

Abstract
In studies of HIV-1, virus production is normally monitored by either a reverse transcriptase assay or a p24 antigen capture ELISA. However, these assays are costly and time-consuming for routine handling of a large number of HIV-1 samples. For example, sample dilution is always required in the ELISA procedure to determine p24 protein levels because of the very narrow range of detectable concentrations in this assay. Here, we establish a novel HIV-1 production assay system to solve the aforementioned problems by using a recently developed small peptide tag called HiBiT. This peptide is a fragment of NanoLuc luciferase and generates a strong luminescent signal when complemented with the remaining subunit. To employ this technology, we constructed a novel full-length proviral HIV-1 DNA clone and a lentiviral packaging vector in which the HiBiT tag was added to the C terminus of the integrase. Tagging the integrase with the HiBiT sequence did not impede the resultant virus production, infectivity, or susceptibility to an integrase inhibitor. EM revealed normal morphology of the virus particles. Most importantly, by comparing between ELISA and the HiBiT luciferase assay, we successfully obtained an excellent linear correlation between p24 concentrations and HiBiT-based luciferase activity. Overall, we conclude that HiBiT-tagged viruses can replace the parental HIV-1 and lentiviral vectors, which enables us to perform a super-rapid, inexpensive, convenient, simple, and highly accurate quantitative assay for HIV-1/lentivirus production. This system can be widely applied to a variety of virological studies, along with screening for candidates of future antiviral drugs.
Keywords: HIV-1, luminescent peptide tag, quantitation, integrase, Vif, virology, human immunodeficiency virus (HIV), viral protein, viral replication, infection
The HIV-1 proviral DNA clone pNL4-3 (1) has been frequently used for in vitro HIV-1 studies worldwide for the last 34 years. Therefore, the resultant NL4-3 viruses are extraordinarily well characterized through a considerable amount of virological research, providing a solid knowledge base for data acquisition and/or interpretation of interexperimental data. Quantitation of viral supernatants from cells transfected with proviral DNA clones, including pNL4-3, is commonly and routinely performed by conducting a standard reverse transcriptase (RT) assay (2) or p24 antigen capture ELISA (3), both of which are time-consuming, taking up to 4 or 5 h or more. Particularly in the case of commercially available ELISA kits for HIV-1 detection, the major disadvantages are the high cost of the assays, and an intrinsic limitation in the linear ranges that require extensive dilution. In this study, we constructed a novel pNL4-3–based full-length proviral HIV-1 DNA clone and a lentiviral vector with a small luminescent peptide tag called HiBiT. When this 11 amino acid peptide tag binds to the larger counterpart protein called LgBiT, the full-length structure of the smallest luciferase protein called NanoLuc is formed (4) (Fig. 1A, upper panel). Because of its small size, HiBiT can be easily fused to the N or C terminus of the protein of interest. After expression of a HiBiT-tagged protein, the assay is performed by simply adding a lytic detection reagent containing LgBiT with the substrate, and the luminescence level of the HiBiT-tagged protein is quickly determined by a luminometer (Fig. 1A, lower panel). Using this system, we established a much faster (∼15 min), equally accurate, and inexpensive method, comparable to the recently established SG-PERT assay (5, 6) (as described under “Discussion”), demonstrating that this molecular tool would largely provide a cost/time-effective routine quantification of HIV-1 and lentiviral vectors.
Figure 1.
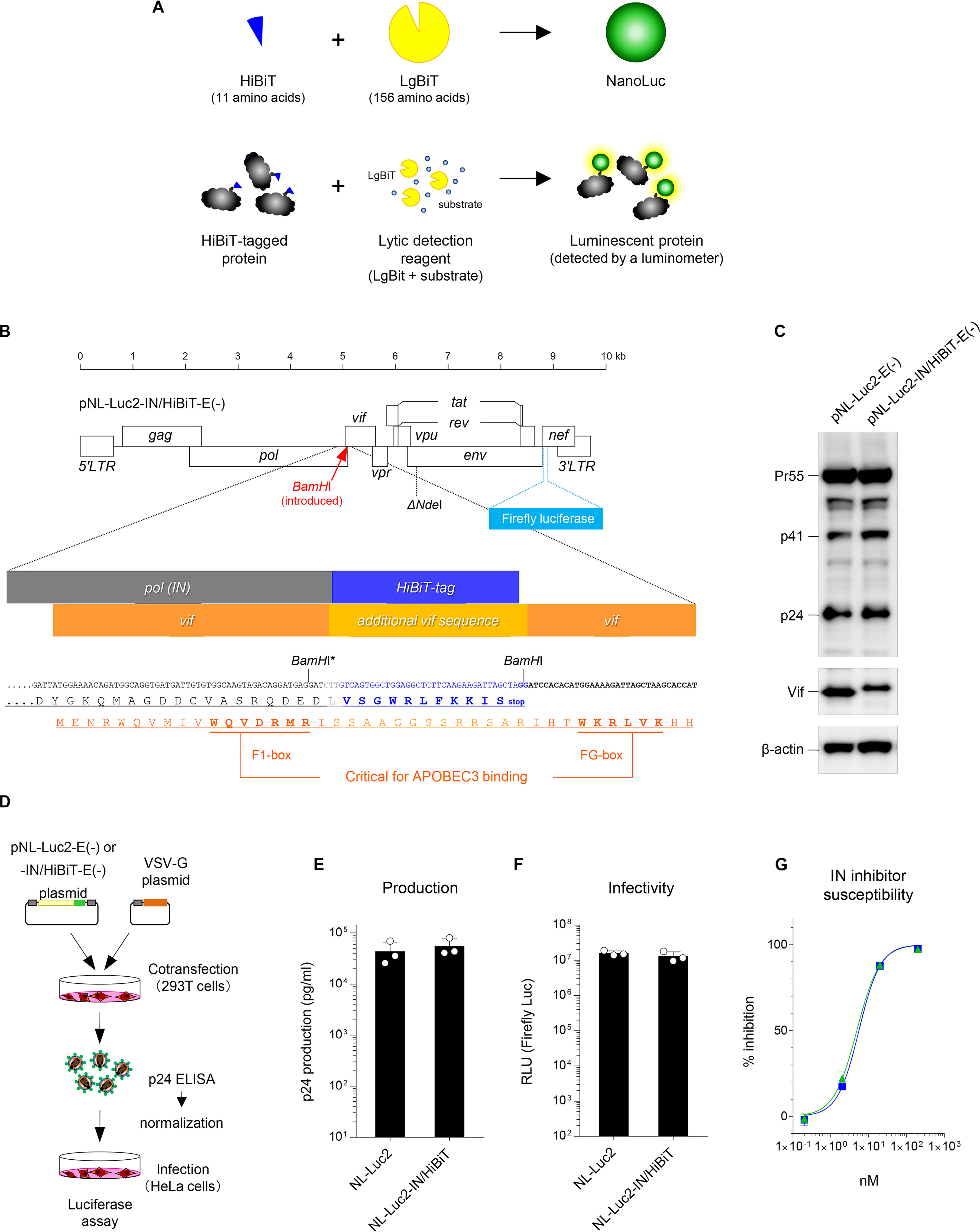
Luciferase reporter HIV-1 proviral DNA with a HiBiT tag maintains the expected levels of production, infectivity, and susceptibility to the IN inhibitor. A, schematic diagram of the HiBiT luciferase assay system (adapted from Promega). B, construction of the HiBiT-tagged HIV-1 proviral DNA designated pNL-Luc2-IN/HiBiT-E(−). An HiBiT tag (11 amino acid sequence shown in blue) was inserted into the C terminus of the integrase of pNL-Luc2-E(−) in which BamHI was newly introduced (the upstream BamHI site with an asterisk was disrupted by inserting the HiBiT tag). The amino acid sequence shown in light orange represents the vif gene with an unrelated sequence created by the HiBiT tag insertion. C, Western blot analysis performed by using extracts from 293T cells transfected with either pNL-Luc2-E(−) or pNL-Luc2-IN/HiBiT-E(−). Antibodies specific for p24 (upper), Vif (middle), and β-actin (lower) were used. Data shown are representative of two independent experiments. D, schematic flowchart of the experimental procedure for HIV-1 virion production and infectivity assays. 293T cells were transfected with either pNL-Luc2-E(−) or pNL-Luc2-IN/HiBiT-E(−) together with pC-VSVg, and 48 h later, the viral supernatants were harvested and subjected to HIV-1 p24 ELISA to determine the level of virion production. Transfection efficiencies were normalized to the activity of firefly luciferase. Equivalent amounts of p24 antigen of VSV-G-pseudotyped viruses were used for the infection of HeLa cells. After 48 h, cells were lysed, and firefly luciferase activities were measured to determine viral infectivity. E and F, comparison of virion production (E) and viral infectivity (F) of NL-Luc2 and NL-Luc2-IN/HiBiT viruses (mean ± S.D. from three independent experiments). RLU, relative light units. G, susceptibility of NL-Luc2 (blue square) and NL-Luc2-IN/HiBiT (green triangle) viruses to the integrase inhibitor dolutegravir. Data from two independent experiments are shown as percent inhibition (mean ± S.D., n = 3 technical replicates). Image prepared based on Promega Corporation product information, with permission.
Results
Viruses carrying the HiBiT-tagged integrase retain the WT level of viral production, infectivity, and susceptibility to the integrase inhibitor dolutegravir
To generate a HiBiT-tagged HIV-1 proviral clone, we first introduced a restriction enzyme site of BamHI at the C-terminal end of the integrase (IN) ORF of the HIV-1 luciferase reporter virus construct pNL-Luc2-E(−)R(+) and used this as a cassette for insertion of the HiBiT tag (Fig. 1B). Then, we transfected 293T cells with the resultant construct pNL-Luc2-IN/HiBiT-E(−) or the parental construct pNL-Luc2-E(−), subjected the cell lysates to Western blot analyses, and found that the HiBiT tag sequence did not affect the expression of the HIV-1 Gag, Vif, and IN proteins (Fig. 1C and Fig. S1A). This proviral clone was cotransfected into 293T cells with the vesicular stomatitis virus G-glycoprotein (VSV-G) expression plasmid, and the levels of p24 antigen in the viral supernatants were measured by ELISA (Fig. 1D). To normalize the transfection efficiency, transfected cells were lysed and subjected to assays for firefly luciferase activity. The luciferase reporter virus carrying the HiBiT-tagged IN (Luc2-IN/HiBiT virus) showed a WT level of virion production (Fig. 1E). Equal amounts of VSV-G-pseudotyped Luc2-WT and IN/HiBiT viruses were inoculated into 293T cells, and luciferase activities were measured to determine viral infectivity (Fig. 1D). The Luc2-IN/HiBiT virus was found to be equally infectious as the WT virus (Fig. 1F). Also in the case of HIV-1 Env-pseudotyped viruses (Fig. S2A), the Luc2-IN/HiBiT virus showed the WT levels of virion production (Fig. S2B) and infectivity (Fig. S2C). Moreover, we assessed whether the susceptibility of IN to its inhibitor was affected by the C-terminal tagging with HiBiT. To test this, we performed drug susceptibility assays using the IN inhibitor dolutegravir. The WT and Luc2-IN/HiBiT viruses were equally sensitive to dolutegravir (Fig. 1G). Thus, tagging the C-terminal end of the IN ORF with the HiBiT sequence does not interfere with the HIV-1 productivity, infectivity, or susceptibility to the IN inhibitor.
Insertion of the N-terminal Vif sequence into the C terminus of IN confers intact anti-A3G activity to the resultant virus, leading to efficient replication in CD4-positive T-cells
To enable us to perform replication assays in primary cells that express the restriction factor APOBEC3G (A3G), the intact function of Vif in the Luc2-IN/HiBiT virus needed to be verified, because an unrelated amino acid sequence derived from the HiBiT tag was inserted in the N-terminal sequence of Vif that overlaps with the C terminus of the IN ORF (although critical domains of Vif for APOBEC3 binding (7–10) were unchanged) (Fig. 1A). To test whether anti-A3G activity remained intact in the Vif protein of the Luc2-IN/HiBiT virus, we cotransfected 293T cells with the VSV-G expression plasmid and either the parental pNL-Luc2, Vif-deficient pNL-Luc2-F(−)E(−), or pNL-Luc2-IN/HiBiT-E(−) construct, together with either a control plasmid or a plasmid encoding A3G, and used viruses obtained from the transfected cells for infection. A3G expression resulted in a marked reduction in infectivity of the Luc2-IN/HiBiT virus to the level of the NL-Luc2-F(−) virus (Fig. 2C), indicating that the Vif protein of the Luc2-IN/HiBiT virus lacks anti-A3G activity. To rescue the lost function of Vif whose sequence was interrupted by the HiBiT tag, we first disrupted the initiation codon of Vif overlapping with the C terminus of the IN ORF. After introducing the BlpI site, we inserted an oligo DNA linker corresponding to an N-terminal vif sequence immediately downstream of the stop codon of the HiBiT tag and codon-optimized the C terminus of the IN (to avoid homologous recombination during reverse transcription) (Fig. 2A). We prepared cell lysates from 293T cells transfected with this construct designated pNL-Luc2-IN/HiBiT-E(−)Fin, performed Western blot analyses and confirmed the expression of Vif (Fig. 2B) as well as that of IN (Fig. S1B) at the expected size. The VSV-G-pseudotyped Luc2-IN/HiBiT-Fin virus produced from cells transfected as described above was tested for infectivity and was found to be fully resistant against A3G (Fig. 2C), demonstrating that the resultant virus gained intact Vif activity, as expected. It should be noted that because the vif sequence is completely conserved in this construct, its function in degrading not only A3G but also cellular PPP2R5 to trigger cell cycle arrest (11), is likely unaffected.
Figure 2.

Rescued Vif activity results in intact anti-APOBEC3G activity and replication ability. A, reconstruction of the HiBiT-tagged HIV-1 proviral DNA that rescues Vif function. After mutating the vif initiation codon (indicated by the red arrowhead), the fragment encoding the 25 amino acid sequence (shown in red) was inserted immediately downstream of the HiBiT tag into BamHI and BlpI sites, the latter of which was newly introduced at the N terminus of the Vif sequence (both sites with asterisks were disrupted by inserting the fragment). B, anti-p24 (upper), anti-Vif (middle), and anti–β-actin (lower) immunoblots of cell extracts transfected with either pNL-Luc2-E(−) or pNL-Luc2-IN/HiBiT-E(−)Fin. Data shown are representative of two independent experiments. C, infection of HeLa cells by VSV-G-pseudotyped luc-reporter WT (NL-Luc2), Vif-deficient NL-Luc2 (NL-Luc2-F(−)), HiBiT-tagged NL-Luc2 (NL-Luc2-IN/HiBiT), and HiBiT-tagged/Vif-inserted NL-Luc2 (NL-Luc2-IN/HiBiT-Fin), produced from cells expressing a vector control (black) or APOBEC3G (gray). Data from two experiments are shown (mean ± S.D., n = 3 technical replicates). RLU, relative light units. D, a representative transmission EM image of NL4-3 and NL-IN/HiBiT-Fin virions accumulated at the surface. Bars, 0.1 μm. E, multiple rounds of virus replication in peripheral blood mononuclear cells. PHA-IL-2-stimulated cells (2.5 × 105) were infected with 25 ng of p24 antigen of either the NL4–3 (filled circle) or NL-IN/HiBiT-Fin (filled square) virus. Supernatants were harvested at the indicated times, and virus replication was monitored using p24 ELISA. The data shown are representative of three independent experiments.
To determine whether an Env-intact full-length version of the viruses could normally replicate in CD4-positive T-cells, we generated a pNL4-3-based IN/HiBiT-Fin proviral clone (pNL-IN/HiBiT-Fin). We first performed transmission EM analyses using M8166 cells infected with either the resultant IN/HiBiT-Fin virus or the parental NL4-3 virus. Electron microscopic images revealed that mature particles of the IN/HiBiT-Fin virus were morphologically normal and indistinguishable from those of the parental NL4-3 virus, demonstrating that the insertion of the HiBiT tag has no influence on virion morphogenesis (Fig. 2D). We infected PHA-stimulated primary CD4-positive T-cells with the WT or IN/HiBiT-Fin virus. Then, we monitored virus replication by measuring the levels of p24 production in the supernatants and found that the replication kinetics of the IN/HiBiT-Fin virus was only slightly delayed compared with that of the parental virus, confirming that the IN/HiBiT-Fin virus is replication competent (Fig. 2E). Taken together, the results indicate that the addition of the N-terminal vif sequence into the C terminus of IN confers anti-A3G activity, resulting in efficient viral replication in primary CD4-positive T-cells.
HiBiT-mediated luciferase activity strongly correlates with p24 concentrations in viral supernatants
Finally, we assessed whether HiBiT-tagged viruses could be normally detected by HiBiT-based luciferase assays and whether a correlation between HiBiT-mediated luciferase activity and the p24 concentration could be observed. We prepared a high-titer virus stock by propagating the IN/HiBiT-Fin virus produced by M8166 cells and measured p24 by a commercially available ELISA kit. Then, we serially diluted the samples as standard viruses in 2-fold increments and subjected them to HiBiT luciferase assays as follows: we added equal amounts of samples and reagents, mixed them well, incubated them for 10 min, and performed measurements for 1 to 10 s per well by using a luminometer (Fig. 3A). HiBiT-mediated luciferase activity strongly correlated with p24 values in viral supernatants diluted on the order of microgram (Fig. 3B), as well as highly diluted samples on the order of picogram (Fig. 3C), with an excellent correlation between p24 levels and HiBiT luciferase activity (R2 = 1 and R2 = 0.9994, respectively). This indicates that the HiBiT activities can be directly translated to p24 antigen levels, routinely by this quick and easy method. In other words, we prepared the viral supernatants with known levels of p24 antigen and serially diluted them, and we could readily create a standard curve of HiBiT luciferase activity and determine the p24 levels of the samples of interest by measuring the activity in this assay. Overall, we conclude that the IN/HiBiT-Fin virus, in which the HiBiT tag and Vif linker were inserted into the C terminus of IN, is completely analogous to the parental HIV-1 strain and will be a highly convenient research tool for future HIV-1 studies.
Figure 3.

HiBiT-based luciferase assays show a strong correlation with p24 values in viral supernatants. A, schematic flowchart of the experimental procedure for HiBiT-based HIV-1 virion production assays. B and C, correlation between known levels of p24 antigen (B, high doses, 15.6 ng to 1 μg; C, low doses, 15.6 pg to 1 ng) measured with p24 antigen ELISA and HiBiT-derived luciferase activity measured with luciferase assays by using supernatants containing replication-competent NL-IN/HiBiT-Fin virus prepared from a human CD4-positive T-cell line. The equation and the coefficient of determination (R2) are shown as indicators of the linearity of this relationship (mean ± S.D. from at least three independent experiments). RLU, relative light units.
Discussion
In this study, we established a novel system for the highly accurate, convenient, and super-rapid detection of the well-analyzed HIV-1 strain pNL4-3 carrying a luminescent peptide-tagged IN. This method provides an efficient routine for quantitating the virus production of full-length infectious HIV-1. Several research groups have developed different real-time RT-PCR methods for virion quantitation of lentiviral vectors or whole HIV-1 viruses, which target various HIV-1 viral genomes (reviewed in Ref. 12) and transgene mRNA (13). These methods require RNA extraction and DNase treatment steps as well as a real-time RT-PCR procedure, taking a total of more than 4-5 h. More recently, Vermeire et al. (5) reported a real-time PCR-based, accurate, fast, and relatively inexpensive method for retroviral quantification designated SG-PERT (SYBR Green I-based PCR-enhanced reverse transcriptase), directly determining RT activity in viral supernatants. According to the report, the reagent cost of the SG-PERT assay is ∼10 times lower per retroviral quantification compared with the measurement of p24 antigen levels with a commercial ELISA kit, and the assay itself only requires less than 2 h of hands-on time. In our study, the cost of the HiBiT-based assay reagents per well to quantify viral supernatants is almost equivalent to that of the SG-PERT assay. In addition, because of the wide range of detectable HiBiT activity, this assay does not require sample dilution similar to the SG-PERT assay, whereas p24 ELISA frequently requires three different dilutions that make the assay more time-consuming. Notably, whereas the whole ELISA procedure normally takes 4-5 h (including sample preparation, incubation, and washing), that of the HiBiT-based assay (a one-step procedure) only takes up to ∼15 min, which is even faster than the aforementioned SG-PERT assay duration.
While our paper was in revision, a different type of HIV-1 reporter vector was reported by another group (14). In their paper, the NanoLuc luciferase itself was used to develop an HIV-1 bioluminescent reporter virus that can simplify measurement of virus production. The resultant virus indeed enabled sensitive quantitation of virion release, but exhibited a defect in viral infectivity and therefore required co-expression with WT Gag to reach WT levels of infectivity. This is probably because of the insertion of NanoLuc between Gag p17 and p24, leading to virion incorporation of this 19-kDa luciferase protein that might have affected Gag conformation and processing. In this context, our replication-competent IN/HiBiT-Fin virus will be a suitable tool for experiments that need to be performed based on multiple replication cycles, e.g. virological characterization of mutant viruses, analysis of host-virus interaction, high-throughput screening of antiviral drugs, and so on.
One clear limitation of our study needs to be mentioned: Our system cannot be applied for bulk virus samples directly obtained from patients unless they are molecularly cloned and reconstructed into HiBiT-harboring proviral DNAs. Because the C-terminal domain of the IN is involved in DNA binding (15), we might have to consider that the typical HIV-1 integration profiles could be altered by the HiBiT tagging at the C terminus. Moreover, with the use of our proviral DNA in studies in vivo (e.g. humanized mice), we cannot rule out the possibility that the HiBiT insertion could affect immunological responses. Therefore, it is likely that this method is specific for molecular clone-based virological studies. By taking advantage of this technique, we also constructed a lentiviral packaging vector carrying the HiBiT tag sequence, which is also fused to the C terminus of the integrase gene, because this could make lentiviral work more convenient. Consistent with the aforementioned observation using the proviral DNA, its viral protein expression (Fig. 4A and Fig. S1C), virion production (Fig. 4B), and transduction (Fig. 4C), including firefly luciferase activity/p24 ratio (Fig. S3), were comparable to those of the WT lentiviral vector.
Figure 4.

The lentiviral system using a lentiviral packaging vector carrying the HiBiT tag sequence exhibits WT levels of virion production and transduction. A, Western blot analysis performed by using pelleted virions derived from either the parental psPAX2 or the psPAX2-IN/HiBiT lentiviral packaging vector. Antibodies specific for p24 (upper) and integrase (lower) were used. Data shown are representative of two independent experiments. B and C, comparison of lentiviral production (B) and transduction (C) resulting from lentiviruses produced from cells transfected with the firefly luciferase–expressing transfer vector pWPI-Luc2 and VSV-G expression plasmid together with either the psPAX2 or the psPAX2-IN/HiBiT lentiviral packaging vector (mean ± S.D. from three independent experiment). RLU, relative light units.
During the submission of this manuscript, we utilized this HiBiT lentiviral pseudotyping system to examine cell entry mediated by spike proteins of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and found that the mutant spike protein from the globally dominant D614G variant showed ∼3.5-fold higher entry than that of the prototypic spike protein (16). This finding is particularly important because such a small but detectable difference in cell entry can be amplified in multiple virus replication cycles, which might reflect the difference in viral transmissibility. It should be noted that our HiBiT lentiviral system allowed us to precisely normalize input virus doses (as shown in Fig. 3, B and C), and therefore provided an enhanced experimental accuracy in the comparison of cell entry between SARS-CoV-2 spike proteins.
Overall, our HiBiT-based assay offers the following advantages: 1) super-rapid (∼15 min); 2) inexpensive (∼ 0.5 U. S. dollars/sample); 3) practical and convenient (no dilution required); 4) extremely simple (one-step procedure); and 5) highly accurate (linear scale over six orders of magnitude ranging from 10 pg to 1 μg of p24 antigen). Overall, we conclude that the HiBiT-tagged proviral HIV-1 DNA and lentiviral vector can be used to replace their parental vectors, making them powerful and reliable molecular tools for future studies of virology.
Experimental Procedures
DNA construction
The VSV-G expression vector pC-VSVg, the HIV-1 Env expression vector pC-NLenv, the HIV-1 proviral construct pNL4-3, its indicator construct pNL-Luc2-E-R+ (referred hereafter as pNL-Luc2-E(−)), pNL-Luc2-F(−)E(−) and the A3G expression plasmid pC-hA3G-HA have been described previously (17, 18). To generate a luciferase reporter HIV-1 proviral DNA carrying HiBiT tag, the SbfI/EcoRI fragment of pNL-Luc2-E(−) was subcloned into PstI/EcoRI-digested pUC19 (termed pUNL-PE), and the BamHI site was introduced by QuikChange mutagenesis (Stratagene) into pUNL-PE using the following specific oligonucleotides (restriction enzyme sites underlined); 5′-GAC AGG ATG AGG ATC CAC ACA TGG AAA AG-3′ and its antisense. The resultant plasmid was digested with BamHI and used for the insertion of an oligonucleotide linker corresponding to HiBiT-tag (5′-GAT CTT GTC AGT GGC TGG AGG CTC TTC AAG AAG ATT AGC TAG-3′ and 5′-GAT CCT AGC TAA TCT TCT TGA AGA GCC TCC AGC CAC TGA CAA-3′). The PstI/EcoRI fragment of the resultant vector pUNL-PE-IN/HiBiT was cloned back into SbfI/EcoRI-digested pNL-Luc2-E(−), and the generated plasmid was designated pNL-Luc2-IN/HiBiT-E(−). The Vif-fixed version of IN/HiBiT virus construct was created as follows. The initiation codon of the vif gene was mutated, and the C terminus of the IN was codon-optimized by QuikChange mutagenesis in pUNL-PE-IN/HiBiT using the following specific oligonucleotides: 5′-CAT CAG GGA TTA CGG TAA ACA AAT GGC TGG AGA CGA TTG TGT TGC TAG CAG ACA AGA CGA GGA TCT-3′ and its antisense (the resultant construct was designated pUNL-PE-IN/HiBiT-ΔF). The BlpI site was introduced by QuikChange mutagenesis into pUNL-PE-IN/HiBiT-ΔF using the following specific oligonucleotides (restriction enzyme sites underlined): 5′-GAA AAG ATT AGC TAA GCA CCA TAT G-3′ and its antisense. The resultant plasmid was digested with BamHI/BlpI and used to insert an oligonucleotide linker encoding the N-terminal Vif sequence (5′-GAT CAT GGA AAA CAG ATG GCA GGT GAT GAT TGT GTG GCA AGT AGA CCG CAT GCG GAT TAA CAC ATG GAA AAG ATT AGT-3′ and 5′-TTA ACT AAT CTT TTC CAT GTG TTA ATC CGC ATG CGG TCT ACT TGC CAC ACA ATC ATC ACC TGC CAT CTG TTT TCC AT-3′) downstream of the HiBiT-tag sequence. The PstI/EcoRI fragment of the resultant vector pUNL-PE-IN/HiBiT-Fin was cloned back into SbfI/EcoRI-digested pNL-Luc2-E(–) or pNL4-3, and the generated plasmids were designated pNL-Luc2-IN/HiBiT-E(–)Fin or pNL-IN/HiBiT-Fin, respectively. To create a lentiviral packaging vector carrying HiBiT-tag, the SbfI/SacI fragment of psPAX2 (19) was subcloned into PstI/SacI-digested pBluescript II SK(+) (termed pBSPAX-PS), and the BamHI site was introduced by QuikChange mutagenesis into pBSPAX-PS using the following specific oligonucleotides (restriction enzyme sites underlined): 5′-GGA TGA GGA TCC ACA CAT G-3 and its antisense. The resultant plasmid was digested with BamHI/BlpI and used for the insertion of an oligonucleotide linker corresponding to HiBiT-tag (5′-GAT CTC GTC AGC GGC TGG CGG CTC TTC AAG AAG ATT AGC TAG-3′ and 5′-GAT CCT AGC TAA TCT TCT TGA AGA GCC GCC AGC CGC TGA CGA-3′; slightly different from the aforementioned HiBiT nucleotide sequence). The PstI/SacI fragment of the resultant vector pBSPAX-IN/HiBiT was cloned back into SbfI/SacI-digested psPAX2, and the generated plasmid was designated psPAX2-IN/HiBiT. To create a lentiviral transfer vector expressing firefly luciferase, the SalI/SalI fragment of pWPI (19) was subcloned into SalI-digested pcDNA3.1 (Invitrogen). The resultant plasmid was digested with EcoRI to remove the internal ribosome entry site and EGFP and further digested with PmeI to insert a PCR-amplified/PmeI-digested codon-optimized luciferase (Luc2) fragment. Then, the SalI/SalI fragment of the plasmid was cloned back into SalI-digested pWPI, and the generated plasmid was designated pWPI-Luc2. All constructs were verified by DNA sequencing.
Cell maintenance, transfection, virion production assays, and protein analyses
293T, HeLa, MAGIC5 (HeLa-CD4 derivative (17)), H9 and M8166 cells were maintained under standard conditions. 293T cells (1.1 × 105) were cotransfected with either 1 μg of pNL-Luc2-E(−), pNL-Luc2-IN/HiBiT-E(−), or pNL-Luc2-IN/HiBiT-E(−)Fin using FuGENE6 (Promega) according to the manufacturer's instructions. For lentivirus production, 293T cells were cotransfected with 500 ng of either psPAX2 or psPAX2-IN/HiBiT and 500 ng of pWPI-Luc2 by using FuGENE6. Sixteen h later, the cells were washed with PBS, and then 1 ml of fresh complete medium was added. After 24 h, the supernatants were harvested and subjected to an HIV-1 p24-antigen capture ELISA (ELISA; XpressBio) to determine the level of virion production. Cells were separated and lysed in 75 μl of cell culture lysis reagent (Promega) or in 75 μl of RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, and cOmplete Protease Inhibitor Mixture (Roche Applied Science)). The former cell lysates were subjected to a luciferase assay to normalize the transfection efficiency using a firefly Luciferase Assay System (Promega) with a Centro LB960 luminometer (Berthold). The latter lysates were subjected to Western blot analysis using an anti-p24 mAb (1:1000; Nu24) (20), an anti-Vif peptide polyclonal antibody (1:200; made in-house; used only for NL viruses), an anti-integrase peptide polyclonal antibody (1:1000; made in-house), and an anti–β-actin mouse mAb (1:5000; Sigma-Aldrich, A5316). Supernatants containing either psPAX2- or psPAX2-IN/HiBiT–derived lentiviral particles were layered onto 20% (w/v) sucrose cushions and subjected to ultracentrifugation (90,000 rpm for 10 min) using an Optima TLX Ultracentrifuge (Beckman Coulter). Pelleted virions were resuspended in SDS-sample buffer and subjected to Western blot analysis using the anti-p24 mAb (1:1000; Nu24) and the anti-integrase peptide polyclonal antibody (1:1000; made in-house). Reacted proteins were visualized by chemiluminescence using an ECL Western blotting detection system (GE Healthcare) and monitored using a LAS-3000 imaging system (FujiFilm).
Virus preparation and infectivity assays
For NL4-3–based virus preparation, 293T cells (1.1 × 105) were cotransfected with 500 ng of pNL-Luc2-E(−), pNL-Luc2-IN/HiBiT-E(−), or pNL-Luc2-IN/HiBiT-E(−)Fin, and 20 ng of either pC-VSVg or pC-NLenv, with or without 10 ng of pC-hA3G-HA and an empty vector up to 1 μg by using FuGENE6. For lentivirus preparation, 293T cells were cotransfected with 475 ng of either psPAX2 or psPAX2-IN/HiBiT, 475 ng of pWPI-Luc2, and 50 ng of pC-VSVg by using FuGENE6. Sixteen h later, the cells were washed with PBS, and then 1 ml of fresh complete medium was added. After 24 h, the supernatants were harvested and treated with 37.5 units/ml DNase I (Roche) at 37°C for 30 min. The viral supernatants were subjected to HIV-1 p24 ELISA to measure the p24 antigen. To determine infectivity, HeLa or MAGIC5 cells (1 × 104) were incubated with the p24 antigen (1 ng) of each viral supernatant. After 48 h, cells were lysed in 100 μl of One-Glo Luciferase Assay Reagent (Promega), and firefly luciferase activities were determined as described above.
Drug susceptibility assays
The integrase inhibitor dolutegravir (Abcam, ab231511) was 10-fold serially diluted and mixed with an equal volume of 1 ng of 24 antigens of the VSV-G-pseudotyped Luc2-WT and IN/HiBiT viruses. The mixture was added to HeLa cells (1 × 104). After 48 h, cells were lysed and subjected to luciferase assays, as described above, to determine the levels of drug susceptibility.
Primary cell culture
Experiments using human samples were approved by the Medical Research Ethics Committee of the National Institute of Infectious Diseases, Japan (approval number 887) and the approved studies abided by the Declaration of Helsinki principles. Peripheral blood mononuclear cells were obtained from healthy volunteer donors who signed an informed consent form. Briefly, peripheral blood mononuclear cells were isolated by Ficoll-Hypaque gradient centrifugation. The CD4-positive T lymphocytes were purified using a Dynabeads CD4-positive isolation kit (Invitrogen) to obtain CD4-positive T cells, and purified cells were cultured in the presence of 3 μg/ml PHA (Sigma-Aldrich) and 10 units/ml IL-2 (Peprotech) for 72 h.
Virus replication assays
CD4-positive T cells (2.5 × 105) were infected for 3 h with either NL4-3 or NL-IN/HiBiT-Fin virus (25 ng of p24 antigen), washed extensively with serum-free medium, and then cultured in fresh complete medium. Supernatants were sampled every 3 days, and p24 antigen production was quantified by ELISA.
HiBiT luciferase assays
A high-titer virus stock as a standard sample was prepared by propagating IN/HiBiT-Fin virus in infected M8166 cells and measuring p24 by ELISA. After determining the levels of p24 antigen, the viral supernatant was aliquoted and stored for future use in different HiBiT-based assays, to avoid repeated freeze-thaw cycles. To create a standard curve, the viral stock with known levels of p24 antigen derived from the IN/HiBiT-Fin virus was serially diluted. Either the standards or the samples of interest (25 μl) and LgBiT protein (1:100)/HiBiT Lytic Substrate (1:50) in Nano-Glo HiBiT Lytic Buffer (25 μl) (Nano-Glo HiBiT Lytic Detection System; Promega) were mixed and incubated for 10 min at room temperature according to the modified manufacturer's instructions. Luciferase activity was determined with a Centro LB960 luminometer. Note that HiBiT assays using target cell lysates were not performed to determine the levels of cell entry because of the background caused by nonspecific attachment of virions to the cell surface.
Transmission EM
H9 cells infected with either the WT NL4-3 or NL-IN/HiBiT-Fin virus were harvested and washed twice with ice-cold PBS. Cells were then pre-fixed with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, for 2 h at room temperature, post-fixed in 1% osmium tetroxide, and embedded in Epon 812 (TAAB Laboratories). Ultrathin sections were stained with uranyl acetate and lead citrate and then observed under a transmission electron microscope (HT7700; Hitachi) at 80 kV.
Statistical analyses
Column graphs that combine bars and individual data points were created with GraphPad Prism version 8.04, and coefficient of determination (R2) and regression equations were also calculated with GraphPad Prism.
Data Availability
All the data described in the article are contained within the manuscript and the supporting information.
Supplementary Material
Acknowledgments
We thank D. Trono (Ecole Polytechnique Fédérale de Lausanne, Switzerland) for providing psPAX2 and pWPI.
This article contains supporting information.
Author contributions—S. O., Y. Z., and K. T. data curation; S. O., Y. Z., M. T., and K. T. formal analysis; S. O., Y. Z., and K. T. validation; S. O., Y. Z., M. T., and K. T. investigation; M. T. and K. T. visualization; S. K. and K. T. resources; K. T. conceptualization; K. T. software; K. T. supervision; K. T. funding acquisition; K. T. methodology; K. T. writing-original draft; K. T. project administration; K. T. writing-review and editing.
Funding and additional information—This work was supported by a grant from the Japan Society for the Promotion of Science KAKENHI, 18K07156 (to K. T.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- IN
- integrase
- PHA
- phytohemagglutinin
- RLU
- relative light units
- VSV-G
- vesicular stomatitis virus G-glycoprotein.
References
- 1. Adachi A., Gendelman H. E., Koenig S., Folks T., Willey R., Rabson A., and Martin M. A. (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284–291 10.1128/JVI.59.2.284-291.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barré-Sinoussi F., Chermann J., Rey F., Nugeyre M., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vézinet-Brun F., Rouzioux C., Rozenbaum W., and Montagnier L. (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 10.1126/science.6189183 [DOI] [PubMed] [Google Scholar]
- 3. Goudsmit J., Lange J. M., Paul D. A., and Dawson G. J. (1987) Antigenemia and antibody titers to core and envelope antigens in AIDS, AIDS-related complex, and subclinical human immunodeficiency virus infection. J. Infect. Dis. 155, 558–560 10.1093/infdis/155.3.558 [DOI] [PubMed] [Google Scholar]
- 4. Dixon A. S., Schwinn M. K., Hall M. P., Zimmerman K., Otto P., Lubben T. H., Butler B. L., Binkowski B. F., Machleidt T., Kirkland T. A., Wood M. G., Eggers C. T., Encell L. P., and Wood K. V. (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 10.1021/acschembio.5b00753 [DOI] [PubMed] [Google Scholar]
- 5. Vermeire J., Naessens E., Vanderstraeten H., Landi A., Iannucci V., Van Nuffel A., Taghon T., Pizzato M., and Verhasselt B. (2012) Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS One 7, e50859 10.1371/journal.pone.0050859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pizzato M., Erlwein O., Bonsall D., Kaye S., Muir D., and McClure M. O. (2009) A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J. Virol. Methods 156, 1–7 10.1016/j.jviromet.2008.10.012 [DOI] [PubMed] [Google Scholar]
- 7. Dang Y., Wang X., Zhou T., York I. A., and Zheng Y. H. (2009) Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 83, 8544–8552 10.1128/JVI.00651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamashita T., Kamada K., Hatcho K., Adachi A., and Nomaguchi M. (2008) Identification of amino acid residues in HIV-1 Vif critical for binding and exclusion of APOBEC3G/F. Microbes Infect. 10, 1142–1149 10.1016/j.micinf.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 9. Russell R. A., and Pathak V. K. (2007) Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 81, 8201–8210 10.1128/JVI.00395-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tian C., Yu X., Zhang W., Wang T., Xu R., and Yu X. F. (2006) Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J. Virol. 80, 3112–3115 10.1128/JVI.80.6.3112-3115.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salamango D. J., Ikeda T., Moghadasi S. A., Wang J., McCann J. L., Serebrenik A. A., Ebrahimi D., Jarvis M. C., Brown W. L., and Harris R. S. (2019) HIV-1 Vif triggers cell cycle arrest by degrading cellular PPP2R5 phospho-regulators. Cell Rep. 29, 1057–1065.e4 10.1016/j.celrep.2019.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delenda C., and Gaillard C. (2005) Real-time quantitative PCR for the design of lentiviral vector analytical assays. Gene Ther. 12, Suppl. 1, S36–S50 10.1038/sj.gt.3302614 [DOI] [PubMed] [Google Scholar]
- 13. Geraerts M., Willems S., Baekelandt V., Debyser Z., and Gijsbers R. (2006) Comparison of lentiviral vector titration methods. BMC Biotechnol. 6, 34 10.1186/1472-6750-6-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kirui J., and Freed E. O. (2020) Generation and validation of a highly sensitive bioluminescent HIV-1 reporter vector that simplifies measurement of virus release. Retrovirology 17, 12 10.1186/s12977-020-00521-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J. C.-H., Krucinski J., Miercke L. J. W., Finer-Moore J. S., Tang A. H., Leavitt A. D., and Stroud R. M. (2000) Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. U. S. A. 97, 8233–8238 10.1073/pnas.150220297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ozono S., Zhang Y., Ode H., Tan T. S., Imai K., Miyoshi K., Kishigami S., Ueno T., Iwatani Y., Suzuki T., and Tokunaga K. (2020) Naturally mutated spike proteins of SARS-CoV-2 variants show differential levels of cell entry. bioRxiv
- 17. Tada T., Zhang Y., Koyama T., Tobiume M., Tsunetsugu-Yokota Y., Yamaoka S., Fujita H., and Tokunaga K. (2015) MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 21, 1502–1507 10.1038/nm.3956 [DOI] [PubMed] [Google Scholar]
- 18. Kinomoto M., Kanno T., Shimura M., Ishizaka Y., Kojima A., Kurata T., Sata T., and Tokunaga K. (2007) All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 35, 2955–2964 10.1093/nar/gkm181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wiznerowicz M., and Trono D. (2003) Conditional suppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 77, 8957–8961 10.1128/jvi.77.16.8957-8951.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsunetsugu-Yokota Y., Akagawa K., Kimoto H., Suzuki K., Iwasaki M., Yasuda S., Häusser G., Hultgren C., Meyerhans A., and Takemori T. (1995) Monocyte-derived cultured dendritic cells are susceptible to human immunodeficiency virus infection and transmit virus to resting T cells in the process of nominal antigen presentation. J. Virol. 69, 4544–4547 10.1128/JVI.69.7.4544-4547.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data described in the article are contained within the manuscript and the supporting information.