

Abstract
Diabetic nephropathy (DN) is a complication of diabetes that is increasing in prevalence in China. Extracellular vesicles (EVs) carrying microRNAs (miRs) may represent a useful tool in the development of therapies for DN. Here, we report that EVs released by adipose-derived mesenchymal stem cells (ADSCs) during DN contain a microRNA, miR-26a-5p, that suppresses DN. Using bioinformatic analyses, we identified differentially expressed miRs in EVs from ADSCs and in DN and predicted downstream regulatory target genes. We isolated mesenchymal stem cells (MSCs) from adipose tissues and collected EVs from the ADSCs. We exposed mouse glomerular podocytes and MP5 cells to high glucose (HG), ADSC-derived EVs, miR-26a-5p inhibitor/antagomir, Toll-like receptor 4 (TLR4) plasmids, or the NF-κB pathway activator (phorbol-12-myristate-13-acetate, or PMA). We used the cell counting kit-8 (CCK-8) assay and flow cytometry to investigate the impact of miR-26a-5p on cell viability and apoptosis and validated the results of these assays with in vivo experiments in nude mice. We found that in DN, miR-26a-5p is expressed at very low levels, whereas TLR4 is highly expressed. Of note, EVs from ADSCs ameliorated the pathological symptoms of DN in diabetic mice and transferred miR-26a-5p to HG-induced MP5 cells, improving viability while suppressing the apoptosis of MP5 cells. We also found that miR-26a-5p protects HG-induced MP5 cells from injury by targeting TLR4, inactivating the NF-κB pathway, and downregulating vascular endothelial growth factor A (VEGFA). Moreover, ADSC-derived EVs transferred miR-26a-5p to mouse glomerular podocytes, which ameliorated DN pathology. These findings suggest that miR-26a-5p from ADSC-derived EVs protects against DN.
Keywords: MicroRNA-328-3p, extracellular vesicles, adipose-derived mesenchymal stem cells, Toll-like receptor 4 (TLR4), NF-κB, vascular endothelial growth factor, diabetic nephropathy, metabolic disorder, inflammation, diabetes, cell biology, cell cycle, cell differentiation, cell culture
Diabetic nephropathy (DN), a common microvascular complication of diabetes, is one of the major causes of death among patients with diabetes (1). It is also the leading cause of both chronic kidney and end-stage renal diseases worldwide (2). The incidence of DN is increasing in developing countries, including China (3). DN is characterized by albuminuria and progressive loss of kidney function (4), and risk factors include hyperglycemia and genetic polymorphisms (5). Moreover, the dysfunction of podocytes contributes to the development of DN (6), and DN progression can be slowed by reducing podocyte injury (7). Current treatment methods for DN mainly rely on the management of hyperglycemia and blood pressure (8). Evidence suggests that extracellular vesicles (EVs) slow the development of DN by protecting podocytes from injury (9). Elucidating mechanisms by which EVs regulate podocyte injury may represent a therapeutic target for the treatment of DN.
EVs are nano-sized membrane vesicles that include exosomes and microvesicles, and they are released by many cells types, including mesenchymal stem cells (MSCs) (10, 11). MSCs are multipotent stem cells that differentiate into a variety of lineages and exert important functions in bone regeneration and repair (12). A recent study highlighted the role of exosomes derived from adipose-derived mesenchymal stem cells (ADSCs) in protecting against the development of DN (13). EVs are known to contain mRNA, microRNAs (miRs), and long noncoding RNAs, thereby transferring genetic information from one cell to the next (14). Interestingly, miR-26a-5p serves as a candidate biomarker for DN by a meta-analysis of profiling studies (15). Our results also showed that miR-26a-5p is highly expressed in EVs derived from MSCs. Loss of miR-26a led to the development of DN in both cultured podocytes and streptozotocin-induced diabetic mice (16). Additionally, miR-26a-5p was suggested to be one of the therapeutic mediators in DN (15). Based on these findings, we hypothesized that EV-derived miR-26a-5p could regulate DN. Toll-like receptor 4 (TLR4), which serves as a signaling receptor for lipopolysaccharides and as a crucial regulator of the innate immunity system, was identified as a downstream target gene of miR-26a-5p (17). One study used M4200 cells to show that TLR4 also participates in the pathogenesis of DN (18). Moreover, downregulation of TLR/NF-κB (NF-κB) using umbelliferone ameliorated renal function in a rat model of DN (19). Notably, the activation of NF-κB is thought to be related to the inflammation and disease development that are associated with DN (20). Upregulation of NF-κB by TLR4 was previously reported in rat pineal glands and human umbilical vein endothelial cells (21, 22). In addition, the NF-κB pathway could promote the expression of vascular endothelial growth factor A (VEGFA) (23), which contributes to glomerular endothelial cell dysfunction as well as albuminuria in DN (24). Taking these findings into consideration, we hypothesized that ADSC-derived EVs transferred miR-26a-5p to other cells regulating the progression of DN by targeting TLR4 and modulating NF-κB and VEGFA. Thus, this study may help to identify a novel strategy for the control of DN.
Results
Bioinformatics analyses for the expression of miR-26a-5p and TLR4 in DN
Analysis of the EVmiRNA database revealed that the expression of miR-26a-5p in the EVs derived from MSCs was significantly upregulated (Fig. 1A). Furthermore, the miRanda database and Starbase database were used to predict the downstream target genes of differentially expressed miRNAs. These results were then compared with a diabetes-related gene expression data set (GSE21340). Five genes were identified at the intersection of these analyses (SMAD1, CELF2, SMAD6, PYGL, and TLR4) (Fig. 1B). Because the miRanda database predicted a binding site between miR-26a-5p and TLR4 (Fig. 1C), we chose to investigate the expression of TLR4 in the GSE21340 data set. We found that TLR4 expression was significantly higher in DN (Fig. 1D). These results suggest that miR-26a-5p delivered by ADSC-derived EVs protect against DN by regulating TLR4.
Figure 1.
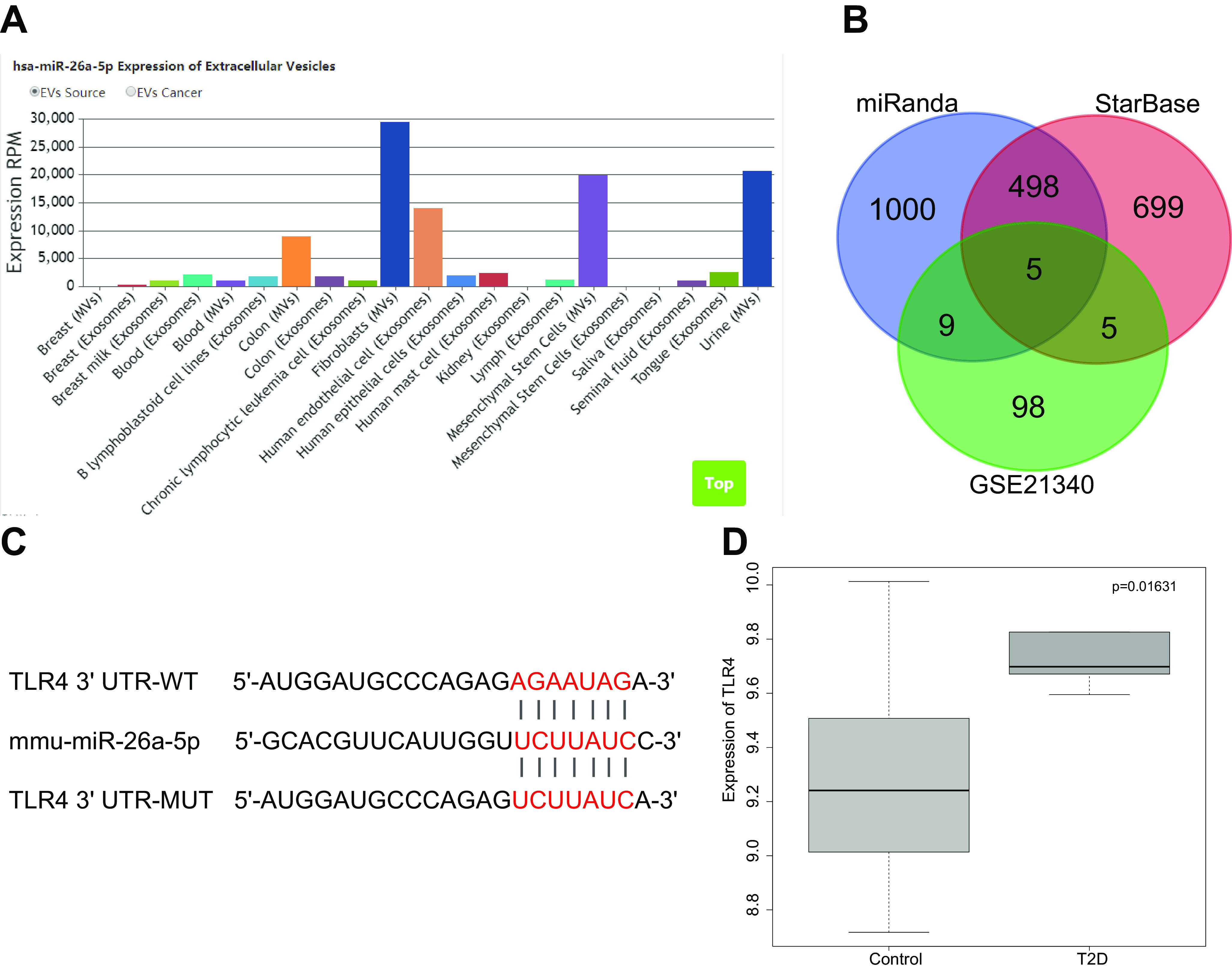
DN is characterized by poor miR-26a-5p expression and high TLR4 expression. A, the expression of miR-26a-5p in EVs. B, the intersection between predicted miR-26a-5p downstream target genes identified by the MiRanda and SatrBase databases and differentially expressed genes from the GSE21340 data set. The number represents the predicted number of target genes. C, a map of the predicted binding site of miR-26a-5p to TLR4 and mutant site. D, the box map for the differential expression of TLR4 gene in diabetes and normal samples. Paired t test was used to compare normal sample and diabetic samples.
EVs were successfully isolated from ADSCs
ADSCs were isolated from the subcutaneous adipose tissues of C57BL/KsJ db/m mice. The surface markers associated with ADSCs were detected by flow cytometry after the third passage. ADSCs were positive for CD29 (96.7%), CD44 (95.2%), CD73 (99.1%), CD90 (98.4%), and HLA-A, -B, and -C (98.5%) but negative for CD14 (4.2%), CD19 (0.26%), CD34 (1.9%), CD45 (1.2%), and HLA-DR (0.89%) (Fig. 2A). The pluripotency of isolated ADSCs was tested. After isolation and culture, ADSCs could be differentiated into osteogenic, lipogenic, and chondrogenic cell types (Fig. 2B). The above results demonstrated that ADSCs isolated from adipose tissues had the property of pluripotent stem cell differentiation.
Figure 2.
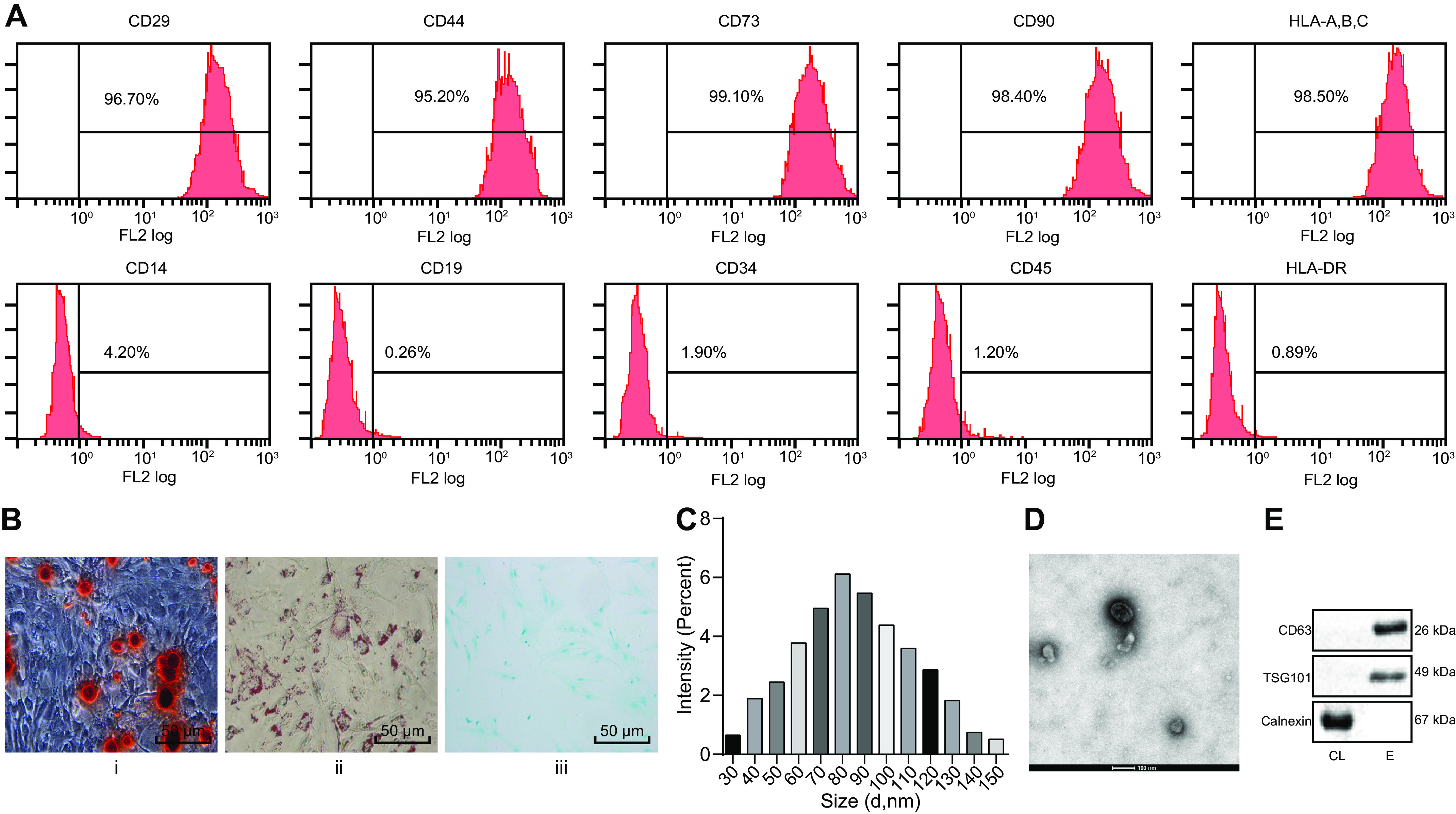
EVs are successfully isolated from ADSCs. A, identification of ADSC surface markers by flow cytometry. B, i, detection of osteogenic differentiation by Alizarin Red S staining. ii, detection of lipogenic differentiation by oil red O staining. iii, detection of chondrogenic differentiation by Alcian blue staining (scale bar, 50 μm). C, size distribution of ADSC-derived EVs measured by DLS. D, observation of morphology of EVs by transmission EM (scale bar, 100 nm). E, CD63, TSG101, and calnexin detected by Western blotting analysis.
Next, EVs isolated from medium used to grow ADSCs were characterized. Dynamic light scattering (DLS) analysis indicated that EVs range in diameter from 30–150 nm. Transmission EM revealed the morphology of EVs to be cup-shaped or spherical. In addition, Western blot analysis of marker proteins of EVs confirmed the expression of CD63 and TSG101, whereas calnexin was absent, indicating that EVs had been successfully isolated (Fig. 2, C–E). The above results demonstrated the successful isolation of EVs from ADSCs.
EVs from ADSCs alleviated the pathological symptoms and suppressed glomerular podocyte apoptosis in a mouse model that spontaneously develops diabetes
To evaluate the mouse model of spontaneous diabetes, we compared the levels of protein found in the urine (within 24 h), serum creatinine (Scr), and blood urea nitrogen (BUN) in C57BL/KsJ db/db spontaneous diabetic mice and C57BL/KsJ db/m control mice. The spontaneously diabetic mice had twice the amount of urine protein and 3 times as much Scr and BUN as the control mice (Fig. 3, A–C). The histopathological phenotype was characterized by periodic acid Schiff (PAS) staining of DN tissues. Spontaneously diabetic mice showed an accumulation of extracellular matrix in their kidney tissues, viability of mesangial cells, and a thickening of basement membrane. In addition, there was increased proteinuria in the lumen, hyaline degeneration, and severe vacuolar degeneration of renal tubular epithelial cells in these mice compared with that of control mice, indicating the successful establishment of the DN cell model (Fig. 3D).
Figure 3.
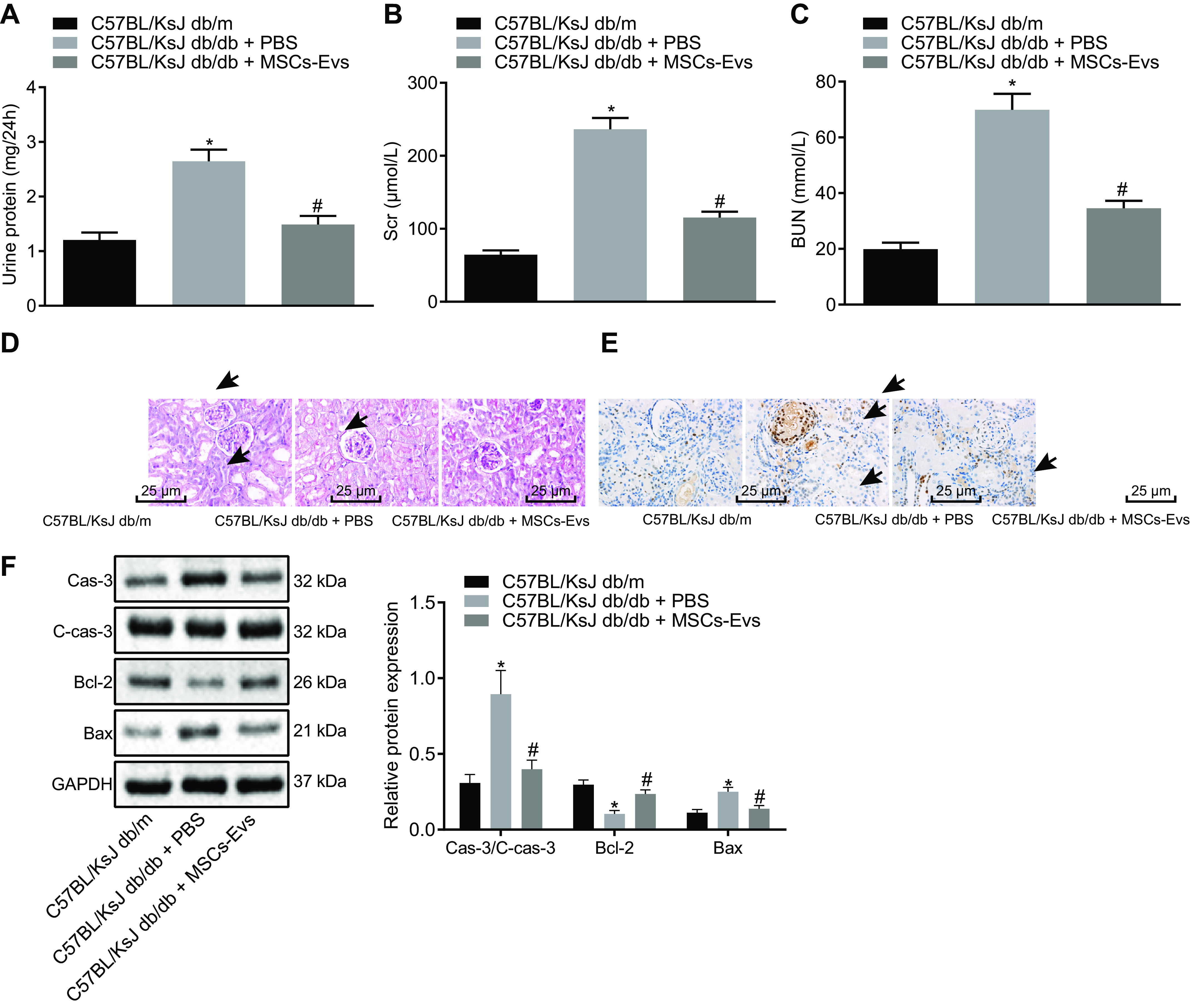
ADSC-derived EVs alleviate the pathological symptoms and suppress glomerular podocyte apoptosis in spontaneous diabetic mice (n = 6 mice per group). A–C, the levels of urine protein (within 24 h), Scr, and BUN of mice. D, the histopathological characterization of DN after PAS staining (the black arrows indicate accumulation of extracellular matrix and thickening of basement membrane) (scale bar, 25 μm). E, the apoptosis of glomerular podocytes detected by TUNEL staining. The black arrow indicates apoptotic cells (the brownish-yellow cells) (scale bar, 25 μm). F, the protein expression of apoptosis-related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) in renal tissues of mice as detected by Western blotting analysis, normalized to GAPDH. Scale bar, 25 μm. Data are expressed as mean ± standard deviation. Data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. The experiment was repeated three times. *, p < 0.05 versus C57BL/KsJ db/m mice. #, p < 0.05 versus C57BL/KsJ db/db mice treated with PBS.
To investigate the effect of EVs from ADSCs on spontaneous diabetic mice, we injected ADSC-derived EVs or PBS into mice via the tail vein, and then changes of urine protein, Scr, and BUN levels were subsequently measured, followed by PAS staining to detect histopathological changes. The results illustrated that treatment with EVs derived from ADSCs notably reduced the levels of urine protein, Scr, and BUN in the DN mouse model and alleviated the histopathological changes associated with DN (Fig. 3, A–D).
Next, TUNEL staining was used to detect the apoptosis of glomerular podocytes in mice. Glomerular podocytes from spontaneously diabetic mice (C57BL/KsJ db/db) exhibited a significant increase in apoptosis compared with control mice (C57BL/KsJ db/m mice), and this was reduced by injection of EVs from ADSCs (Fig. 3E). In addition, apoptosis-related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) from diabetic mice (C57BL/KsJ db/db) and control (C57BL/KsJ db/m) mice were compared. Bcl-2 protein was significantly reduced in diabetic mice compared with control mice, whereas the protein expression of Bax, cleaved caspase-3, and caspase-3 was increased. Injection of EVs from ADSCs reversed these trends (Fig. 3F). These results demonstrated that ADSC-derived EVs are capable of alleviating the pathological symptoms and inhibiting glomerular podocyte apoptosis in DN mice.
EVs from ADSCs inhibited apoptosis of MP5 cells inducedby HG
The ability of mouse glomerular podocytes, MP5 cells, to internalize EVs from ADSCs was assessed using green fluorescent (PKH67)-labeled EVs. The presence of green fluorescence in MP5 cells was observed by fluorescence microscopy, which indicated that MP5 cells were able to internalize ADSC-derived EVs (Fig. 4A). To simulate the DN cell model in vitro, MP5 cells were induced by HG and treated with ADSC-derived EVs (25 μg/ml). Cell viability was detected by CCK-8 assay at 24 h, 48 h, and 72 h, respectively. Although there was no difference in cell viability at 24 h in response to each treatment, at 48 h, high-glucose (HG) treatment markedly decreased MP5 cell viability relative to treatment with either normal glucose levels (NG) or mannitol (MA). Viability could be restored by cotreatment with HG and ADSC-derived EVs. No significant differences in MP5 cell viability were seen between the 48- and 72-h time points in response to treatment (Fig. 4B); therefore, the 48-h time point was chosen for all subsequent experiments.
Figure 4.
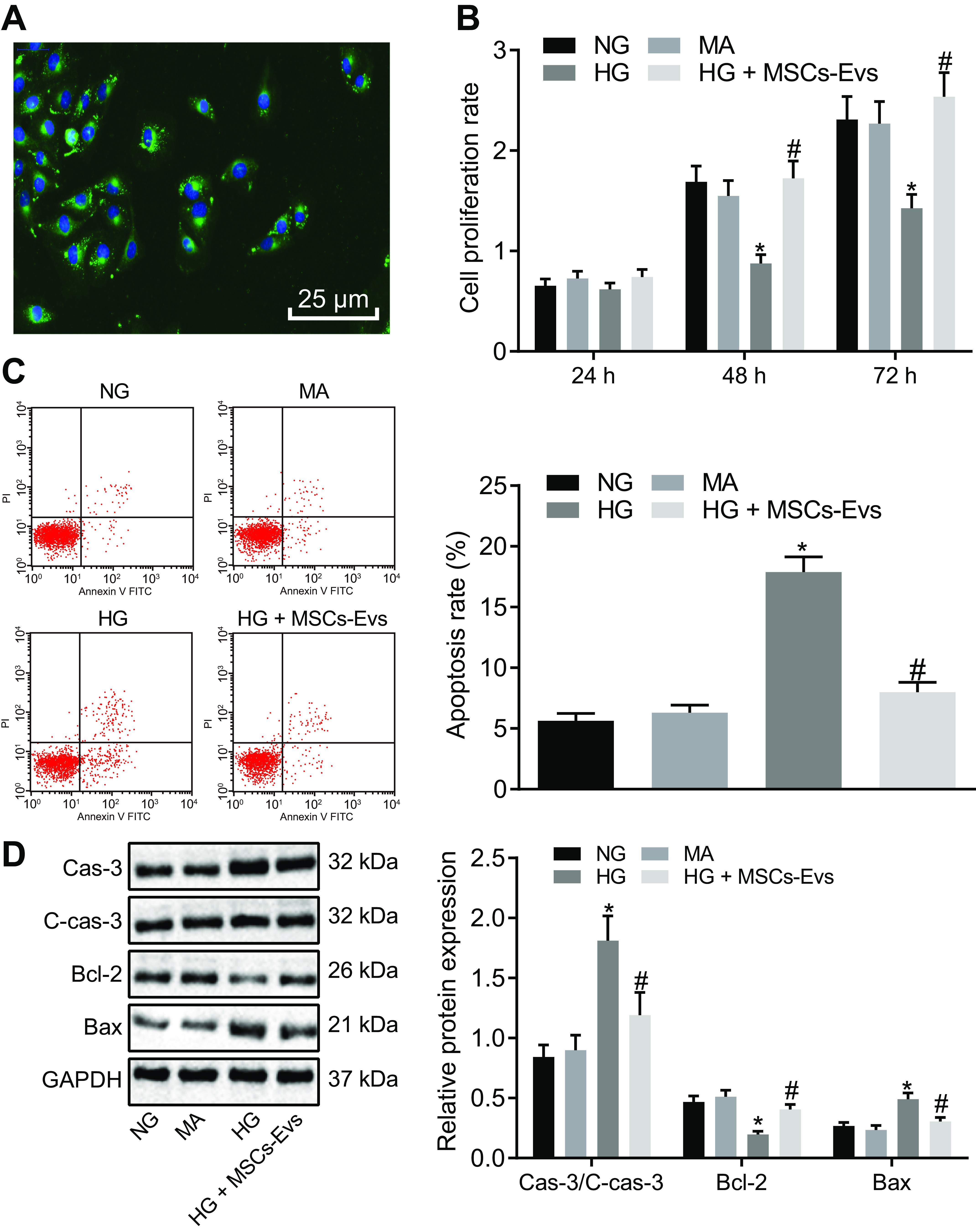
ADSC-derived EVs inhibit apoptosis of MP5 cells induced by HG. A, the PKH67-labeled ADSC-derived EVs observed under a fluorescence microscope (green fluorescence, PKH67; blue fluorescence, 4′,6-diamidino-2-phenylindole) (scale bar, 25 μm). B, viability of MP5 cells after 24 h, 48 h, and 72 h of treatment as detected by CCK-8 assay. C, apoptosis of MP5 cells after treatment at 48 h, as detected by flow cytometry. D, the protein expression of apoptosis-related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) in MP5 cells as detected by Western blotting analysis, normalized to GAPDH. Data are expressed as mean ± standard deviation. B–D, data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. A, repeated-measures ANOVA was used to compare data among multiple groups at different time points, followed by Bonferroni post hoc test. The experiment was repeated three times. *, p < 0.05 versus NG or MA. #, p < 0.05 versus HG.
Flow cytometry was used to detect apoptosis at 48 h after the same treatments described above. The results showed that compared with treatment with either NG or MA, HG induction markedly increased MP5 cell apoptosis, whereas cotreatment of HG and ADSC-derived EVs exhibited reduced cell apoptosis versus HG treatment alone (Fig. 4C). In addition, Western blot analysis was used to detect the expression of apoptosis-related proteins in MP5 cells after 48 h of each treatment. Treatment with HG significantly decreased the protein expression of Bcl-2, whereas increasing that of caspase-3, cleaved caspase-3, and Bax compared with either NG or MA and cotreatment of HG and ADSC-derived EVs increased Bcl-2 protein expression and de-creased that of caspase-3, cleaved caspase-3, and Bax (Fig. 4D). These results suggested that EVs from ADSCs are able to suppress HG-mediated injury of podocytes in vitro.
EVs from ADSCs delivered miR-26a-5p, which inhibited apoptosis in HG-treated MP5 cells
The expression of miR-26a-5p in HG-induced MP5 cells was measured by RT-qPCR after treatment with ADSC-derived EVs or PBS. miR-26a-5p expression was significantly lower after induction with HG than with treatment with NG or MA, but it was notably increased by treatment with ADSC-derived EVs containing miR-26a-5p (Fig. 5A). To confirm that ADSC-derived EVs can transfer miR-26a-5p into MP5 cells, Cy3-miR-26a-5p mimic was transfected into ADSCs and then added to MP5 cell media. Fluorescence microscopy revealed that MP5 cells became noticeably red after treatment (Fig. 5B). These results demonstrate that ADSC-derived EVs transfer miR-26a-5p into MP5 cells.
Figure 5.
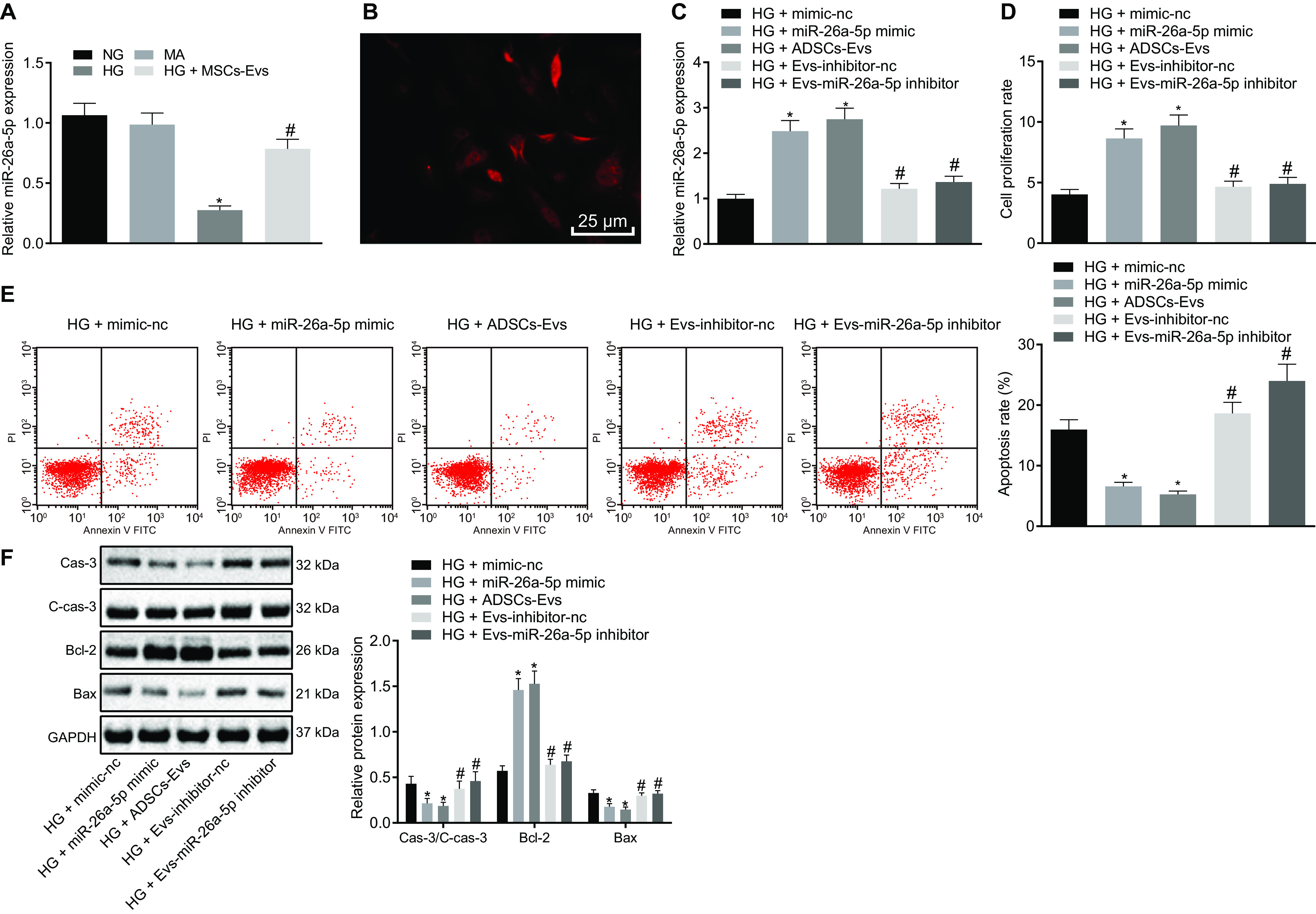
ADSC-derived EVs inhibit HG-induced apoptosis of MP5 cells by transferring miR-26a-5p. A, the expression of miR-26a-5p in MP5 cells as detected by RT-qPCR. B, Cy3-miR-26a-5p mimic was transfected into ADSCs and then cocultured with MP5 cells. The red fluorescence was observed under a fluorescence microscope (scale bar, 25 μm). C, RT-qPCR detection of miR-26a-5p expression in MP5 cells. D, MP5 cell viability as detected by CCK-8 assay. E, the apoptosis of MP5 cells as detected by flow cytometry. F, the protein expression of apoptosis-related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) in MP5 cells as detected by Western blot analysis, normalized to GAPDH. In panel A, *, p < 0.05 versus NG or MA; #, p < 0.05 versus HG + MSCs-Exo. In panels B–F, *, p < 0.05 versus HG + mimic-NC; #, p < 0.05 versus HG + miR-26a-5p mimic or HG + Exo-inhibitor-NC. Data are expressed as mean ± standard deviation. Data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. The experiment was repeated three times.
To determine the effect of miR-26a-5p transferred by ADSC-derived EVs on HG-treated MP5 cells, HG-treated MP5 cells were transfected with either miR-26a-5p mimic, ADSC-derived EVs, or ADSC-derived EVs treated with miR-26a-5p inhibitor. Addition of either miR-26a-5p mimic or ADSC-derived EVs increased the expression of miR-26a-5p in HG-treated MP5 cells, whereas treatment of MP5 cells with ADSC-derived EVs containing miR-26a-5p inhibitor caused a decline in miR-26a-5p expression (Fig. 5C).
Next, MP5 cell viability was assessed using the CCL-8 assay, and apoptosis was measured by flow cytometry. Under HG conditions, treatment with either miR-26a-5p mimic or ADSC-derived EVs markedly improved cell viability and suppressed apoptosis of MP5 cells, whereas ADSC-derived EVs treated with miR-26a-5p inhibitor led to decreased cell viability and increased apoptosis (Fig. 5, D and E).
In addition, Western blot analysis was used to detect apoptosis-related proteins in MP5 cells after each treatment. Treatment with miR-26a-5p mimic or ADSC-derived EVs resulted in increased Bcl-2 protein expression accompanied by a notable decrease in Bax, cleaved caspase-3, and caspase-3 expression. In contrast, ADSC-derived EVs treated with miR-26a-5p inhibitor exhibited markedly decreased Bcl-2 protein expression, whereas the expression of Bax, cleaved caspase-3, and caspase-3 was significantly elevated (Fig. 5F). These results indicate that delivery of miR-26a-5p into MP5 cells by ADSC-derived EVs inhibits HG-induced apoptosis.
miR-26a-5p transferred by ADSC-derived EVs inhibited HG-induced apoptosis of MP5 cells by targeting TLR4
The website TargetScan (RRID:SCR_010845) was able to predict specific binding sites between TLR4 and miR-26a-5p at nucleotides 4477–4484 (Fig. 6A). A Dual-Luciferase reporter gene assay was used to confirm that TLR4 was a target of miR-26a-5p. Cotransfection of miR-26a-5p mimic and TLR4-3'UTR-WT resulted in a marked decrease in fluorescence intensity compared with the mimic negative control (NC) + TLR4-3'UTR-WT cotransfection; however, fluorescence intensity was unchanged in the presence of mimic NC + TLR4-3'UTR-MUT or miR-26a-5p mimic + TLR4-3'UTR-MUT (Fig. 6B).
Figure 6.
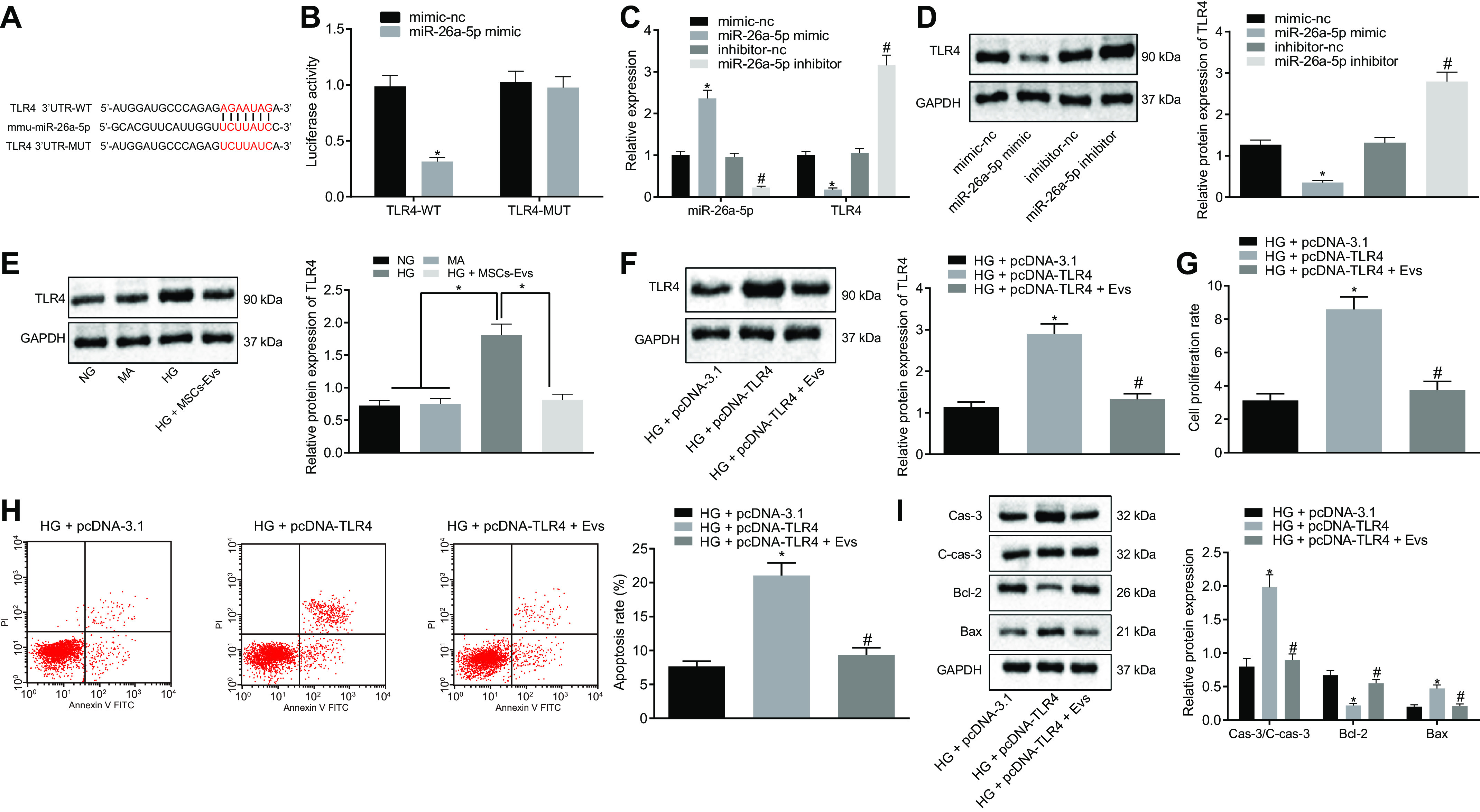
MiR-26a-5p transferred by ADSCs-derived EVs targets TLR4 to inhibit apoptosis of MP5 cells induced by HG. A, the specific binding sites of TLR4 and miR-26a-5p predicted by the TargetScan website (RRID:SCR_010845). *, p < 0.05. B, Dual-Luciferase reporter gene assay confirms TLR4 as a target of miR-26a-5p. *, p < 0.05 versus mimic-NC + TLR4 3'UTR-WT. C, the expression of miR-26a-5p and TLR4 in MP5 cells as detected by RT-qPCR. *, p < 0.05 versus mimic-NC. #, p < 0.05 versus inhibitor-NC. D–F, the protein expression of TLR4 in MP5 cells as detected by Western blot analysis, normalized to GAPDH. G, MP5 cell viability as detected by CCK-8 assay. H, the apoptosis of MP5 cells as detected by flow cytometry. I, the protein expression of apoptosis related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) in MP5 cells as detected by Western blot analysis, normalized to GAPDH. Data are expressed as mean ± standard deviation. Comparisons between two groups were analyzed using unpaired t test. Data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. The experiment was repeated three times. In panel D, *, p < 0.05 versus mimic-NC. #, p < 0.05 versus inhibitor-NC. In panel E, *, p < 0.05 versus NG or MA or HG + MSCs-Exo. In panels F–I, *, p < 0.05 versus HG + pcDNA-3.1. #, p < 0.05 versus HG + pcDNA-TLR4.
RT-qPCR of MP5 cells transfected with miR-26a-5p mimic or inhibitor resulted in a noticeably increased miR-26a-5p expression and a significantly decreased TLR4 expression, whereas opposite effects were detected after treatment with miR-26a-5p inhibitor (Fig. 6, C and D). Western blot analysis revealed that HG treatment of MP5 cells significantly increased TLR4 protein expression compared with treatment with NG or MA treatment, and this could be reversed by treatment with EVs from ADSCs (Fig. 6E). This suggests that miR-26a-5p transferred by ADSC-derived EVs could directly target the TLR4 gene.
To detect the effect of the TLR4 gene regulated by ADSC-derived EVs on HG-induced MP5 cells, HG-treated MP5 cells were overexpressed with TLR4 and then treated with ADSC-derived EVs. First, Western blot analysis was used to detect the cellular protein expression of TLR4 upon each treatment. It was found that under HG conditions, compared with pcDNA-3.1 treatment, pcDNA-TLR4 significantly upregulated the protein expression of TLR4, which was reversed by treatment of pcDNA-TLR4- and ADSC-derived EVs (Fig. 6F).
Subsequently, CCK-8 assay was performed to detect MP5 cell viability, and flow cytometry was used to detect cell apoptosis. The results displayed that under HG conditions, relative to pcDNA-3.1 treatment, pcDNA-TLR4 significantly inhibited the protein cell viability while promoting cell apoptosis; however, promoted cell viability and inhibited cell apoptosis were detected after treatment with pcDNA-TLR4- and ADSC-derived EVs (Fig. 6G and H).
In addition, Western blot analysis was used to detect apoptosis-related proteins in HG-treated MP5 cells after treatment with each plasmid. pcDNA-TLR4 treatment markedly decreased Bcl-2 protein expression while significantly increasing cleaved caspase-3, caspase-3, and Bax protein expression relative to that of pcDNA-3.1 treatment. The opposite results were found after the treatment of HG-induced MP5 cells transfected with pcDNA-TLR4 and exposed to EVs from ADSCs containing miR-26a-5p (Fig. 6I). Combined with the above results, we concluded that miR-26a-5p delivered by ADSC-derived EVs targets TLR4, inhibiting HG-induced MP5 cell apoptosis.
EVs from ADSCs inhibited HG-induced apoptosis of MP5 cells by downregulating TLR4 and the NF-κB/VEGFA signaling pathway
Subsequently, we explored the role of ADSC-derived EVs in the progression of DN via the regulation of the NF-κB pathway and VEGFA. We first investigated whether miR-26a-5p carrying EVs from ADSCs could inhibit the NF-κB pathway by downregulating TLR4 expression. After TLR4 overexpression, MP5 cells induced by HG were further treated with ADSC-derived EVs. Immunofluorescence staining revealed that HG treatment of MP5 cells resulted in p65 localization to the nucleus. EVs from ADSCs were able to reduce p65 nuclear localization, keeping more p65 in the cytoplasm. The effects of EVs could be negated by the overexpression of TLR4, which led to the nuclear localization of p65 protein again (Fig. 7A).
Figure 7.
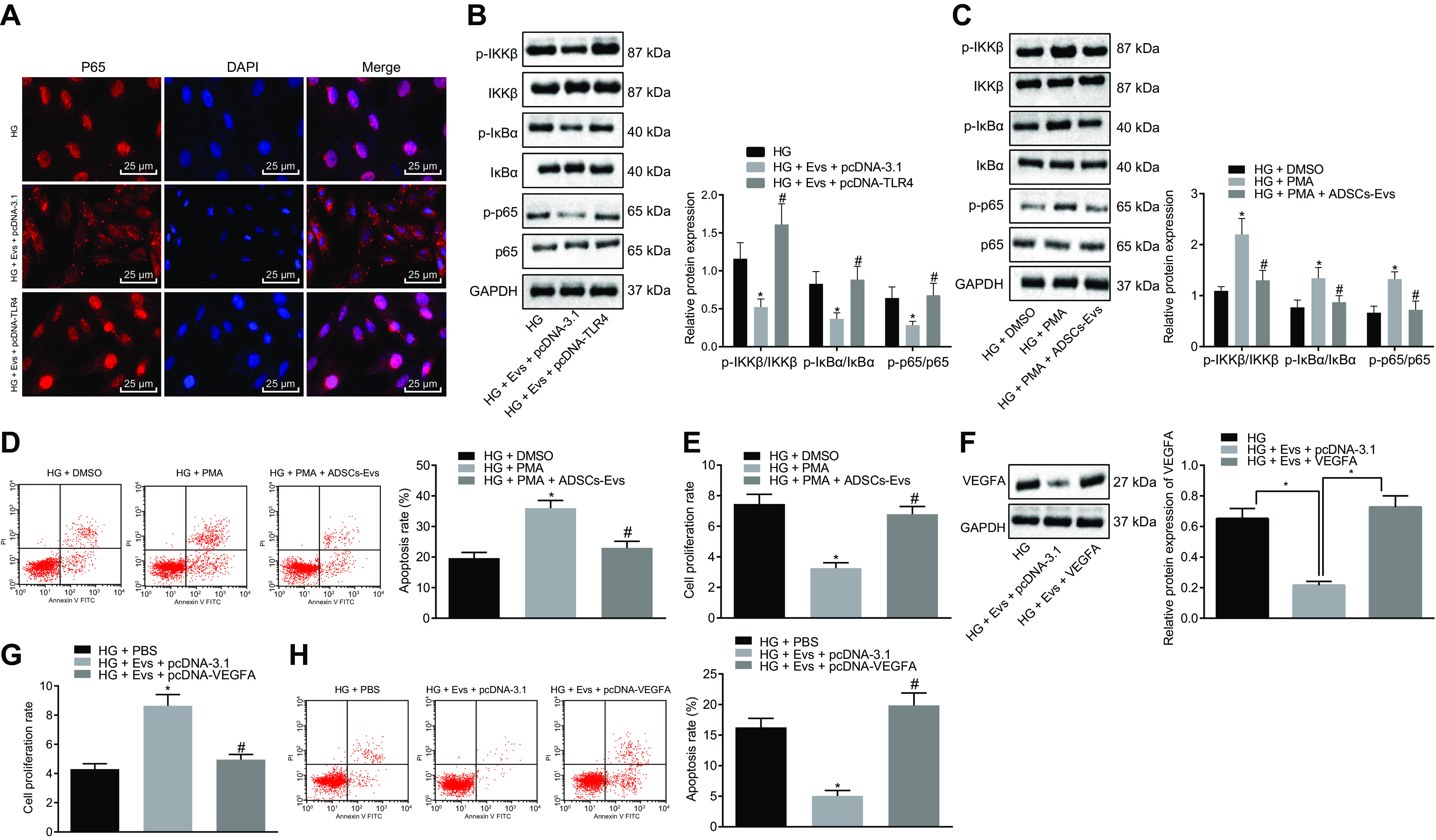
EVs derived from ADSCs deliver MiR-26a-5p, which targets TLR4, downregulates NF-κB/VEGFA, and inhibits the apoptosis of MP5 cells induced by HG. A, immunofluorescence staining of p65 protein in MP5 cells (scale bar, 25 μm). B, the protein expression of NF-κB pathway-related proteins IKKβ, IκBα, and p65 as well as the extent of IKKβ, IκBα, and p65 phosphorylation in MP5 cells as detected by Western blot analysis, normalized to GAPDH. *, p < 0.05 versus HG. #, p < 0.05 versus HG + pcDNA-TLR4 + Exo. C, the protein expression of the NF-κB pathway-related proteins IKKβ, IκBα, p65, and VEGFA as well as the extent of IKKβ, IκBα, and p65 phosphorylation in HG-induced MP5 cells treated with ADSC-derived EVs and the NF-κB pathway activator, PMA, as detected by Western blot analysis, normalized to GAPDH. D, the apoptosis of HG-induced MP5 cells treated with ADSC-derived EVs and the NF-κB pathway activator, PMA, as detected by flow cytometry. E, cell viability in HG-induced MP5 cells treated with ADSC-derived EVs and the NF-κB pathway activator, PMA, as detected by CCK-8 assay. *, p < 0.05 versus HG + DMSO. #, p < 0.05 versus HG + PMA. F, the protein expression of VEGFA in HG-induced MP5 cells after treatment with ADSC-derived EVs and overexpression of VEGFA as detected by Western blot analysis, normalized to GAPDH. *, p < 0.05 versus HG + Exo + pcDNA-3.1. G, cell viability in HG-induced MP5 cells after treatment with ADSC-derived EVs overexpressing VEGFA, as detected by CCK-8 assay. H, cell apoptosis in HG-induced MP5 cells after treatment with ADSC-derived EVs overexpressing VEGFA as detected by flow cytometry. Data are expressed as mean ± standard deviation. Data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. The experiment was repeated three times. *, p < 0.05 versus HG. #, p < 0.05 versus HG + Exo + pcDNA-3.1.
Western blot analysis compared the protein expression of NF-κB pathway-related proteins IKKβ, IκBα, and p65 and the extent of their phosphorylation in the cells after each treatment. Treatment with EVs from ADSCs and pcDNA-3.1 control plasmid led to significantly decreased protein expression of IKKβ, IκBα, and p65 and the extent of their phosphorylation during HG induction, whereas each of these proteins was upregulated by ADSC-derived EVs cotreated with pcDNA-3.1-TLR4 (Fig. 7B).
To verify whether EVs from ADSCs can inhibit apoptosis associated with HG treatment by inhibiting the NF-κB pathway, HG-induced MP5 cells were treated with ADSC-derived EVs and the NF-κB pathway activator, phorbol-12-myristate-13-acetate (PMA; 1 μg/ml). Western blot analysis indicated that PMA treatment significantly increased the protein expression of IKKβ, IκBα, p65, and VEGFA, as well as the extent of their phosphorylation in HG-induced MP5 cells compared with that of treatment with DMSO. Combined treatment with PMA and ADSC-derived EVs significantly decreased all these indicators compared with PMA treatment alone (Fig. 7C). PMA treatment also notably decreased cell viability, as measured by CCK-8 assay, and increased cell apoptosis, as measured by flow cytometry, relative to treatment with DMSO. This effect was reversed by treatment with PMA and ADSC-derived EVs combined (Fig. 7, D and E).
HG-induced MP5 cells were further treated with ADSC-derived EVs after overexpression of VEGFA. VEGFA protein expression was significantly decreased after treatment with ADSC-derived EVs, whereas further overexpression of VEGFA upregulated the VEGFA protein expression (Fig. 7F). HG-induced MP5 cells treated with ADSC-derived EVs plus pcDNA-3.1 displayed increased viability and decreased apoptosis of MP5 cells compared with that of cells treated with PBS. This was reversed by treatment with ADSC-derived EVs and pcDNA-3-VEGFA (Fig. 7G). These data suggest that ADSC-derived EVs inhibit TLR4 expression, resulting in the downregulation of NF-κB/VEGFA and inhibition of HG-induced MP5 cell injury.
Delivery of miR-26a-5p by ADSC-derived EVs reduced the pathological symptoms and cell apoptosis in spontaneous diabetic mice
To explore the effects of miR-26a-5p delivered by ADSC-derived EVs in a mouse model of DN, ADSC-derived EVs and/or miR-26a-5p antagomir were injected into C57BL/KsJ db/db mice via the tail vein. Mice were then observed for changes in urine protein, Scr, and BUN levels, and the histopathological changes of mice were detected using PAS staining. As illustrated, diabetic mice treated with PBS and antagomir NC displayed accumulated extracellular matrix in their kidney tissues, viability of mesangial cells, thickening of basement membrane, and nuclear lamina dispersion, with increased proteinuria in the lumen, hyaline degeneration, and severe vacuolar degeneration of renal tubular epithelial cells. ADSC-derived EVs and antagomir NC treatment led to decreased levels of urine protein, Scr, and BUN in DN modeled mice and alleviated histopathological changes compared with those of PBS and antagomir NC treatment. Treatment with ADSC-derived EVs and miR-26a-5p antagomir treatment markedly increased the levels of urine protein, Scr, and BUN while aggravating the histopathological changes in the mice compared with treatment with ADSC-derived EVs and antagomir NC (Fig. 8, A and B). TUNEL staining showed that treatment with both ADSC-derived EVs and antagomir NC significantly reduced cell apoptosis compared with that of DN mice treated with PBS and antagomir NC. Apoptosis was notably increased in DN mice treated with both ADSC-derived EVs and miR-26a-5p antagomir compared with treatment with ADSC-derived EVs and antagomir NC (Fig. 8C).
Figure 8.
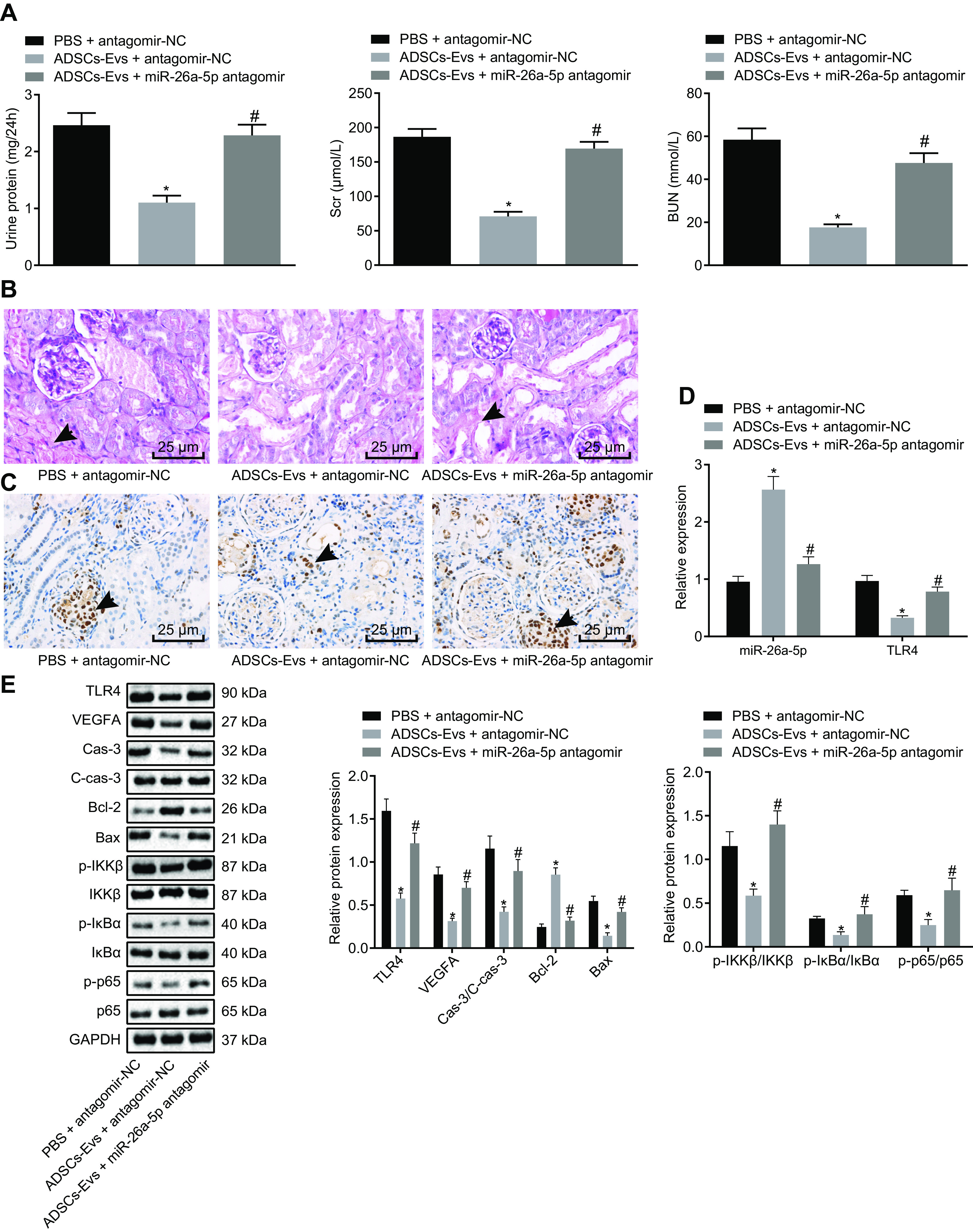
Delivery of miR-26a-5p by ADSC-derived EVs reduces the pathological symptoms and cell apoptosis in spontaneously diabetic mice (n = 6 mice per group). A, the levels of urine protein, Scr, and BUN of mice. B, histopathological characterization of DN determined by PAS staining (the black arrows indicate accumulation of extracellular matrix and thickening of basement membrane) (scale bar, 25 μm). C, the apoptosis of glomerular podocytes detected by TUNEL staining. The black arrow indicates apoptotic cells (the brownish-yellow cells) (scale bar, 25 μm). D, the expression of miR-26a-5p and TLR4 in MP5 cells as detected by RT-qPCR. E, the protein expression of apoptosis-related proteins (caspase-3, cleaved caspase-3, Bcl-2, Bax) in renal tissues of mice as detected by Western blot analysis, normalized to GAPDH. Data are expressed as mean ± standard deviation. Data among multiple groups were compared using one-way ANOVA, followed by Tukey's post hoc test. The experiment was repeated three times. *, p < 0.05 versus PBS + antagomir-NC. #, p < 0.05 versus ADSCs-Exo + antagomir-NC.
RT-qPCR was used to analyze miR-26a-5p and TLR4 expression in the renal tissues of mice in response to different treatments. Treatment with ADSC-derived EVs and antagomir NC significantly increased miR-26a-5p expression and decreased TLR4 expression compared with PBS plus antagomir NC treatment; however, treatment with ADSC-derived EVs plus miR-26a-5p antagomir decreased miR-26a-5p expression and increased TLR4 expression compared with treatment with ADSC-derived EVs and antagomir NC (Fig. 8D).
Western blot analysis was used to detect the levels of apoptosis-related proteins TLR4 and VEGFA as well as NF-κB pathway-related proteins. Treatment with ADSC-derived EVs and antagomir NC led to marked decreases in Bax, cleaved caspase-3, caspase-3, IKKβ, IκBα, p65, and VEGFA protein expression as well as in the extent of IKKβ, IκBα, and p65 phosphorylation, accompanied by an increase in the protein expression of Bcl-2 compared with PBS and antagomir NC treatment. These changes were reversed upon treatment with ADSC-derived EVs and miR-26a-5p antagomir (Fig. 8E). Overall, delivery of miR-26a-5p by ADSC-derived EVs significantly reduces the pathological symptoms and cell apoptosis in spontaneously diabetic mice.
Discussion
DN is a fatal complication of diabetes characterized by metabolic disorders and chronic inflammation (25). Increased diabetic podocyte dysfunction aggravates diabetes-induced kidney injury (26). Because EVs have been shown to confer protection against renal diseases (27), we studied the effects of miR-26a-5p delivery by EVs derived from ADSCs on the pathological symptoms of DN. Delivery of miR-26a-5p by ADSC-derived EVs alleviated several pathological symptoms associated with DN.
Initially, we found that miR-26a-5p was highly expressed in EVs obtained from adipose tissue-derived MSCs; however, miR-26a-5p was poorly expressed and TLR4 was highly expressed in DN. As previously reported, miR-26a-5p is highly expressed in exosomes secreted from ADSCs (28). Similarly, a previous study found that miR-26a expression was low in biopsy samples from patients with DN (29). Moreover, meta-analysis revealed that miR-26a-5p expression was downregulated in DN (15), consistent with our results. In addition, high expression of TLR4 was detected in the glomerular basement membrane, proximal convoluted tubule, and the renal interstitial area of the kidney from rats with DN (30). Finally, another study also observed upregulation of TLR4 in db/db mice and SV40-MES-13 cells compared with that of the normal controls (31).
We subsequently demonstrated that EVs derived from ADSCs were able to alleviate the pathological symptoms of DN, as evidenced by a reduction in protein urine, Scr, and BUN, and suppress glomerular podocyte apoptosis in spontaneous diabetic mice. EVs also prevented apoptosis in HG-induced MP5 cells characterized by a reduction in Bax, cleaved caspase-3, and caspase-3 and increased Bcl-2. Transplantation of ADSCs has been highlighted as a potential therapy for DN (32). Consistent with our results, stem cell-derived EVs were capable of suppressing fibrosis in a mouse model of DN (33). In addition, ADSC-derived EVs alleviated streptozotocin-induced DN in rats (34). Furthermore, this study found that ADSC-derived EVs alleviated signs of DN by delivering miR-26a-5p to nearby cells. In line with our findings, exosomal miR-26a inhibited muscle wasting and renal fibrosis in a mouse model of obstructive kidney disease (35) and suppressed cardiac fibrosis in chronic kidney disease (36). Notably, ADSC-derived exosomes transferred miR-181-5p to injured liver cells, preventing liver fibrosis (37). Urinary protein, blood urea nitrogen, and serum creatinine levels were measured to determine the kidney function, and attenuation of DN was associated with reduced levels of Scr, BUN, and urine protein in the streptozotocin-induced DN rat (38, 39). At the same time, accelerated apoptosis and renal injury in diabetic rats were associated with decreased Bcl-2 and increased BAX cleaved caspase-3 levels (40, 41). These reports support our findings that delivery of miR-26a-5p into MP5 cells by ADSC-derived EVs suppressed the progression of DN.
Furthermore, we found that miR-26a-5p delivered by ADSC-derived EVs targeted TLR4 in HG-induced MP5 cells, inactivating the NF-κB/VEGFA pathway. Interestingly, upregulated TLR4 increases the susceptibility of diabetic rats to acute kidney injury following myocardial infarction (42). Moreover, reduced expression of TLR4 and NF-κB nuclear translocation caused by catalpol expression suppressed HG-induced podocyte injury (43). DN progression is characterized by the activation of NF-κB in tubular epithelial cells (44), and silencing of VEGA attenuated podocyte injury induced by HG (9). Together, these reports support our findings that TLR4 plays a role in the pathological progression of DN. Additionally, inhibition of TLR4 expression downregulated NF-κB in vitro, thereby alleviating the development of atherosclerosis (45). Therefore, it could be concluded that miR-26a-5p, when delivered by ADSC-derived EVs into MP5 cells, targets TLR4, thereby inactivating the NF-κB/VEGFA signaling pathway and regulating MP5 cell injury.
Our findings demonstrated that delivery of miR-26a-5p by ADSC-derived EVs alleviates the pathological symptoms of DN by targeting TLR4, which subsequently inactivates the NF-κB/VEGFA signaling pathway (Fig. 9). This suggests a protective role for miR-26a-5p-containing EVs derived from ADSCs during DN, providing a novel therapeutic target for the prevention of DN in the clinical setting. Moreover, this study was the first to discover the effect of the miR-26a-5p/TLR4 axis in DN. Nevertheless, further studies based on human specimens are required to confirm the underlying molecular mechanism.
Figure 9.
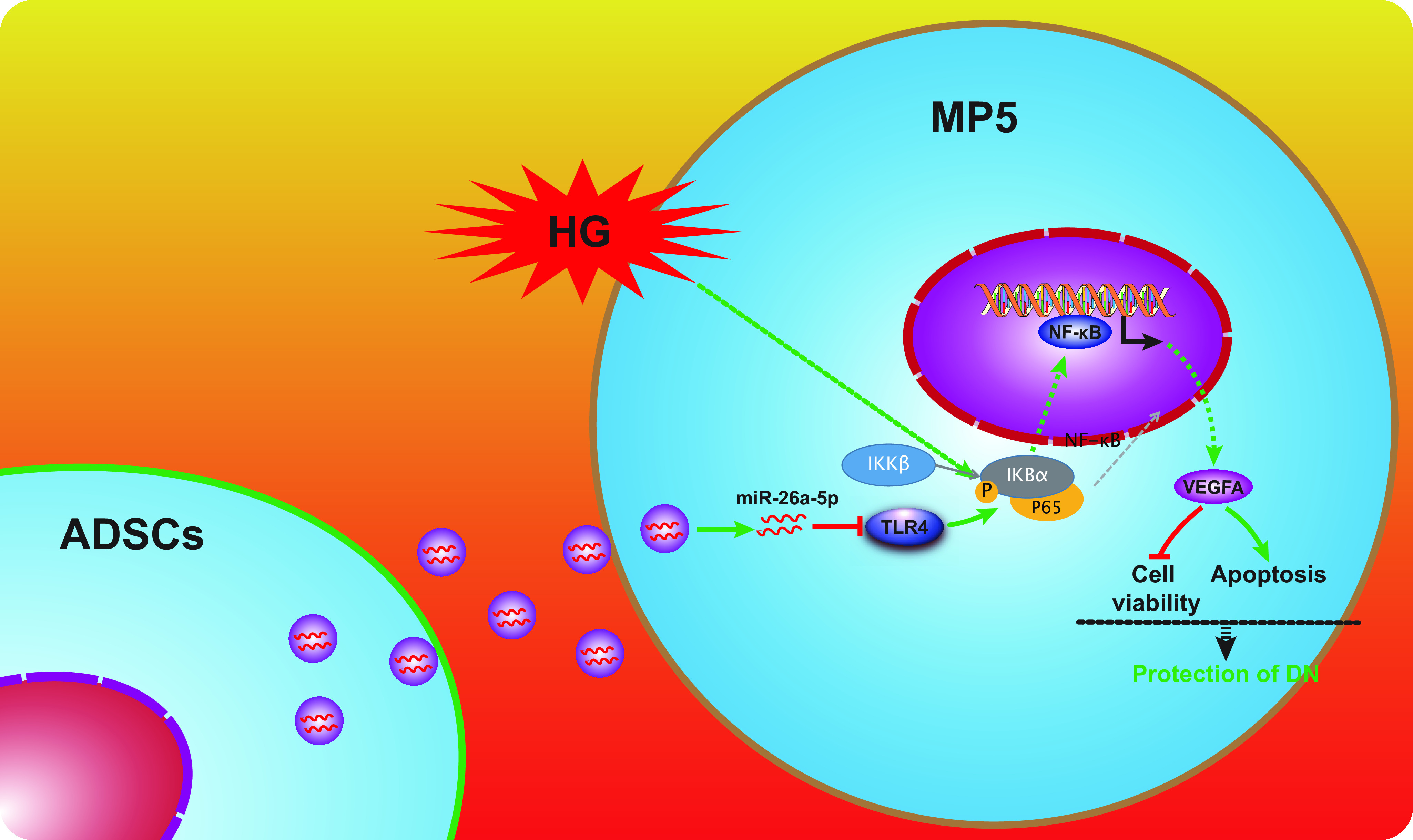
The molecular mechanism by which ADSC-derived EVs alleviate injury caused by DN. EVs derived from ADSCs enter mouse glomerular podocytes, MP5 cells. MiR-26a-5p contained in the ADSC-derived EVs targets and inhibits TLR4, inactivating the NF-κB/VEGFA pathway in HG-induced MP5 cells, characterized by decreased IKKβ, IκBα, and p65 protein expression and a decrease in the extent of the phosphorylation of those proteins. This leads to enhanced viability and suppresses apoptosis of MP5 cells.
Materials and methods
Ethics statement
This study was approved by the medical ethics committee of Huaihe Hospital, Henan University. All animal experiments were performed according to the provisions of the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (46).
Bioinformatic analysis
MiRs present in MSC-derived EVs were obtained from the EVmiRNA database (RRID:SCR_018795). The diabetes-related gene expression data set GSE21340 was downloaded from the GEO database (RRID:SCR_005012), which included 10 normal samples and 5 diabetes samples. Differential expression analysis was conducted using the R language “limma” package (47) using normal samples as controls. The threshold for differential expression analysis was set at a log2 fold change of >1 and p value of <0.05. Downstream targets genes of different miRs were predicted using the miRanda database (RRID:SCR_017496) and the Starbase database (RRID:SCR_016303), and the intersecting genes were overlaid with the differential expression results from the GSE21340 data set. A map for binding sites between miRs and genes was delineated after searching the above miR database.
Experimental materials
Mannitol (MA) (SM8120) was purchased from Solarbio (Beijing, China), and Roswell Park Memorial Institute 1640 (SH30809.01B), Dulbecco's modified Eagle's medium/nutrient mix F12 (DMEM/F12) (SH30023.01B), and BSA (SH30574.03) were purchased from HyClone Company (Logan, UT, USA). Collagenases I (17100-017), II (17101-015), and IV (17104-019) and 0.25% trypsin (15050065) were purchased from Gibco Company (Grand Island, NY, USA). Propidium iodine (25535-16-4), RNase A (9001-99-4), dexamethasone (D4902), β-glycerophosphate (G9422), ascorbic acid (A7562), insulin (I5500), ninhydrin (I7378), and 3-isobutyl-1-methylxanthine (IBMX; I7018) were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA). Triton X-100 (1%) (SBJ-1141) was purchased from SBJbio Selleck Uscn Life Science Inc. (Shanghai, China), and phorbol-12-myristate-13-acetate (PMA) (TLRL-PMA) was from Invitrogen (Carlsbad, CA, USA). The primers for miR-26a-5p inhibitor, miR-26a-5p mimic, and pCDNA-TLR4 were designed and synthesized by GenePharma (Shanghai, China), whereas the miR-26a-5p antagomir was purchased from Biomics Biotechnologies (Jiangsu, China).
Animal treatment
C57BL/KsJ db/m male mice aged 7 weeks and weighing 18–22 g (control mice, n = 6) and C57BL/KsJ db/db mice (a total of 18 spontaneous diabetic mice assigned into three groups, n = 6) were purchased from Cavens Laboratory Animal (SCX, 2016-0010, Changzhou, China). Three to five mice were placed in each cage, with controlled, alternating 12-h day/night cycles and temperature of 22 ± 2 °C, with free access to food and water. When reared to the age of 12 weeks, mice were subjected to characterization of DN phenotype by histopathological examination using periodic acid Schiff (PAS) staining according to the published literature (13). At 13 weeks of age, mice underwent injection with either phosphate-buffered saline (PBS), ADSC-derived EVs, or miR-26a-5p antagomir via tail vein for a 12-week therapy.
Cell isolation and culture
The mice were anesthetized with pentobarbital (200 mg/kg i.p.), and the subcutaneous adipose tissues of the groin were extracted. ADSCs were isolated after collagenase digestion. In short, the fat tissues were cut and washed twice with PBS buffer and centrifuged at 1200 rpm for 10 min. Following the removal of the supernatant, prepared collagenase was added to the precipitates. Detachment was allowed to proceed at 37 °C for 40 min and was terminated by the addition of complete medium. Cells were then filtered using a 40-mm cell filter, centrifuged at 1500 rpm for 8 min, resuspended with complete Roswell Park Memorial Institute 1640 medium, and finally cultured in a humidified incubator at 37 °C with 5% CO2.
In the miR-26a-5p inhibitor experiment, ADSCs were transfected with an miR-26a-5p inhibitor or NC for 48 h, and then ADSC-derived EVs were isolated for further experimentation. Mouse glomerular podocytes, MP5 cells, were purchased from the Institute of Basic Medicine, China Academy of Chinese Medical Sciences (Beijing, China) (3111C0001CCC000230). MP5 cells were cultured in DMEM containing 10% fetal bovine serum (FBS) in a humidified incubator at 37 °C with 5% CO2. MP5 cells were treated with one 5.5 mm d-glucose (NG), 5.5 mm d-glucose + 24.5 mm MA, 30 mm d-glucose (HG), or HG treatment with an increasing concentration of ADSC-derived EVs over 24 h, 48 h, and 72 h. Finally, MP5 cells were treated with NG, MA, HG, or HG plus ADSC-derived EVs for 48 h.
In vitro differentiation of pluripotent ADSCs
Osteoblast differentiation was induced as follows. ADSCs (1 × 105 cells/well) were inoculated into a 6-well culture plate, and once they were adherent to the wall, the cells were cultured with osteoblast culture medium (prepared with DMEM supplemented with 0.17 mm vitamin C, 0.5% FBS, 10 mm β-glycerophosphate, 100 nm dexamethasone, and 1% penicillin/streptomycin, all purchased from Sigma-Aldrich Chemical Company [St. Louis, MO, USA]). The cells were cultured at 37 °C with 5% CO2 for 21–28 days, with the medium being replaced every 2 days. After calcified nodules were observed under a light microscope, the cells were washed twice with PBS, fixed with 4% paraformaldehyde for 15 min, washed twice with deionized water, and stained with Alizarin Red S for 30 min. After staining, cells were washed twice with deionized water and observed under an optical microscope.
Adipocyte differentiation was induced as follows. ADSCs (1 × 105 cells/well) were inoculated into a 6-well culture plate, and the cells were cultured with adipogenic differentiation culture medium composed of DMEM supplemented with glutamine, 10% FBS, 1 μm rosiglitazone, 1 μm dexamethasone, 0.5 mm 3-isobutyl-1-methylxanthine, 10 μg/ml insulin, 0.2 mm indomethacin, and 1% penicillin/streptomycin. After 3 days, the medium was replaced every other day with low-glucose DMEM containing glutamine, 10% FBS, 1% penicillin/streptomycin, 1 mm rosiglitazone, and 10 mg/ml insulin. The cells were cultured at 37 °C with 5% CO2 for 21–28 days. When the fat droplets were obviously visible under optical microscopy, the cells were washed with deionized water, fixed with 4% paraformaldehyde for 15 min, rinsed with PBS, and then incubated with 65% isopropanol for 4 min. The cells were then stained with new, filtered oil red O for 4 min, washed with deionized water, counterstained with hematoxylin, and observed using an optical microscope.
Chondroblast differentiation was subsequently induced. ADSCs (2 × 106 cells/tube) were cultured in a 15-ml centrifuge tube at 37 °C with 5% CO2 for 24 h and then incubated in chondrogenic medium composed of DMEM (4.5 g/liter glucose) supplemented with 100 nm dexamethasone, 0.35 mm proline, 0.17 mm vitamin C, 1 mm sodium pyruvate, 1% insulin-transferrin-selenium, 10 ng/ml transforming growth factor beta 3 (TGFβ-3), and 1% penicillin/streptomycin (Sigma-Aldrich Chemical Company, St. Louis, MO, USA) at 37 °C with 5% CO2 for 21–28 days, during which the medium was replaced every 2 days. After the cells were cultured into spheres with a diameter of 1.5–2 mm, the cells were sliced and stained with Alcian blue. Briefly, the cells are washed twice with deionized water, fixed with 4% paraformaldehyde for 15 min, and cut into 10-μm frozen sections. Finally, the sections were stained with 0.5% Alcian blue dye for 30 min and observed under an optical microscope.
Isolation of EVs
At the third passage, ADSCs were inoculated into a 6-cm culture dish and cultured at 37 °C with 5% CO2. When the cell density reached 70–80%, the cells were cultured in fresh medium containing EVs-free FBS for 24 h, followed by ultracentrifugation at 100,000 × g for 18 h and filtration using a 0.22-μm filter to obtain EV-free FBS. After 24 h of culture, conditioned medium was collected and EVs were extracted by ultracentrifugation. In short, the cell culture medium was centrifuged at 300 × g for 10 min, at 2000 × g for 15 min, and at 12,000 × g for 30 min, respectively, to remove floating cells and cell fragments. Following filtration using a 0.22-μm filter, the supernatant was collected and ultracentrifuged at 100,000 × g at 4 °C for 2 h, washed with PBS, ultracentrifuged for the second time under the same conditions, and resuspended with PBS. ADSC-derived EVs were immediately used in subsequent experimentation or stored at −80 °C (48).
Characterization of EVs
The size distribution of isolated EVs was measured by dynamic light scattering (DLS) using a NanosizerTM instrument (Malvern Instruments, Malvern, UK). The morphology of EVs was observed using a Hitachi H-7650 transmission electron microscope (TEM; Hitachi, Tokyo, Japan). EVs were dissolved in radioimmune precipitation assay buffer and quantified using a bicinchoninic acid (BCA) protein analysis kit (Thermo Fisher Scientific, Rockford, IL, USA). CD63 (ab216130, 1:1000), tumor susceptibility gene 101 (Tsg101) (ab30871, 1:1000), and calnexin (ab22595, 1:1000) were detected by Western blot analysis.
Uptake test of EVs
To determine the uptake of ADSC-derived EVs by MP5 cells, EVs were labeled with green fluorescent dye (PKH67, Sigma-Aldrich Chemical Company [St. Louis, MO, USA]) and incubated with MP5 cells at 37 °C for 3 h. Following several PBS rinses, the cells were fixed with 4% paraformaldehyde for 15 min, washed with PBS, and then stained with 4′,6-diamidino-2-phenylindole (0.5 μg/ml; Invitrogen, Carlsbad, CA, USA). The green fluorescence in MP5 cells was observed under a fluorescence microscope. To detect whether miRs can be transferred from EVs into MP5 cells, MP5 cells treated with ADSC-derived EVs for 3 h were collected for RNA extraction, and miR expression was detected by RT-qPCR.
ADSCs were transfected with Cy3-miR-26a-5p mimic (Ribobio, Guangzhou, China) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and then cocultured with MP5 cells. The uptake of Cy3-miR-26a-5p mimic by MP5 cells was observed via fluorescence microscopy.
Determination of urine protein, Scr, and BUN levels
The urine of mice was collected after 12 weeks of treatment and then analyzed using a BCA kit. In short, BCA working solution was mixed with urine, followed by incubation at 37 °C for 30 min. Optical density values were detected at 562 nm. After treatment with EVs, the plasma was collected via the abdominal aorta, kept at room temperature for 2 h, and centrifuged at 3500 rpm for 5 min. The levels of Scr and BUN were measured using detection kits (Scr, C011-1, Jiang Cheng Biotechnology, Nanjing, China; BUN, C013-2, Jiang Cheng Biotechnology, Nanjing, China).
Cell transfection
ADSCs were transfected with inhibitor NC, miR-26a-5p inhibitor, pcDNA-3.1, pcDNA-TLR4, and pcDNA-VEGFA. miR-26a-5p inhibitor and inhibitor NC were synthesized by Invitrogen (Carlsbad, CA, USA); pcDNA-3.1/TLR4/VEGFA plasmids were purchased from GenePharma (Shanghai, China). The experiment was carried out according to the instructions of the Lipofectamine 2000 kit (Invitrogen, Carlsbad, CA, USA). In brief, cells were inoculated into a 6-well plate 24 h before transfection. After 6 days of culture, cells were resuspended in conditioned medium and then seeded into a new 6-well plate, followed by transfection according to the above grouping. After transfection, the cells were incubated at 37 °C with 5% CO2 for 6–8 h and further cultured for 24–48 h with complete medium after centrifugation.
Dual-Luciferase reporter gene assay
The synthetic TLR4 3'UTR gene fragment was introduced into the psiCHECK-2 vector (Promega Corporation, Madison, WI, USA). The complementary sequence mutation site of the seed sequence was designed based on the TLR4 WT and then inserted into the psiCHECK-2 vector reporter plasmids. The correctly sequenced luciferase reporter plasmids of TLR4 3'UTR-WT (100 ng) and TLR4 3'UTR-mutant type (MUT) (100 ng) were cotransfected into HEK-293T cells with either miR-26a-5p mimic or mimic-NC (2 nm, Dharmacon Inc., Chicago, IL, USA, and CRL-1415, Xinyu Biotechnology Co., Ltd., Shanghai, China), respectively. After 48 h of transfection, cells were collected and lysed. The luciferase activity was detected using a luciferase detection kit (RG005, Beyotime Biotechnology Co., Shanghai, China) on a GloMax 20/20 luminometer fluorescence detector (Promega Corporation, Madison, WI, USA).
Cellular immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 5 min after they were half-dried. Next, the formaldehyde was removed, and 0.5% Triton X-100 prepared with PBS was used to clear the cells for 15 min. Next, the cells were sealed with 1% BSA at room temperature for 30 min. After removal of the sealing solution, the cells were incubated on a coverslip along with the primary antibody against p65 (no. 8242, 1:200, Cell Signaling Technologies [CST], Beverly, MA, USA) at 4 °C overnight and then with the fluorescence-labeled secondary antibody (ab250115, Abcam Inc., Cambridge, MA, USA) at room temperature for 1 h in the dark. Finally, blocking agent (no. MM1402-5ML, MK Bio Science Co., Ltd., Suwon-si, Gyeonggi-do, Korea) containing 4′,6-diamidino-2-phenylindole was used to seal the coverslip, and the cells were observed under a fluorescence microscope (Olympus Inc., Tokyo, Japan).
Flow cytometry
Flow cytometry was used to detect the representative surface markers of ADSCs. In short, ADSCs were detached with trypsin to form a single-cell suspension, washed by PBS (without calcium and magnesium), and then blocked with 10% normal goat serum to prevent nonspecific binding. Afterward, a series of monoclonal antibodies to CD14, CD19, CD29, CD34, CD44, CD45, CD73, CD90, HLA-A, -B, and -C, and HLA-DR (1:100, BioLegend, San Diego, CA, USA) labeled with FITC dye were incubated along with the cells for 30 min. A blank control and an FITC single-step staining control were set up, followed by a PBS rinse. Finally, the cells were resuspended with 10% normal goat serum and analyzed with a CyAn ADP analyzer (Beckman Coulter Life Sciences, Brea, CA, USA).
To assess apoptosis, cells were detached with 0.25% EDTA-free trypsin, centrifuged at 1500 rpm for 5 min, resuspended to precipitate, and washed with PBS. After centrifugation at 1500 rpm for 5 min, the cells were resuspended again with 300 μl binding buffer, followed by incubation with 5 μl annexin V-FITC at room temperature in the dark for 5 min. Subsequently, the cells were stained with 10 μl propidium iodine for 10 min, after which the apoptosis was detected using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
CCK-8 assay
MP5 cells (3000 cells/well) treated with HG or ADSC-derived EVs were inoculated into 96-well plates, followed by incubation for 24, 48, and 72 h. Next, 10 μl CCK-8 reagent was added into each well and incubated for 2 h. The optical density at 450 nm was detected using a microplate reader (Multiskan MK3; Thermo Fisher Scientific, Rockford, IL, USA).
TUNEL staining
In the detection of apoptosis, tissue sections were evaluated according to the reagent specifications (Biobox, BA27). In short, the samples were labeled with DNA fragments in TUNEL solution containing terminal deoxynucleotidyltransferase and streptavidin-FITC. After washing, cells were incubated with paraformaldehyde-conjugated anti-FITC in a dark room at 37 °C for 30 min, followed by addition of DAB reaction solution to the slide and 4′,6-diamidino-2-phenylindole staining. Following hydrochloric acid alcohol differentiation and ethanol dehydration, the samples were incubated with xylene and sealed with neutral resin. The images were processed and analyzed under a microscope (IX53, Olympus Inc., Tokyo, Japan).
RT-qPCR
Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from cultured cells or tissues. Complementary DNA (cDNA) was synthesized from 1 μg total RNA using a Revert Aid first-strand cDNA synthesis kit (Fermentas, Burlington, Ontario, Canada). RT-qPCR analysis then was carried out with the use of the SYBR Premix ExTaqTM II in an ABI PRISM® 7900HT system (Takara Holdings Inc., Kyoto, Japan). GAPDH was used as an internal reference for determining relative mRNA expression using relative quantification (2−ΔΔCt method). The PCR primers used in this study are shown in Table 1. MiRs delivered by EVs were isolated using a SeraMir exosome RNA purification kit (System Biosciences, Mountain View, CA, USA). By following the instructions provided by the TaqMan microRNA assist kit (Applied Biosystems, Foster City, CA, USA), miR was converted to cDNA. The FastStart Universal SYBR Green master mix (Roche, Indianapolis, IN, USA) was used for the RT-qPCR reaction, along with the miR-specific forward primers provided by Sangon Biotechnology (Shanghai, China) and general reverse primers provided by the TaqMan microRNA assay kit. The expression of miRs was normalized to U6.
Table 1.
Primer sequences for RT-qPCRa
| Gene | Primer sequence (5′–3′) |
|---|---|
| miR-26a-5p | F, GCGCGCGTAACAGTGTACAGC |
| R, GTCGTATCCAGTGCAGGGTCC | |
| U6 | F, CTCGCTTCGGCAGCACA |
| R, AACGCTTCACGAATTTGCGT | |
| TLR4 | F, CACTGTTCTTCTCCTGCCTGAC |
| R, TGGTTGAAGAAGGAATGTCATC | |
| GAPDH | F, AGACAGCCGCATCTTCTTGT |
| R, TGATGGCAACAATGTCCACT |
aRT-qPCR, reverse transcription-quantitative PCR; miR, microRNA; TLR4, Toll-like receptor 4; F, forward; R, reverse.
Western blot analysis
Proteins were resolved by SDS-PAGE, transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), and blocked with 5% milk and 0.1% TBS containing Tween 20 for 1 h. Membranes were immunoblotted with primary antibodies to TLR4 (ab13867, 1:1000), VEGFA (ab46154, 1:1000), Bcl-2 (ab196495, 1:1000), Bax (ab53154, 1:500), caspase-3 (ab44976, 1:500), and cleaved caspase-3 (ab2302, 1:500), which were all purchased from Abcam Inc. (Cambridge, MA, USA), and to phosphorylated (p)-IκB kinase β (IKKβ) (no. 2078, 1:1000), IKKβ (no. 8943, 1:1000), p-IκBα (no. 2859, 1:1000), IκB kinase α (IκBα) (no. 4812, 1:1000), p-p65 (no. 3031, 1:1000), and p65 (no. 8242, 1:1000), which were purchased from Cell Signaling Technology (Danvers, MA, USA). Next, the membranes were probed with horseradish peroxidase-conjugated anti-rabbit IgG secondary antibodies purchased from Cell Signaling Technology (Danvers, MA, USA). We developed blots using enhanced chemiluminescence reagent (Thermo Fisher Scientific, Rockford, IL, USA), captured the images using the ChemiDoc XRS Plus luminescent image analyzer (Bio-Rad Laboratories, Hercules, CA, USA), and quantified band area using Image J software. The relative expression of the target protein was normalized to the band intensity of GAPDH.
Statistical analysis
Data analysis was performed utilizing SPSS 21.0 (IBM Corp., Armonk, NY, USA). Data are expressed as mean ± standard deviation. The data between two groups obeying normal distribution and homogeneous variance in paired design were compared using a paired t test and those in unpaired design using unpaired t test. Data among multiple groups were compared using one-way analysis of variance (ANOVA), with a Tukey's test conducted for post hoc analysis. Data among multiple groups at different time points were compared by repeated-measures ANOVA, followed by Bonferroni post hoc test. Pearson's correlation analysis was applied for correlation analysis. The Kaplan-Meier method was used to calculate the survival rate of patients, and log-rank test was employed for single-factor analysis. A p value of <0.05 indicated a statistically significant difference.
Data Availability
All the data are in the manuscript.
Acknowledgments
We give our sincere gratitude to the reviewers for their valuable suggestions.
Author contributions—Y. D. and Q. L. conceptualization; Y. D. and X. Z. supervision; Y. D., Q. L., and X. Z. validation; Y. D., Q. L., and X. Z. visualization; Y. D., X. Z., and J. S. writing-original draft; Y. D., Q. L., Y. W., Y. M., and F. C. writing-review and editing; Q. L. and X. Z. investigation; Y. W., Y. M., F. C., and J. S. resources; Y. W., Y. M., F. C., and J. S. data curation; Y. W., Y. M., F. C., and J. S. software; Y. W., Y. M., F. C., and J. S. formal analysis; Y. W., Y. M., and F. C. methodology; J. S. funding acquisition.
Funding and additional information—This study was supported by the Key Scientific Research Project of Colleges and Universities in Henan Province (16A320031).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- DN
- diabetic nephropathy
- EVs
- extracellular vesicles
- miRs
- microRNAs
- ADSCs
- adipose-derived mesenchymal stem cells
- MSCs
- mesenchymal stem cells
- HG
- high glucose
- PMA
- phorbol-12-myristate-13-acetate
- VEGFA
- vascular endothelial growth factor A
- NG
- normal glucose
- MA
- mannitol
- BUN
- blood urea nitrogen
- Scr
- serum creatinine
- PAS
- periodic acid Schiff
- NC
- negative control
- TEM
- transmission electron microscope
- CCK-8
- cell counting kit-8
- ANOVA
- analysis of variance
- DLS
- dynamic light scattering.
References
- 1. Wang X., Xu Y., Chu C., Li H., Mi J., Wen Z., Zhang S., Wang Q., and Quan S. (2019) Effect of safflower yellow on early type II diabetic nephropathy: a systematic review and meta-analysis of randomized controlled trials. J. Pediatr. Endocrinol. Metab. 32, 653–665 10.1515/jpem-2018-0425 [DOI] [PubMed] [Google Scholar]
- 2. De la Cruz-Cano E., Jimenez-Gonzalez C. D. C., Morales-Garcia V., Pineda-Perez C., Tejas-Juarez J. G., Rendon-Gandarilla F. J., Jimenez-Morales S., and Diaz-Gandarilla J. A. (2019) Arg913Gln variation of SLC12A3 gene is associated with diabetic nephropathy in type 2 diabetes and Gitelman syndrome: a systematic review. BMC Nephrol. 20, 393 10.1186/s12882-019-1590-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Misra A., and Shrivastava U. (2016) Obstructive sleep apnea and diabetic nephropathy. Diabetes Technol. Ther. 18, 405–407 10.1089/dia.2016.0147 [DOI] [PubMed] [Google Scholar]
- 4. Ioannou K. (2017) Diabetic nephropathy: is it always there? Assumptions, weaknesses and pitfalls in the diagnosis. Hormones 16, 351–361 10.14310/horm.2002.1755 [DOI] [PubMed] [Google Scholar]
- 5. Moţa E., Panduru M. N., Popa S. G., and Moţa M. (2009) Risk factors for diabetic nephropathy: intrinsic or extrinsic renal? Rom. J. Intern. Med. 47, 397–401 [PubMed] [Google Scholar]
- 6. Tung C. W., Hsu Y. C., Shih Y. H., Chang P. J., and Lin C. L. (2018) Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology 23(Suppl 4), 32–37 10.1111/nep.13451 [DOI] [PubMed] [Google Scholar]
- 7. Zhong F., Chen H., Xie Y., Azeloglu E. U., Wei C., Zhang W., Li Z., Chuang P. Y., Jim B., Li H., Elmastour F., Riyad J. M., Weber T., Chen H., Wang Y., et al. (2018) Protein S protects against podocyte injury in diabetic nephropathy. J. Am. Soc. Nephrol. 29, 1397–1410 10.1681/ASN.2017030234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y., Wang C., Zhang X., Gu H. F., and Wu L. (2018) Common drugs for stabilization of renal function in the progression of diabetic nephropathy and their relations with hypertension therapy. Curr. Diabetes Rev. 14, 149–161 10.2174/1573399813666170214112115 [DOI] [PubMed] [Google Scholar]
- 9. Duan Y. R., Chen B. P., Chen F., Yang S. X., Zhu C. Y., Ma Y. L., Li Y., and Shi J. (2019) Exosomal microRNA-16-5p from human urine-derived stem cells ameliorates diabetic nephropathy through protection of podocyte. J. Cell Mol. Med. 10.1111/jcmm.14558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gangadaran P., and Ahn B. C. (2020) In vivo tracking of tumor-derived bioluminescent extracellular vesicles in mice. Methods Mol. Biol. 2081, 203–210 10.1007/978-1-4939-9940-8_14 [DOI] [PubMed] [Google Scholar]
- 11. Fiedler T., Rabe M., Mundkowski R. G., Oehmcke-Hecht S., and Peters K. (2018) Adipose-derived mesenchymal stem cells release microvesicles with procoagulant activity. Int. J. Biochem. Cell Biol. 100, 49–53 10.1016/j.biocel.2018.05.008 [DOI] [PubMed] [Google Scholar]
- 12. Yang Q., Jia L., Li X., Guo R., Huang Y., Zheng Y., and Li W. (2018) Long noncoding RNAs: new players in the osteogenic differentiation of bone marrow- and adipose-derived mesenchymal stem cells. Stem Cell Rev. Rep. 14, 297–308 10.1007/s12015-018-9801-5 [DOI] [PubMed] [Google Scholar]
- 13. Jin J., Shi Y., Gong J., Zhao L., Li Y., He Q., and Huang H. (2019) Exosome secreted from adipose-derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte. Stem Cell Res. Ther. 10, 95 10.1186/s13287-019-1177-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tetta C., Ghigo E., Silengo L., Deregibus M. C., and Camussi G. (2013) Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 44, 11–19 10.1007/s12020-012-9839-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gholaminejad A., Abdul Tehrani H., and Gholami Fesharaki M. (2018) Identification of candidate microRNA biomarkers in diabetic nephropathy: a meta-analysis of profiling studies. J. Nephrol. 31, 813–831 10.1007/s40620-018-0511-5 [DOI] [PubMed] [Google Scholar]
- 16. Koga K., Yokoi H., Mori K., Kasahara M., Kuwabara T., Imamaki H., Ishii A., Mori K. P., Kato Y., Ohno S., Toda N., Saleem M. A., Sugawara A., Nakao K., Yanagita M., et al. (2015) MicroRNA-26a inhibits TGF-beta-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia 58, 2169–2180 10.1007/s00125-015-3642-4 [DOI] [PubMed] [Google Scholar]
- 17. Kolz M., Baumert J., Muller M., Khuseyinova N., Klopp N., Thorand B., Meisinger C., Herder C., Koenig W., and Illig T. (2008) Association between variations in the TLR4 gene and incident type 2 diabetes is modified by the ratio of total cholesterol to HDL-cholesterol. BMC Med. Genet. 9, 9 10.1186/1471-2350-9-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu Z. M., Zheng H. Y., Chen L. H., Li Y. L., Wang Q., Liao C. F., and Li X. W. (2018) Low expression of miR-203 promoted diabetic nephropathy via increasing TLR4. Eur. Rev. Med. Pharmacol. Sci 22, 5627–5634 10.26355/eurrev_201809_15828 [DOI] [PubMed] [Google Scholar]
- 19. Wang H. Q., Wang S. S., Chiufai K., Wang Q., and Cheng X. L. (2019) Umbelliferone ameliorates renal function in diabetic nephropathy rats through regulating inflammation and TLR/NF-kappaB pathway. Chin. J. Nat. Med. 17, 346–354 10.1016/S1875-5364(19)30040-8 [DOI] [PubMed] [Google Scholar]
- 20. Li M., Guo Q., Cai H., Wang H., Ma Z., and Zhang X. (2020) miR-218 regulates diabetic nephropathy via targeting IKK-beta and modulating NK-kappaB-mediated inflammation. J. Cell. Physiol. 235, 3362–3371 10.1002/jcp.29224 [DOI] [PubMed] [Google Scholar]
- 21. da Silveira Cruz-Machado S., Carvalho-Sousa C. E., Tamura E. K., Pinato L., Cecon E., Fernandes P. A., de Avellar M. C., Ferreira Z. S., and Markus R. P. (2010) TLR4 and CD14 receptors expressed in rat pineal gland trigger NFKB pathway. J. Pineal Res. 49, 183–192 10.1111/j.1600-079X.2010.00785.x [DOI] [PubMed] [Google Scholar]
- 22. Ni H., Zhao W., Kong X., Li H., and Ouyang J. (2014) Celastrol inhibits lipopolysaccharide-induced angiogenesis by suppressing TLR4-triggered nuclear factor-kappa B activation. Acta Haematol. 131, 102–111 10.1159/000354770 [DOI] [PubMed] [Google Scholar]
- 23. Liang S., Chen Z., Jiang G., Zhou Y., Liu Q., Su Q., Wei W., Du J., and Wang H. (2017) Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-kappaB/IL-6 signals. Cancer Lett. 386, 12–23 10.1016/j.canlet.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 24. Onions K. L., Gamez M., Buckner N. R., Baker S. L., Betteridge K. B., Desideri S., Dallyn B. P., Ramnath R. D., Neal C. R., Farmer L. K., Mathieson P. W., Gnudi L., Alitalo K., Bates D. O., Salmon A. H. J., et al. (2019) VEGFC reduces glomerular albumin permeability and protects against alterations in VEGF receptor expression in diabetic nephropathy. Diabetes 68, 172–187 10.2337/db18-0045 [DOI] [PubMed] [Google Scholar]
- 25. Wang S., Li Y., Fan J., Zhang X., Luan J., Bian Q., Ding T., Wang Y., Wang Z., Song P., Cui D., Mei X., and Ju D. (2017) Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 8, e2937 10.1038/cddis.2017.292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin C. L., Hsu Y. C., Huang Y. T., Shih Y. H., Wang C. J., Chiang W. C., and Chang P. J. (2019) A KDM6A-KLF10 reinforcing feedback mechanism aggravates diabetic podocyte dysfunction. EMBO Mol. Med. 11, e9828 10.15252/emmm.201809828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Erdbrugger U., and Le T. H. (2016) Extracellular vesicles in renal diseases: more than novel biomarkers? J. Am. Soc. Nephrol. 27, 12–26 10.1681/ASN.2015010074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baglio S. R., Rooijers K., Koppers-Lalic D., Verweij F. J., Perez Lanzon M., Zini N., Naaijkens B., Perut F., Niessen H. W., Baldini N., and Pegtel D. M. (2015) Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res. Ther. 6, 127 10.1186/s13287-015-0116-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang L. P., Gao Y. Z., Song B., Yu G., Chen H., Zhang Z. W., Yan C. F., Pan Y. L., and Yu X. Y. (2019) MicroRNAs in the progress of diabetic nephropathy: a systematic review and meta-analysis. Evid. Based Complement. Alternat. Med. 2019, 3513179 10.1155/2019/3513179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu S. Y., Nie X. Z., Zhou W. Y., and Chen J. (2013) Expression and effect of TLR4 in rats with diabetic nephropathy. Asian Pac. J. Trop. Med. 6, 635–639 10.1016/S1995-7645(13)60109-1 [DOI] [PubMed] [Google Scholar]
- 31. Qi H., Yao L., and Liu Q. (2020) NORAD affects the progression of diabetic nephropathy through targeting miR-520h to upregulate TLR4. Biochem. Biophys. Res. Commun. 521, 190–195 10.1016/j.bbrc.2019.10.102 [DOI] [PubMed] [Google Scholar]
- 32. Takemura S., Shimizu T., Oka M., Sekiya S., and Babazono T. (2020) Transplantation of adipose-derived mesenchymal stem cell sheets directly into the kidney suppresses the progression of renal injury in a diabetic nephropathy rat model. J. Diabetes Invest. 11, 545–553 10.1111/jdi.13164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grange C., Tritta S., Tapparo M., Cedrino M., Tetta C., Camussi G., and Brizzi M. F. (2019) Stem cell-derived extracellular vesicles inhibit and revert fibrosis progression in a mouse model of diabetic nephropathy. Sci. Rep. 9, 4468 10.1038/s41598-019-41100-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang Y., Tian X., Bai S., Fan J., Hou W., Tong H., and Li D. (2012) Autologous transplantation of adipose-derived mesenchymal stem cells ameliorates streptozotocin-induced diabetic nephropathy in rats by inhibiting oxidative stress, pro-inflammatory cytokines and the p38 MAPK signaling pathway. Int J. Mol. Med. 30, 85–92 10.3892/ijmm.2012.977 [DOI] [PubMed] [Google Scholar]
- 35. Zhang A., Wang H., Wang B., Yuan Y., Klein J. D., and Wang X. H. (2019) Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. FASEB J. 33, 13590–13601 10.1096/fj.201900884R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang B., Zhang A., Wang H., Klein J. D., Tan L., Wang Z. M., Du J., Naqvi N., Liu B. C., and Wang X. H. (2019) miR-26a limits muscle wasting and cardiac fibrosis through exosome-mediated microRNA transfer in chronic kidney disease. Theranostics 9, 1864–1877 10.7150/thno.29579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qu Y., Zhang Q., Cai X., Li F., Ma Z., Xu M., and Lu L. (2017) Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell Mol. Med. 21, 2491–2502 10.1111/jcmm.13170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian H., Yang J., Xie Z., and Liu J. (2018) Gliquidone alleviates diabetic nephropathy by inhibiting notch/snail signaling pathway. Cell Physiol. Biochem. 51, 2085–2097 10.1159/000495827 [DOI] [PubMed] [Google Scholar]
- 39. Li T., Hua Q., Li N., Cui Y., and Zhao M. (2019) Protective effect of a polysaccharide from Dipsacus asper wall on streptozotocin (STZ)-induced diabetic nephropathy in rat. Int J. Biol. Macromol. 133, 1194–1200 10.1016/j.ijbiomac.2019.04.069 [DOI] [PubMed] [Google Scholar]
- 40. Zhao T., Fu Y., Sun H., and Liu X. (2018) Ligustrazine suppresses neuron apoptosis via the Bax/Bcl-2 and caspase-3 pathway in PC12 cells and in rats with vascular dementia. IUBMB Life 70, 60–70 10.1002/iub.1704 [DOI] [PubMed] [Google Scholar]
- 41. Ying C., Zhou X., Chang Z., Ling H., Cheng X., and Li W. (2016) Blood glucose fluctuation accelerates renal injury involved to inhibit the AKT signaling pathway in diabetic rats. Endocrine 53, 81–96 10.1007/s12020-016-0867-z [DOI] [PubMed] [Google Scholar]
- 42. Ohno K., Kuno A., Murase H., Muratsubaki S., Miki T., Tanno M., Yano T., Ishikawa S., Yamashita T., and Miura T. (2017) Diabetes increases the susceptibility to acute kidney injury after myocardial infarction through augmented activation of renal Toll-like receptors in rats. Am. J. Physiol. Heart Circ. Physiol. 313, H1130–H1142 10.1152/ajpheart.00205.2017 [DOI] [PubMed] [Google Scholar]
- 43. Chen Y., Liu Q., Shan Z., Zhao Y., Li M., Wang B., Zheng X., and Feng W. (2019) The protective effect and mechanism of catalpol on high glucose-induced podocyte injury. BMC Complement. Altern. Med. 19, 244 10.1186/s12906-019-2656-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mezzano S., Aros C., Droguett A., Burgos M. E., Ardiles L., Flores C., Schneider H., Ruiz-Ortega M., and Egido J. (2004) NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol. Dial. Transplant. 19, 2505–2512 10.1093/ndt/gfh207 [DOI] [PubMed] [Google Scholar]
- 45. Geng H., Wang A., Rong G., Zhu B., Deng Y., Chen J., and Zhong R. (2010) The effects of ox-LDL in human atherosclerosis may be mediated in part via the toll-like receptor 4 pathway. Mol. Cell Biochem. 342, 201–206 10.1007/s11010-010-0484-8 [DOI] [PubMed] [Google Scholar]
- 46. National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 47. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qin X., Yu S., Zhou L., Shi M., Hu Y., Xu X., Shen B., Liu S., Yan D., and Feng J. (2017) Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int J. Nanomed. 12, 3721–3733 10.2147/IJN.S131516 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data are in the manuscript.