

Abstract
The carbohydrate response element binding protein (ChREBP) is a glucose-responsive transcription factor that plays a critical role in glucose-mediated induction of genes involved in hepatic glycolysis and lipogenesis. In response to fluctuating blood glucose levels ChREBP activity is regulated mainly by nucleocytoplasmic shuttling of ChREBP. Under high glucose ChREBP binds to importin α and importin β and translocates into the nucleus to initiate transcription. We have previously shown that the nuclear localization signal site (NLS) for ChREBP is bipartite with the NLS extending from Arg158 to Lys190. Here, we report the 2.5 Å crystal structure of the ChREBP-NLS peptide bound to importin α. The structure revealed that the NLS binding is monopartite, with the amino acid residues K171RRI174 from the ChREBP-NLS interacting with ARM2–ARM5 on importin α. We discovered that importin α also binds to the primary binding site of the 14-3-3 proteins with high affinity, which suggests that both importin α and 14-3-3 are each competing with the other for this broad-binding region (residues 117–196) on ChREBP. We screened a small compound library and identified two novel compounds that inhibit the ChREBP-NLS/importin α interaction, nuclear localization, and transcription activities of ChREBP. These candidate molecules support developing inhibitors of ChREBP that may be useful in treatment of obesity and the associated diseases.
Keywords: bZIP transcription factors, carbohydrate binding modules, carbohydrate metabolism, importin alpha, protein structure
Introduction
The liver is the primary site for de novo fatty acid and triglyceride synthesis from excess carbohydrate. The carbohydrate response element binding protein (ChREBP), discovered in this laboratory in 2001, is the glucose-responsive transcription factor responsible for the activation of the liver pyruvate kinase gene and all lipogenesis genes in the liver. ChREBP is an insulin-independent transcription factor [1]. In response to circulating glucose levels, ChREBP is regulated at two levels, nucleus-cytoplasm localization and transcription. When the glucose levels are low, inactive ChREBP mainly localizes in the cytoplasm, and as glucose levels rise, ChREBP localizes into the nucleus to initiate transcription [2,3].
ChREBP is a large transcription factor of 96 kDa and contains several functional domains, including two nuclear export signals, NES1 and NES2, an extended nuclear localization signal (NLS), a proline-rich low-complexity region, and a DNA binding bHLH/ZIP domain (Figure 1A) [1,4,5]. ChREBP senses and responds to changing levels of glucose, termed ‘glucose signaling', in which glucose metabolites regulate intracellular localization, and transcriptional activity [6]. Thus, ChREBP plays an essential role in tightly coupling enhanced glycolysis and fatty acid synthesis following ingestion of high carbohydrate meals.
Figure 1. ChREBP domain organization including phosphorylation sites and subcellular localization under low and high glucose concentrations.

(A) Domain organization and the sequences for the N-terminal glucose-responsive regulatory region of ChREBP. The ChREBP function of residues 100–200 in the N-terminal regulatory region, including the α2, and α3 helices, and the NLS (nuclear localization signal) site are depicted. (B) Proposed model of the regulation of ChREBP subcellular localization by 14-3-3 and importin α binding in response to glucose. In response to high glucose, phosphorylated (inactive) ChREBP, complexed with 14-3-3 in the cytosol, is first dephosphorylated and releases 14-3-3 and CRM1. Dephosphorylated (active) ChREBP forms a complex with importin α and β and is translocated into the nucleus. It is possible that 14-3-3 may remain bound to ChREBP during import. A decrease in glucose results in inactivation of ChREBP by phosphorylation by PKA that occurs within the nucleus and complex formation with 14-3-3 and CRM1 followed by translocation to the cytosol. Note that for clarity, 14-3-3 proteins (27 kD) and importin α are not presented to scale. Modified from ref. [5].
The N-terminal region of ChREBP (amino acid residues 1–250) is necessary and sufficient for nuclear import and export to the cytosol (Figure 1B) [5]. The shuttling of ChREBP between nucleus and cytoplasm requires nuclear transport factors including importin α/importin β binding to the highly conserved NLS and CRM-1 binding to the nuclear export signal (NES) sites for nuclear import and export, respectively. This region contains three α-helices [2]. The α1 corresponds to NES2 (residues 85–95), and α2 helix (residues 117–137) is identified as a primary 14-3-3 binding site. We found that both NES1 and NES2 sites are essential for binding of CRM-1 (Exportin) for ChREBP export to the cytoplasm [7]. We also determined that 14-3-3 binding is facilitated by phosphorylation of Ser196 residue. 14-3-3 proteins are a family of regulatory proteins acting as phospho-serine, phospho-threonine binding modules and regulate the function of a large number of different substrate proteins [8]. We have characterized the ChREBP importin α binding site, an extended α3 helix acting as a classical bipartite nuclear localization signal (NLS) that encompasses ChREBP residues 158–190 [5] (Figure 1A).
The subcellular localization of ChREBP in response to changing levels of circulating blood glucose involves multiple steps, and the regulation is complex. Previous studies have demonstrated that phosphorylation/dephosphorylation is a major mechanism by which ChREBP activity is regulated, and it is mediated through multiple phosphorylation sites, including sites for cAMP-dependent protein kinase (PKA) and AMP-dependent kinase (AMPK) [3,9]. When glucose levels are low and glucagon is elevated, the phosphorylated inactive pool of ChREBP (ChREBP-Ser(P)) binds to 14-3-3 proteins and CRM-1, leading to ChREBP transit out of the nucleus and into the cytoplasm [2,3] (Figure 1B). After a meal, as the glucose levels rise, the concentration of xylulose 5-P (Xu5P) concentration increases, activating a specific protein phosphatase (PP2ABδC), which leads to dephosphorylation of ChREBP. The dephospho-ChREBP (active) then binds to importin α and importin β for nuclear translocation to facilitate transcription of target genes [3,5]. 14-3-3 proteins play a dual role under low glucose levels by binding not only to the primary binding site of ChREBP but also to the phosphorylated Ser196 site near the NLS site, blocking importin α from binding to the NLS site [2]. The competitive binding of 14-3-3 and importin α to the NLS region of ChREBP provides a mechanism for glucose sensing and the role of PKA-mediated phosphorylation of Ser196 in the glucose-dependent regulation of nuclear/cytoplasm localization of ChREBP. In this communication, we present evidence that under high glucose levels importin α also plays a dual role by binding to both the NLS site and the primary binding site of 14-3-3 proteins.
In mice with targeted disruption of the ChREBP gene (ChREBP−/−), de novo fatty acid synthesis and overall adiposity are significantly decreased compared with wild type (WT) mice [10]. Liver triglyceride storage is reduced significantly in ChREBP−/− mice fed a high carbohydrate diet compared with WT mice fed the same diet. We intercrossed ChREBP−/− and ob/ob mice to make ob/ob-ChREBP−/− mice [11]. These doubly deficient mice show reduced overall weight gain and adiposity, which are largely attributable to reduced food intake. Gluconeogenesis is also reduced in the doubly deficient mice. These results suggest that inactivation of ChREBP not only reduces fat synthesis and obesity but also improves glucose tolerance and appetite control.
There is compelling biochemical and genetic evidence that nuclear localization of ChREBP serves as an important step in lipogenesis induction in vivo. Importantly, the disruption of the ChREBP gene is not only safe in mice, but it also ameliorates obesity by reducing food intake and fat synthesis, as ob/ob-ChREBP−/− mice demonstrate [10,11]. These in vivo findings, taken together with the extensive in vitro data describing the roles of ChREBP in carbohydrate metabolism and lipogenesis regulation, provide a strong rationale for the development of new strategies to inhibit ChREBP activities, including nuclear localization, in order to prevent obesity.
First, we investigated the crystal structure of the NLS site of ChREBP complexed with importin α. A major difficulty in the investigation of ChREBP's structure is that it is impossible to isolate full length ChREBP protein due to its susceptibility to protease digestion. Recently, we succeeded in isolating a N-terminal fragment of ChREBP (residues 81–196) complexed with 14-3-3β and determined the crystal structure of the ChREBP-14-3-3β heterodimer [12]. The 3D structure revealed that the only structure visible in the crystals was α2 helix of ChREBP (residues 117–137), the primary binding site of 14-3-3, complexed with 14-3-3 in a novel phosphate-independent manner. The dimer interface between the proteins consists of both electrostatic and van der Waals interactions and surprisingly the binding was partially mediated by free sulfate [12].
Here, we report the crystal structure of the NLS site peptide of ChREBP bound to importin α. We discovered that importin α not only binds to the NLS site but also interacts with the primary binding site of the 14-3-3 proteins. These results shed new light into how these two proteins compete over a much broader region of ChREBP than previously thought and reciprocally regulate nuclear import/export of ChREBP in response to glucose. We also report identification of small molecules that are specifically targeted towards the ChREBP-NLS–importin α interaction.
Materials and methods
Chemicals and vectors
All chemicals were purchased from Sigma unless otherwise indicated. Bacterial expression vector for the human 14-3-3β was a gift from Dr. Steven L. McKnight (University of Texas Southwestern Medical Center), and that for GST-tagged importin α was a gift from Dr. Yoshihiro Yoneda (Osaka University, Osaka, Japan). Both 14-3-3β protein and importin α were purified as described previously [9]. Flag-tagged and GFP-fusion ChREBP proteins were described previously [5,7,9].
Interactions of ChREBP with 14-3-3β or importin α
Isothermal titration calorimetry (ITC) experiments were carried out using a VP-ITC microcalorimeter (MicroCal) as described previously [5].
Protein expression and purification
The procedure for the bacterial expression and purification of GST-tagged and His-tagged ΔIBB form of importin α were performed as described previously [13,14].
Luciferase reporter activity of ChREBP
HepG2 cells were plated in collagen-coated six-well tissue culture plates (BD Bioscience) at a density of 5 × 105 cells/well in Dulbecco's modified Eagle's medium supplemented with 100 units/ml penicillin, 100 µg/ml streptomycin, 10% dialyzed fetal bovine serum albumin. The cells were subjected to transfection with pGL-LPK, pRL-TK, and wild-type or mutated FLAG-ChREBP using Lipofectamine 2000 (Life Technologies, Inc.) according to the manufacturer procedure and incubated for 4 h. The medium containing the liposome–DNA complex was removed and replaced with the medium containing 27.5 mM glucose (HG medium) for 20 h. Luciferase reporter activity was measured sequentially using a Dual-Luciferase reporter assay system (Promega, Fitchburg, WI) using a model TD-20/20 Luminometer (Turner Design, Sunnyvale, CA), as described [2].
Nuclear localization of ChREBP in HepG2 cells
HepG2 cells were placed in 35 mm glass-bottomed plates (MatTek, Ashland, MA), and 3.2 µg of expression plasmid, encoding wild-type or mutated GFP-ChREBP, was transfected into the cells by lipofection and incubated for 4 h, as described above. The medium containing the liposome–DNA complex was removed and replaced with HG medium for 20 h. The cells were fixed with 4% paraformaldehyde at room temperature for 15 min, which was then replaced by phosphate buffered saline. The subcellular localization of GFP-ChREBP was determined as described previously [7,15].
Fluorescence polarization
FP experiments were performed in black 384 well plates with a non-binding surface (Corning; catalog # 3655). Rhodamine-ChREBP peptide (30 nM) and GST-Importin protein (0.3 µM) mix were added to each well from column 2 to 24 in FP assay buffer (50 mM Tris–HCl, pH 7.5, EDTA 0.2 mM) for a final assay volume of 50 µl, and peptide with buffer only to first column using Biotek Multiflo. Small molecules were added to assay plate by Beckman FX with the final concentration of 5 µM. Assay is incubated at room temperature for 1.5 hr. The FP signal in millipolarization (mP) units was measured at an excitation wavelength of 531 nm and emission wavelength of 595 nm in an Envision microplate reader (PerkinElmer).
Crystallization, data collection, structure determination of ChREBP NLS peptides bound importin α ΔIBB
Crystals of the ChREBP NLS peptide (residues 158–190) bound to importin α ΔIBB were grown using the hanging drop vapor diffusion technique. The complex was prepared by mixing 50 µl of importin α ΔIBB (10 mg/ml) with ChREBP NLS peptide (50 µM) and incubated on ice for 20 min. A 1.5 µl aliquot of the mixture was then mixed with 1.5 µl of well solution containing 500–1000 mM sodium citrate, 10 mM DTT, and HEPES pH 6.5–7.5 at 20°C. Diffraction quality crystals were formed in 1–2 weeks, and rod-shaped crystals grew to a maximum size of ∼0.2 mm in 2–3 weeks.
Crystals were harvested, cryo protected with 2.0 M lithium sulfate, and flash-frozen in liquid nitrogen. Diffraction data were collected at beamline 19-ID, Advanced Photon Source, Argonne National Laboratory, Argonne, IL.
Crystals from a droplet were directly transferred to the cryo-protectant of 10% glycerol for one minute, and then transferred to 20% glycerol. The crystals were mounted on nylon loops and flash frozen in a liquid nitrogen stream at 120 K before data collection. All the data were collected at beamline 19-ID, Advanced Photon Source (APS), Argonne National Laboratory, Argonne, IL. The data was reduced using HKL3000 [16], and the intensities were scaled with SCALEPACK from the CCP4 suite [17]. Initial phases were obtained by the molecular replacement method using MOLREP [18]. Nucleoplasmin NLS bound mouse importin α structure (3UL1) excluding NLS peptide was used as a search model. The extra density for the ChREBP NLS peptide was shown after the structure was refined against the data using PHENIX [19]. The ChREBP NLS peptide including side chains was manually built into the density using COOT [20]. The crystal data and final refinement statistics are summarized in Table 1. Structure figures were generated using Chimera [21].
Table 1. Crystallographic data collection and refinement statistics of ChREBP NLS bound structures of importin α.
| PDB CODE | Imp-α:ChREBP(6MJL) |
|---|---|
| Data Collection | |
| space group | P212121 |
| Cell Constants | |
| a (Å) | 77.434 |
| b | 89.691 |
| c | 96.332 |
| α (°) | 90.00 |
| Β | 90.00 |
| γ | 90.00 |
| Resolution (Å)a | 50.00–2.50 (2.54–2.50) |
| Rmergeb (%) | 0.049 (0.357) |
| <I/σ> | 13.2 (1.8) |
| CC1/2 | 0.750 |
| completeness (%) | 97.6 (88.6) |
| Redundancy | 3.9 (3.5) |
| Refinement | |
| Rworkc/Rfreed (%) | 19.8/24.4 |
| unique reflections | 23 621 |
| Mean B Factor (Å2) | |
| Protein | 34.15 |
| Ligand (peptide) | 41.68 |
| Solvent | 32.08 |
| Ramachandran Plot | |
| most favored (%) | 96.7 |
| add. allowed (%) | 3.1 |
| RMSD | |
| bond lengths (Å) | 0.008 |
| bond angles (degree) | 0.967 |
Values in parentheses are for the highest resolution shell;
Rmerge = Σ|I–|/ΣI where I is the integrated intensity of a given reflection;
Rwork = Σ|F(obs)–F(calc)|/ΣF(obs);
Rfree = Σ|F(obs)–F(calc)|/ΣF(obs), calculated using 5% of the data.
Statistics
GraphPad Prism (v. 8.4) software was used for the statistical analyses except for small molecule screening protocol (see below). Results were expressed as mean ± SD. Comparisons among groups were made by factorial analysis of variance or repeated-measure analysis of variance using Tukey-Kramer. Differences were considered statistically significant if P value was smaller than 0.05. Significance was indicated as P < 0.05 and P < 0.01. The Genedata Screener® software (version 13, Genedata, Inc. Basel, Switzerland) was used to process and analyze the screening data for small molecules. For analysis of the data from the primary screen of the UTSW chemical library, experimental results (mP values) obtained from Envision Microplate Reader were processed using the Analyzer module of the Genedata Screener® Suite.
Results
Crystal structure of ChREBP-NLS bound to importin α
The integrated and scaled data indicated that the crystals of ChREBP-NLS peptide bound to importin α indexed in the P212121 space group with cell dimensions a = 77.43 Å, b = 89.69 Å, c = 96.33 Å, alpha = beta = gamma = and 90.0°. The model was refined to 2.50 Å with Rwork = 19.8 and Rfree = 24.4. The crystal data and final refinement statistics are summarized in Table 1. The overall conformation of the ChREBP NLS bound importin α ΔIBB is similar to those reported previously with differences. The ChREBP NLS motif containing a cluster of basic residues (K171/R172/R173/I174) binds to the ARM 2–5 domains of importin α [22,23]. These polar interactions not only orient the ChREBP NLS backbone but the NLS side chains also provide the specificity [24,25] (Figure 2B,C). Figure 2C includes an unbiased omit map to reduce model bias. For example, Lys171 of ChREBP generates hydrogen bond interactions with Thr155 (2.55 Å) of importin α, and Arg172 of ChREBP interacts with both the oxygen atom of Asn228 (3.03 Å) and the aromatic ring of Trp184 (3.38 Å) of importin α. A number of prominent hydrogen bonding and van der Waals contacts are also observed between the NLS site and importin α. In particular, Arg173 of ChREBP NLS has multiple interactions with the main chain carbonyl groups of Leu104, Arg106, and Glu107 of importin α. It also exhibits van der Waals interactions with Glu175 (2.68 Å) of the NLS. Ile174 of ChREBP interacts with the aromatic rings of Trp142 (3.01 Å) and Trp184 (3.13 Å) of importin α via pi stacking interactions. Close inspection of the structure of the NLS peptide:importin α complex reveals that the other residues of the peptide also have some hydrogen bonding and/or van der Waals interactions. For example, Asn168 of ChREBP interacts with Asp270 (2.79 Å) and the aromatic ring of Trp273 (3.76 Å) of importin α. Similar to other known nuclear localization signals, the clusters of the basic residues surrounded by acidic and neutral residues on ChREBP position the backbone of the peptide and enable hydrophobic interactions with non-polar regions of the side chains. The specificity is provided by ionic interactions with charged NLS side chains.
Figure 2. Overview of the structures of importin α bound with ChREBP NLS 158–190.

(A) Ribbon diagram of the ChREBP-NLS peptide (pink) bound to importin α. (B) Major interactions between ChREBP NLS 158–190 and importin α. P2–P5 pockets of importin α were occupied by K171–I174 of ChREBP NLS 158–190, and the interacting residues of importin α are all shown. (C) Close-up view of ChREBP NLS 158–190 in the active site of importin α with omit map (Fo–Fc) contoured at 3σ around ChREBP peptide. (D) Close-up view of ChREBP NLS 158–190 in the active site of importin α with electro density map (2Fo–Fc) contoured at 1σ around ChREBP peptide.
Effect of structure-based mutation of the NLS site K171–I174 of ChREBP on the activities
To validate the observed major binding site of ChREBP NLS for importin α, we made a series of site-directed mutations of K171A, R172A, R173A and I174A in the ChREBP. However, glucose-stimulated, ChREBP-dependent transcriptional activity in HepG2 cells was not significantly affected by these mutations, except for the activity of R173A, which was significantly decreased (33%) (Figure 3A). Previously, we have demonstrated that double mutation of Lys171 and another NLS site including K159A/K171A and K171A/K183A resulted in diminished ChREBP nuclear localization as well as transcriptional activities by ∼30% to 20%, respectively, in hepatocytes [5]. It is unclear why the series of mutations failed to inactivate the transcriptional activity, even when the nuclear localization activity was decreased to the same extent (∼26%) as that of the individual K171A mutation. These differences could be due to the use of the NLS site mutated peptides in the crystallographic studies, while the full-length ChREBP proteins mutated on the same site were used in the biological assays.
Figure 3. Effects of mutations (K171R172R173I174) on the major binding-site of NLS on transcriptional activity and nuclear localization of ChREBP.

(A) Transcriptional activity of DNA constructs expressing the indicated FLAG-ChREBP proteins using firefly luciferase under control of LPK promoter, and Renilla luciferase (an internal control) were co-transfected into HepG2 cells during a 4-h incubation in DMEM containing 5.5 mM glucose. Fresh medium containing 27.5 mM glucose was added and the cells were incubated for an additional 20 h. Luciferase activities were measured and expressed as firefly luciferase activity relative to Renilla luciferase activity. The values presented are the mean ± S.D. of the results of eight independent experiments. * P < 0.05; *** P < 0.001. (B) Nuclear localization of ChREBP in HepG2 cells. HepG2 cells were transfected with DNA constructs expressing GFP-WT ChREBP or indicated GFP-ChREBP mutants during a 4 h incubation in DMEM with 5.5 mM glucose. Fresh medium containing 27.5 mM glucose was added, and the cells were incubated an additional 20 h. The values presented are the means ± S.D. of four sets of 100 fluorescent cells. * P < 0.05; ** P < 0.01; *** P < 0.001.
Glucose-induced nuclear localization activity of the GFP-ChREBP R173A mutant was also inhibited to the same extent, similar to the effects on the transcription activity (Figure 3B). The Lys171 mutation only slightly inhibited nuclear localization (20%) but had no effect on the transcriptional activity.
These results indicate that the alteration of Lys171–Ile174 results in only a modest inhibition of ChREBP activity and furthermore suggests that there are multiple other binding sites that contribute to the importin α binding, which are not detectable by the crystallography experiments.
Importin α binds to the primary 14-3-3 binding site
Unexpectedly, we discovered through ITC measurements that importin α binds to the α2 helix peptide (residues 117–140). This sequence is also the high affinity primary binding site for the 14-3-3 protein, exhibiting KD values of ∼0.82 µM (SD ± 0.64) (Figure 4 and Table 2). On the other hand, 14-3-3β binds to the α2 peptides of ChREBP with the KD values of 2.8 µM as measured by ITC. These results suggest that importin α competes effectively with 14-3-3 proteins for the primary binding site. Previously, we assumed that 14-3-3 proteins were always bound to ChREBP, even in the nucleus, but these results suggest that is an unlikely scenario.
Figure 4. ITC measurement of interaction between ChREBP-α2 helix and importin α.

ITC measurement for binding of importin α (25 µM) to the primary binding site (ChREBP-α2 helix) peptide. Error bars represent the standard deviations from triplicate measurements.
Table 2. Affinity constants of 14-3-3β and importin α binding to phospho-(+P) and dephospho-(−P) α2 helix and NLS peptides of ChREBP.
| KD values (µM) | KD values (µM) | ||
|---|---|---|---|
| Peptides | α2 (117–140) | NLS (170–198) | |
| −P | −P | +P | |
| 14-3-3B | 2.8 | 6.3 | 0.62 |
| Importin α | 0.82 | 3.2 | NB |
ITC experiments were performed to measure the binding constant between ChREBP peptides containing the α2 helix (residues 117–140) and the NLS (residues 170–198) to 14-3-3β [12] and importin α.
Primary and secondary screens for inhibitors of NLS/importin α
We have developed a fluorescence polarization (FP) assay as a tool for high throughput screening (HTS) in order to identify compounds that disrupt the interaction between a Rhodamine-tagged ChREBP-NLS peptide (158–190) and importin α. We performed a primary HTS with a chemical library comprising 8000 compounds at a concentration of 10 µM in a 384-well plate format (single replicate per compound). We identified a dozen compounds that inhibit the interaction between the NLS peptide and importin α. Those compounds were assayed manually in a dose-response format using FP assay as the readout. Of those initial hits, compound 1 exhibited the strongest inhibition in a lower throughput version of the FP assay.
We resynthesized hit compound 1 and prepared an additional five analogs for a preliminary Structure-Activity Relationship (SAR) study. Details of the chemical syntheses are mentioned in the Supplementary Information section. The effect of these six compounds on ChREBP transcriptional activity, as assessed by a ChREBP-LPK promoter driven luciferase reporter construct [1], was determined in HepG2 cells under high glucose conditions (Table 3). Our results indicate that all six candidates (10 µM) antagonized the transcriptional activity of ChREBP to varying degrees, with compounds 1 and 2 showing the strongest inhibition. Removal of the bottom aromatic ring (R1) as in analogs 4 and 5 slightly reduced activity, whereas replacement with the heteroaromatic thiophene (as in analog 3) lowered activity even further. The low % inhibition with analog 6 indicates that the top aminothiazole substituent (R2) cannot be replaced with a simple dimethylamino group.
Table 3. Inhibition of ChREBP transcriptional activity with small molecules.
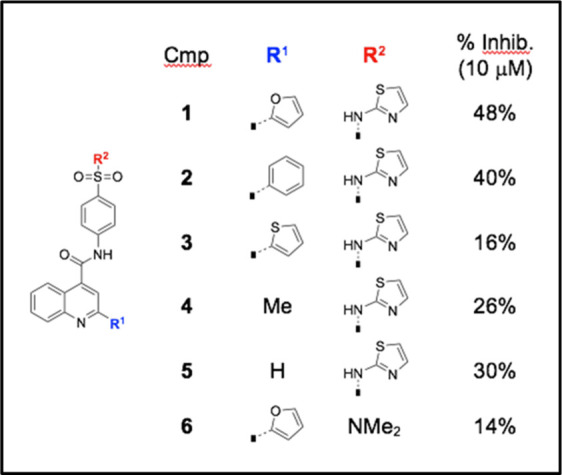 |
The 8000 compound subset is a plate-based diversity subset that is representative of the chemical ‘space’ covered by the larger UT Southwestern institutional library (∼200 000compounds). This subset was constructed in collaboration with Chemical Diversity (ChemDiv, Inc.) with a proprietary algorithm. Like the larger library, the compounds in the subset satisfy a relaxed set of Lipinsky's rules, with 99% having a molecular mass less than 550 (average 250–300), which is optimal for drug-like starting points for developing chemical tools and/or building lead compounds through medicinal chemistry efforts. The HTS Core has a substantial historical data record for this subset (>100 screens), which is very useful in selecting compounds to move forward.
Small-molecule compounds inhibit the NLS–importin α interaction
Since those compounds are inhibitors of ChREBP-NLS–importin α interaction, one would expect the nuclear localization activity of ChREBP is also inhibited. To characterize those inhibitor compounds Furthermore, we selected the compounds 1 and 2 for a dose-dependent inhibition of the NLS peptide- importin α interaction using the FP assay. Compound 1 exhibited a biphasic inhibition curve, suggesting a high affinity (IC50 = 0.7 µM) and low affinity binding site, while compound 2 showed a hyperbolic saturation curve (IC50 = 0.6 µM) (Figure 5A).
Figure 5. Small compounds inhibit importin α-ChREBP-NLS peptides interaction.

FP measurements were carried out as described under Experimental procedure. (A) Dose response curves of compounds 1 (blue), and 2 (red). The double reciprocal plots of the FP reaction between importin α-ChREBP-NLS in the presence of varying concentrations (0 µM (blue ◇), 0.6 µM(red □) and 2 µM (green △) of compound 1 (B), and compound 2 in the presence of (0 µM (blue ◇), 0.6 µM (red □) and 2 µM (green △) (C).
To gain insight into the mechanism of inhibition of the NLS/importin α interaction, we performed the FP assay in varied concentrations of the compounds in the presence of constant concentrations of importin α and the NLS peptide. Double reciprocal plots of these data showed a series of parallel lines indicating that the inhibition is uncompetitive, which suggest these compounds are likely binding to a site other than the importin α/NLS interacting site (Figure 5B,C).
Compounds 1 and 2 inhibit the nuclear localization and transcription activities of ChREBP
To assess whether compounds 1 and 2 inhibit the glucose-stimulated nuclear localization of ChREBP, we expressed GFP-ChREBP in HepG2 cells and determined the nuclear localization activity using a confocal microscope. Our results indicate that, at a concentration of 3 µM, both compounds inhibited the nuclear localization of GFP-ChREBP ∼75% under these conditions (Figure 6A).
Figure 6. Compounds 1 and 2 inhibit the nuclear localization and the transcriptional activities of ChREBP.

(A) The nuclear localization of ChREBP was determined in the reaction mixture containing GFP-ChREBP in the presence of compound 1 (3 µM; O) and compound 2 (3 µM; ◇), under the conditions described under Experimental Procedure. (B) The transcriptional activity of ChREBP was measured in the presence of varying concentrations of compound 1 and compound 2.
Next, we determined that both the compounds 1 and 2 (at 3 µM) inhibited the transcriptional activity of ChREBP ∼30% in HepG2 cells under high glucose (Figure 6B), and the dose response inhibition curves of compound 1 and 2 showed an IC50 value of ∼1 µM, comparable to the ITC binding measurement (Figure 4).
Discussion
Obesity and its associated diseases are among the most serious public health problems facing the U.S. population. Overeating may be the single most significant cause of the current epidemic of obesity. When carbohydrate-intake exceeds short-term requirements for energy, carbohydrates are converted to long term storage as fat. It is now firmly established that ChREBP is the major transcription factor responsible for converting excess carbohydrates to storage fat in the liver. In response to nutrients, especially carbohydrates, ChREBP up-regulates transcription of glycolytic and all lipogenesis enzyme genes, independent of insulin. ChREBP function is regulated at two levels, nuclear localization and transcriptional activity. The first step to activate ChREBP via nuclear localization after dephosphorylation of phospho-ChREBP is the interaction with importin α. By inhibiting this interaction, we may be able to block ChREBP activity, thereby limiting lipogenesis.
Crystal structure of ChREBP-NLS:importin αcomplex
Previously we identified the ChREBP NLS as an extended classical bipartite NLS site minimally encompassing residues Lys158–Lys190 (5). Replacing Lys159/Lys190 residues of ChREBP with alanines resulted in the loss of glucose-stimulated transcriptional activity and nuclear localization. However, in the work described here, we failed to detect any of the C-terminal residues K189KRLR193 of the NLS bound to importin α in the crystal structure, suggesting that the ChREBP NLS (158–190) interacts with importin α as a classical mono-partite signal rather than bipartite NLS [5]. Based on the crystal structure, we suggest that the ChREBP NLS binds to importin α in a mono-partite mode. However, we cannot rule out the possibility of a bipartite ChREBP NLS:importin α interaction as some of the local interactions may be unstable under the crystallization conditions. In fact, secondary structure analyses suggest that the NLS of ChREBP is a flexible and extended α helix, which may explain why the crystal structure failed to reveal the C-terminal region binding to importin α.
Binding of ChREBP NLS to importin α in the major binding site (K171RRI174) is generally similar to analogous structures reported in literature, with the highly conserved lysine on the P2 pocket, arginine/lysine residues in P3 pocket, and arginine residues in P4 pocket of the importin α. A significant difference is that Ile174 of ChREBP displays pi-stacking interactions with the nearby tryptophan residues (Trp142 and Trp184) of importin α, which is unique to the ChREBP NLS interactions.
Among the major binding site residues (Lys171–Ile174) of ChREBP, Arg173 has the most interactions with importin α compared with the other three residues. The guanidino group of the arginine interacts with the main chain carbonyl groups of Leu104, Arg106, and Glu107 of importin α through four hydrogen-bonds. In addition, Arg173 also interacts with P110 of importin α with van der Waals force.
However, mutation of the major binding site residues resulted in only a partial loss of the ChREBP activity, suggesting that the rest of the broad N-terminal region of ChREBP seems to be interacting with importin α in vivo.
In contrast with the ChREBP-NLS/ importin α structure, the prototypical, NLS Simian virus 40 (SV-40) structure [26] occupies both the major and minor importin α binding sites. Both proteins bound to the major sites very closely (Figure 7A). However, Arg172 and Ile174 of ChREBP NLS localized at the P3 and P5 pockets, which is significantly different from that of SV-40 (Figure 7B). Unlike Arg172 of ChREBP, Lys129 of SV-40 was not positioned to enable hydrogen-bonding interactions with Asn228 of importin α. Furthermore, the Lys131 of SV-40 interacts with the importin α Gln181 residue, which is different from ChREBP Ile174. According to published results [23,27], P2, P3 and P5 pockets have highly conserved preferences for long basic-side chains, while P4 has a more diverse interaction. In contrast, the Arg173 and Ile174 residues of ChREBP NLS are localized in the P4 and P5 pockets, respectively. Having a neutral residue in P5 rather than in P4 may be beneficial due to the possibility of pi-stacking interactions within the Trp142.
Figure 7. Comparison between ChREBP NLS and importin α interaction with those of SV-40 and AR.

(A) The superposed structure of importin α bound with ChREBP NLS 158–190 (pink) and SV-40 (tan, 1EJL). (B) Hydrogen bonding interactions of ChREBP (pink) and SV-40 (tan) at the P2–P5 pockets of major binding site of importin α. (C) Superimposed structures of AR (blue) and ChREBP NLS158–190 (pink) bound with importin α. (D) Close-up view of the superimposed structures of AR (blue) and ChREBP NLS 158–190 (pink) in the major site of importin α.
Figure 8. Proposed models for competition between importin α and 14-3-3 proteins in the regulation of nuclear/cytosol localization of ChREBP.

Under low glucose, increased glucagon and cAMP activates PKA leading to phosphorylation of ChREBP-Ser196, which leads to 14-3-3 binding then localization of the ChREBP-14-3-3 complex to the cytoplasm (top). In response to high glucose, the phosphorylated inactive ChREBP is dephosphorylated by Xu5P-activated PP2A, releasing 14-3-3 from the Ser196/NLS site (middle). Importin α also displaces the 14-3-3 protein from its primary binding site at the α2 helix and thereby localizes the ChREBP into the nucleus (bottom).
Similar to ChREBP-NLS, the androgen receptor (AR)-NLS only binds to the major importin α site [22,28]. As shown in Figure 7C, the major importin α binding site sequences reveal that the ChREBP-NLS (KRRI) and AR-NLS (KLKK) each contain a single hydrophobic branched chain amino acid and exhibit 10× lower affinity (or higher KD) than SV40 and nucleoplasmin which contains four basic residues and no hydrophobic amino acid (Table 4). The physiological significance of the lower affinity NLS may suggest that the imported ChREBP–importin α-importin β complex is more rapidly dissociated than the high affinity NLS site in the nucleus in order to aid in the recycling of the import carrier proteins for next rounds of the import process. Another explanation for the weaker affinity of ChREBP and AR is that these proteins have longer linker regions, which are not bound to importin α, and these disordered regions likely contribute to a weaker affinity. In contrast, the NLSs of SV40, which lacks a linker region is able to bind to both major and minor sites. These structural differences are consistent with ChREBP NLS exhibiting a weaker affinity, because of binding only to the major binding site, in contrast with SV40 and nucleoplasmin (Table 4).
Table 4. Comparison of nuclear localization sequences.
| NLS | Sequence | KD (µM) (Ref.) |
|---|---|---|
| ChREBP 158–190 | R158KPEAVVLEGNYWKRRIEVVMREYHKWRIYYKK190 | 3.03 ± 0.95 [5] |
| Nucleoplasmin | K155RPAATKKAGQAKKKK170 | 0.19 ± 0.02 [23] |
| SV40 | P126KKKRKV132 | 0.31 ± 0.15 [26] |
| AR | R617KCYEAGMTLGARKLKKL634 | 5 ± 0.1 [22] |
Key basic residues of NLS for the major binding site are presented in red. The KD value of ChREBP-NLS — importin α was determined with ITC measurement.
Two novel compounds (1 and 2), identified by HTS screening of a small-subset compound library, demonstrate the inhibition of ChREBP-NLS/importin α interaction, nuclear localization, and transcription activities of ChREBP. These candidate small molecules support developing inhibitors of ChREBP that may be useful for the treatment of obesity, diabetes and their associated diseases.
Importin α regulates export of ChREBP from nucleus
We have previously shown that importin α binds to the NLS peptide (residues 158–190) of ChREBP, while 14-3-3β also binds to the adjacent phosphorylated Ser196 [5]. Based on these observations, we concluded that under low glucose conditions, 14-3-3 proteins have a duel function in regulation of cytosol localization of ChREBP: (a) exporting inactive ChREBP(P) out of nucleus, and (b) inhibiting nuclear import by blocking importin α from binding to the NLS site of ChREBP. However, we discovered rather unexpectedly that importin α binds to the α2 helix peptide (residues 117–140) with a KD value of 0.82 µM (Table 2), which is the primary binding site for 14-3-3. 14-3-3β binds to the α2 peptide of ChREBP with the KD values of 2.8 µM (Table 2). These results suggest a new mechanism in which importin α competes effectively with 14-3-3 proteins for its primary binding site (α2 helix) with comparable affinity. Thus, these results also strongly suggest that importin α and 14-3-3 proteins bind competitively to a much broader region of ChREBP than previously thought, which includes the glucose sensing and responsive region (residues 117–196) encompassing α2 and α3 helices in the N- terminal of ChREBP. Furthermore, the ITC and the FP results provide additional support to the idea that the phosphorylation state of Ser196 of ChREBP plays the major role in regulating the subcellular localization in response to glucose [3,29]. Under low glucose conditions, the 14-3-3 protein stimulates not only export of inactive ChREBP into the cytosol, but also inhibits the nuclear localization of ChREBP by blocking importin α from binding to the NLS when Ser196 is phosphorylated. Under high glucose conditions, the current results demonstrated that importin α also plays a dual role in which it not only stimulates nuclear import of ChREBP, but also inhibits 14-3-3 from binding to its primary binding site.
Small compounds inhibitors of nucleus importin of ChrEBP
We report finding two inhibitor compounds (1 and 2) which inhibit the interaction between a ChREBP-NLS peptide and importin α, as demonstrated by FP measurements. However, the inhibitors did not bind to importin α alone suggesting that they bind only to the NLS–importin α-complex. The inhibition appeared to be uncompetitive, which suggest that these compounds may be binding to a site near the interface of the NLS and importin α. Our attempts to crystallize these compounds in an importin α-NLS-inhibitor complex did not succeed, partly due to insolubility of these compounds in aqueous media at concentrations needed for crystallization. In addition to the FP measurements, these compounds also inhibited the nuclear localization and transcriptional activities of ChREBP under high glucose, and the extent of the inhibition was comparable in all three assay methods. Based on these results, we suggest that the ChREBP-NLS is a potential target for drug discovery in treatment of obesity and the associated diseases. Ahn et al. [30] recently reported that a compound termed SBI-477 inhibits intramuscular lipid accumulation by inhibiting triacylglceride synthesis and activated glucose uptake in human skeletal muscle. They obtained evidence that SBI-477 inhibits MondoA. MondoA is a paralog of ChREBP and is expressed mostly in non-hepatic tissues, but its biological function is less well characterized than ChREBP [31]. Since most of the functional domains including NES, NLS and bHLH are highly conserved between those transcription factors, it is not surprising that MondoA is involved in glucose metabolism and fat synthesis in non-hepatic tissues such as skeletal muscle [32]. In contrast with ChREBP, MondoA is known to respond to insulin and suppresses glucose uptake in myocytes by up-regulating the expression of thioredoxin interacting protein (TXNIP) and arrestin domain-containing protein (ARRDC4) [32–34]. Ahn et al. [30] reported that SBI-477 also inhibits the nuclear localization of Mondo A in skeletal myoblasts in response to insulin, but the precise molecular mechanism is unknown. There is no structural similarity between SBI-477 and compounds 1 or 2. They concluded that SBI-477 inhibits MondoA leading to down-regulation of TXNIP and ARRDC4. The subsequent increase in glucose uptake in myocytes results in increased insulin sensitivity [30].
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
The authors would like to acknowledge support from VA Merit Review Grant (K.U.) and the Welch Foundation Grant I-1720 (K.U.).
Open Access
Open access for this article was enabled by the participation of the University of Texas Southwestern Medical Centre in an all-inclusive Read & Publish pilot with Portland Press and the Biochemical Society under a transformative agreement with EBSCO.
Author Contributions
H.J. and K.U. designed the experiments and performed most of the experiments and data analysis. H.J. performed the X-ray crystallography and structure analysis. K.S.M. and J.D. designed and synthesized all intermediates and final compounds presented with the exception of the initial HTS hits, S.W. and B.P. supported and guided assay optimization and screen execution in the HTS Core facility. B.P. and A.Z. analyzed the data from the 8K primary screen and the confirmation screen and provided interpretation of the results. T.N., H.W. and H.S. performed the ChREBP activity assays, T.T. and H.J. performed the fluorescence polarization measurements, Y.L. developed the fluorescence polarization assay for the HTS, R.M.W. made the ITC measurements, and K.U., H.J., R.K.B. and R.M.W. wrote and edited the manuscript.
Data Availability
All data are contained within the manuscript except for the X-ray crystallography data, which is housed at the PDB (6MJL). The model co-ordinates and structural factor amplitudes have been deposited in the Protein Data Bank for structures of the ChREBP and NLS (6MJL).
Abbreviations
- ChREBP
carbohydrate response element binding protein
- NLS
nuclear localization
- TXNIP
thioredoxin interacting protein
Supplementary Material
References
- 1.Yamashita H., Takenoshita M., Sakurai M., Bruick R.K., Henzel W.J., Shillinglaw W. et al. (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl Acad. Sci. U.S.A. 98, 9116–9121 10.1073/pnas.161284298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakiyama H., Wynn R.M., Lee W.R., Fukasawa M., Mizuguchi H., Gardner K.H. et al. (2008) Regulation of nuclear import/export of ChREBP; Interaction of an alpha -helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 283, 24899–24908 10.1074/jbc.M804308200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawaguchi T., Takenoshita M., Kabashima T. and Uyeda K. (2001) Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl Acad. Sci. U.S.A. 98, 13710–13715 10.1073/pnas.231370798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukasawa M., Ge Q., Wynn R.M., Ishii S. and Uyeda K. (2009) Coordinate regulation/localization of the carbohydrate responsive binding protein (ChREBP) by two nuclear export signal sites: discovery of a new leucine-rich nuclear export signal site. Biochem. Biophys. Res. Commun. 391, 1166–1169 10.1016/j.bbrc.2009.11.115 [DOI] [PubMed] [Google Scholar]
- 5.Ge Q., Nakagawa T., Wynn R.M., Chook Y.M., Miller B.C. and Uyeda K. (2011) Importin-alpha protein binding to a nuclear localization signal of carbohydrate response element-binding protein (ChREBP). J. Biol. Chem. 286, 28119–28127 10.1074/jbc.M111.237016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uyeda K. and Repa J.J. (2006) Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110 10.1016/j.cmet.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 7.Fukasawa M., Tsuchiya T., Takayama E., Shinomiya N., Uyeda K., Sakakibara R. et al. (2004) Identification and characterization of the hypoxia-responsive element of the human placental 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene. J. Biochem. (Tokyo) 136, 273–277 10.1093/jb/mvh137 [DOI] [PubMed] [Google Scholar]
- 8.Yang X., Lee W.H., Sobott F., Papagrigoriou E., Robinson C.V., Grossmann J.G. et al. (2006) Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl Acad. Sci. U.S.A. 103, 17237–17242 10.1073/pnas.0605779103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawaguchi T., Osatomi K., Yamashita H., Kabashima T. and Uyeda K. (2002) Mechanism for fatty acid ‘sparing’ effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 277, 3829–3835 10.1074/jbc.M107895200 [DOI] [PubMed] [Google Scholar]
- 10.Iizuka K., Bruick R.K., Liang G., Horton J.D. and Uyeda K. (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl Acad. Sci. U.S.A. 101, 7281–7286 10.1073/pnas.0401516101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iizuka K., Miller B. and Uyeda K. (2006) Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 291, E358–E364 10.1152/ajpendo.00027.2006 [DOI] [PubMed] [Google Scholar]
- 12.Ge Q., Huang N., Wynn R.M., Li Y., Du X., Miller B. et al. (2012) Structural characterization of a unique interface between carbohydrate response element-binding protein (ChREBP) and 14-3-3beta protein. J. Biol. Chem. 287, 41914–41921 10.1074/jbc.M112.418855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontes M.R., Teh T., Jans D., Brinkworth R.I. and Kobe B. (2003) Structural basis for the specificity of bipartite nuclear localization sequence binding by importin-alpha. J. Biol. Chem. 278, 27981–27987 10.1074/jbc.M303275200 [DOI] [PubMed] [Google Scholar]
- 14.Yang S.N., Takeda A.A., Fontes M.R., Harris J.M., Jans D.A. and Kobe B. (2010) Probing the specificity of binding to the major nuclear localization sequence-binding site of importin-alpha using oriented peptide library screening. J. Biol. Chem. 285, 19935–19946 10.1074/jbc.M109.079574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eilers A.L., Sundwall E., Lin M., Sullivan A.A. and Ayer D.E. (2002) A novel heterodimerization domain, CRM1, and 14-3-3 control subcellular localization of the mondoA-Mlx heterocomplex. Mol. Cell. Biol. 22, 8514–8526 10.1128/MCB.22.24.8514-8526.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minor W., Cymborowski M., Otwinowski Z. and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- 17.Potterton E., Briggs P., Turkenburg M. and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 10.1107/S0907444903008126 [DOI] [PubMed] [Google Scholar]
- 18.Vagin A. and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 19.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N. et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emsley P. and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 21.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. et al. (2004) UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 22.Cutress M.L., Whitaker H.C., Mills I.G., Stewart M. and Neal D.E. (2008) Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 121, 957–968 10.1242/jcs.022103 [DOI] [PubMed] [Google Scholar]
- 23.Marfori M., Mynott A., Ellis J.J., Mehdi A.M., Saunders N.F., Curmi P.M. et al. (2011) Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 1813, 1562–1577 10.1016/j.bbamcr.2010.10.013 [DOI] [PubMed] [Google Scholar]
- 24.Chook Y.M. and Blobel G. (2001) Karyopherins and nuclear import. Curr. Opin. Struct. Biol. 11, 703–715 10.1016/S0959-440X(01)00264-0 [DOI] [PubMed] [Google Scholar]
- 25.Kobe B. (1999) Autoinhibition by an internal nuclear localization signal revealed by the crystal structure of mammalian importin alpha. Nat. Struct. Biol. 6, 388–397 10.1038/7625 [DOI] [PubMed] [Google Scholar]
- 26.Kalderon D., Richardson W.D., Markham A.F. and Smith A.E. (1984) Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature 311, 33–38 10.1038/311033a0 [DOI] [PubMed] [Google Scholar]
- 27.Hodel M.R., Corbett A.H. and Hodel A.E. (2001) Dissection of a nuclear localization signal. J. Biol. Chem. 276, 1317–1325 10.1074/jbc.M008522200 [DOI] [PubMed] [Google Scholar]
- 28.Chen M.H., Ben-Efraim I., Mitrousis G., Walker-Kopp N., Sims P.J. and Cingolani G. (2005) Phospholipid scramblase 1 contains a nonclassical nuclear localization signal with unique binding site in importin alpha. J. Biol. Chem. 280, 10599–10606 10.1074/jbc.M413194200 [DOI] [PubMed] [Google Scholar]
- 29.Kabashima T., Kawaguchi T., Wadzinski B.E. and Uyeda K. (2003) Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl Acad. Sci. U.S.A. 100, 5107–5112 10.1073/pnas.0730817100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahn B., Soundarapandian M.M., Sessions H., Peddibhotla S., Roth G.P., Li J.L. et al. (2016) Mondoa coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J. Clin. Invest. 126, 3567–3579 10.1172/JCI87382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Billin A.N., Eilers A.L., Coulter K.L., Logan J.S. and Ayer D.E. (2000) Mondoa, a novel basic helix-loop-helix-leucine zipper transcriptional activator that constitutes a positive branch of a max-like network. Mol. Cell. Biol. 20, 8845–8854 10.1128/MCB.20.23.8845-8854.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoltzman C.A., Peterson C.W., Breen K.T., Muoio D.M., Billin A.N. and Ayer D.E. (2008) Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl Acad. Sci. U.S.A. 105, 6912–6917 10.1073/pnas.0712199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaadige M.R., Looper R.E., Kamalanaadhan S. and Ayer D.E. (2009) Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating mondoA transcriptional activity. Proc. Natl Acad. Sci. U.S.A. 106, 14878–14883 10.1073/pnas.0901221106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patwari P., Chutkow W.A., Cummings K., Verstraeten V.L., Lammerding J., Schreiter E.R. et al. (2009) Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J. Biol. Chem. 284, 24996–25003 10.1074/jbc.M109.018093 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript except for the X-ray crystallography data, which is housed at the PDB (6MJL). The model co-ordinates and structural factor amplitudes have been deposited in the Protein Data Bank for structures of the ChREBP and NLS (6MJL).