarthropathy, arthralgia, arthritis, swollen joints, decreased joint mobility, osteopenia, osteoporosis Cardiovascular: Congenital heart disease, patent ductus arteriosus Other: Marfanoid habitus, coarse facial features, furrowed forehead, ptosis, thickened eyelids, turtle-backed nails, digital clubbing |
AD Autosomal dominant, AR Autosomal recessive, SSc systemic sclerosis
Myhre syndrome is caused by mutations in SMAD encoding for SMAD4 protein, a transducer mediating transforming growth factor β (TGF-β) signalling [2–4]. Skin fibroblasts from patients with Myhre syndrome show increased SMAD4 expression, impaired matrix deposition, and altered expression of genes encoding matrix metalloproteinases and related inhibitors. Losartan, an angiotensin-II type 1 receptor blocker but also a (lesser-known) TGF-β antagonist has been shown in vitro to normalize metalloproteinase and related inhibitor transcript levels, and to correct the extracellular matrix (ECM) deposition defect in fibroblasts from these patients [30]. Some patients with aortic pathology associated with Myhre syndrome have already been treated with losartan, with reports of stabilisation of their vasculopathy; but the effect on skin fibrosis has never been described [30–34]. We suggest that further studies could explore losartan (or other therapies acting on the SMAD4 pathway) as a potential targeted therapeutic option for cutaneous fibrosis associated with this rare genetic disease. At the time of writing this report losartan therapy is being considered for the patient described herein.
Several other conditions may also mimic juvenile scleroderma (Table 1). Skin thickening is common to all of these disorders, and may be localized (morphoea-like), or widespread (like diffuse scleroderma) [35–45]. Vasculopathy is frequently observed and should be actively screened for. We highlight for the first time in this case the abnormal nailfold capillaroscopy with similar findings to those observed in SSc. Degenerative cardiac or pulmonary manifestations may also exhibit a secondary inflammatory component, thus posing considerable diagnostic challenges and making it more likely that such patients could be exposed to ineffective but toxic immunosuppression, as illustrated by our case [1, 2, 4, 46–49]. On occasions, autoimmunity has also been described [50–53]. The management and long term outcome of these genetic scleroderma mimics is, however, entirely different and immunosuppression may not be required or may in fact be harmful in some cases [54, 55]. We therefore suggest that genetic testing should be considered in all patients with sclerodermatous skin disease of very young onset (infancy) and recommend screening for vasculopathy (including congenital heart disease and aortopathy) with echocardiography, and non-invasive angiography. Genetic screening for monogenic diseases should also be considered in older patients with scleroderma with atypical clinical course; and in those not responding to conventional immunosuppression.
Regarding the methodology of genetic screening, our case again illustrates the importance of next-generation sequencing (NGS) methodologies in this context. Mainly due to lack of routine NGS methods, initial routine genetic testing of candidate genes by Sanger was performed for this patient. This was a time consuming, costly, and mainly “clinician best guess” driven approach, which resulted in diagnostic delay of several months. Whole exome and genome sequencing and targeted gene panels now allow rapid, simultaneous detection of multiple genes, and are increasingly being used as diagnostic tools and to explore the pathogenesis of monogenic diseases [56–61]. These techniques are particularly useful for screening diseases with overlapping phenotypes. For instance, we (and many others) have used NGS to extensively study monogenic systemic inflammation, with significant diagnostic and therapeutic impact [60, 61]. Similarly, we anticipate that application of NGS genetic screening to cohorts of patients with juvenile scleroderma (in all its forms) may identify a proportion with monogenic disease, and that evidence of tissue inflammation and autoimmunity should not preclude the possibility of a genetic diagnosis for the reasons discussed above.
Understanding the genetic basis of these genetic diseases with sclerodermatous features is not only crucial to secure diagnoses, improve prognostication and to facilitate genetic counselling but may also provide clues to the pathogenesis of sporadic cases. For instance, several of the genetic mimics of scleroderma involve the TGF-β pathway [2, 62–64]. At the cellular level, TGF-β plays potent roles in proliferation, differentiation and apoptosis of many cell types, and therefore unsurprisingly germline mutations in the TGF-β signalling pathway cause various phenotypes affecting the skeletal, muscular, and/or cardiovascular systems [2, 62–65]. TGF-β has also been identified as a regulator of pathological fibrogenesis in juvenile and adult onset systemic sclerosis [64–68]. A wide range of drugs targeting the TGF-β signalling pathways are now available [69–73], and need to be tested for their ability to modulate the phenotypes of both these inherited scleroderma mimics but possibly also for efficacy in addition to anti-inflammatory medication in sporadic systemic sclerosis, given their overlapping pathomechanisms.
Conclusion
Myhre syndrome is a rare genetic disorder that causes skin thickening and joint contractures, and may be misdiagnosed as juvenile scleroderma (systemic sclerosis). Many other genetic conditions can similarly mimic the clinical manifestations of juvenile scleroderma and should be considered in the differential diagnosis of juvenile scleroderma. Onset in infancy and comorbidities such as structural heart disease, large vessel vasculopathy, dysmorphic features, developmental delay, and hearing loss are important clues to a genetic diagnosis. Clinical application of NGS is likely to transform the genetic diagnostic approach to young patients with scleroderma-like diseases and suggest targeted therapies for some cases. Therapeutic targets for sporadic cases of juvenile scleroderma are also likely to emerge, given the overlapping disease mechanisms for all these conditions leading to vasculopathy, skin and organ fibrosis.
Supplementary information
Additional file 1 Supplemental Table 1. Features of juvenile localised scleroderma, juvenile systemic sclerosis and Myhre syndrome.
Acknowledgments
Dr. Eleftheriou, Professor Sebire and Professor Brogan acknowledge the National Institute of Health Research (NIHR) Biomedical Research Centre at GOSH.
Disclaimer
The views expressed are those of the authors and not necessarily those of the NHS, NIHR, or Department of Health.
Abbreviations
- ANA
Anti-nuclear antibodies
- ECM
Extracellular Matrix
- dsDNA
Double-stranded DNA
- NGS
Next generation sequencing
- SSc
Systemic sclerosis
- TGF-β
Transforming growth factor type beta
Authors’ contributions
BJ, RJ, PB and DE conceived the study, obtained and analysed data and drafted the manuscript. YH, EO, CP, NS and KH obtained and analysed data and drafted the manuscript. All authors read and approved the final manuscript.
Funding
No funding sources. Dr. Jensen was supported by GOSH Children’s Charity Grant (CP_RSRCH_003) and Rosetrees Trust Grant (A2584) Dr. Eleftheriou and Dr. Hong were supported by Versus Arthritis (grants 20164, 21593, and 21791). Professor Brogan is supported by the Great Ormond Street Hospital (GOSH) Children’s Charity.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed.
Ethics approval and consent to participate
Formal written consent for publication was obtained from the patient’s mother and is available on request.
Consent for publication
Formal written consent for publication was obtained from the patient’s mother.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Barbara Jensen, Email: barbara.jensen@ucl.ac.uk.
Despina Eleftheriou, Email: d.eleftheriou@ucl.ac.uk.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12969-020-00466-1.
References
- 1.Myhre SA, Ruvalcaba RH, Graham CB. A new growth deficiency syndrome. Clin Genet. 1981;20(1):1–5. doi: 10.1111/j.1399-0004.1981.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 2.Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, et al. Mutations at a single codon in mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2012;44(1):85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 3.Caputo V, Bocchinfuso G, Castori M, Traversa A, Pizzuti A, Stella L, et al. Novel SMAD4 mutation causing Myhre syndrome. Am J Med Genet A. 2014;164(7):1835–1840. doi: 10.1002/ajmg.a.36544. [DOI] [PubMed] [Google Scholar]
- 4.Caputo V, Cianetti L, Niceta M, Carta C, Ciolfi A, Bocchinfuso G, et al. A restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre syndrome. Am J Hum Genet. 2012;90(1):161–169. doi: 10.1016/j.ajhg.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin AE, Michot C, Cormier-Daire V, L’Ecuyer TJ, Matherne GP, Barnes BH, et al. Gain-of-function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. Am J Med Genet A. 2016;170(10):2617–2631. doi: 10.1002/ajmg.a.37739. [DOI] [PubMed] [Google Scholar]
- 6.Garavelli L, Maini I, Baccilieri F, Ivanovski I, Pollazzon M, Rosato S, et al. Natural history and life-threatening complications in Myhre syndrome and review of the literature. Eur J Pediatr. 2016;175(10):1307–1315. doi: 10.1007/s00431-016-2761-3. [DOI] [PubMed] [Google Scholar]
- 7.Starr LJ, Grange DK, Delaney JW, Yetman AT, Hammel JM, Sanmann JN, et al. Myhre syndrome: clinical features and restrictive cardiopulmonary complications. Am J Med Genet A. 2015;167(12):2893–2901. doi: 10.1002/ajmg.a.37273. [DOI] [PubMed] [Google Scholar]
- 8.McGowan R, Gulati R, McHenry P, Cooke A, Butler S, Keng WT, et al. Clinical features and respiratory complications in Myhre syndrome. Eur J Med Genet. 2011;54(6):e553–e559. doi: 10.1016/j.ejmg.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Michot C, Le Goff C, Mahaut C, Afenjar A, Brooks AS, Campeau PM, et al. Myhre and LAPS syndromes: clinical and molecular review of 32 patients. Eur J Hum Genet. 2014;22(11):1272–1277. doi: 10.1038/ejhg.2013.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Steensel MAM, Vreeburg M, Steijlen PM, de Die-Smulders C. Myhre syndrome in a female with previously undescribed symptoms: further delineation of the phenotype. Am J Med Genet A. 2005;139A(2):127–130. doi: 10.1002/ajmg.a.30988. [DOI] [PubMed] [Google Scholar]
- 11.Titomanlio L, Marzano MG, Rossi E, D’armiento M, De Brasi D, Vega GR, et al. Case of Myhre syndrome with autism and peculiar skin histological findings. Am J Med Genet. 2001;103(2):163–165. doi: 10.1002/ajmg.1517. [DOI] [PubMed] [Google Scholar]
- 12.Starr LJ, Lindor NM, Lin AE. GeneReviews®. Seattle: University of Washington; 2017. Myhre syndrome. [Google Scholar]
- 13.Torres JE, Sánchez JL. Histopathologic differentiation between localized and systemic scleroderma. Am J Dermatopathol. 1998;20(3):242–245. doi: 10.1097/00000372-199806000-00003. [DOI] [PubMed] [Google Scholar]
- 14.McNiff JM, Glusac EJ, Lazova RZ, Carroll CB. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21(4):315–319. doi: 10.1097/00000372-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 16.Allanson JE. Noonan syndrome. J Med Genet. 1987;24(1):9–13. doi: 10.1136/jmg.24.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of Noonan syndrome. Arch Dis Child. 1992;67(2):178–183. doi: 10.1136/adc.67.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sznajer Y, Keren B, Baumann C, Pereira S, Alberti C, Elion J, et al. The spectrum of cardiac anomalies in Noonan syndrome as a result of mutations in the PTPN11 gene. Pediatrics. 2007;119(6):e1325–e1331. doi: 10.1542/peds.2006-0211. [DOI] [PubMed] [Google Scholar]
- 19.Ferrero GB, Baldassarre G, Delmonaco AG, Biamino E, Banaudi E, Carta C, et al. Clinical and molecular characterization of 40 patients with Noonan syndrome. Eur J Med Genet. 2008;51(6):566–572. doi: 10.1016/j.ejmg.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 20.DeBusk FL. The Hutchinson-Gilford progeria syndrome: report of 4 cases and review of the literature. J Pediatr. 1972;80(4):697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 21.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in Lamin a cause Hutchinson–Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao H, Hegele RA. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090) J Hum Genet. 2003;48(5):271–274. doi: 10.1007/s10038-003-0025-3. [DOI] [PubMed] [Google Scholar]
- 23.Winchester P, Grossman H, Lim WN, Danes BS. A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. Am J Roentgenol. 1969;106(1):121–128. doi: 10.2214/ajr.106.1.121. [DOI] [PubMed] [Google Scholar]
- 24.Hollister DW, Rimoin DL, Lachman RS, Cohen AH, Reed WB, Westin GW. The Winchester syndrome: a nonlysosomal connective tissue disease. J Pediatr. 1974;84(5):701–709. doi: 10.1016/s0022-3476(74)80012-0. [DOI] [PubMed] [Google Scholar]
- 25.Prapanpoch S, Jorgenson RJ, Langlais RP, Nummikoski PV. Winchester syndrome: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol. 1992;74(5):671–677. doi: 10.1016/0030-4220(92)90363-u. [DOI] [PubMed] [Google Scholar]
- 26.Evans BR, Mosig RA, Lobl M, Martignetti CR, Camacho C, Grum-Tokars V, et al. Mutation of membrane Type-1 metalloproteinase, MT1-MMP, causes the multicentric Osteolysis and arthritis disease Winchester syndrome. Am J Hum Genet. 2012;91(3):572–576. doi: 10.1016/j.ajhg.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin AE, Alali A, Starr LJ, Shah N, Beavis A, Pereira EM, et al. Gain-of-function pathogenic variants in SMAD4 are associated with neoplasia in Myhre syndrome. Am J Med Genet A. 2020;182(2):328–337. doi: 10.1002/ajmg.a.61430. [DOI] [PubMed] [Google Scholar]
- 28.Online Mendelian Inheritance in Man Database, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD).. Available from: https://omim.org/. [cited 2019 Jun 4].
- 29.Genetics Home Reference Database. National Library of Medicine (US). Bethesda (MD). Available from: https://ghr.nlm.nih.gov/. [cited 2019 Jun 4].
- 30.Piccolo P, Mithbaokar P, Sabatino V, Tolmie J, Melis D, Schiaffino MC, et al. SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. Eur J Hum Genet. 2014;22(8):988–994. doi: 10.1038/ejhg.2013.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., III Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358(26):2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groenink M, den Hartog AW, Franken R, Radonic T, de Waard V, Timmermans J, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491–3500. doi: 10.1093/eurheartj/eht334. [DOI] [PubMed] [Google Scholar]
- 33.Araujo AQ, Arteaga E, Ianni BM, Buck PC, Rabello R, Mady C. Effect of losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am J Cardiol. 2005;96(11):1563–1567. doi: 10.1016/j.amjcard.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 34.Shimada YJ, Passeri JJ, Baggish AL, O’Callaghan C, Lowry PA, Yannekis G, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013;1(6):480–487. doi: 10.1016/j.jchf.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N Engl J Med. 2008;358(6):592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rork JF, Huang JT, Gordon LB, Kleinman M, Kieran MW, Liang MG. Initial cutaneous manifestations of Hutchinson-Gilford Progeria syndrome. Pediatr Dermatol. 2014;31(2):196–202. doi: 10.1111/pde.12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, et al. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27(6):558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonullu E, Bilge NŞY, Kaşifoğlu T, Korkmaz C. Werner’s syndrome may be lost in the shadow of the scleroderma. Rheumatol Int. 2013;33(5):1309–1312. doi: 10.1007/s00296-012-2564-4. [DOI] [PubMed] [Google Scholar]
- 39.Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001;102(1):11–17. doi: 10.1002/1096-8628(20010722)102:1<11::aid-ajmg1413>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 40.Shastry S, Simha V, Godbole K, Sbraccia P, Melancon S, Yajnik CS, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192–E197. doi: 10.1210/jc.2010-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senniappan S, Hughes M, Shah P, Shah V, Kaski JP, Brogan P, et al. Pigmentary hypertrichosis and non-autoimmune insulin-dependent diabetes mellitus (PHID) syndrome is associated with severe chronic inflammation and cardiomyopathy, and represents a new monogenic autoinflammatory syndrome. J Pediatr Endocrinol Metab. 2013;26(9–10):877–82. [DOI] [PubMed]
- 42.Freyschmidt J. Melorheostosis: a review of 23 cases. Eur Radiol. 2001;11(3):474–479. doi: 10.1007/s003300000562. [DOI] [PubMed] [Google Scholar]
- 43.Ehrig T, Cockerell CJ. Buschke-ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J Am Acad Dermatol. 2003;49(6):1163–1166. doi: 10.1016/s0190-9622(03)00481-x. [DOI] [PubMed] [Google Scholar]
- 44.Liu T, McCalmont TH, Frieden IJ, Williams ML, Connolly MK, Gilliam AE. The stiff skin syndrome: case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144(10):1351–1359. doi: 10.1001/archderm.144.10.1351. [DOI] [PubMed] [Google Scholar]
- 45.Muñoz-Santos C, Guilabert A, Moreno N, To-Figueras J, Badenas C, Darwich E, et al. Familial and sporadic Porphyria Cutanea Tarda: clinical and biochemical features and risk factors in 152 patients. Medicine (Baltimore) 2010;89(2):69–74. doi: 10.1097/MD.0b013e3181d50928. [DOI] [PubMed] [Google Scholar]
- 46.Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR scleroderma trials and research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi: 10.1136/ard.2009.114264. [DOI] [PubMed] [Google Scholar]
- 47.Rubio-Rivas M, Royo C, Simeón CP, Corbella X, Fonollosa V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44(2):208–219. doi: 10.1016/j.semarthrit.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007;66(7):940–944. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med. 2016;129(3):324–331. doi: 10.1016/j.amjmed.2015.10.037. [DOI] [PubMed] [Google Scholar]
- 50.Yan X, Jin J. Primary cutaneous amyloidosis associated with autoimmune hepatitis-primary biliary cirrhosis overlap syndrome and Sjögren syndrome: A case report. Medicine (Baltimore). 2018;97(8):e0004. [DOI] [PMC free article] [PubMed]
- 51.Kogure A, Ohshima Y, Watanabe N, Ohba T, Miyata M, Ohara M, et al. A case of Werner’s syndrome associated with systemic lupus erythematosus. Clin Rheumatol. 1995;14(2):199–203. doi: 10.1007/BF02214944. [DOI] [PubMed] [Google Scholar]
- 52.Fritsch S, Wojcik AS, Schade L, Machota MM, Brenner FM, Paiva ES. Increased photosensitivity? Case report of porphyria cutanea tarda associated with systemic lupus erythematosus. Rev Bras Reum. 2012;52(6):965–970. [PubMed] [Google Scholar]
- 53.Kostik MM, Chikova IA, Avramenko VV, Vasyakina LI, Le Trionnaire E, Chasnyk VG, et al. Farber lipogranulomatosis with predominant joint involvement mimicking juvenile idiopathic arthritis. J Inherit Metab Dis. 2013;36(6):1079–1080. doi: 10.1007/s10545-012-9573-z. [DOI] [PubMed] [Google Scholar]
- 54.Orteu CH, Ong VH, Denton CP. Scleroderma mimics – clinical features and management. Best Pract Res Clin Rheumatol. 2020;p101489. [DOI] [PubMed]
- 55.Foti R, Leonardi R, Rondinone R, Di Gangi M, Leonetti C, Canova M, et al. Scleroderma-like disorders. Autoimmun Rev. 2008;7(4):331–339. doi: 10.1016/j.autrev.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Rusmini M, Federici S, Caroli F, Grossi A, Baldi M, Obici L, et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann Rheum Dis. 2016;75(8):1550–1557. doi: 10.1136/annrheumdis-2015-207701. [DOI] [PubMed] [Google Scholar]
- 57.Ortega-Moreno L, Giráldez BG, Soto-Insuga V, Losada-Del Pozo R, Rodrigo-Moreno M, Alarcón-Morcillo C, et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS One. 2017;12(11):e0188978. doi: 10.1371/journal.pone.0188978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: A novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133(2):529–534.e1. doi: 10.1016/j.jaci.2013.08.032. [DOI] [PubMed] [Google Scholar]
- 59.Mak ACY, Tang PLF, Cleveland C, Smith MH, Kari Connolly M, Katsumoto TR, et al. Brief report: whole-exome sequencing for identification of potential causal variants for diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2016;68(9):2257–2262. doi: 10.1002/art.39721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCreary D, Omoyinmi E, Hong Y, Mulhern C, Papadopoulou C, Casimir M, et al. Development and validation of a targeted next-generation sequencing gene panel for children with neuroinflammation. JAMA Netw Open. 2019;2(10):e1914274. doi: 10.1001/jamanetworkopen.2019.14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Omoyinmi E, Standing A, Keylock A, Price-Kuehne F, Gomes SM, Rowczenio D, et al. Clinical impact of a targeted next-generation sequencing gene panel for autoinflammation and vasculitis. PLoS One. 2017;12(7):e0181874. [DOI] [PMC free article] [PubMed]
- 62.Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-β bioavailability regulation. Nat Genet. 2008;40(9):1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med. 2010;2(23):23ra20. doi: 10.1126/scitranslmed.3000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bader-Meunier B, Bonafé L, Fraitag S, Breton S, Bodemer C, Baujat G. Mutation in MMP2 gene may result in scleroderma-like skin thickening. Ann Rheum Dis. 2016;75(1):e1. doi: 10.1136/annrheumdis-2015-208182. [DOI] [PubMed] [Google Scholar]
- 65.Mori Y, Chen S-J, Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum. 2003;48(7):1964–1978. doi: 10.1002/art.11157. [DOI] [PubMed] [Google Scholar]
- 66.Kawakami T, Ihn H, Xu W, Smith E, LeRoy C, Trojanowska M. Increased expression of TGF-β receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-β signaling to scleroderma phenotype. J Invest Dermatol. 1998;110(1):47–51. doi: 10.1046/j.1523-1747.1998.00073.x. [DOI] [PubMed] [Google Scholar]
- 67.Kubo M, Ihn H, Yamane K, Tamaki K. Up-regulated expression of transforming growth factor β receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum. 2001;44(3):731–734. doi: 10.1002/1529-0131(200103)44:3<731::AID-ANR124>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 68.Sargent JL, Milano A, Bhattacharyya S, Varga J, Connolly MK, Chang HY, et al. A TGFβ-responsive gene signature is associated with a subset of diffuse scleroderma with increased disease severity. J Invest Dermatol. 2010;130(3):694–705. doi: 10.1038/jid.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mead AL, Wong TTL, Cordeiro MF, Anderson IK, Khaw PT. Evaluation of anti-TGF-β2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Investig Opthalmol Vis Sci. 2003;44(8):3394–3401. doi: 10.1167/iovs.02-0978. [DOI] [PubMed] [Google Scholar]
- 70.Lim D-S, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103(6):789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamada H, Tajima S, Nishikawa T. Tranilast inhibits collagen synthesis in normal, scleroderma and keloid fibroblasts at a late passage culture but not at an early passage culture. J Dermatol Sci. 1995;9(1):45–47. doi: 10.1016/0923-1811(94)00355-i. [DOI] [PubMed] [Google Scholar]
- 72.Soria A, Cario-André M, Lepreux S, Rezvani HR, Pasquet JM, Pain C, et al. The effect of Imatinib (Glivec®) on scleroderma and normal dermal fibroblasts: a preclinical study. Dermatology. 2008;216(2):109–117. doi: 10.1159/000111507. [DOI] [PubMed] [Google Scholar]
- 73.Pannu J, Asano Y, Nakerakanti S, Smith E, Jablonska S, Blaszczyk M, et al. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58(8):2528–2537. doi: 10.1002/art.23698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Supplemental Table 1. Features of juvenile localised scleroderma, juvenile systemic sclerosis and Myhre syndrome.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analysed.