

Abstract
Hydrogen/deuterium exchange (HDX) is used in protein biophysics to probe folding dynamics, intermolecular interactions, epitope and other mapping [1, 2]. A typical procedure often involves HDX in buffered D2O solution followed by pepsin digestion, and liquid chromatography/electrospray ionization Mass Spectrometry (LC-ESI-MS) analysis. In this work, HDX of protein ions was conducted in the ESI source. Both native electrospray (ESI) droplets of ubiquitin and denatured myoglobin were exposed to D2O vapor in the source region of a Bruker SolariX 12T FTICR-mass spectrometer. Electron Capture Dissociation (ECD) was used to assess deuterium incorporation at the residue level. This in-source HDX, on the millisecond-timescale, exchanges side-chain hydrogens and fast-exchanging amides compared to conventional-minutes-to-hours HDX of backbone hydrogens in solution with less sample preparation (i.e., no D2O/protein mixing and incubation, no quenching, protein digestion, or LC separation).
Graphical Abstract
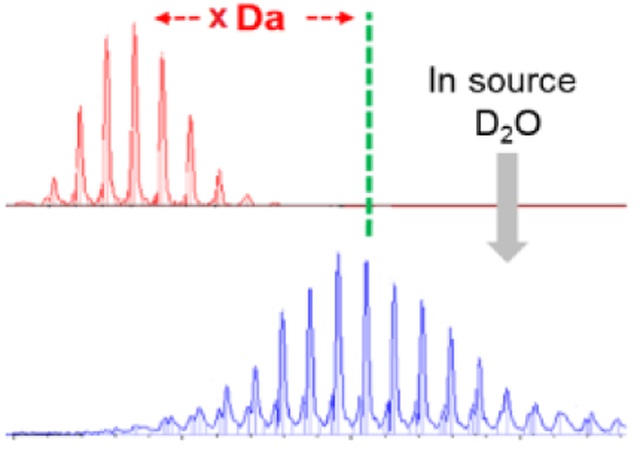
Although hydrogen/deuterium exchange (HDX) has become a mature approach for protein footprinting, there are opportunities for further development. HDX is used in protein biophysics to examine protein-drug binding and protein-protein interactions and follow changes in conformation and measure protein-ligand affinities [3–6]. HDX also can follow folding dynamics and map epitopes and other protein interfaces [3–6]. The typical solution HDX procedures are time-consuming and require multi-step sample preparations. To shorten the exchange time, investigators developed theta capillary spray [7] and gas-phase HDX [8–10] that allow exchange times as short as 20 μs to be followed.
We are motivated to conduct HDX in the electrospray ionization (ESI) source because ESI has the ability to introduce native or near-native protein ions in the gas phase. The native protein is indicated by its low charge-state distributions and collision cross section (CCS) corresponding to the folded structure [11]. HDX-MS in a three-syringe, continuous-flow setup with top-down ECD was used to study native holo-myoglobin [12].
Recent publications demonstrated the use of a pseudo gas-phase approach for desalting ionized biological molecules and crude oil in the source region of a mass spectrometer [13]. Further, gas-phase HDX in an external ion source of the 7 T FT-ICR probed the molecular structures of sodiated glycine oligomers [14]. Applications of in-source HDX were pursued for interfaces of protein complexes [8] and for carbohydrate molecules [15]. HDX in the traveling wave ion guide [9] and in a hexapole collision cell [16] allowed the study of protein conformations in the gas phase.
In this work, we exposed electro sprayed droplets containing the proteins ubiquitin and myoglobin under both denaturing and native conditions to D2O vapors in the source region of a Bruker SolariX 12 T Fourier Transform Ion Cyclotron (FTICR) mass spectrometer. This HDX is considered to be a combination of gas-phase and solution, as previously described [17]. Thus, information from this experiment will likely be different from that obtained from solution HDX [14]. We describe a preliminary, fast HDX method with medium-sized proteins as proof of principle. We chose ubiquitin as a test, owing to its small size (MW 8.5 kDa) and its many previous studies, and apomyoglobin, a medium-size protein (16.9 kDa).
Experimental
All proteins (ubiquitin, myoglobin) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Chemicals, solvents, and gases were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Praxair (St. Louis, MO, USA). Model proteins were prepared at 10–15 μM in both denatured (50:50:0.1 of H2O:MeOH:FA) and native (100 mM ammonium acetate buffer, NH4OAc) solution.
All experiments were performed with a Bruker SolariX 12 T FTICR and Bruker MaXis II Quadrupole Time-of-Flight (Q-TOF) mass spectrometers in the positive-ion mode. Source conditions, D2O temperature, dry-gas flow rate, and dry-gas temperature were optimized in triplicate, error bars in Fig. 1–4 represent standard deviations. Set up for in-source HDX was adapted from that of Chanthamontri, et al. [13]. Briefly, the source region of the FTICR mass spectrometer was exposed to D2O vapor by using the modified gas-inlet system located between the 250 L liquid nitrogen Dewar and Bruker Apollo ESI Source. This allowed the transfer of D2O vapor to the HDX zone in the ESI chamber, located behind the spray shield and in front of the heated capillary. Gas-flow rates were controlled by using Bruker software. Quantifoil BioShake iQ High-Speed Microplate Thermoshaker Lab Mixer 001891 was used as the temperature control unit for maintaining the temperature of the D2O reservoir. Temperatures of D2O vapor were assumed to be the same as those set with the control unit where the D2O reservoir was situated (see Supporting Information).
Figure 1.
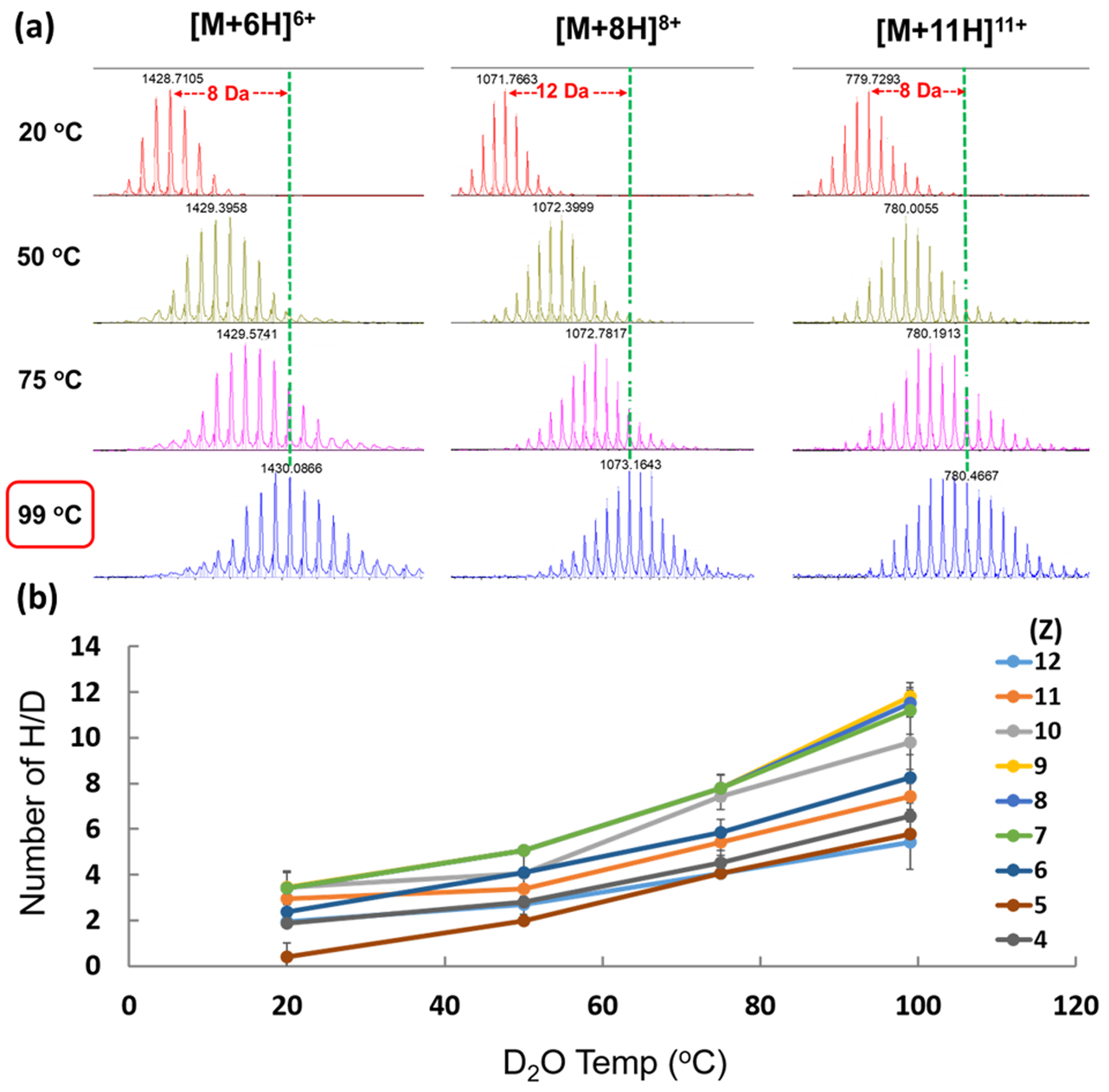
(a) Effect of D2O temperature on HDX of +6, +8, and +11 charge states of ubiquitin in H2O:MeOH:FA. (b) Dependence of HDX of denatured ubiquitin ions on temp.
Figure 4.
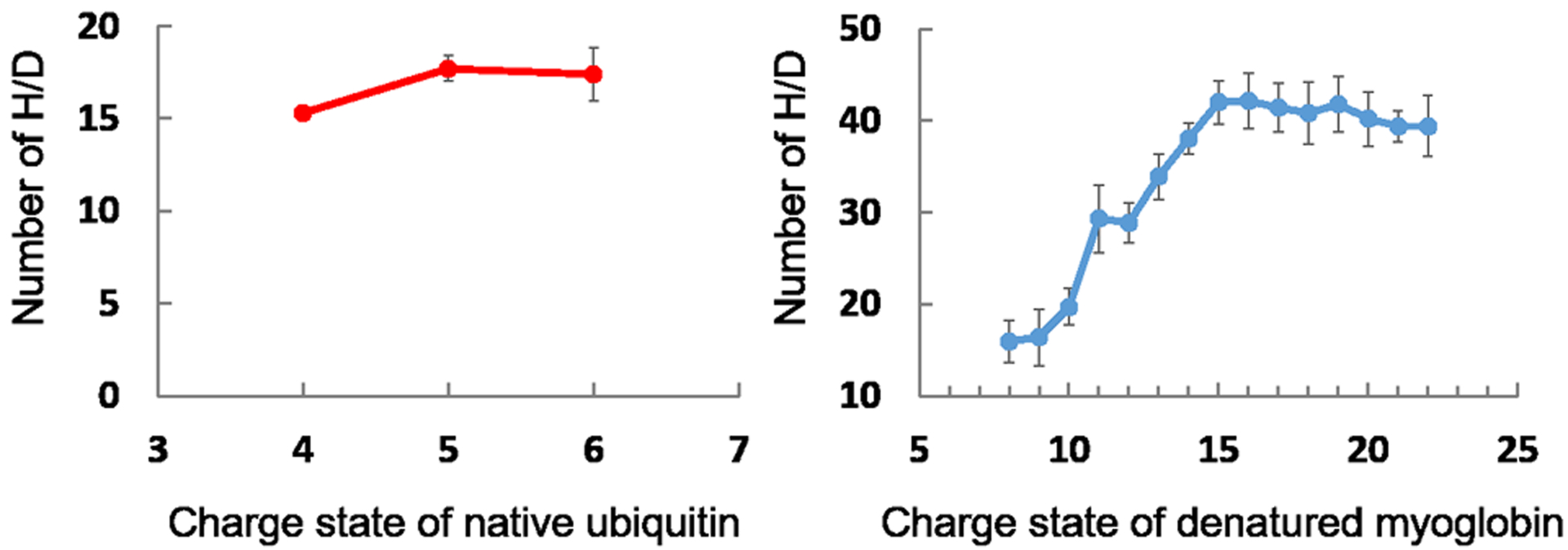
Deuterium uptake as a function charge state for native ESI of ubiquitin (a) and denatured myoglobin (b).
Electron-capture dissociation (ECD) [18] was used for top-down sequencing to minimize hydrogen/deuterium scrambling induced by collisional induced dissociation (CID) [19]. Ionization was accomplished via direct infusion electrospray with a flow rate of 3 μL/min. HDX extent was determined from centroids of c- and z- ion peaks and were taken as a measure of the solvent accessibility of the protein in its native form.
Results and Discussion
We used denatured ubiquitin as a model protein to optimize D2O temperatures, dry-gas (N2) flow rates, and temperature. The D2O temperature was varied from room temperature (20 °C) to 100 °C by using the temperature control unit. The amount of HDX for all charge states increased as the D2O temperature was increased (Fig. 1).
Because the temperature provided by the control unit for the D2O reservoir was limited, the temperature to obtain the highest HDX was 100 °C, as shown in Figure 1.
The dry gas (N2) flow rates were then investigated from 1.5 to 7 L/min as shown in Figure 2. The lower flow rate of dry gas results in a greater extent of HDX as the slower rate allowed enough time for the gas to purge the spray chamber with D2O. If the dry gas flow rates were too large, the gas did not have enough time to purge the spray chamber, or it likely bypassed the D2O reservoir resulting in a lower extent of HDX. Therefore, the dry gas flow rates in the range of 2.0–2.5 L/min were optimized for most of the charge states.
Figure 2.
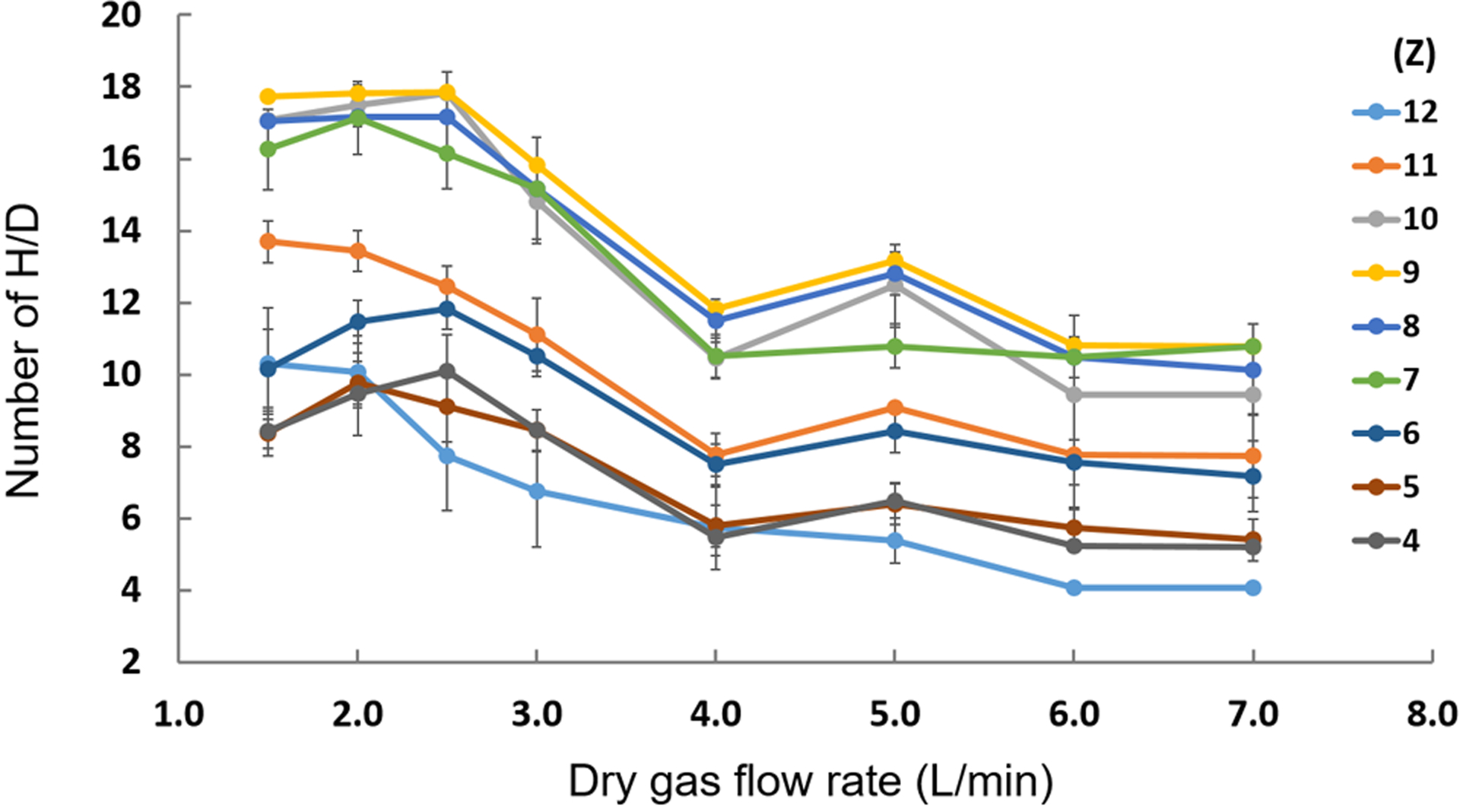
Effect of dry gas (N2) flow rate on HDX of ubiquitin in H2O, MeOH, and FA.
The dry-gas temperatures were also varied from 50–350 °C (Fig. 3). The dry gas temperature does not have much effect on the extent of HDX. Therefore, a dry-gas temperature of 150–200 °C was chosen to avoid denaturation of the native protein for later applications and to avoid D2O condensation.
Figure 3.
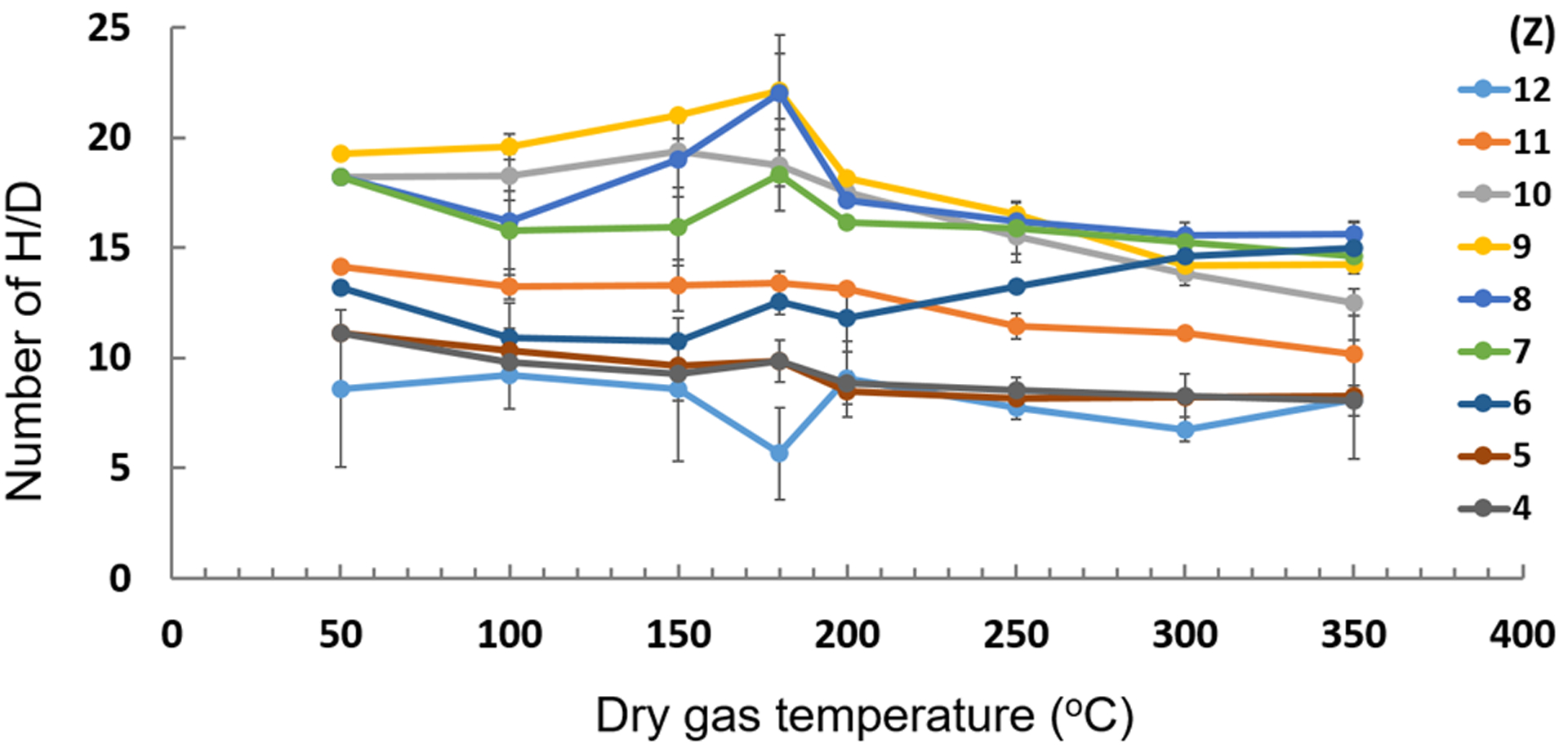
Effect of dry-gas (N2) temperature on HDX of ubiquitin in H2O, MeOH, and FA.
After establishing the optimum conditions for in-source HDX, we measured the extent of exchange for ubiquitin and myoglobin ionized in both native ESI (100–150 mM NH4OAc buffer) and denatured (50:50:0.1 H2O:MeOH:FA solutions), (Fig. 4). We measured the extent of HDX as a function of charge state for native ESI of ubiquitin and denatured ESI of myoglobin. The number of deuteriums taken up was calculated by using the following equations.
where Ii is the intensity of ith peak, (m/z)i is its mass to charge ratio, is the mass after HDX, is the mass of the native or denatured proteins.
The ESI spectrum of ubiquitin in NH4OAc buffer shows a charge-state distribution (CSD) spanning +4, +5, and +6, strongly suggesting the protein is native. After exposure of the ESI droplets under native conditions to D2O vapor in the source region, the exchange extents for ubiquitin were 17, 18, and 15 for the +6, +5, and +4 charged species, respectively. Thus, there is only small structural variation in the native form of ubiquitin as a function of charge in this regime. These results agree with conclusions from a study of collisional cross sections (CCS) of the same charge states of native ubiquitin ions [20].
The mass spectrum of denatured myoglobin after in-source HDX shows that the number of Ds incorporated increases with an increase in charge state, confirming that the higher the charge state, the more open is the structure owing to electrostatic repulsion. This increase continues until it reaches a plateau (i.e., at +15 charge state). Data from charge states +8 to +15 matched the CCS measured previously [21]. In the same study, however, CCS of apomyoglobin increased until the +22-charge state. It is possible that H/D back exchange occurred during our experiments for denatured protein ions, and further investigation is needed.
Collisional-Induced Dissociation (CID) has been widely used for structural characterization and identification of proteins, peptides, and other molecules. Several research groups have attempted to use CID for analyzing proteins after deuterium labeling by using triple-quadrupole MS/MS, Q-TOF MS/MS, and nozzle-skimmer CID FT-ICR-MS. Theoretical and experimental studies indicate that CID is accompanied by extensive intramolecular H/D “scrambling” [22, 23]. This phenomenon can lead to partial or complete randomization of the HDX pattern, causing loss of site-specific information.
Owing to the concerns of H/D scrambling, we used electron-based dissociation techniques (e.g., ECD) to probe protein conformation in top-down HDX-MS. We also investigated residue-level HDX using ECD. An ECD product-ion spectrum of denatured ubiquitin showed approximately 83% sequence coverage, whereas the ECD-product ion spectrum of native ubiquitin showed ~ 40% coverage. The numbers of incorporated Ds measured from centroids of c- and z- (cleavages at N-Cα bonds) ion peaks were used to determine those areas of solvent accessibility in the native protein. Higher resolution D-uptake information at the residue level was obtained by using top-down ECD (Fig. 5). Unfortunately, there are not enough fragments representing the middle region of ubiquitin because the protein, in its native form, is difficult to fragment, causing loss of HDX information at the residue level in that region. Another ion activation method, such as UVPD, might provide more fragment information if H/D scrambling can be avoided.
Figure 5.
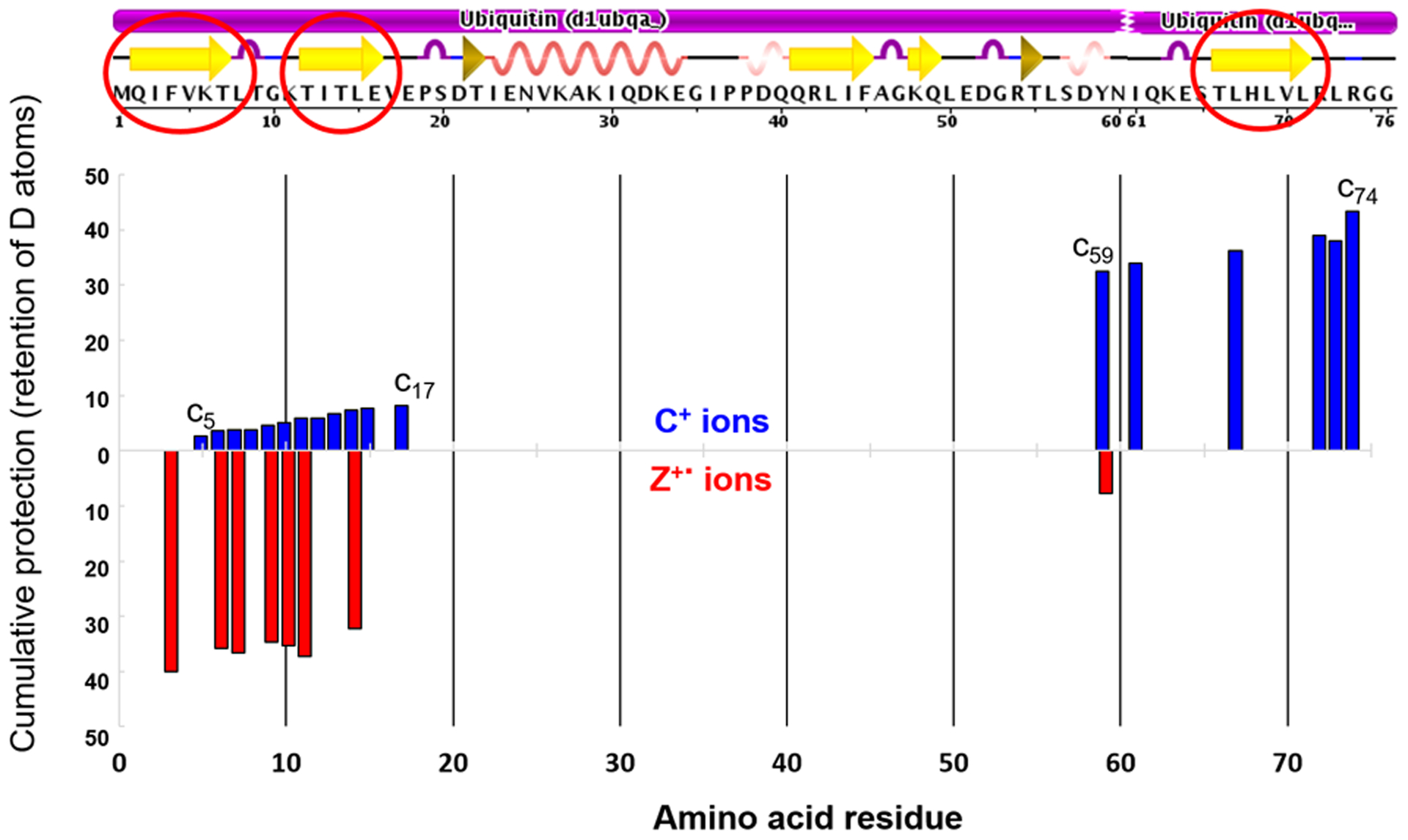
Higher spatial resolution D-uptake (residue level) of native ubiquitin obtained by using top-down ECD.
Conclusions
In-source HDX of native protein ions provides structural information at low and sub μM concentrations. Only small sample quantities are required prior to analysis (i.e., no D2O/protein mixing and incubation, no quenching, no protein digestion, no LC separation). The main advantage of in-source HDX experiments is they provide sub-sec HDX kinetics of side-chain hydrogens compared to conventional min-to-hours HDX of backbone Hs in solution [24]. Information obtained from this type of study is likely to be different from that obtained from solution HDX because exchange kinetics and exchange sites are different [14].
This in-source HDX approach may be useful for studying protein-drug interactions on a global level (intact molecule level). ECD may help locate the binding site of the drug even at the residue level if H/D scrambling is avoided. The top-down ECD experiment affords higher spatial resolution than regular LC-MS experiments in conventional HDX because most of those approaches give data at the peptide level. Here, top-down ECD of native proteins undergoing in-source HDX was demonstrated. High-coverage applications await the development of ECD for larger proteins.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIGMS (P41GM103422 and R24GM136766) to MLG and the Faculty Research Incentive Fund, St. Louis College of Pharmacy (Grant #20-158-9770-6660) to NS.
References
- 1.Wales TE, Engen JR: Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 25, 158–170 (2006) [DOI] [PubMed] [Google Scholar]
- 2.Chance M Wiley, Hoboken, N.J. (2008) [Google Scholar]
- 3.Hoofnagle Andrew N., Resing Katheryn A., Ahn NG: Protein Analysis by Hydrogen Exchange Mass Spectrometry. Annual Review of Biophysics and Biomolecular Structure. 32, 1–25 (2003) [DOI] [PubMed] [Google Scholar]
- 4.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR: Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 8, 43–59 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engen JR: Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Analytical Chemistry. 81, 7870–7875 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konermann L, Pan J, Liu YH: Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 40, 1224–1234 (2011) [DOI] [PubMed] [Google Scholar]
- 7.Jansson ET, Lai YH, Santiago JG, Zare RN: Rapid Hydrogen-Deuterium Exchange in Liquid Droplets. J Am Chem Soc. 139, 6851–6854 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mistarz UH, Brown JM, Haselmann KF, Rand KD: Probing the Binding Interfaces of Protein Complexes Using Gas-Phase H/D Exchange Mass Spectrometry. Structure. 24, 310–318 (2016) [DOI] [PubMed] [Google Scholar]
- 9.Rand KD, Pringle SD, Murphy JP 3rd, Fadgen KE, Brown J, Engen JR: Gas-phase hydrogen/deuterium exchange in a traveling wave ion guide for the examination of protein conformations. Analytical chemistry. 81, 10019–10028 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uppal SS, Beasley SE, Scian M, Guttman M: Gas-Phase Hydrogen/Deuterium Exchange for Distinguishing Isomeric Carbohydrate Ions. Analytical Chemistry. 89, 4737–4742 (2017) [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Cui W, Gross ML: Native electrospray ionization and electron-capture dissociation for comparison of protein structure in solution and the gas phase. Int J Mass Spectrom. 354–355, (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan J, Han J, Borchers CH, Konermann L: Hydrogen/Deuterium Exchange Mass Spectrometry with Top-Down Electron Capture Dissociation for Characterizing Structural Transitions of a 17 kDa Protein. Journal of the American Chemical Society. 131, 12801–12808 (2009) [DOI] [PubMed] [Google Scholar]
- 13.Chanthamontri CK, Stopford AP, Snowdon RW, Oldenburg TB, Larter SR: On-line desalting of crude oil in the source region of a Fourier transform ion cyclotron resonance mass spectrometer. J Am Soc Mass Spectrom. 25, 1506–1510 (2014) [DOI] [PubMed] [Google Scholar]
- 14.Cox HA, Julian RR, Lee S-W, Beauchamp JL: Gas-Phase H/D Exchange of Sodiated Glycine Oligomers with ND3: Exchange Kinetics Do Not Reflect Parent Ion Structures. Journal of the American Chemical Society. 126, 6485–6490 (2004) [DOI] [PubMed] [Google Scholar]
- 15.Kostyukevich Y, Kononikhin A, Popov I, Nikolaev E: In-ESI source hydrogen/deuterium exchange of carbohydrate ions. Anal Chem. 86, 2595–2600 (2014) [DOI] [PubMed] [Google Scholar]
- 16.Pan J, Heath BL, Jockusch RA, Konermann L: Structural interrogation of electrosprayed peptide ions by gas-phase H/D exchange and electron capture dissociation mass spectrometry. Anal Chem. 84, 373–378 (2012) [DOI] [PubMed] [Google Scholar]
- 17.Liyanage OT, Brantley MR, Calixte EI, Solouki T, Shuford KL, Gallagher ES: Characterization of Electrospray Ionization (ESI) Parameters on In-ESI Hydrogen/Deuterium Exchange of Carbohydrate-Metal Ion Adducts. J Am Soc Mass Spectrom. 30, 235–247 (2019) [DOI] [PubMed] [Google Scholar]
- 18.Zubarev RA, Kelleher NL, McLafferty FW: Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. Journal of the American Chemical Society. 120, 3265–3266 (1998) [Google Scholar]
- 19.Jørgensen TJD, Gårdsvoll H, Ploug M, Roepstorff P: Intramolecular Migration of Amide Hydrogens in Protonated Peptides upon Collisional Activation. Journal of the American Chemical Society. 127, 2785–2793 (2005) [DOI] [PubMed] [Google Scholar]
- 20.Salbo R, Bush MF, Naver H, Campuzano I, Robinson CV, Pettersson I, Jorgensen TJ, Haselmann KF: Traveling-wave ion mobility mass spectrometry of protein complexes: accurate calibrated collision cross-sections of human insulin oligomers. Rapid Commun Mass Spectrom. 26, 1181–1193 (2012) [DOI] [PubMed] [Google Scholar]
- 21.Shelimov KB, Jarrold MF: Conformations, Unfolding, and Refolding of Apomyoglobin in Vacuum: An Activation Barrier for Gas-Phase Protein Folding. Journal of the American Chemical Society. 119, 2987–2994 (1997) [Google Scholar]
- 22.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA: Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom. 20, 1514–1517 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson PL, Pan J, Wilson DJ, Dempsey B, Lajoie G, Shilton B, Konermann L: Hydrogen/deuterium scrambling during quadrupole time-of-flight MS/MS analysis of a zinc-binding protein domain. Anal Chem. 79, 153–160 (2007) [DOI] [PubMed] [Google Scholar]
- 24.Kharlamova A, DeMuth JC, McLuckey SA: Vapor Treatment of Electrospray Droplets: Evidence for the Folding of Initially Denatured Proteins on the Sub-Millisecond Time-Scale. Journal of The American Society for Mass Spectrometry. 23, 88–101 (2012) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.