

Abstract
Myeloid derived suppressor cells (MDSCs) have gained significant attention for their immunosuppressive role in cancer and their ability to contribute to tumor progression and metastasis. Understanding the role of MDSCs in driving cancer cell migration, a process fundamental to metastasis, is essential to fully comprehend and target MDSC-tumor cell interactions. This study employs micro-fabricated platforms which simulate the structural cues present in the tumor microenvironment (TME) to elucidate the effects of MDSCs on the migratory phenotype of cancer cells at the single cell level. The results indicate that the presence of MDSCs enhances the motility of cancer-epithelial cells when directional cues (either topographical or spatial) are present. This behavior appears to be independent of cell-cell contact and driven by soluble byproducts from heterotypic interactions between MDSCs and cancer cells. Moreover, MDSC cell-motility is also impacted by the presence of cancer cells and the cancer cell secretome in the presence of directional cues. Epithelial dedifferentiation is the likely mechanism for changes in cancer cell motility in response to MDSCs. These results highlight the biochemical and bio-structural conditions under which MDSCs can support cancer cell migration, and could therefore provide new avenues of research and therapy aimed at stemming cancer progression.
Keywords: Myeloid derived suppressor cells (MDSC), cancer, cell-migration, aligned-substrates, micro/nano platforms
Summary:
This study explores the reciprocal interaction between the Myeloid Derived Suppressor Cells (MDSCs) and cancer cells, mediated by structural/directional cues from the environment, leading to enhanced migration not only in cancer cells but also MDSCs. It combines biomimetic platforms with one of the major players of cancer immunology to uncover synergies that could potentially contribute to disease progression.
Graphical Abstract
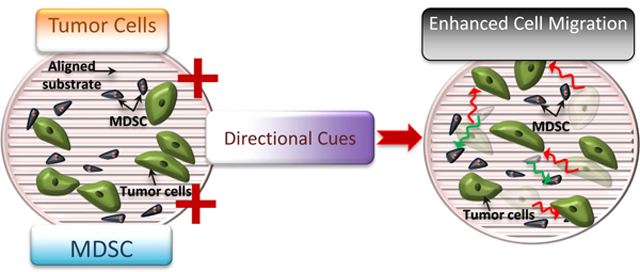
1. Introduction:
Metastasis is the primary cause of cancer-associated morbidity and mortality. During this process, highly motile cancerous cells leave the primary tumor, enter the circulation, and eventually extravasate to colonize distant organs and tissues. Cancer cell motility is driven in part by a milieu of signaling and structural changes in the tumor microenvironment (TME) that accompany tumor progression. These micro-environmental changes stem from cellular components including cancerous cells, stromal fibroblasts, tumor vasculature, immune cells, as well as non-cellular factors, including the extracellular matrix (ECM) structure and composition. All of these components evolve over time to support tumor progression and metastasis,1, 2 and crosstalk between the various TME compartments is critical for tumor growth and survival.3 For instance, stromal cells interacting with the tumor, adopt a highly reactive phenotype and remodel the ECM to facilitate tumorigenesis and metastasis.4 Moreover, tumor cell survival is highly dependent on the evasion of anti-tumor immunity via recruitment and activation of “tumor-protecting” immune cells.5, 6 A central mechanism driving these immunosuppressive responses is the recruitment of myeloid derived suppressor cells (MDSCs) to the TME.7
MDSCs are a heterogeneous population of immature, myeloid-derived cells that exhibit strong T-cell immunosuppression and include precursors of macrophages, granulocytes and dendritic cells.8, 9 MDSCs exhibit marked expansion in tumor bearing hosts, and their suppression of naïve T-cell activation in the lymph nodes and effector T-cell function in the tumor is a key factor driving tumorigenesis.10–17 MDSC accumulation has also been shown to be directly correlated to cancer stage, tumor burden, and poor prognosis in patients.18 The presence of MDSCs adversely affects the patient’s response to immuno-therapy, while their depletion in cancer patients augments the anti-tumor immune response and improves clinical outcomes.19, 20 The presence of MDSCs in the TME has also been associated with increased cancer cell metastasis, in part via the promotion of epithelial to mesenchymal transitions (EMT),21–23 and matrix-metalloproteinase-9 (MMP-9)-driven ECM remodeling.24–26 However, relatively little is known about how MDSCs affect the dynamics of cancer cell migration at single cell level or if these effects are dependent on the structural properties of TME.
Migration of malignant cells out of the primary tumor is critical to metastatic progression. Cancer cells change their phenotype and employ different modes of migration to escape the primary tumor.27, 28 Moreover, structural changes to the tumor ECM are also important in promoting increased cancer cell motility and dissemination,29, 30 including remodeling and alignment of ECM fibers (~1–20 μm in size) at the invasive front.31–34 A number of micro- and nanoscale platforms have been used to better understand the dynamics of cancer cell dissemination, at the single-cell level, in ex vivo models of aligned fiber matrices.35–39 Previous studies by our group have shown that aligned polydimethylsiloxane (PDMS) substrates with feature sizes in the 1.5–2μm range are able to modulate the migratory behavior of cancer cells.35, 36 Recently we also found that mouse and patient-derived MDSCs are responsive to PDMS microstructures in the same size range.40 However, to the best of our knowledge, no study has looked at the dynamics and synergistic impact of MDSCs and the microstructure on cancer cell motility with single-cell resolution. Here we use three different platforms to investigate the synergistic interactions between MDSCs and cancer cells. First, standard tissue-culture plastic (TCPS) is used as a baseline. Second, micro-fabricated aligned PDMS surfaces are used to recapitulate the aligned fiber networks that have been observed in the TME during tumor development.34, 41 Finally, in addition to these single cell assays, we also used a cell culture insert to recapitulate the tumor margin and the directional dissemination of tumor cells away from this margin. As a result, we have extensively investigated how cancer cells and MDSCs interact and modulate each other’s migratory phenotype under various conditions.
2. Results:
2.1. Co-culture of MDSCs with cancer cells on standard culture surfaces is insufficient to modulate cancer cell migration.
MDSCs accumulate in the tumor vicinity where they shield cancerous cells from host immunity. In order to understand how the intermixing of the two cell populations affects the migratory ability of cancer cells, the widely used mouse spleen derived MDSC cell line, MSC2,40, 42, 43 and well established clonal mouse breast cancer cell lines, Py230 (epithelial phenotype) and Py8119 (mesenchymal phenotype), 44-46were employed in our studies. The MDSCs and either of the cancer cell lines were intermixed and seeded on standard tissue culture polystyrene (TCPS) surfaces. The migration of individual cells was recorded and analyzed to identify potential changes in cell motility as a result of the interaction between MDSCs and cancer cells. As shown in Figure 1 (a, b), the two cell populations were uniformly interspersed and identified through fluorescent cell labeling (Py230 in red, Py8119 in green, and MSC2 unlabeled). The migration patterns of individual cells were tracked for 24 hours using time-lapse microscopy (Figure 1 b). Based on individual cell tracking under control monoculture conditions, Py8119 (mesenchymal) cells were more migratory than the Py230 (epithelial) cells (Figure 1 c, d), as indicated by an accumulated distance of 604.26 (IQR 499.48–711.98) μm vs 394.91 (IQR 318.15– 507.01) μm and velocity of 0.42 (IQR 0.35– 0.49) μm/min vs 0.27 (IQR 0.22–0.35) μm/min (*p<0.001). In addition to increased motility, the Euclidean distance (i.e., net displacement between the initial and final position of the cell) travelled by the Py8119 cells [95.09 (IQR 54.84–163.56) μm] was also significantly higher than that of the Py230 cells [56.34 (IQR 25.56– 88.35) μm] (Figure 1 e, *p<0.001). This observation provides functional confirmation of a more aggressive and migratory phenotype for the mesenchymal cells compared to their epithelial counterparts. However, as shown in Figure 1 (c–f), the presence of MDSCs (Py230+MSC2 and Py8119+MSC2) did not lead to statistically significant changes in motility for either Py230 or Py8119 cells. These results suggest that under standard cell culture conditions (i.e., flat cell culture surfaces), MDSCs do not significantly impact cancer cell motility.
Figure 1. Cancer cell migration in MDSC–PyMT cancer cell co-culture on TCPS.
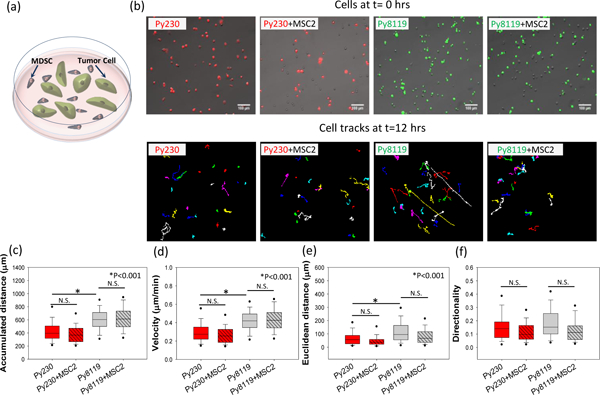
(a) Schematic diagram of the cell migration setup for PyMT mouse cancer epithelial (Py230) and mesenchymal (Py8119) cells in co-culture with mouse MDSCs (MSC2). (b) For the migration studies, tumor cells were labeled with a lipophilic dye to differentiate them from the MSC2 cells in co-culture (Py230 - red and Py8119 – green), and individual tumor cells were tracked over 24 hours. While the Py8119 cells are more migratory than the Py230 cells in monoculture conditions, as highlighted by the (c) accumulated distance and (d) cell velocity, the presence of MSC2s in co-culture with the tumor cells causes no significant changes in cancer cell (c) accumulated distance, (d) velocity, (e) Euclidean distance, and (f) cell directionality. One Way ANOVA on ranks was used to establish statistical significance. (n=3 samples, 60–100 cells per group), scale bar =100μm.
2.2. Co-culture of MDSCs with cancer cells on aligned matrices leads to increased motility in epithelial but not mesenchymal tumor cells.
Flat cell culture surfaces fail to recapitulate some of the key structural components of the TME responsible for driving cancer cell migration and invasion. Specifically, alignment of ECM fibers has been shown to enhance cancer cell migration/invasion and we therefore modified our co-culture configurations to better simulate and understand how these structural cues impact cancer cell motility in the presence of MDSCs. In order to provide these structural cues, cells were cultured on micro-patterned surfaces that partially mimic the ECM fiber alignment observed during tumor progression.35, 36, 40, 41 These surfaces consisted of linear patterns ~2 μm wide, ~1.5 μm tall, separated by a 2 μm gap, and were fabricated from polydimethylsiloxane (PDMS) via replica molding from a photolithographically patterned silicon master and the topography was confirmed using scanning electron microscopy (Figure 2 a) .35–37 These patterned PDMS substrates were employed to probe epithelial and mesenchymal tumor cell motility in the presence and absence of MDSCs. MDSCs and cancer cells (Py230 and Py8119) were seeded on patterned PDMS as a polydispersed mono- or co-cultures, and time lapse microscopy was used to monitor cancer cell motility at the single-cell level. Similar to studies on flat cell culture surfaces (Figure 1), monocultures of Py8119 cells exhibited increased single-cell accumulated distance [663.6 (IQR 537. 8–899.9) μm] and velocity [0.46 (IQR 0.37–0.63) μm/min] compared to monocultures of Py230 cells [467.7 (IQR 390.3–540.3) μm and 0.33 (IQR 0.27–0.37) μm/min respectively] (Figure 2 d, e, *p<0.001). However, there was no significant difference in Euclidean distance or directionality between the two cell types (Figure 2 f, g).
Figure 2. Cancer cell migration in MDSC –PyMT cancer cell polydispersed co-culture on patterned PDMS substrates.
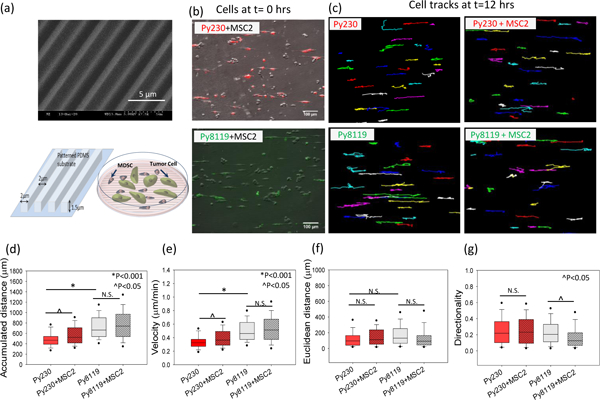
(a) Scanning electron micrograph (SEM) and schematic diagram of patterned PDMS substrates and cell migration setup for PyMT mouse cancer epithelial (Py230) and mesenchymal (Py8119) cells in polydispersed co-culture with mouse MDSCs (MSC2) on these substrates. For the migration studies, (b) tumor cells were labeled with live cell tracker fluorescent dye to differentiate them from the MSC2 cells in co-culture and (c) individual tumor cells were tracked over 24 hours to evaluate migration parameters. (d, e) A significant increase in accumulated distance and cell velocity was observed for the Py230 cells in the presence of MSC2s, while no significant difference was found in cell velocity for mesenchymal cells (Py8119) under co-culture conditions compared to monoculture. (f, g) The Euclidean distance and migration directionality do not change for Py230 cells while the directionality drops significantly for the Py8119 cells in the presence of MSC2 cells. One Way ANOVA on ranks was used to establish p-value. (n=3 samples, 60–100 cells per group), scale bar =100μm.
In contrast to studies on TCPS surfaces, when aligned surfaces were utilized, the co-culture of cancer cells with MDSCs resulted in a statistically significant increase in Py230 cell migration distance and velocity as compared to monocultures of Py230 cells (Figure 2 d, e, ^p<0.05). However, co-cultures of Py8119 cells with MDSCs on aligned surfaces resulted in no significant differences in migration distance or velocity (Figure 2 d, e). Despite a significant gain in velocity by Py230 cells in the presence of MDSCs, this increase was not sufficient to surpass the mesenchymal phenotype Py8119 velocity (both in monoculture and co-culture, *p<0.001). Moreover, we noticed that the Euclidean distance covered by both Py230 and Py8119 cells, did not show any significant change in co-culture with MDSCs despite the increase in cell velocity (Figure 2 f). This indicates that while the Py230 cells on aligned surfaces attain higher motility in the presence of MDSCs, in a polydispersed co-culture, there is no net gain in directionality. In fact, the mesenchymal Py8119 cells exhibit a statistically significant drop in the directionality of cell migration (^p<0.05) in polydispersed co-cultures with MDSCs (Figure 2 g), which could be due to the presence of conflicting directionality cues that are impacting the migrational persistence of cancer cells.
To further corroborate the changes observed in the migratory behavior of mouse cancer cells, especially epithelial cancer cells, migration studies on aligned matrices were carried out with a human mammary epithelial cell line (MCF10A) in the presence of patient-derived MDSCs. Consistent with results in Py230 cells, MCF10A cells, which typically do not exhibit an inherently migratory phenotype, displayed a significant increase in motility when co-cultured with patient-derived MDSCs (Figure S1). Specifically, we observed a statistically significant increase in accumulated distance from 203.58 (IQR 153.77–346.73) μm to 374.41 (IQR 280.27–546.08) μm when MCF10As were co-cultured with patient derived MDSCs (Figure S1 a, p<0.001). Similarly, we also observed a statistically significant increase in velocity from to 0.32 (IQR 0.24–0.55) μm/min to 0.59 (IQR 0.44– 0.86) μm/min (Figure S1 b, p<0.001), Euclidean distance from 18.47 (IQR 6.82–33.87) μm to 83.58 (IQR 26.77–177.40) μm (Figure S1 c, p<0.001) and directionality from 0.084 (0.038–0.15) to 0.19 (IQR 0.08–0.43) (Figure S1 d, p<0.001). These results emphasize a potential role for human MDSCs in driving cancer cell migration. However, further studies with MDSCs from multiple patients are needed to fully evaluate the extent of these outcomes.
2.3. MDSCs drive directional migration in both epithelial and mesenchymal tumor cells, and enhanced motility in epithelial tumor cells, independent of direct MDSC-cancer cell contact.
Once we established the influence of MDSCs on single-clone velocity and displacement in polydispersed co-cultures with cancer cells, we proceeded to investigate whether MDSCs can also trigger directional migration in compartmentalized co-cultures. For these studies, cancer cells and/or MDSCs were plated in independent compartments of an Ibidi insert (ibidi® GmbH, Munich, Germany) separated by a 500 μm gap on plain TCPS substrates. Plain TCPS substrates were used instead of patterned PDMS to allow for the study of MDSC-driven migration directionality in the absence of directional structural cues from the substrate. For monocultures, the two compartments were plated with the same cell type, while for co-culture; one side was seeded with cancer cells and the other side with MDSCs (Figure 3 a, b). Cancer cell migration towards the 500μm gap was recorded over 24 hours (Supplementary Videos S1–S4). To evaluate if changes in cell migration were due to the presence of chemotactic cues emanating from the MDSCs, the migration parameters in the horizontal/MDSC (x-direction) direction were quantified. Unlike the observation for polydispersed co-cultures on TCPS, the compartmentalized co-culture on TCPS resulted in a statistically significant increase in both single-clone velocity [from 0.15 (IQR 0.13–0.18) μm/min to 0.22 (IQR 0.18–0.27) μm/min] (p<0.001) and accumulated distance [from 214.84 (IQR 180.32– 258.07) μm to 313.87 (IQR 251.94 – 386.29) μm] (p<0.001) for the epithelial Py230 cell population, in the horizontal/MDSC direction (Figure 3 c, e). It is important to note that this MDSC driven increase in Py230 migration occurred even in the absence of an aligned textured surface, suggesting that directional cues, not only in the form of structural patterns but also in the form of chemotactic gradients possibly associated with MDSC-derived soluble factors, can drive cancer cell migration. However, for the mesenchymal Py8119 cell population, no changes in accumulated distance or migration velocity were detected in the compartmentalized co-culture on TCPS (Figure 3 c, e). Nevertheless, we found a significant increase in the Euclidean distance for both Py230 [172.26 (IQR 142.10–210.16) μm to 284.19 (IQR 225.65– 342.42) μm] (*p<0.001) and Py8119 cells [109.36 (IQR 60.48– 152.74) μm to 132.90 (IQR 83.55–198.39) μm] (^p<0.005) (Figure 3 d). Moreover, this also resulted in a significant increase in the directionality of migration towards the MDSCs for both cancer cell types (Figure 3 f). These results suggest that MDSCs can promote driving directional cancer cell migration away from the tumor margin, and as such, future in vivo studies are warranted to evaluate the extent to which circulating, tumor-, or tissue-resident MDSCs modulate cancer cell dissemination and metastasis.
Figure 3. Cancer cell migration in MDSC–PyMT cancer cell compartmentalized co-culture.
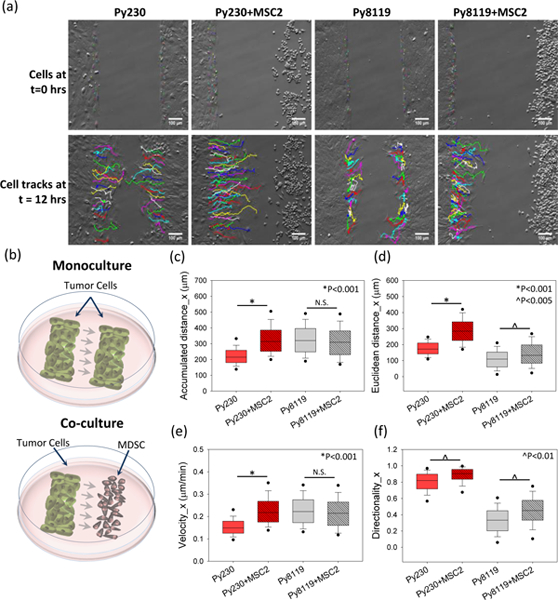
Mouse cancer cells (PyMT, mesenchymal and epithelial) with mouse MDSCs (MSC2) in compartmentalized co-culture as shown in (b)schematic setup at time t=0. (a) Individual cell tracks at time 0 and 12 hours for cancer cells in monoculture and co-culture conditions. A significant increase in (c) accumulated distance, (d) Euclidean distance, (e) velocity and (f) directionality is observed for Py230 cells under co-culture conditions compared to monoculture. The mesenchymal (Py8119) cells show increased (d) Euclidean distance and (f) directionality, but do not show a change in the (c) accumulated distance and (e) velocity. One Way ANOVA on ranks was used to establish p-value. (n=3 samples, 60–100 cells per group).
2.4. MDSC migration patterns are reciprocally impacted by the presence of cancer cells.
In addition to investigating the role MDSCs play in the modulating cancer cell migration at the single-clone level, we also evaluated how the presence of Py230 or Py8119 cancer cells influence MDSC motility under polydispersed and compartmentalized co-culture settings (Figure 4 a, b). Single-cell tracking analysis revealed that on aligned textured surfaces MDSCs show a significant increase in accumulated distance and velocity in the presence of epithelial Py230 cells in polydispersed co-culture configurations (*p<0.001) but no statistically significant changes in any MDSC migration parameter were observed in the presence of mesenchymal Py8119 cells (Figure 4 c-d). MDSCs also showed a significant increase in Euclidean distance under polydispersed co-cultures with epithelial Py230 cells on aligned textured surfaces (Figure 4 d, *p<0.001). The presence of epithelial Py230 cells also led to a significant increase in MDSC accumulated distance in the direction of cancer cells under compartmentalized co-culture conditions (Figure 4 e, ^p<0.05). Interestingly, the Euclidean distance for MDSCs were negatively impacted under compartmentalized co-culture conditions (Figure 4 f, *p<0.001) in the presence of Py8119 cells. This suggests an increase in short-ranged, back and forth motions in the MDSC population, presumably driven by cues derived from the Py8119 cell monolayers.
Figure 4. MDSC cell migration in MDSC–PyMT cancer cell polydispersed on patterned PDMS co-culture and compartmentalized co-culture.
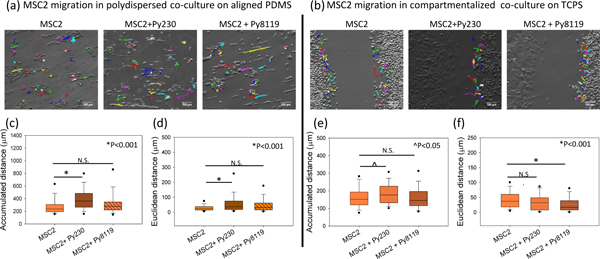
MSC2 cell migration tracks at 12 hours under (a) poly-dispersed co-culture on textured PDMS, and (b) compartmentalized co-culture on TCPS. In polydispersed culture, the presence of Py230 cells leads to a significant increase in (c) accumulated distance and (d) Euclidean distance, while no change is observed in the presence of Py8119 cells. In the compartmentalized configuration, a significant increase in (e) accumulated distance is noted for MSC2 cells in the presence of Py230 cells, but not with Py8119 cells. (f) The Euclidean distance of MSC2 cells drops significantly in the presence of Py8119 cells. One Way ANOVA on ranks was used to establish p-value. (n=3 samples, 60–100 cells per group), scale bar =100μm.
2.5. Enhanced cancer cell motility is driven by soluble factors derived from the interaction between MDSCs and cancer cells, while enhanced MDSC migration is primarily driven by tumor cell-secreted factors.
Our observations with the co-culture migration studies indicate a reciprocal interaction between PyMT cells and MDSCs, which modulate the migratory behavior of both MDSC and cancer cells. To probe the mechanisms of this interaction, we investigated the role of secreted factors in driving the cancer cell and MDSC migration. First, PyMT cells were cultured on the aligned textured surface and treated with conditioned media derived from MDSCs cultured independently on an aligned patterned surface (Figure 5 a). Unlike polydispersed co-culture conditions, single-clone migration velocities for epithelial Py230 cells and mesenchymal Py8119 cells treated with MDSC conditioned media were significantly lower than that of the control cells (Figure 5 b). This reduction in migration velocities for the cancerous cells is in stark contrast to their increased motility when co-cultured with MDSCs (Figure 2) and suggests that the MDSC secretome alone may not be the primary driver of enhanced cancer cell motility under co-culture conditions. To evaluate whether MDSC-cancer cell interactions are needed to drive a pro-migratory secretome, we employed conditioned media derived from the co-culture of PyMT and MDSC cells and used it to study cancer cell migration on aligned textured surfaces (Figure 5 c). The results indicate that conditioned media derived from the PyMT-MDSC co-cultures promoted a dramatic increase in cancer cell migration velocities for epithelial Py230 cells, but not for mesenchymal Py8119 cells, closely resembling the results from polydispersed co-cultures on aligned matrices. These observations confirm the significance of the interaction between MDSCs and tumor cells in order to drive a pro-migratory behavior in cancer cells.
Figure 5. Cancer and MDSC cell migration in the presence of monoculture or co-culture derived conditioned media.
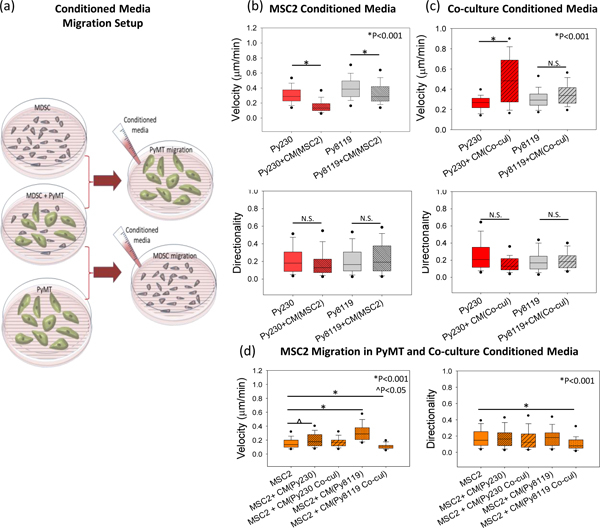
(a) Schematic setup for cell migration for mesenchymal (Py8119) and epithelial (Py230) cancer cells or mouse MDSCs (MSC2) on patterned substrates in the presence of conditioned media derived from a monoculture (MDSCs for PyMT migration and vice-versa) or a co-culture of MDSCs with the respective cancer cell (Py230 or Py8119). (b) Cell velocity drops significantly for both Py230 cells and Py8119 cells in the presence of MDSC monoculture-derived conditioned media, with no change in directionality. (c) In the presence of co-culture derived conditioned media, cell velocity increases for Py230 cells, while no statistically significant difference is noticed in Py8119 cell velocity and directionality of both cell types. (d) Cell velocity for the MDSCs show an increase in the presence of PyMT-derived conditioned media but not in the presence of co-culture derived conditioned media. Directionality does not change except a drop in the presence of co-culture conditioned media. Statistical significance was established by one-way ANOVA on ranks. (n=3 samples, 60–100 cells per group).
Similarly, in order to understand the driving force behind the enhanced migration of MDSCs in co-culture with cancer cells, we exposed MDSCs to conditioned media derived from either monoculture of PyMT cells, or from the co-culture of PyMT and MDSCs. Our results indicate that MDSCs showed enhanced migration velocities in the presence of both Py230 and Py8119 monoculture-derived conditioned media but not in the presence of co-culture conditioned media (Figure 5 d, ^p<0.05 and *p<0.001 respectively). No significant differences in the cell directionality were observed except a drop in the directionality when treated with Py8119 co-culture conditioned media. While further studies are needed to fully elucidate the composition and impact of the MDSC, PyMT and MDSC/cancer cell secretome on cell motility, these results indicate the presence of complex and dynamic interactions between the two cell types that affect their secretomes and eventually cell migration.
2.6. Epithelial dedifferentiation underlies increased cancer cell motility in response to MDSCs.
The changes in the migratory phenotype of the epithelial cancer cells are indicative of metastatic transformation. As discussed in prior studies, these changes in migration point to the possibility of epithelial dedifferentiation in cancerous cells.21, 22 E-cadherin is a calcium dependent adhesion protein that mediates tight homotypic interactions between epithelial cells. Loss of E-cadherin in cancer cells is associated with epithelial dedifferentiation, which is conducive to increased cancer cell motility and metastasis47. To evaluate if increased cancer cell motility is partly driven by epithelial dedifferentiation, flow cytometry, immunofluorescence and qRT-PCR were used to document changes in E-cadherin expression in response to the presence of MDSCs (Figure 6). Our flow cytometry results indicate that ~98% of Py230 cells in monoculture were positive for E-cadherin (Figure 6 a, d). However, when co-cultured with MDSCs, only ~75% of the cancer cell population (i.e., negative for the myeloid CD11b marker) was positive for E-cadherin (Figure 6 b, d). Furthermore, the PCR data showed a ~10 fold drop in the E-cadherin gene expression levels in Py230 cells in the presence of MDSCs (Figure 6 f). These observations were also corroborated with the lower intensity of E-cadherin staining for Py230 cells in co-culture with MSC2s (Figure 6 e). This confirms that in the presence of MDSCs, changes in the motility patterns of epithelial Py230 cells are presumably driven to some extent by epithelial dedifferentiation.
Figure 6. E-cadherin and TGF-β expression levels of Py230 (epithelial) cancer cells in monoculture or co-culture with MDSC as a marker of EMT.
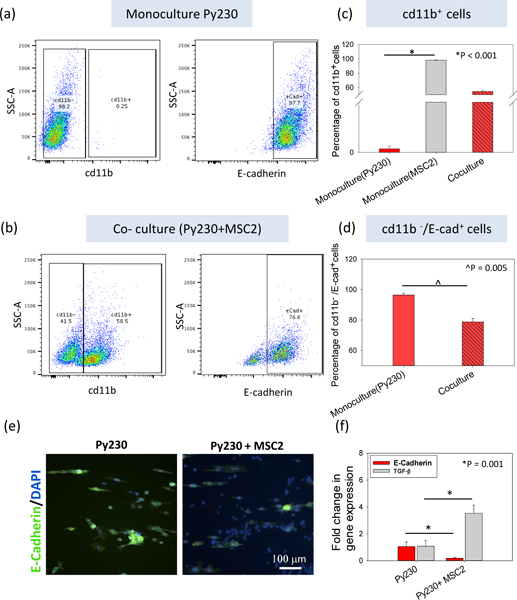
(a) A flow cytometry analysis of a Py230 cell monoculture and (b) co-culture with MSC2 cells was performed to measure the expression of proteins involved in EMT, namely E-cadherin. CD11b was used as the marker for the MSC2 cells (myeloid origin). (c) For Py230 monoculture, CD11b+ cells were virtually absent, while in co-culture, ~50% of the cells were CD11b+. (d) Among the CD11b- cell population in the co-culture, ~75% of the cells are E-cadherin+ compared to ~98% under monoculture. (e) Immunofluorescent staining and (f) PCR for E-cadherin also show a decrease in expression in the presence of MDSCs. (f) Gene expression of TGF-β shows an increase in the presence of MDSCs. One Way ANOVA and t-test were used to establish statistical significance. (n=3 samples).
Prior studies on morphological changes in the epithelial cells have indicated that transforming growth factor –beta (TGF-β) is one of the major factors driving the process.48 Moreover, it is also established that epithelial cells can undergo the transformation process via autocrine production of TGF-β. 49 We found that co-culture of Py230 (epithelial) cancer cells with MDSCs lead to significantly higher gene expression for TGF-β (~3.5 fold, Figure 6f,*p<0.001) in the Py230 cells. This increase indicates that TGF-β driven epithelial dedifferentiation could be one of the possible signaling pathways responsible for increased migration of cancer cells in the presence of MDSCs. However, further studies are needed to do a comprehensive characterization of the cytokine profiles in MDSC- cancer cell interaction.
3. Discussion
Cell migration is an integral part of cancer progression and metastasis. However, the mechanisms and drivers of cancer cell migration remain elusive. The dynamic nature of cancer cells is driven by both intrinsic genotypic/phenotypic changes, and cues from the surrounding microenvironment. The interaction of cancer cells with various components of the TME is critical to disease progression and is driven by feedback loop signaling between cancerous cells and other cellular components. Immune cells recruited to TME are critical components of these reciprocal interactions where instead of destroying the tumor cells, the cancer associated immune cells provide protection from immune surveillance. MDSCs, a heterogeneous immature cell population of myeloid origin, have garnered significant attention within context of cancer immuno-suppression. While the role of MDSCs in cancer is mostly studied in terms of surveillance or evasion, more recent studies have highlighted the complexity of their role within the TME. Studies by Toh et al, for example, indicated that the MDSCs drive cancer metastasis through EMT.21 In this study, we furthered this knowledge with findings that support the role of MDSC-cancer cell interactions in driving both MDSC and cancer cell motility, at the single-clone level. In addition to exploring changes cancer cell motility when exposed to MDSCs or conditioned media, this study also provides a more thorough perspective of the reciprocal signaling between the two cell types as a function of their spatial disposition and the structural cues in the TME. Most prior in vitro studies on the interaction of MDSCs and cancer cells, to the best of our knowledge, do not include the role of structural cues from the ECM in driving the behavior of and interaction between the two cell types. Results from this study demonstrate that while MDSCs can augment epithelial cancer cell motility, these effects require the presence of a directional cue such as alignment of the underlying substrate or compartmentalized positioning of the two cell types (Figure 1–3). Moreover, either of those two factors, aligned surface cues or compartmentalized cell positioning, is independently sufficient to drive the interactive migratory behavior between MDSCs and tumor cells. These results highlight the multitude and interdependence of not only cellular but also structural components in the TME that drive the evolution and progression of the tumor. Interestingly, unlike previous studies, this work draws contrast between the interactions of epithelial versus mesenchymal cancer cells with MDSCs, wherein epithelial cells are far more responsive to the presence of MDSCs in terms of motility (Figure 2, 3). While the mesenchymal cells maintain a migratory dominance over epithelial cells in all monoculture and co-culture settings, the epithelial cells show significantly enhanced motility in the presence of MDSCs. We documented these enhanced migratory epithelial phenotypes not only in mouse cells but also with established human breast epithelial cell lines (MCF10A) co-cultured with patient-derived MDSCs (Figure S1). The differences in response of the epithelial versus mesenchymal phenotype indicate the active role of MDSCs in driving a relatively quiescent cell type towards a more aggressive behavior, while leaving the already aggressive phenotype “unaltered”. Furthermore, while it is well-known that tumor cells secrete cytokines to recruit MDSCs to the TME, our results with compartmentalized co-cultures indicate that MDSCs may also provide directional chemotactic signaling to drive cancer cell migration towards MDSCs (Figure 3). Cancer cells not only migrate faster but also follow a consistent path towards the MDSCs in compartmentalized culture. These findings align well with earlier studies demonstrating the role of MDSCs in establishing a pre-metastatic niche where accumulation of MDSCs precedes and leads to formation of secondary tumors.50 Furthermore, the results from the conditioned media experiments (Figure 5) suggest that the secretome resulting from MDSC-cancer cell interactions can drive cancer cell motility remotely, potentially independent of their physical location in the body. In addition, it is interesting to note that we observed a decreased cancer cell motility when exposed to MDSC monoculture-derived conditioned media (Figure 5 b). One potential explanation for this observation is that MDSCs in the absence of cancer cells or any other stimulating factor (e.g., nitric oxide, LPS) are in an inactive state,42, 43, 51 which could presumably favor a secretome that is more prone to hindering cancer cell motility. Future studies are warranted to comprehensively examine differences in the MDSC secretome under inactive vs. active conditions within the context of cancer cell motility.
In addition to the migratory phenotype of the cancer cells, this work also demonstrates reciprocity from the MDSC population. Specifically, in the presence of aligned/structural or positional cues, MDSCs show enhanced migration in response to the presence of cancer epithelial cells (Figure 4). Furthermore, our results indicate that the interactions between the two cell types are mediated by soluble byproducts, and do not necessarily require physical cell-cell interactions (Figure 5). These results are consistent with prior reports on the reciprocal signaling between MDSCs and cancer cells, and indicate that not only do MDSCs play a role in cancer metastasis, but the signaling from the tumor is instrumental in mobilizing and recruiting MDSCs to the TME.23, 52 However, it is important to note that while the effects are independent of cell-cell contact, the feedback signaling between cancer cells and MDSCs induced through co-culture is critical for these effects to unfold. Specifically, we found that the MDSC secretome alone is not sufficient to drive the enhanced cancer cell migration while the secreted soluble factors derived from MDSC-cancer cell co-cultures can drive enhanced cancer cell migration (Figure 5 b,c). Interestingly, we found that the secretome from cancer cells alone is sufficient to drive enhanced MDSC migration (Figure 5 d). In conjunction with prior work, our studies confirm that epithelial dedifferentiation likely underlies enhanced cancer migration in response to MDSCs, and that TGF-β signaling may be a critical mechanism that mediates this interaction (Figure 6).21 Although, these studies provide firm groundwork establishing the role of MDSCs in cancer cell migration, we recognize that additional studies with primary MDSCs from normal and cancer patients, and/or three-dimensional models of the TME, are needed to further improve our understanding of the cross-talk between these cell populations, and the role it plays in disease progression or relapse. We also note that the goal of these studies was to understand how reciprocal signaling between MDSCs and cancer cells influenced cell motility on a variety of substrates that mimic certain aspects of the TME (aligned matrix and tumor margin). However, we note that there are important differences between TCPS and PDMS surfaces and that these studies were not designed to compare the behavior of cancer cells and MDSCs on these different substrates.
4. Conclusions:
Overall, this study demonstrates that MDSCs, in conjunction with the structural and directional cues from the microenvironment, play a significant role in driving enhanced cancer cell migration and dissemination. Our results provide further evidence that the complex feedback signaling between the two cell types could be driving enhanced cellular motility that could potentially be conducive to changes in the in vivo recruitment as well as dissemination patterns of MDSCs and cancerous cells, respectively. Altogether, micro/nanoscale tools enable single-cell level interrogation that not only add to our understanding of the role MDSCs play beyond immunosuppression, but may also open up new avenues of research for the development of targeted therapies aimed at countering potentially pro-metastatic MDSC-cancer cell interactions.
5. Experimental Section
5.1. Substrate fabrication:
Polydimethylsiloxane (PDMS) substrates with linear patterns of 2μm width and 1.5μm height were fabricated using a replica molding process from a silicon master. The master silicon wafer was fabricated using photolithography with Shipley 1813 photoresist. Sylgard 184™ silicone elastomer was mixed with a curing agent at a 10:1 ratio. The mixture was spin coated on the master wafer followed by degassing and cured at 60◦C for 2 hours. The patterned PDMS sheets were then demolded and sterilized with ethanol prior to cell culture.
5.2. Cell culture:
The mouse mammary cancer cell lines used in this study were derived from MMTV-PyMT transgene-induced mammary tumors in C57BL/6 mice (ATCC, Manassas, VA). The two cell lines we used, Py8119 and Py230, were derived from the same tumor model but have distinct mesenchymal (Py8119) or epithelial-like (Py230) features.44 The cells were kept in culture with F-12/Kaighn’s medium supplemented with 5% fetal bovine serum (FBS) and 0.1% MITO+ Serum Extender (Corning®). The murine MDSC cell line, MSC-2 (gift from Gregoire Mignot), was cultured in RPMI medium (Gibco™, Dublin,IE) containing 10% FBS and 1% sodium pyruvate. All the cells were maintained at 37◦C, 5% CO2 and 95% humidity. For human cell line studies, the human breast epithelial cell line MCF10A was used and these cells were kept in culture in MEGM media kit (Lonza Corporation) further supplemented with 100ng/ml cholera toxin. Primary human MDSCs were derived from peripheral blood from cancer patients with informed consent under IRB-approved protocols for human subject research (IRB protocols 1999C0348 and 2017C0054). RosetteSep HLA-myeloid cell enrichment kit (Stemcell Technologies) followed by Ficoll-Paque centrifugation (GE healthcare) were used for processing of blood samples. MDSC were isolated by subsequent negative selection of HLA-DRneg cells using anti-HLA-DR MicroBeads (Miltenyi Biotec) for 15 minutes at 4◦C and isolated using a MS-MACS column.
5.3. Cell migration:
Cell migration studies were performed using time-lapse live cell microscopy with an Okolab™ (OKOLAB S.R.L. NA, Italy) stage-top incubator installed on an Olympus IX81 inverted widefield microscope (Olympus Corporation, Tokyo, Japan). For poly-dispersed co-culture studies, cancer cells labeled with a live cell stain DiI/Dio (Vybrant™ Multicolor Cell-Labeling Kit, Invitrogen, Carlsbad, CA) and MSC2 cells (1:1) were seeded on either flat TCPS or patterned PDMS substrates, and allowed to adhere for 24 hours. This ratio was selected based on previous reports.53–55 For co-culture experiments, the cells were seeded in the cancer cell culture medium. Cells were then imaged over 24 hours at 10-minute intervals. For spatially separated migration, the cells were seeded into an ibidi insert (ibidi® GmbH, Munich, Germany)) on TCPS at 103 cells/cm2 to create two monolayers separated by a ~500 μm gap. Once the cells adhered, the insert was removed and the cells were imaged over 24 hours, as described above. To quantify migration parameters, 60–100 individual cells per sample were tracked (i.e., the central nucleous region) on the collected image series using the Manual Tracking ImageJ plugin (NIH, Bethesda, MD). In case of cell division during the tracking, only one of the daughter cells from the division was randomly selected to complete the track. Image tracks were subsequently analyzed using a Matlab™ script to compute migration parameters for individual cells as described in prior work.30 Briefly, for each cell
Where, is the position of the cell at time point, is the time interval between consecutive time points and is the total number of time points.
To assess the role of cell-secreted factors on cell migration, conditioned media was collected for MSC2 cells alone, cancer cells alone, or co-culture of MSC2 and cancer cells. The culture medium was collected and centrifuged to remove the cell debris. The supernatant was then added to the cancer cell or MSC2 migration samples, and the migration assay was performed as described above.
5.4. Flow cytometry:
Py230 cells were cultured either alone or in co-culture with MSC2 cells for 24 hours under normal conditions. The cells were then trypsinized and re-suspended in PBS at concentration of 1×106 cells/ml. Cells were then labeled with PE-conjugated E-cadherin antibody and APC-conjugated Anti-CD11b antibody (Biolegend, San-Diego,CA ) as per the manufacturer’s recommendation. The cells were then incubated at 4°C for 30 minutes in the dark. After washing the cells, the fluorescence was read/processed using a Becton Dickinson FACSAria III.
5.5. Gene expression analyses.
Total RNA was extracted using the TRizol reagent (ThermoFisher). Reverse transcription reactions were performed using 500–1000 ng RNA in a 20 μl reaction with the superscript VILO cDNA synthesis kit (ThermoFisher). cDNA was used as a template to measure the expression levels of pro- and anti-inflammatory genes by quantitative real-time PCR using predesigned primers. Real-time PCR reactions were performed using the QuantStudio 3 Real-Time PCR System with TaqMan fast advance chemistry (Thermo Scientific) with the following conditions: 95 °C 10 min, 40 cycles of 95 °C 1 min, 60 °C 1 min, and 72 °C 1 min. Gene expression for TGF-β and E-cadherin (Cdh1) was normalized against the house keeping genes GAPDH and ATP-6.
5.6. Immunofluorescence.
Py230 cells grown on 24 well plates either in monoculture or in co-culture with MSC2 cells. After 24 hours in culture, cells were fixed for 15 min with 10 % formalin (ThermoFisher) at room temperature, washed twice with PBS for 5 min, and permeabilized for 5 min with 0.1% Triton X-100 (in PBS). For immunofluorescence labeling, cells were blocked for 1.5 hours with 2.5 % NGS (S-1000–20, Vector laboratories). Primary antibody E-Cadherin (131900, ThermoFisher) in 2.5% NGS was incubated overnight at 4°C. Antibody binding was visualized by fluorochrome-conjugated secondary antibodies. Nucleic were stained with DAPI (1:5000). All samples were imaged using fluorescent light microscope Nikon Ti-2e.
5.7. Statistical analysis:
Statistical analysis or all the data was done using SigmaPlot (Systat Software, Inc.). For the cell migration studies, 60–100 cells were tracked for each experimental group (combined n=3 samples). Normality of the data distribution was tested using Shapiro-Wilk test. The non-normally distributed data were analyzed using one-way analysis of variance (ANOVA) on ranks and the data are reported as median with intequartile range (IQR). The non-normally distributed data are graphed as box and whisker plots with the median and 5th and 95th percentile points. Statistical significance was accepted at p≤0.05. Pair-wise multiple comparisons were performed using Dunn’s method to document statistically significant differences between groups. For normally distributed data, one-way analysis of variance (ANOVA) was used to establish statistical significance and pairwise post-hoc analysis was done using Holm-Sidak method and data is presented as mean ± standard error. For normally distributed data pair comparisons in flow cytometry and PCR data, two tailed p values were established using Student’s t-test (equal variance).
Supplementary Material
Acknowledgements:
Funding for DGP was partly provided by NINDS/NIH (R21NS099869), NIBIB/NIH (DP2EB028110). Funding for SDS was partly provided by AHA (18PRE34070054) and Colciencias. The MSC-2 cells were a kind donation from Gregoire Mignot to the lab of William E. Carson (OSU).
Contributor Information
Dr. Vasudha C. Shukla, Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University Wexner Medical Center, Columbus, OH 43210 USA Department of Biomedical Engineering, The Ohio State University, Columbus, OH 43210 USA.
Silvia Duarte-Sanmiguel, Department of Biomedical Engineering, OSU Nutrition, The Ohio State University, Columbus, OH, 43210, USA.
Ana Panic, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA.
Abirami Senthilvelan, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA.
Jordan Moore, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA.
Dr. Christopher Bobba, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA
Brooke Benner, Biomedical Sciences Graduate Program, The Ohio State University, Columbus, 43210, USA.
Prof. William E. Carson, III, Department of Surgery, Comprehensive Cancer Center, The Ohio State University Wexner Medical Center, Columbus, OH, 43210, USA.
Prof. Samir N. Ghadiali, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA; Dorothy M. Davis Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA.
Dr. Daniel Gallego-Perez, Department of Biomedical Engineering, The Ohio State University, Columbus, OH, 43210, USA; Dorothy M. Davis Heart and lung Research Institute, Department of Surgery, The Ohio State Wexner Medical Center, Columbus, OH, 43210, USA.
References
- 1.Quail DF and Joyce JA, Nature Medicine, 2013, 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Dijk M, Goransson SA and Stromblad S, Exp Cell Res, 2013, 319, 1663–1670. [DOI] [PubMed] [Google Scholar]
- 3.Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, Coussens LM and DeClerck YA, Cancer Research, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C. and Marini FC, in Breast Cancer Res, 2016, vol. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prendergast GC, Oncogene, 2008, 27, 3889–3900. [DOI] [PubMed] [Google Scholar]
- 6.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara H, Signori E, Honoki K, Georgakilas AG, Amin A, Helferich WG, Boosani CS, Guha G, Ciriolo MR, Chen S, Mohammed SI, Azmi AS, Keith WN, Bilsland A, Bhakta D, Halicka D, Fujii H, Aquilano K, Ashraf SS, Nowsheen S, Yang X, Choi BK and Kwon BS, Semin Cancer Biol, 2015, 35 Suppl, S185–s198. [DOI] [PubMed] [Google Scholar]
- 7.Umansky V, Blattner C, Gebhardt C. and Utikal J, in Vaccines (Basel), 2016, vol. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millrud CR, Bergenfelz C. and Leandersson K, Oncotarget, 2017, 8, 3649–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI and Nagaraj S, Nat Rev Immunol, 2009, 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP and Gabrilovich DI, J Immunol, 2001, 166, 678–689. [DOI] [PubMed] [Google Scholar]
- 11.Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, Zanon A, Rossi CR, Nitti D, Bronte V. and Zanovello P, J Immunol, 2009, 182, 6562–6568. [DOI] [PubMed] [Google Scholar]
- 12.Ochoa AC, Zea AH, Hernandez C. and Rodriguez PC, Clin Cancer Res, 2007, 13, 721s–726s. [DOI] [PubMed] [Google Scholar]
- 13.Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, Gillanders WE, Linehan DC and Goedegebuure P, Cancer Immunol Immunother, 2012, 61, 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Chang EW, Wong SC, Ong SM, Chong DQ and Ling KL, J Immunol, 2013, 190, 794–804. [DOI] [PubMed] [Google Scholar]
- 15.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ and Montero AJ, Cancer Immunol Immunother, 2009, 58, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Younos IH, Dafferner AJ, Gulen D, Britton HC and Talmadge JE, Int Immunopharmacol, 2012, 13, 245–256. [DOI] [PubMed] [Google Scholar]
- 17.Donkor MK, Lahue E, Hoke TA, Shafer LR, Coskun U, Solheim JC, Gulen D, Bishay J. and Talmadge JE, Int Immunopharmacol, 2009, 9, 937–948. [DOI] [PubMed] [Google Scholar]
- 18.Ai L, Mu S, Wang Y, Wang H, Cai L, Li W. and Hu Y, BMC Cancer, 2018, 18, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, Hubert A. and Baniyash M, Cancer Res, 2014, 74, 6022–6035. [DOI] [PubMed] [Google Scholar]
- 20.Califano JA, Khan Z, Noonan KA, Rudraraju L, Zhang Z, Wang H, Goodman S, Gourin CG, Ha PK, Fakhry C, Saunders J, Levine M, Tang M, Neuner G, Richmon JD, Blanco R, Agrawal N, Koch WM, Marur S, Weed DT, Serafini P. and Borrello I, Clin Cancer Res, 2015, 21, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, Kato M, Prevost-Blondel A, Thiery JP and Abastado JP, PLoS Biol, 2011, 9, e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Gu Y, Xue Y, Yuan M, Cao X. and Liu Q, in Oncotarget, 2017, vol. 8, pp. 114554–114567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh K, Lee O-Y, Shon SY, Nam O, Ryu PM, Seo MW and Lee D-S, Breast Cancer Research, 2013, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandau S, Moses K. and Lang S, Semin Cancer Biol, 2013, 23, 171–182. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC and Moses HL, Cancer Cell, 2008, 13, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, Aoki M, Oshima M, Hattori M, Takabayashi A, Minato N. and Taketo MM, Nat Genet, 2007, 39, 467–475. [DOI] [PubMed] [Google Scholar]
- 27.Thiery JP, Journal, 2003, 15. [Google Scholar]
- 28.Huber MA, Kraut N. and Beug H, Curr Opin Cell Biol, 2005, 17, 548–558. [DOI] [PubMed] [Google Scholar]
- 29.Kaushik N, Kim S, Suh Y. and Lee SJ, Arch Pharm Res, 2019, 42, 40–47. [DOI] [PubMed] [Google Scholar]
- 30.Shukla VC, Higuita-Castro N, Nana-Sinkam P. and Ghadiali SN, J Biomed Mater Res A, 2016, 104, 1182–1193. [DOI] [PubMed] [Google Scholar]
- 31.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL and Weaver VM, Cell, 2009, 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silver FH, Freeman JW and Seehra GP, J Biomech, 2003, 36, 1529–1553. [DOI] [PubMed] [Google Scholar]
- 33.Ushiki T, Arch Histol Cytol, 2002, 65, 109–126. [DOI] [PubMed] [Google Scholar]
- 34.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES and Weaver VM, Integr Biol (Camb), 2015, 7, 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallego-Perez D, Chang L, Shi J, Ma J, Kim SH, Zhao X, Malkoc V, Wang X, Minata M, Kwak KJ, Wu Y, Lafyatis GP, Lu W, Hansford DJ, Nakano I. and Lee LJ, Nano Lett, 2016, 16, 5326–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallego-Perez D, Higuita-Castro N, Denning L, DeJesus J, Dahl K, Sarkar A. and Hansford DJ, Lab Chip, 2012, 12, 4424–4432. [DOI] [PubMed] [Google Scholar]
- 37.Gu SQ, Gallego-Perez D, McClory SP, Shi J, Han J, Lee LJ and Schoenberg DR, Nucleic Acids Res, 2016, 44, 5811–5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S-H, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H, Chhipa R, Cho Ju H., Mohyeldin A, Beck S, Kurozumi K, Kuroiwa T, Iwata R, Asai A, Kim J, Sulman Erik P., Cheng S-Y, Lee LJ, Nakada M, Guttridge D, DasGupta B, Goidts V, Bhat Krishna P. and Nakano I, Cancer Cell, 2016, 29, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones CE, Hammer AM, Cho Y, Sizemore GM, Cukierman E, Yee LD, Ghadiali SN, Ostrowski MC and Leight JL, Neoplasia, 2019, 21, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duarte-Sanmiguel S, Shukla V, Benner B, Moore J, Lemmerman L, Lawrence W, Panic A, Wang S, Idzkowski N, Guio-Vega G, Higuita-Castro N, Ghadiali S, Carson WE and Gallego-Perez D, Scientific Reports, 2020, 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG and Keely PJ, BMC Med, 2006, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, Young HA and Zanovello P, J Immunol, 2000, 165, 6723–6730. [DOI] [PubMed] [Google Scholar]
- 43.Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael E, Duggan M, Campbell A, Keller K, Landi I, Zhong Y, Dubovsky J, Howard JH, Yu L, Harrington B, Old M, Reiff S, Mace T, Tridandapani S, Muthusamy N, Caligiuri MA, Byrd JC and Carson WE 3rd, Cancer Res, 2016, 76, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biswas T, Gu X, Yang J, Ellies LG and Sun LZ, Cancer Lett, 2014, 346, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meade KJ, Sanchez F, Aguayo A, Nadales N, Hamalian SG, Uhlendorf TL, Banner LR and Kelber JA, Oncotarget, 2019, 10, 3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bao L, Cardiff RD, Steinbach P, Messer KS and Ellies LG, Breast Cancer Res, 2015, 17, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES and Weinberg RA, Cancer Res, 2008, 68, 3645–3654. [DOI] [PubMed] [Google Scholar]
- 48.Hao Y, Baker D. and ten Dijke P, in Int J Mol Sci, 2019, vol. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alcaraz A, Mrowiec A, Insausti CL, Garcia-Vizcaino EM, Ruiz-Canada C, Lopez-Martinez MC, Moraleda JM and Nicolas FJ, Cell Transplant, 2013, 22, 1351–1367. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Ding Y, Guo N. and Wang S, Front Immunol, 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fionda C, Abruzzese MP, Santoni A. and Cippitelli M, Curr Med Chem, 2016, 23, 2618–2636. [DOI] [PubMed] [Google Scholar]
- 52.Condamine T, Mastio J. and Gabrilovich DI, J Leukoc Biol, 2015, 98, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA and Ostrand-Rosenberg S, J Leukoc Biol, 2014, 96, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma X, Wang M, Yin T, Zhao Y. and Wei X, Front Oncol, 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nam S, Lee A, Lim J. and Lim J-S, Biomolecules & therapeutics, 2019, 27, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.