

Abstract
N6-Methyladenosine (m6A), the methylation of the adenosine base at the nitrogen 6 position, is the most common epitranscriptomic modification of mRNA that affects a wide variety of biological functions. We have previously reported that hepatitis B viral RNAs are m6A-modified, displaying a dual functional role in the viral life cycle. Here, we show that cellular m6A machinery regulates host innate immunity against hepatitis B and C viral infections by inducing m6A modification of viral transcripts. The depletion of the m6A writer enzymes (METTL3 and METTL14) leads to an increase in viral RNA recognition by retinoic acid–inducible gene I (RIG-I), thereby stimulating type I interferon production. This is reversed in cells in which m6A METTL3 and METTL14 are overexpressed. The m6A modification of viral RNAs renders RIG-I signaling less effective, whereas single nucleotide mutation of m6A consensus motif of viral RNAs enhances RIG-I sensing activity. Importantly, m6A reader proteins (YTHDF2 and YTHDF3) inhibit RIG-I–transduced signaling activated by viral RNAs by occupying m6A-modified RNAs and inhibiting RIG-I recognition. Collectively, our results provide new insights into the mechanism of immune evasion via m6A modification of viral RNAs.
Keywords: m6A modification, immune evasion, RIG-I sensing, innate immunity, viral immunology, cellular immune response, hepatitis B virus (HBV, Hep B), hepatitis C virus (HCV, Hep C)
Hepatitis B virus (HBV) and hepatitis C virus (HCV) are diverse viruses that belong to the Hepadnaviridae and Flaviviridae families, respectively, but share common pathologies (1, 2). Although HBV is a DNA virus, it replicates via an RNA intermediate termed pregenomic RNA (pgRNA) to produce viral DNA by the reverse-transcription activity by the encoded viral polymerase (1). HCV is a positive-sense single-stranded RNA virus and encodes an RNA-dependent RNA polymerase (viral polymerase) to replicate viral RNA from a template RNA (2). HBV and HCV infections cause liver inflammation and carry the risk for the development of hepatocellular carcinoma (3). When a virus infects a host cell, an innate immune response, which represents the first line of defense, is triggered within minutes. Viruses, however, escape host innate immunity by subverting these pathways to optimize long-term survival (4). The HBV pgRNA and HCV RNA genomes are recognized by RIG-I for the activation of interferon (IFN) synthesis (5, 6). HCV nonstructural proteins (NS3/4 protease) cleave mitochondrial antiviral-signaling protein (MAVS) to block the innate immune signals (7). However, HCV infection still induces IFN synthesis in primary human hepatocyte and in the chimpanzee model, representing acute infection (8, 9). Although RIG-I recognizes the HBV pgRNA to activate the immune response, HBV successfully suppresses IFN-α/β synthesis (5, 10, 11). One of the mechanisms among others has been attributed to HBx protein, which disrupts RIG-I/MAVS signals by ubiquitination of mitochondria-associated MAVS protein (12–14). The mechanisms by which these viruses suppress innate immunity continue to be the subject of investigations.
The RIG-I signaling pathway is one of the host-defense systems, designed to eliminate the viral infection. RIG-I protein consists of tandem N-terminal caspase activation and recruitment domains (CARDs) that participate in signaling, a DExD/H-box helicase domain with RNA binding and ATP-hydrolysis activity, and a C-terminal domain. The 5′ ppp of double-strand RNA is the ligand of RIG-I (15). In the absence of ligand RNA, RIG-I adopts auto-repressed conformation (16). The conformational change of RIG-I is induced by RNA ligand interacting with a C-terminal domain, which releases CARDs for Lys63-linked polyubiquitination and binding of unanchored Lys63-linked ubiquitin chains (17). The polyubiquitinated CARDs then engage the adaptor, MAVS via CARD–CARD domain interactions, to activate a cascade of downstream signaling resulting in type I IFN gene synthesis (18). Phosphorylation of IRF-3, a transcription factor, is one of the key molecules in the pathway promoting the transcription of IFN genes (19).
The mammalian mRNAs are modified by diverse chemical modifications such as N6-methyladenosine (m6A), 5-methlcytidine, and inosine in addition to N7-methylguanosine, which is part of the 5′-terminal cap (20). Among the diverse internal RNA chemical modifications, m6A RNA methylation is the most prevalent mRNA modification. (21). Over 25% of mammalian transcripts are m6A-modified. m6A methylation has been linked to various biological processes, which include; innate immune response, sex determination, stem cell differentiation, circadian clock, meiosis, stress response, and cancer (21). A methyltransferase complex composed of METT3 (methyltransferase-like 3), METTL14, and WTAP place m6A modification on mRNA co-transcriptionally (21). This modification is typically enriched in 3′-UTR and near the stop codons of cellular mRNA. The m6A-modified mRNA is especially recognized by YTH-domain family proteins (YTHDF1, YTHDF2, and YTHDF3) to regulate mRNA stability, translation, and localization (22, 23). Identification of m6A demethylase FTO (fat mass and obesity-associated protein) and ALKBH5, capable of eliminating m6A methylation, suggests that m6A modification is reversibly catalyzed by methyltransferase and demethylase (24). Recently, the presence of chemical modification has also been demonstrated in many viral RNAs, including HIV, HBV, and HCV (25–30). HBV encodes a single m6A-modified consensus DRACH motif, localized in the ε stem-loop of HBV transcripts. pgRNA carries this motif twice at the 5′ and 3′ termini because of terminal redundancy (26). The m6A at 5′ ε stem-loop is in the vicinity of the RT initiation priming site of pgRNA and aids in the RT activity, whereas m6A at the 3′ ε stem-loop in all viral transcripts affects RNA stability (26). In the case of HCV, the plus polarity RNA genome contains several m6A-modified regions (∼19 regions) (24). YTHDFs interactions with the viral genome have been shown to regulate virus particle production (27). The m6A site in HCV E1 genomic region reduces virus particle production through inhibiting virus assembly by interaction with YTHDF proteins (27). Surprisingly, these modifications are more frequent in viruses than cellular mRNAs (29, 31, 32). Although differences in the role of m6A among viruses can lead to different consequences in their life cycle, these reports reinforce the notion that these RNA modifications might play an important role in various aspects of the viral life cycle and most importantly in disease pathogenesis.
In this study, we define the role of m6A-modified HBV transcripts and the HCV RNA genome in regulating the host innate immune systems. We demonstrate that cellular m6A machinery regulates the RIG-I signaling pathway activated by virus infection. Importantly, m6A modification in the RIG-I–sensing region of viral RNAs reduces innate immune response by inhibition of RIG-I recognition of viral RNAs. Furthermore, we found that YTHDF2 binds m6A-modified viral RNAs preventing their recognition by RIG-I. Taken together, our results reveal a novel mechanism of immune evasion via m6A modification of viral RNA, where YTHDF2 binds m6A-modified motifs of viral RNAs, preventing recognition by RIG-I.
Results
m6A methyltransferase negatively regulates innate immune response
To determine whether m6A modification of viral RNA affects host immune response, we used the previously developed mutants of m6A modification sites (A1907C) of HBV 1.3-mer; in the pgRNA 5′ stem-loop (5′-MT), or the 3′ stem-loop (3′-MT) (Fig. 1A). The p-IRF-3 expression level was up-regulated in HBV 1.3-mer 5′-MT transfected cells compared with WT transfection, whereas mutation of m6A site in the 3′ stem-loop did not affect phosphorylation of IRF-3 (199 ± 25.7% increase of p-IRF-3 levels in 5′-MT transfection; Fig. 1B). Poly(I:C) serves as a positive control. Because the mutation of m6A site in ε structure leads to a base-pair mismatch in lower stem-loop, the secondary structure of lower stem-loop could be distorted by A1907C mutation (Fig. 1C). Because the induced p-IRF-3 by pHBV 1.3-mer 5′-MT transfection could be due to this structure alteration, we analyzed p-IRF-3 levels in cells transfected with compensatory mutant (CM) plasmid, in which U is mutated to G to restore base pairing (Fig. 1, C and D). Importantly, we found that p-IRF-3 levels were similar to the original mutant (5′-MT), suggesting that helical structural change did not cause this effect. Next, we determined whether cellular m6A machinery affects innate immune response activated by the presence of HBV RNA, and we depleted METTL3 and 14 by siRNA in HepG2 hepatoma cells and transfected these cells with HBV 1.3-mer plasmid. The silencing of m6A methyltransferases increased phosphorylation of IRF-3 induced by HBV transfection relative to its level in cells treated with control siRNA (178 ± 20.21% increase of p-IRF-3 level in METTL3 + 14 depletion) (Fig. 1E), whereas METTL3 and 14 overexpression reduced phosphorylation of IRF-3 (49 ± 5.69% decrease of p-IRF-3 in METTL3 and 14 overexpression) (Fig. 1F). Interestingly, HBV transfection in HepG2 cells induces IRF-3 activation but does not induce IFN synthesis (5). Thus, we could analyze only p-IRF-3 levels in HBV transfected cells. Previously, we reported that m6A modification of 5′ ε structure induces reverse-transcription activity, and m6A modification of 3′ ε structure reduces viral RNA stabilities. Thus, our observed effects of the silencing of m6A methyltransferases on IRF-3 activation could be due to the alterations of HBV replication and translation. To exclude this possibility, we depleted m6A machinery in pHBV 1.3-mer 5′-MT or 3′-MT transfected cells and analyzed phosphorylation of IRF-3 levels (Fig. S1, A and B). The knockdown of METTL3 and 14 did not affect p-IRF-3 levels in pHBV 1.3-mer 5′-MT transfected cells, whereas depletion of m6A methyltransferases induced phosphorylation of IRF-3 in pHBV 1.3-mer 3′-MT transfected cells. Collectively, these results suggest that m6A modification of HBV 5′ ε RNA critically determines IRF-3 activation during HBV replication. Because it is known that the 5′ ε structure is recognized by RIG-I to induce the innate immune response (5), our results imply that m6A modification of 5′ ε structure may affect RIG-I signal transduction.
Figure 1.
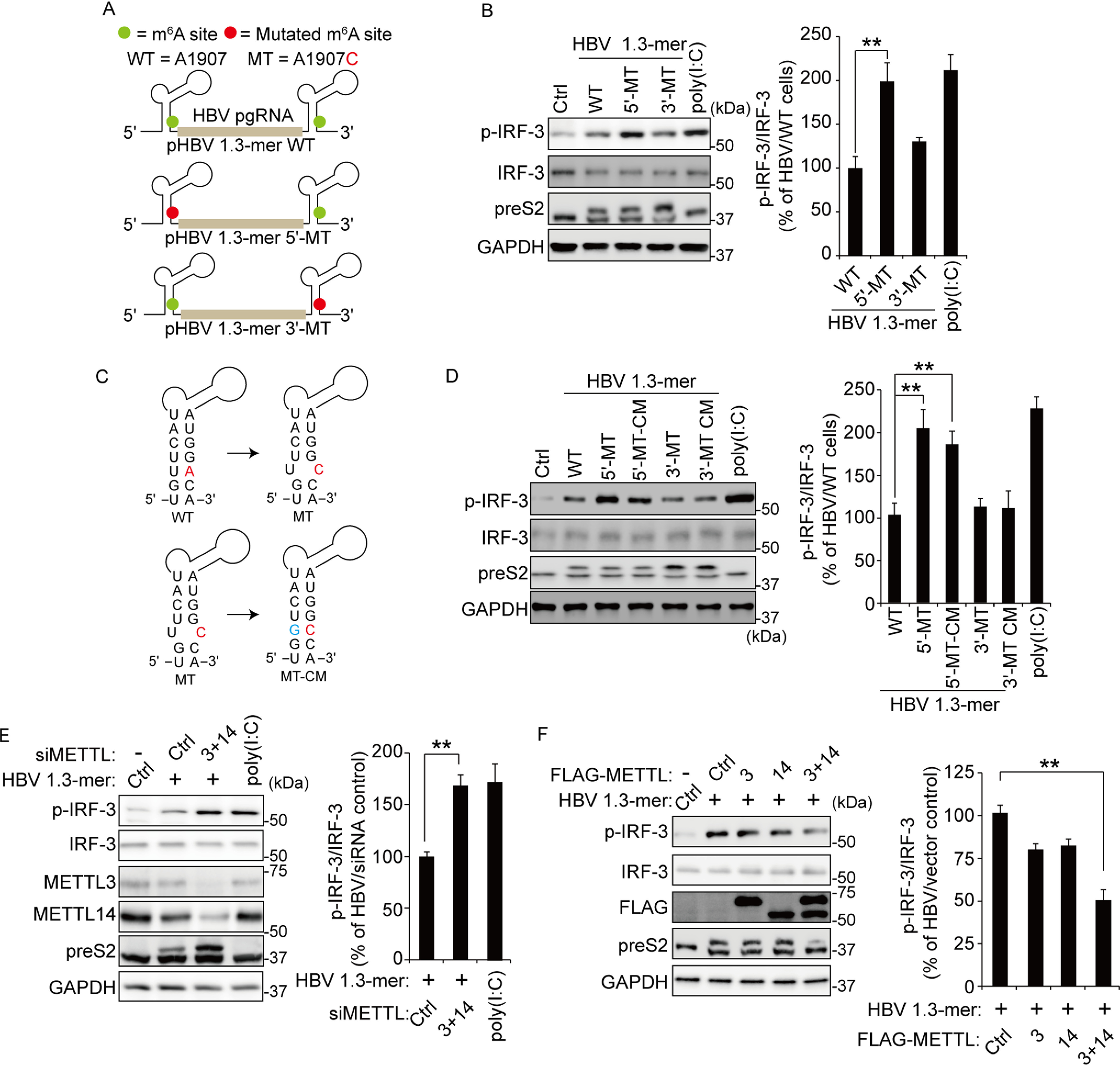
The m6A modification of HBV pgRNA affects activation of IRF-3. A, schematics represent the location of the A1907C mutations of 5′ and 3′ m6A sites in HBV pgRNA. Green circles indicate the m6A site in HBV pgRNA. The pHBV 1.3-mer 5′-MT having the A1907C mutation at the 5′ end and pHBV 1.3-mer 3′-MT at the 3′ end. B, HepG2 cells were transfected with each indicated HBV plasmid. After 72 h, the cells were harvested to assess expression levels of p-IRF-3. Poly(I:C) was transfected in HepG2 cells before harvesting for 16 h. The right panel shows that p-IRF-3 protein levels relative to the IRF-3 from three independent experiments were quantified using ImageJ. C, the mutation of m6A site (red) in the ε structure is predicted to create a bubble. The compensatory U1851G mutation (green) was established either at the 5′ end (pHBV-5′-MT–CM) or the 3′ end (pHBV-3′-MT–CM). D, the indicated plasmids were transfected in HepG2 cells for 72 h. The lysates were extracted from these cells to analyzed p-IRF-3 levels. The p-IRF-3 levels were normalized by IRF-3 using ImageJ (in the right panel). E, relative p-IRF-3 levels in HBV 1.3-mer transfected HepG2 cells 4 h after siMETTL3 + 14 or control siRNA transfection was assessed by immunoblotting. HepG2 cells were stimulated by poly(I:C) for 16 h. The p-IRF-3 levels relative to IRF-3 from three independent experiments were quantified using ImageJ. F, HepG2 cells treated with HBV 1.3-mer were transfected with control or FLAG-METTL3/14 plasmids. After 72 h, the indicated proteins were analyzed by immunoblotting. The levels of p-IRF-3 relative to the IRF-3 from three independent experiments were quantified using ImageJ (in the right panel). In B and D–F, the error bars are the standard deviations of three independent experiments, each involving triplicate assays. Statistical significance of the difference between groups was determined via an unpaired Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Ctrl, control.
We also carried out similar experiments during HCV RNA transfection. We depleted METTL3 and 14 by siRNAs transfection of Huh7 cells and transfected these cells with in vitro transcribed HCV GND RNA from JFH-1 GND plasmid (Fig. 2, A–C). JFH-1 GND contains a point mutation in the viral polymerase region; hence it does not replicate but RIG-I sensing is normal (33). Because HCV NS3/4 protein expression inhibits the immune response by cleaving the MAVS protein (7), we surmised that transfection with the replication-defective mutant HCV genome instead of replication efficient HCV RNA was suitable for immune response studies. We determined IFN-β mRNA and IRF-3 phosphorylation levels in cells harvested at 16 h post-transfected with HCV GND RNA. We observed that METTL3 and 14 depletion significantly increased IFN-β synthesis and IRF-3 activation relative to its level in cells treated with control siRNA (1.6-fold increase of IFN-β mRNA in METTL3 + 14 depleted cells; 177 ± 15.5% increase of p-IRF-3 level in METTL3 + 14 depleted cells; Fig. 2, A and C). Conversely, overexpression of m6A methyltransferases complexes decreased IFN-β mRNA levels and phosphorylation of IRF-3 (2.1-fold decrease of IFN-β mRNA in METTL3 + 14 overexpression; 58 ± 17.1% decrease of p-IRF-3 level in METTL3 + 14 overexpression) (Fig. 2, D and F). These results suggest that host m6A machinery regulates the IRF-3–mediated IFN signaling pathway induced by the HCV RNAs in the early step of HCV infection.
Figure 2.
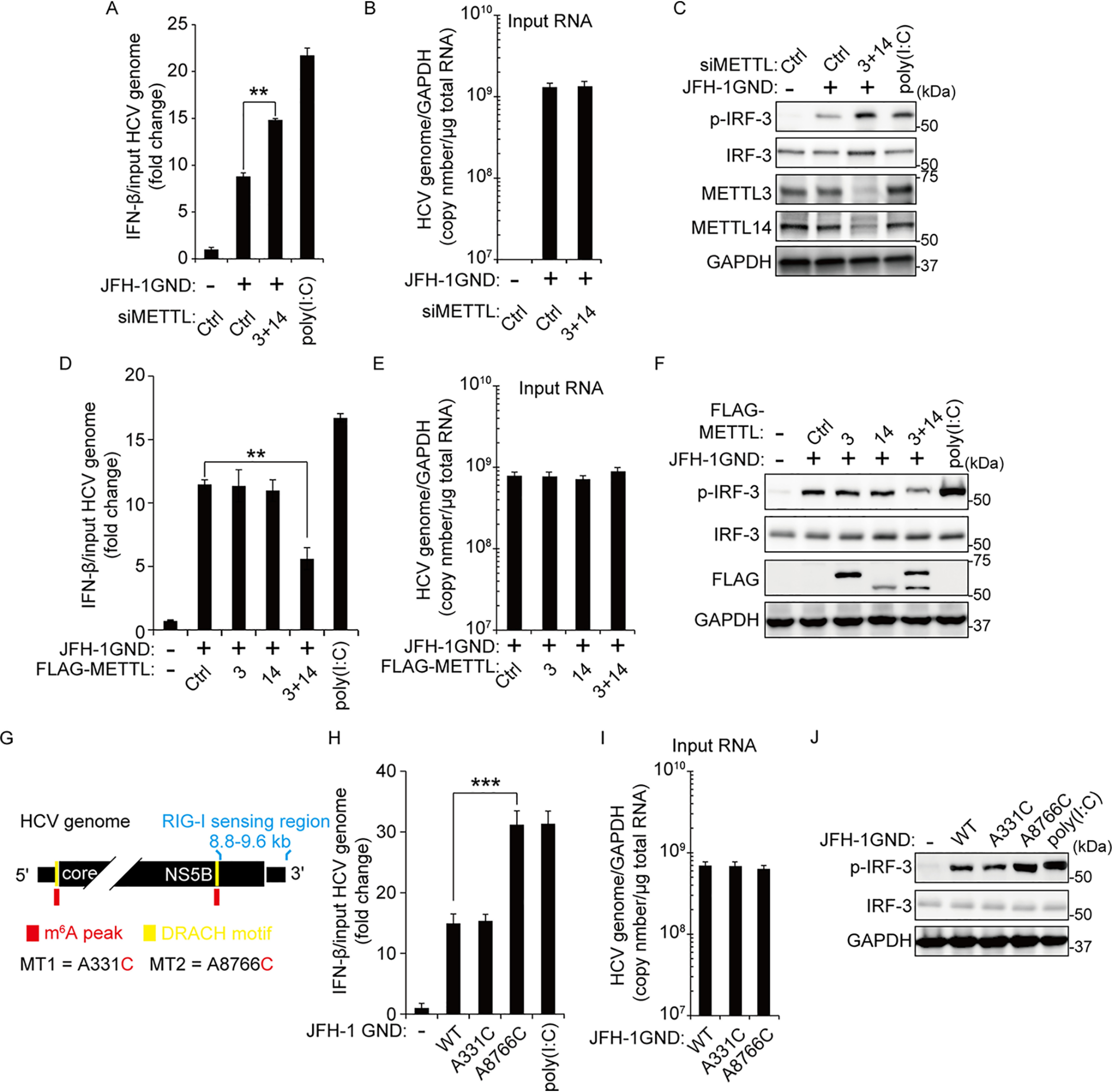
The m6A modification of HCV genome inhibits IRF-3–mediated IFN signals. A–C, Huh7 cells treated with siMETTL3 + 14 or control siRNA were transfected with JFH-1 GND RNA. IFN-β mRNA levels were assessed by qRT-PCR at 16 h post–JFH-1 GND RNA transfection. The HCV genome titers were analyzed by qRT-PCR for normalizing IFN-β mRNA (B). The p-IRF-3 protein levels were analyzed by immunoblotting in whole-cell lysates (C). D–F, FLAG-METLL3/14 transfected with Huh7 cells were treated with JFH-1 GND RNA for 16 h. The cells were harvested to assess expression levels of IFN-β mRNA (D) or the indicated proteins (F). The input HCV genome levels were quantified for calculating IFN-β mRNA (E). G, schematics indicate the location of the A8766C mutation of the m6A site in the NS5B-3′-UTR region of the HCV genome. The A331C mutation is in domain IV of the HCV IRES region. Red indicates m6A peak by MeRIP analysis. Yellow indicates the DRACH motif in the m6A peak. H–J, qRT-PCR analysis of IFN-β mRNA relative to input HCV genome in JFH-1 GND RNA or JFH-1 GND A8766C RNA transfected with Huh7 cells. The cells were harvested at 16 h post-transfection, and the relative levels of IFN-β mRNA (H) or p-IRF-3 (J) were analyzed. The input JFH-1 GND RNA levels were analyzed by qRT-PCR (I). JFH-1 A331C RNA was used as a control. The data for this figure are from three independent experiments, and the bars represent the means ± S.D. Statistical significance of the difference between groups was determined via an unpaired Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Ctrl, control.
After determining that m6A machinery affects IFN signaling pathway activated by the HCV RNA, we next tested whether m6A modification within pathogen-associated molecular patterns (PAMPs) recognized by RIG-I affects IFN-β mRNA level and phosphorylation of IRF-3. Saito et al. (6) identified RIG-I–sensing nucleotides 8872–9616 in the 3′ end of the HCV genome, which acts as a PAMP. There are approximately 19 regions that are m6A-modified within the HCV RNA genome (27). We analyzed whether the HCV PAMP RNA contains m6A-methylated sites. Unexpectedly, the HCV PAMP RNA did not contain any m6A modification (DRACH motif), but an m6A modification site was identified at nucleotide 8766 of HCV RNA ∼100 bp upstream of the PAMP RNA (Fig. 2G) (27). Because m6A methyltransferases regulated IFN signaling pathway activated by HCV RNA transfection, we assessed the possibility that m6A modification of HCV 8766 nucleotide affects the IRF-3–mediated IFN signaling pathway (Fig. 2, H and J). We addressed this question by mutating m6A sites at nucleotides 8766 (A8766C) and 331 (A331C) of HCV GND RNA, respectively. HCV A331C mutant genome was used as a negative control because it lacks an m6A DRACH consensus and is not a RIG-I–sensitive site. Initially, we analyzed IFN-β mRNA levels in A8766C-mutated HCV genome-transfected cells and found that A8766C mutation, but not A331C mutation, substantially increased IFN-β mRNA levels compared with the HCV GND WT infected cells. The A8766C mutated HCV RNA transfection also increased phosphorylation of IRF-3 (2.6-fold increase of IFN-β mRNA level and 253 ± 41.8% increase of p-IRF-3 level in HCV GND A8766C RNA transfected cells; Fig. 2, H and J). We next tested whether the depletion of m6A machinery affects IFN signals activated by HCV GND A8766C because other m6A sites of the HCV genome might affect IFN-β mRNA and IRF-3 activation. Interestingly, the silencing of METTL3 and 14 did not increase IFN-β mRNA and phosphorylation of IRF-3 in HCV GND A8766C transfected cells but induced IRF-3–mediated IFN-β expression in HCV GND A331C transfected cells (Fig. S2). These results illustrate that m6A modification of HCV 8766 nucleotide affects the function of HCV PAMP RNA. In particular, the deficiency of m6A modification in other nucleotides of the HCV genome may not affect the IRF-3–mediated IFN signal pathway.
Because the effect of depletion of m6A machinery can directly regulate IRF-3–mediated IFN signals, we analyzed IFN-β mRNA and phosphorylation of IRF-3 activated by poly(I:C) in METTL3- and METTL14-depleted cells. The depletion of m6A methyltransferases did not affect IFN-β mRNA and IRF-3 activation, suggesting that m6A modification of specific sites of viral RNAs is important in IRF-3–mediated IFN signals (Fig. S3).
m6A modification of viral RNA regulates recognition by RIG-I
Having found that the m6A modification of viral RNAs (HBV pgRNA and HCV RNA genome) regulates phosphorylation of IRF-3, we investigated whether the m6A modification of viral RNAs affects RIG-I signal transduction. We transfected pHBV 1.3-mer or 5′-MT in RIG-I–depleted cells and found that HBV transfection did not induce phosphorylation of IRF-3 in the absence of RIG-I (Fig. 3A). Similarly, HCV genome (GND or GND A8766C) transfection did not increase IFN-β mRNA and phosphorylation of IRF-3 in RIG-I–depleted cells (Fig. 3, B and C). These results reveal that m6A modification of viral RNAs regulates IFN signals which depends on RIG-I protein.
Figure 3.

The mutation on the m6A site of viral RNA increases interaction with RIG-I. A, HepG2 cells treated with siRNA of RIG-I or control siRNA were transfected with pHBV 1.3-mer or pHBV 1.3-mer 5′-MT. The p-IRF-3 levels were assessed by immunoblotting at 72 h post-pHBV 1.3-mer transfection. B and C, relative IFN-β mRNA (B) or p-IRF-3 (C) levels in HCV GND or A8766C transfected Huh7 cells 32 h after siRIG-I or control siRNA transfection were assessed by qRT-PCR or immunoblotting at 16 h post-HCV RNA transfection. Relative IFN-β mRNA levels were normalized by input HCV GND or A8766C RNA levels. The input HCV GND or A8766C RNA levels were analyzed by qRT-PCR, as shown in Fig. S4A. D, RNA immunoprecipitation from the indicated cells using anti-FLAG antibody, with qRT-PCR analysis of HBV pgRNA quantified as relative enrichment RNA levels (in the left panel). The enrichment RNA levels were normalized by input HBV pgRNA levels, as shown in Fig. S4B. Immunoblot analysis of FLAG–RIG-I in the input and IP is shown on the right panels. E, enrichment of HCV genome following immunoprecipitation of FLAG-tagged RIG-I from extracts of Huh7 cells 48 h after transfection. Enriched HCV genome was quantified by qRT-PCR as fold enrichment relative to control. The enrichment HCV RNA levels were normalized by input HCV RNA levels. The input HCV RNA levels are shown in Fig. S4C. FLAG-tagged RIG-I levels in input and IP were assessed by immunoblotting (in the right panel). In B, D, and E, the error bars are the standard deviations of three independent experiments. The P values were calculated using an unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Because the silencing of RIG-I inhibited p-IRF-3 levels induced by HBV and HCV, we tested whether m6A modification affects the interaction between viral RNA and RIG-I. We transfected FLAG-tagged RIG-I protein-expressing cells with plasmids encoding HBV 1.3-mer WT, 5′-MT, or 3′-MT and performed immunoprecipitation using an anti-FLAG antibody. Fig. 3D shows that FLAG–RIG-I immunoprecipitates were enriched with HBV pgRNA relative to control. Interestingly, the mutation in the m6A site of HBV 5′ ε dramatically increased RIG-I recognition compared with the WT and 3′-MT expressing cells, suggesting that the m6A modification of 5′ ε plays a critical role in RIG-I recognition. We also analyzed whether the m6A modification of nucleotide 8766 of the HCV genome affects RIG-I sensing activity. Fig. 3E shows that RIG-I prefers m6A site (A8766C) mutated HCV GND genome rather than the HCV GND WT RNA genome. These results demonstrate that m6A modification in the HBV PAMP RNA and specific sites near the HCV PAMP region render them less sensitive to RIG-I, because the mutations in those sites enhance RIG-I interaction substantially. Overall, these observations suggest that HBV and HCV use m6A modification of their viral RNAs to avoid RIG-I recognition as non–self-ligand.
YTHDF2 and YTHDF3 proteins affect RIG-I signal pathway
Recently, several studies illustrated that the YTHDF (YTH domain-containing protein family) proteins (m6A readers) can bind to m6A-modified mRNA and regulate the stability and translation of target RNAs (22, 23). Therefore, we investigated the possibility that YTHDF proteins affect RIG-I signal transduction induced by HBV and HCV via interacting with m6A-modified viral RNAs. We transfected each HBV 1.3-mer or 5′-MT–expressing cells with plasmids encoding FLAG-YTHDF1, YTHDF2, or YTHDF3 and analyzed p-IRF-3 expression levels (Fig. 4, A and B, and Fig. S5, A and B). Immunoblot analysis shows that YTHDF2 and YTHDF3, but not YTHDF1, reduced p-IRF-3 expression levels induced by HBV 1.3-mer transfection, whereas in HBV 1.3-mer 5′-MT plasmid transfected cells, YTDHF proteins did not affect phosphorylation of IRF-3. Furthermore, the silencing of YTHDF2 and YTHDF3 induced the phosphorylation of IRF-3 in HBV 1.3-mer transfected, whereas no effect of silencing was observed with p-IRF-3 expression levels in HBV 1.3-mer 5′-MT transfected cells (Fig. 4, C and D, and Fig. S5, C and D).
Figure 4.

YTHDF2 and 3 proteins inhibit the IRF-3–mediated immune response activated by HBV and HCV. A and B, FLAG-YTHDFs (1, 2, and 3) plasmids were transfected into HepG2 cells. After 4 h, the cells were transfected with HBV 1.3-mer or HBV 1.3-mer 5′-MT plasmids for 72 h. The indicated protein levels were analyzed by immunoblotting. C and D, siRNAs of YTHDFs (1, 2, and 3) were treated into HepG2 cells. After 4 h, the cells were transfected with HBV 1.3-mer or HBV 1.3-mer 5′-MT plasmids. After 72, the cells were harvested to assess expression levels of p-IRF-3 protein levels. E and F, FLAG-YTHDFs (1, 2, and 3) plasmids were transfected into Huh7 cells. After 32 h, the cells were transfected with JFH-1 GND or A8876C RNA for 16 h. IFN-β mRNA and p-IRF-3 expression levels were analyzed by qRT-PCR and immunoblotting, respectively. G and H, siRNAs of YTHDFs (1, 2, and 3) were treated into Huh7 cells. After 32 h, the cells were transfected with JFH-1 GND or A8876C RNA. After 16 h, the cells were harvested to assess the expression levels of p-IRF-3 protein. In E–H, IFN-β mRNA levels were calculated by input HCV RNA levels. Fig. S5 (I–L) shows the input HCV copy number. The error bars are the standard deviations of three independent experiments. P values were calculated using an unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. n.s., not significant by unpaired Student's t test; Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
After determining that YTHDF proteins regulate phosphorylation of IRF-3 in HBV replicating cells, we next investigated whether YTHDF proteins could similarly affect the IRF-3–mediated IFN signaling pathway in HCV GND RNA transfected cells. We observed that overexpression of YTHDF2 and YTHDF3 significantly reduced p-IRF-3 and IFN-β mRNA levels activated by HCV GND RNA transfection (Fig. 4E and Fig. S5E), and depletion of YTHDF2 and YTHDF3 substantially increased IFN signaling pathway in cells transfected with HCV GND RNA (Fig. 4G and Fig. S5G). However, YTHDF2 and YTHDF3 did not affect p-IRF-3 and IFN-β mRNA levels in cells transfected with HCV GND RNA mutated within the m6A site (A8766C) (Fig. 4, F and H, and Fig. S5, F and H). These results reveal that YTHDF2 and YTHDF3 play a critical role in the RIG-I signal pathway activated by the presence of viral m6A containing RNAs.
In addition, we investigated whether YTHDF2 and YTHDF3 interact with the m6A site-mutated HBV pgRNA. All YTHDF proteins (YTHDF1, 2, and 3) bound to HBV pgRNA (Fig. S6). HBV pgRNA was more enriched by YTHDF2 and YTHDF3 than YTHDF1. We found that mutation on the m6A site of the 5′ ε structure of pgRNA decreased binding affinity with YTHDF2 and YTHDF3 compared with WT, but did not affect interaction with YTHDF1 (Fig. S6A). However, the 5′ ε mutated pgRNA levels enriched by YTHDF2 and YTHDF3 were not dramatically reduced to compare with WT, because YTHDF proteins can still recognize the 5′ ε mutated pgRNA through m6A-modified 3′ ε of pgRNA. We also analyzed whether mutation of HCV nucleotide 8766 m6A site affects interaction with YTHDF proteins, but this mutation did not affect binding affinity between HCV genome and YTHDFs (Fig. S6C). In fact, m6A modification of HCV RNA occurs in several regions (∼19 regions). Because YTHDFs recognize other multiple m6A-modified sites in the RNA genome, we could not observe that mutation of nucleotide 8766 m6A site affect interaction with YTHDFs. Together, these results suggest that m6A reader proteins (YTHDF2 and YTHDF3) inhibit the RIG-I signaling pathway activated by HBV and HCV, raising the possibility that the ability of YTHDF2 and YTHDF3 in regulating immune response is due to interaction with m6A-modified viral RNAs.
m6A modification of viral RNA inhibits RIG-I recognition through recruitment of m6A reader proteins
To verify whether the YTHDF2 protein has any impact on RIG-I recognition of viral RNA, we performed immunoprecipitation experiments using cell lysates from siYTHDF2 (depleted of YTHDF2 expression), FLAG–RIG-I, and HBV 1.3-mer transfected cells. The results presented in Fig. 5A indicated that the siYTHDF2 transfection increased interaction between RIG-I and HBV WT pgRNA. However, siYTHDF2 did not affect RIG-I recognition of 5′ ε m6A site-mutated HBV pgRNA. CREBBP RNA is being used as a positive host RNA control. Similarly, we investigated the effect of YTHDF2 silencing on RIG-I recognition of the HCV RNA genome. In Fig. 5B, RT-qPCR analysis shows that the knockdown of YTHDF2 induced RIG-I sensing activity to the HCV genome, whereas the interaction between mutated nucleotide 8766 m6A site of HCV genome and RIG-I was not affected by YTHDF2 silencing. Although mutation of HCV nucleotide 8766 m6A site could not affect overall interaction between YTHDFs and its viral RNA (Fig. S6C), the data in Fig. 5B demonstrates that interaction of YTHDF2 with HCV nucleotide 8766 is important in regulating RIG-I recognition. Taken together, our results reveal that YTHDF2 inhibits RIG-I recognition of viral RNAs via interacting with m6A-modified viral RNAs, leading to disrupt RIG-I–mediated immune response (Fig. 6).
Figure 5.

The knockdown of YTHDF2 induces RIG-I recognition of viral RNAs. A, HepG2 cells were treated with siYTHDF2, and then the cells were transfected FLAG–RIG-I and HBV expressing vectors (WT or 5′-MT). After 48 h, the cells were harvested. RNA immunoprecipitation from purified lysates using anti-FLAG antibody, with qRT-PCR analysis of HBV pgRNA was quantified as relative enrichment RNA level. The enriched HBV pgRNA levels were calculated by input HBV pgRNA levels. The input HBV pgRNA levels are represented in Fig. S7A. Immunoblot analysis of FLAG–RIG-I in the input and IP is shown in the right panels. B, siYTHDF2 treated into Huh7 cells were transfected with FLAG–RIG-I plasmid and HCV RNA (JFH-1 GND RNA or JFH-1 GND A8876C RNA). After 48 h, the cells were harvested to assess enriched HCV RNA by FLAG-tagged RIG-I. Enriched HCV RNA levels were normalized by input HCV RNA. The input HCV RNA levels have shown in Fig. S7B. The P values were calculated using an unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. n.s., not significant by unpaired Student's t test; Ctrl, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 6.

Model for virus evading immune response via m6A modification of their genomic RNA. The viral genomic RNA is recognized by RIG-I, leading to produce IFN signals. The m6A modification of viral genomic promotes interaction with YTHDF2 and the YTHDF2 binding to viral RNA disrupts RIG-I sensing activity.
Discussion
The new roles of m6A in regulating the biological functions of RNAs are constantly emerging. Recent studies have shown that several RNA viral genomes, as well as the RNA transcripts of DNA viruses, are m6A-modified, and this m6A modification of viral RNAs affects various aspects of the viral infection and associated pathogenesis (25–27, 29, 31, 32, 34). In this study, we examined the mechanism by which antiviral immune response is regulated by cellular m6A machinery. YTHDF proteins are considered reader proteins that bind m6A-modified RNAs and in doing so can block the interaction of RIG-I and affect downstream IFN synthesis (Fig. 6). Here, we demonstrated that m6A modification of HBV pgRNA and HCV genomic RNA permits their interactions with YTHDF proteins and thereby inhibits RIG-I recognition. This is shown by silencing methyltransferases (METTL3/14) and YTHDF proteins, respectively, and by using m6A-deficient mutants.
The host presents multiple immune defense systems against microbes, but viruses have evolved effective mechanisms to evade host immune systems to maintain persistent infection (4). HBV is considered a stealth virus because of its ability to evade host immune response and establish a chronic infection (35). Several studies have shown diverse strategies for HBV in immune evasion. HBV induces Parkin-dependent recruitment of the linear ubiquitin assembly complex to mitochondria to attenuate innate immunity (12–14). During HCV replication, HCV genome is sensed as a PAMP, leading to activation of the innate immune response and adaptive immune response. Despite activated host immune systems, 70–80% of people with acute HCV infection develop chronic infections, because the virus has evolved to escape host immune systems by several mechanisms (36). One of these mechanisms is that HCV NS3/4 protease-induced cleavage of MAVS, which blocks downstream IFN signaling pathway (7). Despite these findings, the immune evasion mechanisms of HBV and HCV are still being actively investigated. Here, we present yet another strategy, utilized by HBV and HCV to evade the host immune system through m6A modification. The deficiency of m6A modification of viral RNA enhanced RIG-I binding affinity and stimulated activation of IRF-3, leading to increased IFN synthesis (Figs. 1–3). Thus, m6A methylation may act as a brake to prevent RIG-I signal transduction. One of the characteristics of m6A modification in distinguishing the self-RNAs from the non–self-RNAs has been recently highlighted, based on the findings that m6A suppresses recognition by Toll-like receptors 3 and 7 and RIG-I (37–39). In view of this, m6A modifications of HBV and HCV RNAs may serve as molecular signatures for distinguishing self- from non–self-RNAs through RNA sensor RIG-I, thus evading RIG-I recognition by mimicking to present as cellular RNA via m6A modification. RNAs containing m6A modification fail to trigger RIG-I conformational change needed for its activation (39). Because a large fraction (70–80%) of HCV infections become chronic, m6A modification may serve as self-RNA in evading immune response to promote chronic hepatitis. m6A modification strategy may additionally contribute to the successful evasion of innate immunity by HCV via MAVS cleavage/inactivation. It remains to be investigated whether m6A modifications of other viral RNAs that are recognized by RIG-I regulate the RIG-I signaling pathway. If proven, the inhibition of RIG-I signals by m6A modification of viral RNA can be one of the common immune evasion pathways used by viruses to maintain persistent viral infection. This hypothesis is supported by the findings that m6A modification frequently occurs in viral RNAs than cellular RNAs (31, 34). Consistent with our observations described here is the report of human metapneumovirus m6A modification, which enables the viral RNA to escape recognition by RIG-I (40). Our results suggest that this immune evasion strategy facilitates the maintenance of chronic infection via regulating m6A modification of viral RNA. In addition to m6A modification, virus RNAs are also modified by several other chemical modifications including 5-methylcytosine, uridine to pseudouridine (U to Ψ), and adenosine to inosine (A to I) editing, and functions of these modifications are being characterized (41–44). If these modifications occur within the PAMP area of viral RNA, viruses can also use these modifications to mimic as self-RNA from non–self-RNA and disrupt host immune systems. This interesting issue is currently under further investigation. Of interest is the observation that RIG-I contains a histidine residue (His830) in its binding pocket that offers a steric exclusion of N1-2′-O-methylated RNAs. Whether such mechanisms exist for m6A-modified RNAs is not known (45).
Generally, m6A is known to function on host cellular RNA to regulate stability, translation, and localization through interaction with YTHDF proteins (22, 23). We found that YTHDF2 inhibits RIG-I recognition of viral RNA via interacting with m6A-modified PAMP RNA (HBV) or specific site near the PAMP region (HCV) (Fig. 5). These results imply that YTHDF2 can regulate innate immunity in two ways, in which YTHDF2 inhibits RIG-I recognition of viral RNA and reduces the stability of viral RNA leading to controlled levels of translation (46). However, it was reported that m6A modification of HCV RNA did not affect its stability (27). Here, we report that the function of m6A modification of HCV RNA in RIG-1 recognition is due to only inhibition of RIG-I sensing. Further, YTHDF2 has no known RNase activity. It recruits either CCR4–NOT complex to regulate target RNA stability or ISG20 to degrade target viral RNAs (22, 47, 48). Many RNA-binding proteins are known to interact with YTHDF proteins (49, 50), suggesting that these interactions could regulate RIG-I access to viral RNAs. Consequently, it will be important in the future to identify the interactome network of YTHDFs during virus infection, which may unravel a mystery of the regulatory network of RNA-binding proteins that affect virus infection–mediated innate immune response.
We have recently demonstrated that HBV and HCV infections reduce host phosphatase and tensin (PTEN) homolog mRNA stability via up-regulating m6A modification (11). Because PTEN protein is related to nuclear localization of p-IRF-3 for activation of IFN signals, decreased PTEN expression by HBV and HCV disrupts innate immune response (51). In light of these results, we propose that HBV and HCV regulate immune response using m6A modification in two ways. First, m6A modification of viral RNA inhibits reduce RIG-I sensing activity, thus down-regulating downstream IFN signals. Next, HBV and HCV infections indirectly affect host immune response via decreasing PTEN expression levels by regulating m6A modification. These results collectively suggest that HBV and HCV attenuate host innate immune response via regulating cellular m6A machinery. Our study may also suggest a reasonable model applicable to the development of live attenuated vaccines. The deficient of m6A in viral RNA may provide a rational application of live attenuated vaccine candidates for viruses recognized by RIG-I. The obvious advantage is that m6A deficiency in HBV and HCV induces a higher IFN synthesis and might in turn enhance adaptive immunity. This study demonstrates a new concept for the development of live attenuated vaccines through the use of m6A modification. Overall, our results provide a novel mechanism by which the innate immune response to viral infection is regulated by cellular m6A machinery.
Experimental procedures
Plasmids, antibodies, and reagents
HBV 1.3-mer was kindly provided by Dr. Wang-Shick Ryu (Yonsei University). HBV 1.3-mer 5′-MT and 3′-MT plasmids were constructed as previously described (26). HBV 1.3-mer 5′-MT–CM and 3′-MT–CM plasmids were generated by site-directed mutagenesis. FLAG-YTHDF1, 2, and 3 plasmids were a kind gift from Dr. Stacy M. Horner (Duke University Medical Center). HCV pJFH-1 and GND plasmids were obtained from Dr. Takaji Wakita (National Institute of Infectious Disease, Japan). HCV pJFH-1 A331C and A8766C were generated by site-directed mutagenesis. The pFlagCMV4–RIG-I plasmid was used to express WT human RIG-I (1–925 amino acids) in cells. Antibodies were obtained as follow: anti-preS2 (catalog no. SC-23944), anti-YTHDF3 (catalog no. SC-379119), anti-glyceraldehyde-3-phosphate dehydrogenase (catalog no. SC-47724) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-METTL3 (catalog no. 15073-1-AP) antibody from Proteintech Group (Rosemont, IL, USA), anti-METTL14 (catalog no. HPA038002) antibody from Sigma–Aldrich, anti-YTHDF1 (catalog no. 86463), anti-YTHDF2 (catalog no. 80014), anti-FLAG (catalog no. 14793), IRF-3 (catalog no. 4302), and anti-phospho-IRF-3 (catalog no. 29047) antibodies from Cell Signaling Technology (Danvers, MA, USA). These antibodies were diluted with a 1:1000 ratio in 5% BSA buffer for immunoblotting. The ON-TARGET plus siRNAs of METTL3 (catalog no. L-005170-02-0005), METTL14 (catalog no. L-014169-02-0005), YTHDF1 (catalog no. L-018095-02-0005), YTHDF2 (catalog no. L-021009-02-0005), and YTHDF3 (catalog no. L-017080-01-0005) were obtained from Dharmacon (Lafayette, CO, USA). Poly(I:C) (catalog no. tlrl-pic) was purchased from InvivoGen (San Diego, CA, USA).
Cell culture and transfection
The Huh7 and HepG2 cells were obtained from ATCC (Manassas, VA, USA). Huh7 and HepG2 cells were transfected with plasmids using TransIT-LT1 (Mirus, Madison, WI, USA). Poly(I:C) was delivered into cells using TransIT-mRNA kit (Mirus). siRNAs were transfected using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol.
qRT-PCR
Total RNA and immunoprecipitated RNA were isolated using TRIzol reagent (Invitrogen). The quantitative PCR was assessed with Ssoadvanced Universal SYBR Green supermix (Bio-Rad) using the following primers: HBV pgRNA (forward primer, 5′-CTCAATCTCGGGAATCTCAATGT-3′; reverse primer, 5′-TGGATAAAACCTAGGAGGCATAAT-3′), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward primer, 5′-CCTGCACCACCAACTGCTTA-3′; reverse primer, 5′-CATGAGTCCTTCCACGATACCA-3′), and IFN-β mRNA (forward primer, 5′-ATGACCAACAAGTGTCTCCTCC-3′; reverse primer, 5′-GCTCATGGAAAGAGCTGTAGTG-3′). All IFN-β mRNA expression levels, normalized to input HCV RNA, were analyzed using the ΔΔCt method. HCV genome levels were quantified using a TaqMan probe specific for the HCV 5′-UTR region, as described previously (52).
Western blotting and immunoprecipitation
The cells were lysed in Nonidet P-40 lysis buffer (1% Nonidet P-40, 50 mm Tris-HCl, pH 8.0, 150 mm NaCl) supplemented with a protease inhibitor and a phosphatase inhibitor (Thermo Scientific). Extracted cell lysates were immunoprecipitated using anti-FLAG M2 magnetic beads (Sigma–Aldrich) for 2 h on a rotator at 4 °C. Purified cell lysates and immunoprecipitates were resolved by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were incubated by using various primary antibodies.
Statistical analysis
All results are presented as means ± S.D. from at least three independent experiments. The p value was calculated using a one-tailed unpaired Student's t test.
Data availability
All data described in the article are contained within the article.
Supplementary Material
This article contains supporting information.
Author contributions—G.-W. K. investigation; G.-W. K. and A. S. writing-original draft; H. I. and M. K. formal analysis; A. S. supervision; A. S. project administration.
Funding and additional information—This work is supported by National Institutes of Health Grants AI125350, AI139234, and AI085087 (to A. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- HBV
- hepatis B virus
- m6A
- N6-methyladenosine
- HCV
- hepatitis C virus
- RIG-I
- retinoic acid–inducible gene I
- pgRNA
- pregenomic RNA
- IFN
- interferon
- MAVS
- mitochondrial antiviral-signaling protein
- CARD
- caspase activation and recruitment domain
- CM
- compensatory mutant
- PAMP
- pathogen-associated molecular pattern
- PTEN
- phosphatase and tensin
- IP
- immunoprecipitate.
References
- 1. Seeger C., and Mason W. S. (2015) Molecular biology of hepatitis B virus infection. Virology 479-480, 672–686 10.1016/j.virol.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suzuki T., Aizaki H., Murakami K., Shoji I., and Wakita T. (2007) Molecular biology of hepatitis C virus. J. Gastroenterol. 42, 411–423 10.1007/s00535-007-2030-3 [DOI] [PubMed] [Google Scholar]
- 3. de Martel C., Maucort-Boulch D., Plummer M., and Franceschi S. (2015) World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 62, 1190–1200 10.1002/hep.27969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braciale T. J., and Hahn Y. S. (2013) Immunity to viruses. Immunol. Rev. 255, 5–12 10.1111/imr.12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sato S., Li K., Kameyama T., Hayashi T., Ishida Y., Murakami S., Watanabe T., Iijima S., Sakurai Y., Watashi K., Tsutsumi S., Sato Y., Akita H., Wakita T., Rice C. M., et al. (2015) The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 10.1016/j.immuni.2014.12.016 [DOI] [PubMed] [Google Scholar]
- 6. Saito T., Owen D. M., Jiang F. G., Marcotrigiano J., and Gale M. (2008) Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454, 523–527 10.1038/nature07106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li X. D., Sun L. J., Seth R. B., Pineda G., and Chen Z. J. J. (2005) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 102, 17717–17722 10.1073/pnas.0508531102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang D., Liu N., Zuo C., Lei S., Wu X., Zhou F., Liu C., and Zhu H. (2011) Innate host response in primary human hepatocytes with hepatitis C virus infection. PLoS One 6, e27552 10.1371/journal.pone.0027552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barth H., Rybczynska J., Patient R., Choi Y., Sapp R. K., Baumert T. F., Krawczynski K., and Liang T. J. (2011) Both innate and adaptive immunity mediate protective immunity against hepatitis C virus infection in chimpanzees. Hepatology 54, 1135–1148 10.1002/hep.24489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang H., and Ryu W. S. (2010) Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog. 6, e1000986 10.1371/journal.ppat.1000986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim G. W., Imam H., Khan M., Mir S. A., Kim S. J., Yoon S. K., Hur W., and Siddiqui A. (2020) HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology 10.1002/hep.31313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan M., Syed G. H., Kim S. J., and Siddiqui A. (2016) Hepatitis B virus–induced Parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 12, e1005693 10.1371/journal.ppat.1005693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X., Li Y., Mao A., Li C., Li Y., and Tien P. (2010) Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-β induction by disrupting the VISA-associated complex. Cell. Mol. Immunol. 7, 341–348 10.1038/cmi.2010.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S. J., Khan M., Quan J., Till A., Subramani S., and Siddiqui A. (2013) Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 9, e1003722 10.1371/journal.ppat.1003722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hornung V., Ellegast J., Kim S., Brzózka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K. K., Schlee M., Endres S., and Hartmann G. (2006) 5'-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 10.1126/science.1132505 [DOI] [PubMed] [Google Scholar]
- 16. Pichlmair A., Schulz O., Tan C. P., Näslund T. I., Liljeström P., Weber F., and Reis e Sousa C. (2006) RIG-I–mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science 314, 997–1001 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 17. Zeng W., Sun L., Jiang X., Chen X., Hou F., Adhikari A., Xu M., and Chen Z. J. (2010) Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330 10.1016/j.cell.2010.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gack M. U., Shin Y. C., Joo C. H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., and Jung J. U. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I–mediated antiviral activity. Nature 446, 916–920 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 19. Lin R., Heylbroeck C., Pitha P. M., and Hiscott J. (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 18, 2986–2996 10.1128/mcb.18.5.2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Flamme M., McKenzie L. K., Sarac I., and Hollenstein M. (2019) Chemical methods for the modification of RNA. Methods 161, 64–82 10.1016/j.ymeth.2019.03.018 [DOI] [PubMed] [Google Scholar]
- 21. Shi H. L., Wei J. B., and He C. (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 10.1016/j.molcel.2019.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Du H., Zhao Y., He J. Q., Zhang Y., Xi H. R., Liu M. F., Ma J. B., and Wu L. G. (2016) YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 10.1038/ncomms12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi H. L., Wang X., Lu Z. K., Zhao B. X. S., Ma H. H., Hsu P. J., Liu C., and He C. (2017) YTHDF3 facilitates translation and decay of N6-methyladenosine–modified RNA. Cell Res. 27, 315–328 10.1038/cr.2017.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yue Y., Liu J., and He C. (2015) RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 10.1101/gad.262766.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lichinchi G., Gao S., Saletore Y., Gonzalez G. M., Bansal V., Wang Y., Mason C. E., and Rana T. M. (2016) Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 1, 16011 10.1038/nmicrobiol.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Imam H., Khan M., Gokhale N. S., McIntyre A. B. R., Kim G. W., Jang J. Y., Kim S. J., Mason C. E., Horner S. M., and Siddiqui A. (2018) N6-Methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl. Acad. Sci. U.S.A. 115, 8829–8834 10.1073/pnas.1808319115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gokhale N. S., McIntyre A. B. R., McFadden M. J., Roder A. E., Kennedy E. M., Gandara J. A., Hopcraft S. E., Quicke K. M., Vazquez C., Willer J., Ilkayeva O. R., Law B. A., Holley C. L., Garcia-Blanco M. A., Evans M. J., et al. (2016) N6-Methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe 20, 654–665 10.1016/j.chom.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tirumuru N., Zhao B. S., Lu W., Lu Z., He C., and Wu L. (2016) N6-Methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife 5, e15528 10.7554/eLife.15528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kennedy E. M., Bogerd H. P., Kornepati A. V. R., Kang D., Ghoshal D., Marshall J. B., Poling B. C., Tsai K., Gokhale N. S., Horner S. M., and Cullen B. R. (2016) Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe 19, 675–685 10.1016/j.chom.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McIntyre W., Netzband R., Bonenfant G., Biegel J. M., Miller C., Fuchs G., Henderson E., Arra M., Canki M., Fabris D., and Pager C. T. (2018) Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res. 46, 5776–5791 10.1093/nar/gky029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gonzales-van Horn S. R., and Sarnow P. (2017) Making the mark: the role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe 21, 661–669 10.1016/j.chom.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hesser C. R., Karijolich J., Dominissini D., He C., and Glaunsinger B. A. (2018) N6-Methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi's sarcoma-associated herpesvirus infection. PLoS Pathog. 14, e1006995 10.1371/journal.ppat.1006995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z. J., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., and Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manners O., Baquero-Perez B., and Whitehouse A. (2019) m6A: widespread regulatory control in virus replication. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 370–381 10.1016/j.bbagrm.2018.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wieland S. F., and Chisari F. V. (2005) Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 79, 9369–9380 10.1128/JVI.79.15.9369-9380.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seeff L. B. (2009) The history of the “natural history” of hepatitis C (1968-2009). Liver Int. 29, 89–99 10.1111/j.1478-3231.2008.01927.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karikó K., Buckstein M., Ni H., and Weissman D. (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 10.1016/j.immuni.2005.06.008 [DOI] [PubMed] [Google Scholar]
- 38. Sioud M., Furset G., and Cekaite L. (2007) Suppression of immunostimulatory siRNA-driven innate immune activation by 2'-modified RNAs. Biochem. Biophys. Res. Commun. 361, 122–126 10.1016/j.bbrc.2007.06.177 [DOI] [PubMed] [Google Scholar]
- 39. Durbin A. F., Wang C., Marcotrigiano J., and Gehrke L. (2016) RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio 7, e00833–16 10.1128/mBio.00833-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu M., Zhang Z., Xue M., Zhao B. S., Harder O., Li A., Liang X., Gao T. Z., Xu Y., Zhou J., Feng Z., Niewiesk S., Peeples M. E., He C., and Li J. (2020) N6-Methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 5, 584–598 10.1038/s41564-019-0653-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. George C. X., John L., and Samuel C. E. (2014) An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interferon Cytokine Res. 34, 437–446 10.1089/jir.2014.0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X., Ma S., and Yi C. (2016) Pseudouridine: the fifth RNA nucleotide with renewed interests. Curr. Opin. Chem. Biol 33, 108–116 10.1016/j.cbpa.2016.06.014 [DOI] [PubMed] [Google Scholar]
- 43. Slotkin W., and Nishikura K. (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med. 5, 105 10.1186/gm508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spenkuch F., Motorin Y., and Helm M. (2014) Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol. 11, 1540–1554 10.4161/15476286.2014.992278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schuberth-Wagner C., Ludwig J., Bruder A. K., Herzner A. M., Zillinger T., Goldeck M., Schmidt T., Schmid-Burgk J. L., Kerber R., Wolter S., Stümpel J. P., Roth A., Bartok E., Drosten C., Coch C., et al. (2015) A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1-2'O-methylated self RNA. Immunity 43, 41–51 10.1016/j.immuni.2015.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhong L., Liao D., Zhang M., Zeng C., Li X., Zhang R., Ma H., and Kang T. (2019) YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 442, 252–261 10.1016/j.canlet.2018.11.006 [DOI] [PubMed] [Google Scholar]
- 47. Imam H., Kim G. W., Mir S. A., Khan M., and Siddiqui A. (2020) Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified hepatitis B virus transcripts. PLoS Pathog. 16, e1008338 10.1371/journal.ppat.1008338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., and He C. (2014) N6-Methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 10.1038/nature12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schwartz S., Mumbach M. R., Jovanovic M., Wang T., Maciag K., Bushkin G. G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D., Sanjana N. E., Freinkman E., Pacold M. E., Satija R., Mikkelsen T. S., et al. (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5' sites. Cell Rep. 8, 284–296 10.1016/j.celrep.2014.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang X., Zhao B. S., Roundtree I. A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., and He C. (2015) N6-Methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 10.1016/j.cell.2015.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li S., Zhu M. Z., Pan R. G., Fang T., Cao Y. Y., Chen S. L., Zhao X. L., Lei C. Q., Guo L., Chen Y., Li C. M., Jokitalo E., Yin Y. X., Shu H. B., and Guo D. Y. (2016) The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nat. Immunol. 17, 241–249 10.1038/ni.3311 [DOI] [PubMed] [Google Scholar]
- 52. Kim G. W., Lee S. H., Cho H., Kim M., Shin E. C., and Oh J. W. (2016) Hepatitis C virus core protein promotes miR-122 destabilization by inhibiting GLD-2. PLoS Pathog. 12, e1005714 10.1371/journal.ppat.1005714 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described in the article are contained within the article.