

Abstract
Cryptozona siamensis, one of the most widespread land snails, is native to Thailand, and plays a key role as an agricultural pest and intermediate host for Angiostrongylus spp. However, its genetic diversity and population structure has not yet been investigated, and are poorly understood. Therefore, a genetic analysis of the C. siamensis population in Thailand was conducted, based mitochondrial 16S rRNA (402 bp) and COI (602 bp) gene fragment sequences. Cryptozona siamensis randomly collected from 17 locations in four populations across Thailand, between May 2017 and July 2018. Fifty-eight snails were used to examine the phylogeny, genetic diversity, and genetic structure. The maximum likelihood tree based on the 16S rRNA and COI fragment sequences revealed two main clades. A total of 14 haplotypes with 44 nucleotide variable sites were found in the 16S rRNA sequences, while 14 haplotypes with 57 nucleotide variable sites were found in the COI sequences. The genetic diversity of C. siamensis in term of the number of haplotypes and haplotype diversity, was found to be high but the nucleotide diversity showed low levels of genetic differentiation for the COI sequence as also noted with the 16S rRNA sequence. The population genetic structure of C. siamensis revealed genetic difference in most populations in Thailand. However, low genetic difference in some populations may be due to high gene flow. This study provides novel insights into the basic molecular genetics of C. siamensis.
Introduction
The land snail Cryptozona siamensis, a terrestrial pulmonate gastropod, belongs to the family Ariophantidae [1, 2]. Cryptozona siamensis have been reported as the intermediate host of Angiostrongylus that cause eosinophilic meningitis in human worldwide especially China, Taiwan, and Thailand [3, 4]. Cryptozona siamensis are important as intermediate hosts that promote the endemicity and transmission of Angiostrongylus cantonensis and A. malaysiensis [3, 5, 6]. The distribution of the snail hosts facilitates establishment of the life cycle of the parasite. In addition, the distribution of the land snails accelerates the spread of A. cantonensis [7, 8]. Ingestions of raw or undercooked infected snails, snails accidentally chopped up in vegetables, vegetable juices, salads, or foods contaminated by the slime of infected snails are highly risk factors for infection with A. cantonensis in human [9]. Cryptozona siamensis is native to Thailand, and is regarded as a cosmopolitan species, being one of the most widespread land snails in Southeast Asia [2, 10], with reports of C. siamensis from areas adjacent to Thailand, such as Malaysia, Singapore, and Laos [6, 11, 12]. Cryptozona siamensis has gained attention as an important agricultural and horticultural pest, in India, the United States of America, and Thailand [13, 14].
In the last few years, DNA sequencing data has been used to study and clarify the evolution of morphological characteristics of ambiguous organisms [15–18] while genetic studies of land snails using DNA sequence data could be valuable for their identification [19–21]. The most commonly used genes for genetic analysis in land snails are the mitochondrial cytochrome c oxidase subunit I (COI) and 16S rRNA genes [20, 22–24]. The COI genes have a greater range of phylogenetic information, compared with other mitochondrial genes, and are considered to be robust evolutionary markers for studies of inter-specific relationships [25, 26]. In addition, 16S rRNA gene has a high level of inter-specific polymorphisms. Hence, the 16S rRNA has been widely used in the genetic studies of snails [25, 27]. The genetic structure of the land snails has been studied from several geographical regions. In Hawaii, analysis of COI and 16S rRNA genes by haplotype networks, gene tree topologies, pairwise molecular divergence, and FST matrices revealed the substantial geographic genetic structuring and complex dispersal patterns of the land snail Succinea caduca [28]. In China, Zhou et al. studied population genetic structure of the land snail Camaena cicatricosa from 20 locations using mitochondrial gene (COI and 16S rRNA) and internal transcribed spacer (ITS2) sequences. This showed significant fixation indices of genetic differentiation and high gene flow among most populations [29]. In Thailand, the genetic variation of the COI in Achatina fulica was found to be low [30]. In Langkawi island of Malaysia, low genetic diversity of family Ariophantidae (Cryptozona siamensis and Sarika resplendens) and Dyakiiaea (Quantula striata) was also noted by using 16S rRNA gene [11].
Molecular population phylogeographic studies can offer information on specific genetic variations, population formations, and genetic structure [31]. Moreover, they can also help to identify how a population has been affected by various factors, including the ecological environment, climate, human activities, and geographical conditions [32–34]. However, the genetic diversity and structure of C. siamensis is currently poorly understood, as it has been the focus of only a limited number of studies. The genetic structure of C. siamensis from 3 populations of Thailand and additional one population from Malaysia was studied on allozyme variation on horizontal starch gel electrophoresis [2]. Although the genetic structure of C. siamensis was examined in previous study, no nucleotide sequencing of this land snail is available in the country. Therefore, the objective of this research was to investigate the genetic diversity and genetic structure of C. siamensis from Thailand, based on mitochondrial DNA-sequence variation at the COI and 16S rRNA loci.
Methods
Ethic and biosafety statement
The experimental protocol for the use of animals (snail intermediate host) in this study was approved by the Center for Animal Research of Naresuan University (Project Ethics Approval No: NU-AQ610711). The biosafety protocol was approved by the Naresuan University Institutional Biosafety Committee (Project Approval No: NUIBC MI 61-08-50).
Collection and preliminary identification of the snails
During a survey for A. cantonensis, which uses snails as an intermediate host, C. siamensis specimens were randomly collected from 17 locations across Thailand, between May 2017 and July 2018 (Fig 1). The snail populations (A, B, C, and D) were defined according to the biogeographical regions (Table 1). The snails were located in several types of natural habitats, such as under or above trunks of fallen trees, under stones, in the pot of flower, and in wall crevices. The snails were collected by hand picking and then put in a net with air for transporting to the Department of Microbiology and Parasitology, Faculty of Medical Science, Naresuan University, Phitsanulok, Thailand. All snails were cleaned with tap water and were preliminary identified according to the previously recorded morphological description of C. siamensis [12]. These included having medium sized shell (> 30 mm in shell width) that was two-toned in color, a surface with fine reticulate sculptures a discoidal shell with a low spire, light straw colored ventral part, light brown dorsal part, a fine surface with fine dense axial grooves, and a smooth ventral surface, and is slightly shiny [12]. The body of each C. siamensis specimen was removed from its shell, and approximately 25 mg of foot tissue was removed and preserved at -20°C for subsequent DNA extraction. To detect the Angiostrongylus larvae, the remaining snail tissue was artificially digested with a 0.7% (w/v) pepsin solution, as previously described [3]. Fortunately, all the C. siamensis samples in this study were found to be negative for Angiostrongylus larvae.
Fig 1. Geographical locations of the 4 populations of C. siamensis in Thailand, with the 14 haplotypes based on the 16S rRNA and COI sequences.

Numbers inside the closed and opened circles indicates the haplotype number.
Table 1. Sampling locations of the Cryptozona siamensis collected from across Thailand, with the 14 haplotypes based on the 16S rRNA and COI sequences.
| Population | Province | Latitude | Longitude | Sample Name | 16S rRNA haplotype | COI haplotype |
|---|---|---|---|---|---|---|
| A | Phitsanulok | 16.821123 | 100.265851 | Phitsanulok 1 | 16S12 | CO3 |
| Phitsanulok 2 | 16S12 | CO3 | ||||
| Phitsanulok 3 | 16S12 | CO3 | ||||
| Phitsanulok 4 | 16S6 | CO9 | ||||
| Phetchabun | 16.301669 | 101.119280 | Phetchabun 1 | 16S5 | CO2 | |
| Phetchabun 2 | 16S5 | CO2 | ||||
| Phetchabun 3 | 16S5 | CO5 | ||||
| Phetchabun 4 | 16S5 | CO2 | ||||
| Pathum Thani | 14.020839 | 100.525027 | Pathum Thani 1 | 16S5 | CO2 | |
| Pathum Thani 2 | 16S5 | CO2 | ||||
| Pathum Thani 3 | 16S5 | CO2 | ||||
| Pathum Thani 4 | 16S5 | CO2 | ||||
| Nakhon Pathom | 13.819920 | 100.062167 | Nakhon Pathom 1 | 16S13 | CO10 | |
| Nakhon Pathom 2 | 16S12 | CO12 | ||||
| Nakhon Pathom 3 | 16S12 | CO12 | ||||
| Nakhon Pathom 4 | 16S12 | CO12 | ||||
| Chon Buri | 13.361143 | 100.984671 | Chon Buri 1 | 16S10 | CO7 | |
| Chon Buri 2 | 16S10 | CO7 | ||||
| Chon Buri 3 | 16S10 | CO13 | ||||
| B | Loei | 17.486023 | 101.722300 | Loei 1 | 16S4 | CO1 |
| Loei 2 | 16S4 | CO1 | ||||
| Loei 3 | 16S12 | CO3 | ||||
| Loei 4 | 16S5 | CO2 | ||||
| Nong Bua Lam Phu | 17.221824 | 102.426036 | Nong Bua Lam Phu 1 | 16S4 | CO1 | |
| Nong Bua Lam Phu 2 | 16S4 | CO1 | ||||
| Nong Bua Lam Phu 3 | 16S4 | CO1 | ||||
| Nong Bua Lam Phu 4 | 16S4 | CO1 | ||||
| Chaiyaphum | 15.806817 | 102.031502 | Chaiyaphum 1 | 16S4 | CO1 | |
| Chaiyaphum 2 | 16S5 | CO2 | ||||
| Chaiyaphum 3 | 16S4 | CO1 | ||||
| Chaiyaphum 4 | 16S5 | CO2 | ||||
| Maha Sarakham | 16.013201 | 103.161516 | Maha Sarakham 1 | 16S8 | CO6 | |
| Maha Sarakham 2 | 16S8 | CO6 | ||||
| Maha Sarakham 3 | 16S4 | CO1 | ||||
| Maha Sarakham 4 | 16S5 | CO2 | ||||
| Maha Sarakham 5 | 16S5 | CO2 | ||||
| Buri Ram | 14.993001 | 103.102919 | Buri Ram 1 | 16S4 | CO1 | |
| Buri Ram 2 | 16S4 | CO1 | ||||
| Buri Ram 3 | 16S8 | CO6 | ||||
| Buri Ram 4 | 16S4 | CO1 | ||||
| C | Chiang Rai | 19.907165 | 99.830955 | Chiang Rai 1 | 16S3 | CO4 |
| Chiang Rai 2 | 16S3 | CO2 | ||||
| Chiang Rai 3 | 16S4 | CO1 | ||||
| Chiang Mai | 18.706064 | 98.981716 | Chiang Mai 1 | 16S14 | CO11 | |
| Chiang Mai 2 | 16S7 | CO5 | ||||
| Nan | 18.775631 | 100.773041 | Nan 1 | 16S3 | CO4 | |
| Nan 2 | 16S3 | CO4 | ||||
| Nan 3 | 16S3 | CO4 | ||||
| Uttaradit | 17.620088 | 100.099294 | Uttaradit 1 | 16S5 | CO2 | |
| D | Chumphon | 10.493049 | 99.180019 | Chumphon 1 | 16S11 | CO8 |
| Chumphon 2 | 16S11 | CO14 | ||||
| Chumphon 3 | 16S4 | CO1 | ||||
| Chumphon 4 | 16S11 | CO8 | ||||
| Chumphon 5 | 16S4 | CO1 | ||||
| Surat Thani | 9.138238 | 99.321748 | Surat Thani 1 | 16S7 | CO5 | |
| Surat Thani 2 | 16S7 | CO5 | ||||
| Surat Thani 3 | 16S7 | CO5 | ||||
| Pattani | 6.761830 | 101.323254 | Pattani 1 | 16S3 | CO4 | |
| Total | 17 | 58 |
Genomic DNA extraction
Genomic DNA from each individual C. siamensis was extracted using a Tissue & Cell Genomic DNA Purification Kit (GMbiolab Co., Ltd. Taichung, Taiwan), according to the manufacturer’s instructions. An aliquot of the DNA solution was checked by running it on a 0.8% (w/v) agarose gel, in a 1 × TBE buffer, at 100 V. The gel was stained with ethidium bromide, followed by a destaining with distilled water, and photographed under UV light. The rest of the DNA solution was kept at -20°C for later use as the PCR template.
Polymerase chain reaction (PCR) and sequencing
The DNA fragment (500 bp) of the 16S rRNA gene was amplified by PCR using the 16Sar (5′-CGCCTGTTTATCAAAAACAT-3′) and 16Sbr (5′-CCGGTCTGAACTCAGATCACGT-3′) primer [35]. The PCR amplifications were performed in a 30 μl total volume, containing 15 μl of EconoTaq® PLUS 2 × Master mix (1×; Lucigen Corporation, Middleton, WI, USA), 1.5 μl of 5 μM of each primer (0.25 μM), 9 μl of distilled water, and 3 μl of the DNA template (20–200 ng). Thermal cycling for the 16S rRNA PCR amplification was performed at 96°C for 2 min, followed by 35 cycles of 94°C for 30 s, 45°C for 1 min, and 72°C for 2 min, and then a final 72°C for 5 min [36]. The 710 bp segment of the COI gene was amplified using the LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAG GGTGACCAAAAAATCA-3′) primer [37] as described for the 16S rRNA except the thermal cycling used annealing and extension time of 40 s and 90 s, respectively. All PCR amplifications were conducted in a Biometra TOne Thermal cycler (Analytik Jena AG, Jena, Germany). The amplified products were analyzed by 1.2% (w/v) agarose gel-electrophoresis at 100 V, stained with ethidium bromide, destained with distilled water, and visualized and photographed under UV light. The PCR products were then purified using a NucleoSpin® Gel and a PCR Clean-up kit (Macherey-Nagel, Germany), according to the manufacturer’s instructions. An aliquot of the purified PCR product was checked by 1.2% (w/v) agarose gel electrophoresis as above, while the rest was used as the template for commercial sequencing (Macrogen Inc., Seoul, Korea) in both the forward and reverse directions using the same primers as in the PCR.
Sequence and phylogenetic analysis
The nucleotide sequences were edited by viewing the peaks of the chromatogram in the SeqMan II software (DNASTAR, Madison, WI, USA). Phylogenetic analysis, including species identification (conversion of molecular operational taxonomic unit to likely species designation) of the Cryptozona was performed by BLASTn searching the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) an aligning the obtained homologous nucleotide sequences using ClustalW. Phylogenies were estimated using maximum likelihood (ML) with the general time reversible model, neighbor joining (NJ) with Kimura two-parameter (K2), and maximum parsimony (MP) with the subtree-pruning-regrafting (SPR) and node support values, based on 1000 bootstrap replicates, in the MEGA Version 7.0 program [38]. In addition, Bayesian inference (BI) analysis was performed using MrBayes version 3.2 [39], where the tree space was explored, using four chains for each run of a Markov chain Monte Carlo algorithm (MCMC). The BI analysis was run for 10 000 000 generations and sampled every 100 generations. The last 10 000 trees were used for the Bayesian posterior probabilities (bpp) with a burnin of 90 001 samples, as previous reported [40]. Although, four methods were used for constructing a phylogeny, only ML topology was showed in the present study. The bootstrap values from three methods and the percentage of Bayesian posterior probabilities were indicated on the branch of ML tree.
Genetic analysis
Haplotype diversity and nucleotide diversity were calculated in ARLEQUIN, version 3.5.1.2 [41]. The relationships among the haplotypes were estimated using the median joining (MJ) network [42]. The MJ network analysis was performed in NETWORK, version 5.0.1.1, based on 65 sequences of 16S rRNA (includes 7 sequences from GenBank) and 58 sequences of COI sequences.
The genetic differentiation among the populations from each region, was calculated in ARLEQUIN, based on pairwise FST. Analysis of molecular variance (AMOVA) performed in ARLEQUIN was used to test the genetic difference among groups. Although the data were generated for 16S rRNA and COI gene fragments, for most of the populations from the different regions, the substitution rate was higher for the COI, making it the more appropriate and informative population level marker. Thus, the COI data were the focus of the population genetic structure analyses.
Results
Molecular identification of Cryptozona siamensis
To identify the Cryptozona species, 58 individual land snails (19 samples from population A, 21 samples from population B, 9 samples from population C, and 9 samples from population D) were randomly selected for genetic studies. PCR-based analysis and sequencing of their 16S rRNA and COI regions, was performed, together with a BLASTN search of the edited sequences. Based on the 402 bp of the 16S rRNA gene, all 58 sequences (GenBank accession nos. MK858467-MK858524) in this study showed high identity (96 to 99%) with those of GenBank accession no. JQ728565, which is in agreement with the identification of the taxa in the present study as C. siamensis.
Phylogeny of Cryptozona siamensis
A phylogenetic tree was constructed using the ML, NJ, MP, and BI methods, and the 58 sequences, based on their 16S rRNA genes, were divided into two main clades. Based on the topology of ML tree (Fig 2), clade 1 (from all populations) contained 55 of the sequences, and they were sourced from Phitsanulok, Phetchabun, Pathum Thani, Nakhon Pathom, Loei, Nong Bua Lam Phu, Chaiyaphum, Maha Sarakham, Buri Ram, Chiang Rai, Chiang Mai, Nan, Uttaradit, Chumphon, Surat Thani, and Pattani Provinces. These sequences were closely related to 7 sequences of C. siamensis from Malaysia with 97% of Bayesian posterior probability. Clade 2 contained 3 sequences from the Chon Buri Province (Fig 2), with 100% bootstrap support values for each ML and NJ method. The intraspecific distances among the samples were 0.0–5.4% (S1 Table).
Fig 2. Maximum likelihood phylogenetic tree generated from 58 sequences of a partial 16S rRNA sequence (402 bp) of C. siamensis, collected from across Thailand.
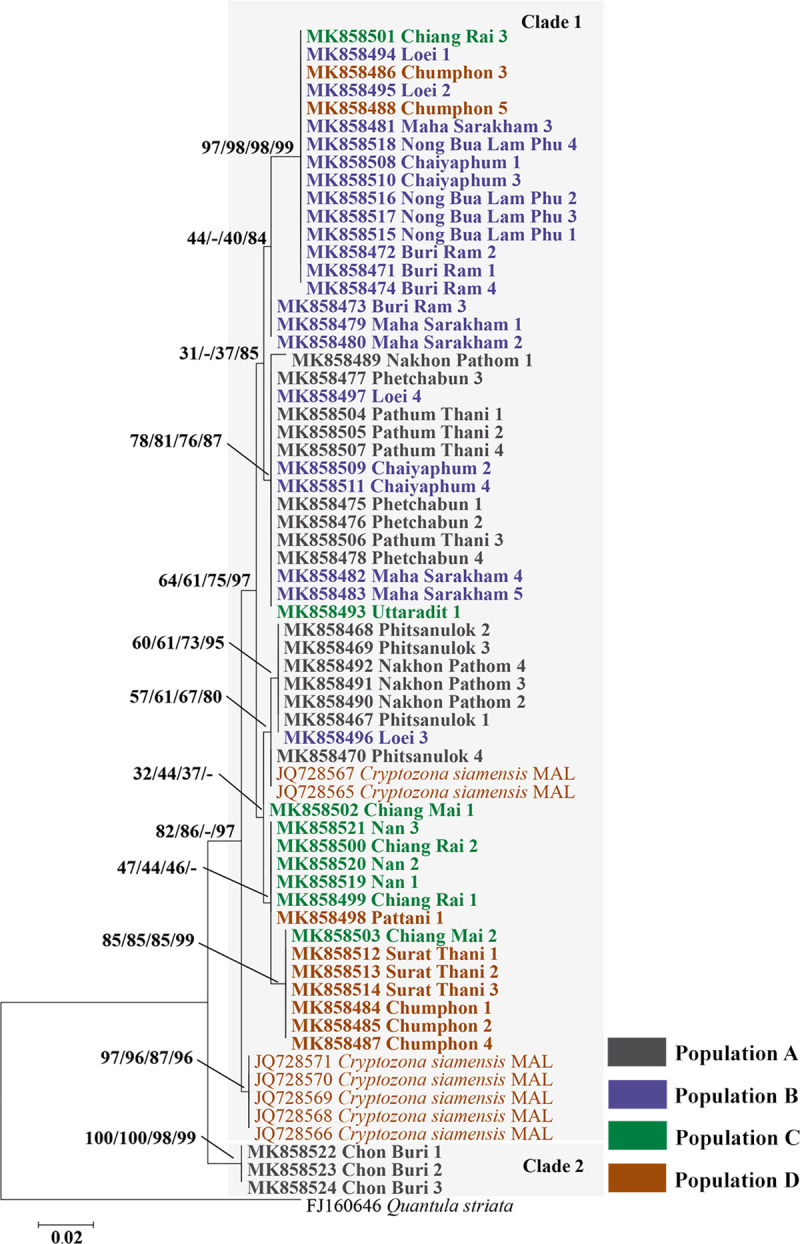
Support values (ML bootstrap/NJ bootstrap/MP bootstrap/Bayesian posterior probabilities) are shown above the branches. Bold letters indicate the sequences obtained in the present study. Quantula striata was used as the out-group. MAL = Malaysia.
The phylogenetic analysis based on the COI sequences (602 bp), for the 58 C. siamensis samples (GenBank accession nos. MK858409-MK858466), revealed 2 main clades (Fig 3). Clade 1 contained 55 sequences with highest bootstrap support values for ML and NJ of 100% and 100%, respectively. Clade 2 contained 3 sequences where the bootstrap support values for ML, NJ, and MP were 100% for each method. Intraspecific distances among the samples were 0.0–7.5% (S2 Table).
Fig 3. Maximum likelihood phylogenetic tree generated from 58 sequences of a partial COI sequence (602 bp) of C. siamensis, collected from across Thailand.

Support values (ML bootstrap/NJ bootstrap/MP bootstrap/Bayesian posterior probabilities) are shown above the branches. Bold letters indicate the sequences obtained in the present study. Ariophanta lahatensis and Helicarion mastersi were used as the out-group.
Mitochondrial DNA sequence variation
The mitochondrial 16S rRNA gene (402 bp) was obtained from 58 sequences of C. siamensis in Thailand and 7 sequences from Malaysia in 4 populations (A, B, C, and D). Fourteen haplotypes (16S1-16S14) were identified with 44 nucleotide variation sites (S3 Table). Of these, 9 haplotypes (16S1, 16S2, 16S6, 16S8, 16S9, 16S10, 16S11, 16S13, and 16S14) were unique, and 5 haplotypes were shared by at least two populations (Table 1 and S4 Table). The geographically wide spread haplotype 16S3, is shared between the populations C and D. Haplotype 16S4 was shared between the populations B, C, and D. Haplotype 16S5 was shared between the populations A, B, and C. Haplotype 16S7 was shared between the populations C and D. Haplotype 16S12 was shared between the populations A and B (Figs 1 and 4). The haplotype diversity in each population ranged from 0.6238 in population B to 0.8833 in population D, with a mean of 0.8779. Nucleotide diversity in each population ranged from 0.0088 in population C to 0.0169 in population A, with a mean of 0.0169 (Table 2).
Fig 4. Mitochondrial DNA genealogy for 65 16S rRNA sequences (58 sequences from Thailand and 7 sequences from Malaysia) from C. siamensis, constructed using a median joining network method.

Each haplotype is represented by a circle. Sizes of the circles are relative to the number of individuals sharing specific haplotypes. Each mutation between the haplotypes is represented by a bar.
Table 2. Haplotype and nucleotide diversity of 4 populations of Cryptozona siamensis identified in Thailand and Malaysia, based on their mitochondrial 16S rRNA sequences.
| Populations | Number of samples | No. haplotype | Haplotype diversity (h), mean ± SD | Nucleotide diversity (π), mean ± SD |
|---|---|---|---|---|
| A | 19 | 5 | 0.7310±0.0663 | 0.0169±0.0093 |
| B | 21 | 4 | 0.6238±0.0899 | 0.0119±0.0067 |
| C | 9 | 5 | 0.7222±0.1592 | 0.0088±0.0056 |
| D | 16 | 7 | 0.8833±0.0452 | 0.0155±0.0087 |
| Total | 65 | 14 | 0.8779±0.0215 | 0.0169±0.0089 |
Fourteen haplotypes (CO1-CO14), from the 58 sequences of the 4 populations (A, B, C, and D) in Thailand, were identified, based on the COI genes (602 bp) with nucleotide variations at 57 sites (S5 Table). Of these, 9 haplotypes (CO6, CO7, CO8, CO9, CO10, CO11, CO12, CO13, and CO14) were unique, and 5 haplotypes were shared by at least two populations (Table 1 and S6 Table). The geographically spread haplotype CO1 was shared between the population B, C, and D. Haplotype CO2 was shared between the populations A, B, and C. Haplotype CO3 was shared between the populations A and B. Haplotype CO4 was shared between the populations C and D. Haplotype CO5 was shared between the populations A, C, and D (Figs 1 and 5). The haplotype diversity in each population ranged from 0.6238 in population B to 0.8611 in population D, with a mean of 0.8609. Nucleotide diversity in each population ranged from 0.0083 in population B to 0.0271 in population A, with a mean of 0.0180 (Table 3).
Fig 5. Mitochondrial DNA genealogy for 58 COI sequences of C. siamensis constructed using a median joining network method.

Each haplotype is represented by a circle. Sizes of the circles are relative to the number of individuals sharing specific haplotypes. Each mutation between the haplotypes is represented by a bar.
Table 3. Haplotype and nucleotide diversity of 4 populations of Cryptozona siamensis in Thailand, based on their mitochondrial cytochrome c oxidase subunit I gene sequences.
| Populations | Number of samples | No. haplotype | Haplotype diversity (h), mean ± SD | Nucleotide diversity (π), mean ± SD |
|---|---|---|---|---|
| A | 19 | 8 | 0.8363±0.0651 | 0.0271±0.0141 |
| B | 21 | 4 | 0.6238±0.0899 | 0.0083±0.0047 |
| C | 9 | 5 | 0.8056±0.1196 | 0.0131±0.0076 |
| D | 9 | 5 | 0.8611±0.0872 | 0.0098±0.0059 |
| Total | 58 | 14 | 0.8609±0.0271 | 0.0180±0.0092 |
Population genetic structure
Population pairwise FST values for the 16S rRNA sequences of the C. siamensis, revealed statistically significant differentiation (P < 0.05), whereas between the populations C and D no genetic differences were identified (Table 4). The population pairwise FST values based on the COI sequences, between the populations A, B, C, and D were significantly different genetically (Table 5).
Table 4. Population pairwise FST, between the 4 populations of Cryptozona siamensis in Thailand and Malaysia, based on the mitochondrial 16S rRNA gene sequences.
* P<0.05.
Table 5. Population pairwise FST between 4 populations of Cryptozona siamensis in Thailand, based on the mitochondrial cytochrome c oxidase subunit I gene sequences.
* P<0.05.
Discussion
We have analyzed the genetics of the land snail C. siamensis, based on its mitochondrial 16S rRNA and COI genes. Cryptozona siamensis showed high haplotype diversity (16S rRNA mean haplotype diversity = 0.8779 and the COI gene mean haplotype diversity = 0.8609). The 65 16S rRNA gene sequences, were classified into 14 haplotypes, 9 that were unique, and 5 that were shared between the different populations. The 58 COI sequences were classified into 14 haplotypes, 9 that were unique and 5 that were shared between the different populations. High haplotype diversity (39 mitochondrial haplotypes) was also reported from Camaena cicatricose in China [29]. In contrast, low haplotype diversity (2 COI haplotypes) was found in A. fulica in Thailand [30]. The genetic diversity of the 16S rRNA and the COI genes of the C. siamensis, were different. These results were similar to those from a previous report on the genetic diversity of the 16S rRNA and COI genes of the land snail Cyclophorus fulguratus [40]; as the genetic variation in the 16S rRNA gene was lower than that of the COI gene. This suggested that the 16S rRNA gene in the land snail was a slowly evolving region of the mtDNA [43]. In terms of the evolution rate, the COI gene was superior to the 16S rRNA gene [44], and thus considered to be a reliable phylogenetic marker for the C. siamensis. Many previous reports in land snails have established that the COI gene is a reliable molecular marker for phylogenetic analysis [44–46]. In contrast with the haplotype diversity, the nucleotide diversity was low for both the 16S rRNA (mean = 0.0169) and COI (mean = 0.0180) genes for this snail. The low nucleotide diversity of C. siamensis in the present study could be due to small sample sizes. This was consistent with the previous research on the Succinea cadusa snail [28]. The pattern of high haplotype diversity and low nucleotide diversity implies that independent founder events have resulted in multiple unique populations, that each persisted in isolation for sufficient amounts of time, as to allow the accumulation of substitutions through drift [28]. The genetic diversity of the species could be affected by mutation and selection. These effects may play an important role in populations with genetic diversity. In addition, the climate may effect on the population size. This could be observed in other snails such as Camaena cicatricosa and Succinea caduca [28, 29]. In addition, the genetic diversity of the Collisella subrugosa snail in Brazil showed variation due to environmental conditions and differing selection pressures [47].
The population genetic structure was examined using the COI and 16S sequence data from 4 populations of Thailand. The substitution rate for the COI gene was high in the C. siamensis from all populations. Previous research has suggested that when the pairwise FST is greater than 0.15, it implies a high level of genetic differentiation among the population. Whereas, if the pairwise FST is between 0 and 0.05, it implies a low level of genetic differentiation between the populations [48]. In this study, the population genetic structure of the pairwise FST values showed a significant genetic differentiation between the most populations, based on the analysis of the 16S rRNA and COI genes. Moreover, the phylogenetic analysis and haplotype network construction showed the lack of clear population genetic structures. This suggests that the gene flow within the C. siamensis population in Thailand might bring about genetic homogeneity [49]. In this study, almost all of the pairwise FST values, based on the 16S rRNA and COI sequences between the populations, were over 0.15. In contrast, the pairwise FST based values on the 16S rRNA sequences, showed different results in some populations compared to the COI sequences. This suggested that the 16S rRNA gene had a relatively slow of evolution rate, and a low variability, compared to the COI gene [44, 50]. This agreed with the study of Feng et al. (2011), who found more variable nucleotide sites in the COI gene than in the 16S rRNA in the mollusk of the family Pectinidae. Genetic differentiation of C. siamensis population may be affected by the ecological environment, climate barriers, and geographic barrier factors [2, 29]. This was supported by previous studies that showed that the ecological environment factor on population differentiation, has been reported in other land snails, such as Camaena cicatricose [29] and Cyclophorus fulguratus [42, 51]. In addition, the migration and expansion repeated of population from different areas lead to continuously mix the populations of land snails. It is likely to affect accumulation of high genetic diversity within the population of the species. This was reported in pulmonate snail, Euhadra quaesita [52]. It may be possible reason leading to the genetic different in population of C. siamensis.
The genetic homogeneity of C. siamensis in Thailand based on 16S rRNA and COI genes was found among some populations. The results suggested that extensive gene-flow was a possibility. In support of this, considerable levels of gene-flow have been reported among C. siamensis populations in Thailand. Populations of C. siamensis in the areas with a low level of genetic differentiation, exhibit higher levels of gene flow than the populations with a high genetic differentiation [53]. Although this particular land snail has a low dispersal ability, the water, wind, anthropochory, and other factors, can lead to a wider distribution, especially for human activities [54]. Recently, Prasankok and Panha (2011) reported on the allozyme variations in the C. siamensis from the three regions of Thailand, and one region in Malaysia. The population of C. siamensis among the 3 geographic regions (north, central, and south) of Thailand as well as the population in Malaysia, showed a high degree of gene flow. This was consistent with the research that the haplotype network of C. siamensis in Thailand was shared and linked to the haplotype from Malaysia with mutation steps. In the present study, gene flow within the C. siamensis population in Thailand could be possible for several reasons. Cryptozona siamensis often occurred in habitats associated with human activities, such as vegetable gardens, in flower pots, and in wall crevices of houses. In particular, the transportation of potted plants or vegetables contaminated with snails across provinces may have promoted the movement of snails. Therefore, the possibility of dispersal of C. siamensis among populations in each area by humans could be plausible. Similarly, the African land snail in China, A. fulica, could be spread through shipments of plants especially pot plants [55], as well as the C. cicatricosa in China could be spread through cargo transportation [29]. Therefore, the dispersal of C. siamensis between populations in each region may arise by human activities [2]. The possibility of gene flow may be associated with the anthropochoric effects of snail dispersal [2, 56]. In addition, birds perhaps, mediated important in the transport of snails [57]. The movements of the eggs or small snails attached to the birds may have been important in the distributions between each area in regions. Thus, it is suggested that gene flow in the C. siamensis from Thailand may be explained using several possibilities. In addition, transportation by human activities may promote the spread of A. cantonensis which is hosted by this land snail. Similarly, Lv et al. (2009) reported the distribution of A. cantonensis in China associated with invasive land snail, A. fulica [8]. Although no C. siamensis infected with A. cantonensis was found in the present study, previous reports found that C. siamensis was infected with A. cantonensis. In Thailand, C. siamensis infected with A. cantonensis was found in Phetchabun, Kalasin, Phitsanulok, and Kamphaeng Phet provinces [3]. In general, food made from C. siamensis are uncommon dish for people in Thailand. Human may get infection with A. cantonensis by consumption of vegetable contaminated with A. cantonensis larvae [58]. Therefore, the C. siamensis is important to maintain the life cycle of A. cantonensis and may transmit to human.
Conclusion
We have reported on the genetic diversity of the 16S rRNA and the COI genes, from the C. siamensis samples taken in Thailand. The maximum likelihood tree based on the 16S rRNA and COI fragment sequences of C. siamensis revealed two main clades. Most of the sequences fell in clade 1 and 3 samples from Chon Buri Province (population A) was placed in clade 2. The genetic diversity of C. siamensis in term of the number of haplotypes and haplotype diversity, was found to be high but the nucleotide diversity among the different populations of Thailand showed low levels of genetic differentiation for the COI sequence as also noted with the 16S rRNA sequences. Population genetic structure of C. siamensis based on FST value in 16S rRNA and COI genes was genetically different among most of population exception of some populations. Genetic differentiation in the populations of the C. siamensis may result from the effects of the ecological environment, climate barriers, and geographic barriers. However, low genetic difference in some populations may be due to high gene flow which may occur from the transportation by human activities. In addition, transportation by human may lead to spread of A. cantonensis which is hosted by C. siamensis. This study shows the genetic diversity of the C. siamensis populations across several regions of Thailand, and their interrelatedness.
Supporting information
(XLS)
(XLS)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
We thank Professor Dr. Pairot Pramual, Faculty of Science, Maha Sarakham University for his guidance in analyzing the genetic diversity of C. siamensis.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Research Council of Thailand through the Annual Research Fund Naresuan University (Grant number R2562B079 and R2562B078).
References
- 1.Panha S. A checklist and classification of the terrestrial pulmonate snail of Thailand. Walkerana. 1996;8: 31–40. [Google Scholar]
- 2.Prasankok P, Panha S. Genetic structure of the common terrestrial pulmonate snail, Cryptozona siamensis (Pfeiffer, 1856), in Thailand. Biochem Sys Ecol. 2011;39: 449–457. [Google Scholar]
- 3.Vitta A, Srisongcram N, Thiproaj J, Wongma A, Polsut W, Fukruksa C, et al. Phylogeny of Angiostrongylus cantonensis in Thailand based on cytochrome c oxidase subunit I gene sequence. Southeast Asian J Trop Med Public Health. 2016;47: 377–386. [PubMed] [Google Scholar]
- 4.Wang QP, Lai DH, Zhu XQ, Chen XG, Lun ZR. Human angiostrongyliasis. Lancet Infect Dis. 2008;8: 621–630. [DOI] [PubMed] [Google Scholar]
- 5.Cowie RH. Biology, systematics, life cycle, and distribution of Angiostrongylus cantonensis, the cause of rat lungworm disease. Hawaii J Med Public Health. 2013;72: 6–9. [PMC free article] [PubMed] [Google Scholar]
- 6.Rodpai R, Intapan PM, Thanchomnang T, Sanpool O, Sadaow L, Laymanivong S, et al. Angiostrongylus cantonensis and A. malaysiensis broadly overlap in Thailand, Lao PDR, Cambodia and Myanmar: a molecular survey of larvae in land snails. PLoS One. 2016;11(8): e0161128 10.1371/journal.pone.0161128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv S, Zhang Y, Wang XH, Liu HX, Zhu D, Yin WG, et al. Experimental study on compatibility of three species of freshwater snails with Angiostrongylus cantonensis. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 2006;24: 277–280. [PubMed] [Google Scholar]
- 8.Lv S, Zhang Y, Liu HX, Hu L, Yang K, Steinmann P, et al. Invasive snails and an emerging infectious disease: results from the first national survey on Angiostrongylus cantonensis in China. PLos Negl Trop Dis. 2009;3(2): e368 10.1371/journal.pntd.0000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asato R, Taira K, Nakamura M, Kudaka J, Itokazu K, Kawanaka M. Changing epidemiology of angiostrongyliasis cantonensis in Okinawa Prefecture. Japan Jpn J Infect Dis. 2004;57: 184–186. [PubMed] [Google Scholar]
- 10.Panha S, Sutcharit C, Tongkerd P, Naggs F. An illustrated guide to the land snails of Thailand. Biodiversity Research and Training Program; 2009. [Google Scholar]
- 11.Abu-Bakar SB, Razali NM, Naggs F, Wade C, Mohd-Nor SA, Aileen-Tan SH. The mitochondrial 16 s rRNA reveals high anthropogenic influence on land snail diversity in a preliminary island survey. Mol Biol Rep. 2014;41: 1799–1805. [DOI] [PubMed] [Google Scholar]
- 12.Tan SK, Chan SY, Nguang LHS, Low MEY. Making its way down the Peninsula: Discovery of the non‐native Cryptozona siamensis (L. Pfeiffer, 1856) in Singapore, with a note on its status in Peninsular Malaysia (Helicarionoidea: Ariophantidae). OMP. 2016;5: 1–9. [Google Scholar]
- 13.Meissner H, Lemay A, Bertone C, Schwartzburg K, Fergusen L, Newton L. Evaluation of pathways for exotic plant pest movement into and within the greater Caribbean region. Caribbean invasive species working group (CISWG) and plant epidemiology and risk analysis laboratory (PERAL), center for plant health science and technology (CPHST): United States department of agriculture (USDA); 2009. [Google Scholar]
- 14.Raut SK, Ghose KC. Pestiferous land snails of India. Zoological survey of India; 1984. [Google Scholar]
- 15.Mouahid G, Clerissi C, Allienne J-F, Chaparro C, Al Yafae S, Mintsa Nguema R, et al. The phylogeny of the genus Indoplanorbis (Gastropoda, Planorbidae) from Africa and the French West Indies. Zool Scr. 2018;47: 558–564. [Google Scholar]
- 16.Standley CJ, Goodacre SL, Wade CM, Stothard JR. The population genetic structure of Biomphalaria choanomphala in Lake Victoria, East Africa: implications for schistosomiasis transmission. Parasit Vectors. 2014;7: 524 10.1186/s13071-014-0524-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogler AP, Monaghan MT. Recent advances in DNA taxonomy. J Zoolog Syst Evol Res. 2007;45: 1–10. [Google Scholar]
- 18.Zeng X, Yiu WC, Cheung KH, Yip HY, Nong W, He P, et al. Distribution and current infection status of Biomphalaria straminea in Hong Kong. Parasit Vectors. 2017;10: 351 10.1186/s13071-017-2285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali RF, Neiber MT, Walther F, Hausdorf B. Morphological and genetic differentiation of Eremina desertorum (Gastropoda, Pulmonata, Helicidae) in Egypt. Zool Scr. 2015;45: 48–61. [Google Scholar]
- 20.Colgan DJ, Ponder WF, Beacham E, Macaranas J. Molecular phylogenetics of Caenogastropoda (Gastropoda: Mollusca). Mol Phylogenet Evol. 2007;42: 717–737. [DOI] [PubMed] [Google Scholar]
- 21.Lee YC, Lue KY, Wu WL. Molecular evidence for a polyphyletic genus Japonia (Architaenioglossa: Cyclophoridae) and with the description of a new genus and two new species. Zootaxa. 2008;1792: 22–38. [Google Scholar]
- 22.Neiber MT, Hausdorf B. Molecular phylogeny and biogeography of the land snail genus Monacha (Gastropoda, Hygromiidae). Zool Scr. 2016;46: 308–321. [Google Scholar]
- 23.Liew T-S, Schilthuizen M, Vermeulen JJ. Systematic revision of the genus Everettia Godwin-Austen, 1891 (Mollusca: Gastropoda: Dyakiidae) in Sabah, northern Borneo. Zool J Linn Soc. 2009;157: 515–550. [Google Scholar]
- 24.Fontanilla IKC, Sta. Maria IMP, Garcia JRM, Ghate H, Naggs F, Wade CM. Restricted Genetic Variation in Populations of Achatina (Lissachatina) fulica outside of East Africa and the Indian Ocean Islands Points to the Indian Ocean Islands as the Earliest Known Common Source. PLoS One. 2014;9(9): e105151 10.1371/journal.pone.0105151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An HS, Jee YJ, Min KS, Kim BL, Han SJ. Phylogenetic analysis of six species of pacific abalone (Haliotidae) based on DNA sequences of 16S rRNA and cytochrome c oxidase subunit I mitochondrial genes. Mar Biotechnol. 2005;7: 373–380. [DOI] [PubMed] [Google Scholar]
- 26.Grande C, Templado J, Cervera JL, Zardoya R. Phylogenetics relationship among Opisthobranchia (Mollusca: Gastropoda) based on mitochondrial cox 1, trnV, and rrnL genes. Mol Phylogenet Evol. 2004;33: 378–78. [DOI] [PubMed] [Google Scholar]
- 27.Holznagel WE, Lydeard C. A molecular phylogeny of north American Pleuroceridae (Gastropoda: Cerithioidea) based on mitochondrial 16S rDNA sequences. J Molluscan Stud. 2000;66: 233–257. [Google Scholar]
- 28.Holland BS, Cowie RH. A geographic mosaic of passive dispersal: population structure in the endemic Hawaiian amber snail Succinea caduca (Mighels, 1845). Mol Ecol. 2007;16: 2422–2435. [DOI] [PubMed] [Google Scholar]
- 29.Zhou W, Yang H, Ding H, Yang S, Lin J, Wang P. Population genetic structure of the land snail Camaena cicatricosa (Stylommatophora, Camaenidae) in China inferred from mitochondrial genes and ITS2 sequences. Sci Rep. 2017;7: 15590 10.1038/s41598-017-15758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumidae A, Janthu P, Subkrasae C, Dekumyoy P, Thanwisai A, Vitta A. Genetic characterization of Angiostrongylus larvae and their intermediate host, Achatina fulica, in Thailand. PLoS One. 2019;14(9): e0223257 10.1371/journal.pone.0223257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avise J. Phylogeography: the history and formation of species. Harvard university press; 2000. [Google Scholar]
- 32.Bohonak AJ. Dispersal, gene flow, and population structure. Q Rev Biol. 1999;74: 21–45. [DOI] [PubMed] [Google Scholar]
- 33.Byrne M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quat Sci Rev. 2008;27: 2576–2585. [Google Scholar]
- 34.Jin YT, Brown RP, Liu NF. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan Plateau. Mol Ecol. 2008;17: 1971–1982. [DOI] [PubMed] [Google Scholar]
- 35.Kessing B, Croom H, Martin A, Mcintosh C, Mcmillan WO, Palumbi S. The simple fool’s guide to PCR. Honolulu: University of Hawaii; 1989. [Google Scholar]
- 36.Wade CM, Mordan PB. Evolution within the gastropod molluscs; using the ribosomal RNA gene cluster as an indicator of phylogenetic relationships. J Molluscan Stud. 2000;66: 565–570. [Google Scholar]
- 37.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Marine Biol Biotechnol. 1994;3: 294–299. [PubMed] [Google Scholar]
- 38.Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61: 539–542. 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nantarat N, Tongkerd P, Sutcharit C, Wade CM, Naggs F, Panha S. Phylogenetic relationships of the operculate land snail genus Cyclophorus Montfort, 1810 in Thailand. Mol Phylogenet Evol. 2014;70: 99–111. [DOI] [PubMed] [Google Scholar]
- 41.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under linux and windows. Mol Ecol Resour. 2010;10: 564–567. [DOI] [PubMed] [Google Scholar]
- 42.Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16: 37–48. [DOI] [PubMed] [Google Scholar]
- 43.Kornilios P, Poulakakis N, Mylonas M, Vardinoyannis K. The phylogeny and biogeography of the genus Zonites Montfort,1810 (Gastropoda: Pulmonata): preliminary evidence from mitochondrial data. J Molluscan Stud. 2009;75: 109–117. [Google Scholar]
- 44.Feng YW, Li Q, Kong LF, Zheng XD. DNA barcoding and phylogenetic analysis of Pectinidae (Mollusca: Bivalvia) based on mitochondrial COI and 16S rRNA genes. Mol Biol Rep. 2011;38: 291–299. [DOI] [PubMed] [Google Scholar]
- 45.Awodiran MO, Arthur MC, Idowu AJ. Molecular phylogeny of three species of land snails (Stylommatophora and Achatinidae), Archachatina marginata (Swainson, 1821), Achatina achatina (Linnaeus, 1758), and Achatina fulica (Bowdich, 1822) in some southern states and north central states in Nigeria. AWUT-SerBio. 2015;18: 95–106. [Google Scholar]
- 46.Liu HP, Hershler R, Thompson PG. Phylogenetic relationships of Cochliopinae (Rissooidea: Hydrobiidae): an enigmatic group of aquatic gastropods. Mol Phylogenet Evol. 2001;21: 17–25. [DOI] [PubMed] [Google Scholar]
- 47.Jose J, Solferini VS. Population genetics of Collisella subrugosa (Patellogastropoda: Acmaeidae): evidence of two scales of population structure. Genetica. 2007;130: 73–82. [DOI] [PubMed] [Google Scholar]
- 48.Jones JS. Ecological genetics and natural selection in molluscs. Science. 1973;182: 546–552. [DOI] [PubMed] [Google Scholar]
- 49.Szalanski AL, Austin JW, McKern JA, Steelman CD, Gold RE. Mitochondrial and ribosomal internal transcribed spacer 1 diversity of Cimex lectularius (Hemiptera: Cimicidae). J Med Entomol. 2008;45: 229–236. [DOI] [PubMed] [Google Scholar]
- 50.Knowlton N, Weigt LA. New dates and new rates for divergence across the Isthmus of Panama. Proc R Soc Lond B. 1998;265: 2257–2263. [Google Scholar]
- 51.Prasankok P, Sutcharit C, Tongkerd P, Panha S. Biochemical assessment of the taxonomic diversity of the operculate land snail, Cyclophorus fulguratus (Gastropoda: Cyclophoridae), from Thailand. Biochem Syst Ecol. 2008;36: 900–906. [Google Scholar]
- 52.Watanabe Y, Chiba S. High within-population mitochondrial DNA variation due to microvicariance and population mixing in the land snail Euhadra quaesita (Pulmonata:Bradybaena). Mol Ecol. 2002;10: 2635–2645. [DOI] [PubMed] [Google Scholar]
- 53.Millar CL, Libby WJ, Falk DA, Holsinger KE. Strategies for conserving clinal, ecotypic, and disjunct population diversity in widespread species. New York: Oxford University Press; 1991. [Google Scholar]
- 54.Chen DN, Gao JX. Economic fauna sinica of China, Terrestrial Mollusca. Beijing: Science Press; 1987. [Google Scholar]
- 55.Mead AR. The Giant African Land Snail: a problem in economic malacology. USA: University of Chicago Press; 1961. pp. 257. [Google Scholar]
- 56.Schilthuizen M, Lombearts M. Population structure and levels of gene flow in the Mediterranean land snail Albinaria corrugata (Pulmonata: Clausiliidae). Evolution. 1994;48: 577–586. [DOI] [PubMed] [Google Scholar]
- 57.Gittenberger E, Groenberg DSJ, Kokshoom B, Preece RC. Molecular trails from hitch-hiking snails. 2006;439: 409. [DOI] [PubMed] [Google Scholar]
- 58.Marquardt WC, Demaree RS, Grieve RB. Parasitology and vector biology. 2nd ed California: Academic Press; 2000. pp. 702. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
(XLS)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.