

Abstract
Bacterial formation of trimethylamine (TMA) from carnitine in the gut microbiome has been linked to cardiovascular disease. During this process, the two-component carnitine monooxygenase (CntAB) catalyzes the oxygen-dependent cleavage of carnitine to TMA and malic semialdehyde. Individual redox states of the reductase CntB and the catalytic component CntA were investigated based on mutagenesis and electron paramagnetic resonance (EPR) spectroscopic approaches. Protein ligands of the flavin mononucleotide (FMN) and the plant-type [2Fe-2S] cluster of CntB and also of the Rieske-type [2Fe-2S] cluster and the mononuclear [Fe] center of CntA were identified. EPR spectroscopy of variant CntA proteins suggested a hierarchical metallocenter maturation, Rieske [2Fe-2S] followed by the mononuclear [Fe] center. NADH-dependent electron transfer via the redox components of CntB and within the trimeric CntA complex for the activation of molecular oxygen was investigated. EPR experiments indicated that the two electrons from NADH were allocated to the plant-type [2Fe-2S] cluster and to FMN in the form of a flavin semiquinone radical. Single-turnover experiments of this reduced CntB species indicated the translocation of the first electron onto the [Fe] center and the second electron onto the Rieske-type [2Fe-2S] cluster of CntA to finally allow for oxygen activation as a basis for carnitine cleavage. EPR spectroscopic investigation of CntA variants indicated an unusual intermolecular electron transfer between the subunits of the CntA trimer via the “bridging” residue Glu-205. On the basis of these data, a redox catalytic cycle for carnitine monooxygenase was proposed.
Keywords: carnitine monooxygenase, CntAB, trimethylamine, gut microbiota, Rieske-type oxygenase, l-carnitine, electron paramagnetic resonance, site-directed mutagenesis, metalloenzyme, iron-sulfur protein, enzyme mechanism, electron paramagnetic resonance (EPR)
The human gut contains trillions of microbes supporting to breakdown otherwise indigestible food constituents (1). There is growing evidence that the human microbiota plays an important role in health and disease (2–5). The gut microbiome was related to obesity (3, 6), to the development of the adaptive immune system (7), and to the emergence of atherosclerosis (8). Accordingly, manipulation of the gut microbiota was considered a future therapeutic strategy to combat disease and improve health (4).
Dietary nutrients containing a trimethylamine (TMA) moiety, inter alia l-carnitine or choline, were related to the development of atherosclerotic heart disease (9). The C-N bond of these compounds is cleaved by bacterial carnitine monooxygenase (CntAB) or choline trimethylamine-lyase (CutCD), leading to the volatile compound TMA, which is further metabolized in the human liver by flavin monooxygenases (FMOs) (Fig. 1). The resulting trimethylamine N-oxide molecule (TMAO) was shown to enhance atherosclerosis development in animal models (10, 11). In multiple human studies, elevated TMAO levels have been associated with prevalent cardiovascular diseases and the risks for myocardial infarction and stroke (12, 13). Only recently, inhibition of the choline lyase CutCD using the substrate analog 3,3-dimethyl-1-butanol was exemplified as an efficient strategy to attenuate choline diet-enhanced atherosclerosis in a mouse model (14) (Fig. 1).
Figure 1.

Gut microbiota and metabolism of dietary nutrients associated with atherosclerosis. The gut microbiota makes use of dietary nutrients l-carnitine and choline as carbon and energy sources. The C-N bond of these compounds is cleaved by TMA lyases. The resulting TMA molecule is further metabolized in the human liver by flavin monooxygenases (FMOs) of the host. The synthesized TMAO molecule was shown to enhance atherosclerosis development. In multiple human studies, elevated TMAO levels have been associated with cardiovascular diseases and the risks for myocardial infarction and stroke. For the TMA lyase CutCD, an efficient targeted inhibitor was identified that prevented the formation of TMA in mouse models.
The two-component carnitine monooxygenase CntAB (EC 1.14.13.239) catalyzes the oxidative cleavage of l-carnitine into TMA and malic semialdehyde (Fig. 2A). The latter compound serves as a carbon and energy source and is channeled into the tricarboxylic acid cycle by virtue of malic semialdehyde dehydrogenase and malate dehydrogenase (15). Despite the structural similarity of the substrates l-carnitine and choline, CntAB catalysis is completely unrelated to CutCD, which utilizes an unusual glycyl radical enzyme mechanism (16).
Figure 2.
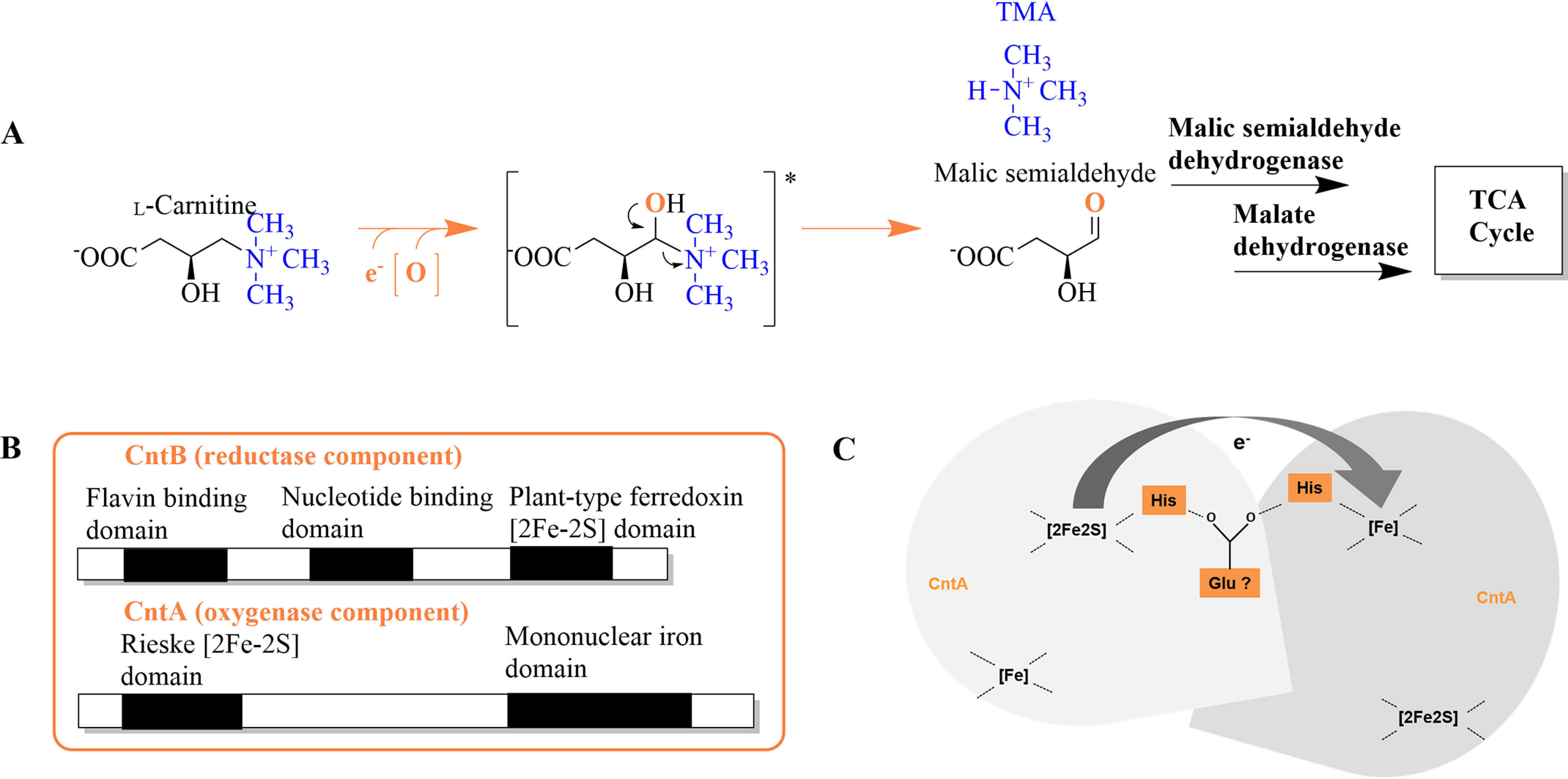
Synthesis of TMA and malic semialdehyde from l-carnitine, postulated domain architecture of CntA and CntB, and proposed electron transfer between subunits of CntA. A, conversion of l-carnitine via a proposed hydroxyl-intermediate (*) into TMA and malic semialdehyde, which is channeled into the tricarboxylic acid (TCA) cycle via malic semialdehyde dehydrogenase and malate dehydrogenase. B, theoretical sequence analyses revealed motifs of a flavin binding domain, nucleotide binding domain, and plant-type ferredoxin [2Fe-2S] domain in sequences of CntB and of a Rieske [2Fe-2S] and a mononuclear iron domain in CntA. C, schematic depiction of the proposed intersubunit electron transfer via a bridging glutamate residue from the Rieske [2Fe-2S] onto the mononuclear iron center of CntA.
Only recently, the clustering of genes cntA and cntB in bacterial reference genomes of the human microbiota has been demonstrated (17, 18). Theoretical sequence analyses for CntB revealed a potential nucleotide binding sequence, a flavin binding domain, and a motif characteristic of the presence of a plant-type [2Fe-2S] cluster. The sequence of CntA shows substantial homology to the family of Rieske-type oxygenases (Fig. 2B) (19). These iron-dependent enzyme systems typically comprise an oxygenase, a specific reductase, and eventually an additional ferredoxin component (20). Numerous Rieske-type oxygenases have been described in the literature, e.g. the first discovered toluene dioxygenase and naphthalene dioxygenase (21, 22), other biotechnologically relevant systems that can break aromatic ring structures (23, 24), and Rieske-type oxygenases that oxidize compounds such as cholesterol (25) or the amine groups of 4-aminobenzylamine, stachydrine, or caffeine (26–28). All highlighted oxygenases share a highly conserved Rieske sequence (-CXHX15-17CXXH-), providing two cysteine and two histidine ligands for the characteristic [2Fe-2S] cluster. Apart from this, CntA also possess a mononuclear iron-binding region. The related [Fe] center of Rieske proteins, typically ligated by two histidine residues and one carboxylate ligand, plays a fundamental role in the sophisticated activation of molecular oxygen (Fig. 2). The oligomeric architecture of Rieske oxygenases is of central importance, because the electron transfer onto the [Fe] center from the Rieske center is facilitated via an intersubunit rather than an intrasubunit redox process (29) (Fig. 2C). As judged from three-dimensional protein structures, a highly conserved aspartate residue (e.g. Asp-205 in naphthalene 1,2-dioxygenase, PDB entry 1NDO) is substantial for the electron transfer between the adjacent subunits (29). Interestingly, this “bridging” aspartate is replaced by a glutamate in all available sequences of CntA.
Recently, carnitine monooxygenase activity of the Acinetobacter baumannii enzyme was reconstituted, and the functional relevance of the abovementioned Glu-205 has been demonstrated by site-directed mutagenesis (18). Furthermore, a closely related substrate specificity of orthologous CntA proteins was demonstrated in vitro. High activity in the presence of l-carnitine and gamma-butyrobetaine, medium activity toward glycine betaine, and very low activity in the presence of choline was shown (14, 18). Apart from this, the underlying bio-inorganic redox chemistry of the unusual Rieske-type carnitine monooxygenase has not been investigated.
Here, we present the first biochemical characterization of the redox-active components of CntB and CntA from A. baumannii. Site-directed mutagenesis in combination with kinetic and electron paramagnetic resonance (EPR) experiments allowed us to resolve individual electron transfer steps required for the oxygenation of the carnitine substrate.
Results
Enzymatic activity of CntAB
Components CntB and CntA of the carnitine monooxygenase from A. baumannii were individually overproduced and purified from Escherichia coli cells. The N-terminally His-tagged CntB protein was affinity purified using Co2+-loaded chelating Sepharose. Approximately 5 mg purified CntB with a relative molecular weight of ∼45,000 (calculated molecular mass, 37,151 Da) was obtained from a 1-liter cell culture (SDS-PAGE) (Fig. 3A, lane 3).
Figure 3.
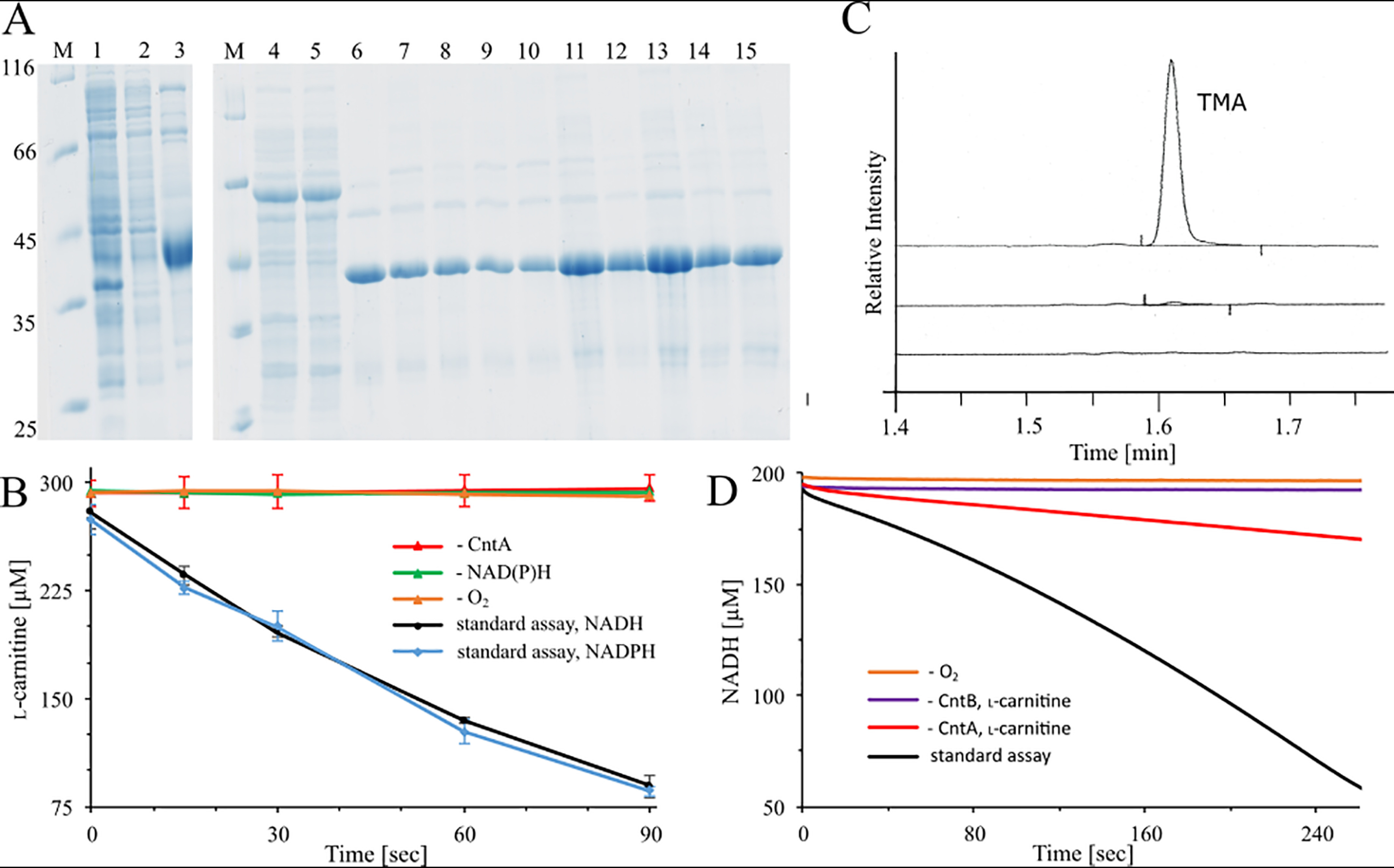
Production and purification of recombinant CntB and CntA proteins, l-carnitine depletion assay, TMA determination, and NADH depletion assay. A, SDS-PAGE analysis of CntB His tag affinity purification using Co2+-loaded chelating Sepharose. Lane 1, E. coli cells after IPTG induction; lane 2, supernatant after ultracentrifugation; lane 3, elution fraction of WT CntB. Affinity purification of GST-tagged CntA was also done: lane 4, E. coli cells after IPTG induction; lane 5, supernatant after ultracentrifugation; lanes 6–15, purified CntA WT and variants C86A, H88A, C106A, H109A, E205Q, E205D, H208A, H213A, and D323A after affinity chromatography on GSH-Sepharose (untagged target proteins eluted after on-column PreScission protease cleavage). Lanes M, molecular mass marker; relative molecular masses (×1,000) are indicated. B, l-carnitine depletion assay. A standard assay containing 5 μm purified CntA, 20 μm purified CntB, and 300 μm l-carnitine at 37 °C was initiated by the addition of 2 mm the electron donor NADH (black trace) or NADPH (blue trace). Samples were prepared at least in triplicates and collected after 15, 30, 60, and 90 s, and l-carnitine concentrations were determined colorimetrically in a coupled carnitine acetyltransferase assay (see Experimental procedures). Control reactions in the absence of CntA (red trace), electron donor (green trace), or O2 (orange trace; proteins purified under strict anaerobic conditions) were performed. C, TMA analysis by GC. Top, a standard assay in the presence of 2 mm l-carnitine and 5 mm NADH was incubated at 37 °C for 2 min. The reaction was stopped in the presence of 1 M perchloric acid and subjected to GC analyses. Control reactions in the absence of CntB (middle) or NADH (bottom) were performed. D, NADH depletion assay in the presence of 2 μm purified CntA and CntB, 300 μm l-carnitine, and 200 μm NADH (black trace). The absorption at 340 nm was continuously monitored (black trace). Control reactions in the absence of O2 (orange trace), CntB (purple trace), and CntA (red trace) were performed.
The catalytic component CntA was overproduced as an N-terminal GST fusion protein and purified using chromatography on GSH-Sepharose. The untagged target protein was eluted after on-column cleavage with PreScission protease. Overall, 6 mg CntA was purified from a 1-liter culture volume. SDS-PAGE analysis (Fig. 3A, lane 6) revealed a relative molecular weight of ∼43,000 (calculated molecular mass, 43,155 Da).
CntAB-dependent conversion of l-carnitine into malic semialdehyde and TMA recently has been exemplified using ion chromatography- and GC-MS-based approaches, respectively (18). Here, we established a microplate reader-based l-carnitine depletion assay. Carnitine monooxygenase activity was reconstituted in the presence of 20 μm CntB, 5 μm CntA, 300 μm l-carnitine, and 2 mm electron donor NADH (or NADPH). l-carnitine concentrations were determined in the presence of acetyl-CoA and carnitine acetyltransferase. Emerging CoA leads to the cleavage of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), and the resulting 2-thio-5-nitrobenzoic acid (TNB) was spectroscopically monitored. A specific CntAB activity of 771 ± 67 nmol min−1 mg−1 was determined (Fig. 3B). The reconstituted enzyme was substantially more active than recently described (80 nmol min−1 mg−1) (18). Parallel emergence of TMA was verified by GC (Fig. 3C). Alternatively, a spectroscopic NADH depletion assay (performed at lower cofactor concentration) revealed a specific activity of 407 ± 11 nmol min−1 mg−1 (Fig. 3D).
Oxygenation of the carnitine substrate must be considered a key step of CntAB catalysis, and molecular oxygen was proposed to be a source of the carbonyl oxygen of the malic semialdehyde reaction product (18). Only recently, in vivo experiments demonstrated higher TMA production in more aerobic environments. However, verification of molecular oxygen as a cosubstrate is still pending.
Accordingly, components CntB and CntA were recombinantly purified under strict anaerobic conditions. A subsequent carnitine depletion assay or an NADH depletion assay (performed in an anaerobic chamber) did not reveal detectable CntAB activity (Fig. 3B and D), in agreement with the reaction scheme depicted in Fig. 2A. Enzymatic activity was effectively restored upon oxygen exposure.
CntB is an FMN-dependent reductase
CntB acts as a carnitine monooxygenase reductase, transferring reduction equivalents from NADH onto CntA. Analytical size exclusion chromatography revealed a native molecular mass of 43,000 Da, indicating that CntB is functional as a monomer. The purified protein showed an orange/yellow color, and UV-visible spectroscopy revealed absorption maxima at 272 nm, 340 nm, 410 nm, and 463 nm, which might be indicative of a prosthetic FAD or FMN cofactor and/or the presence of an iron-sulfur cluster (30) (Fig. 4A).
Figure 4.
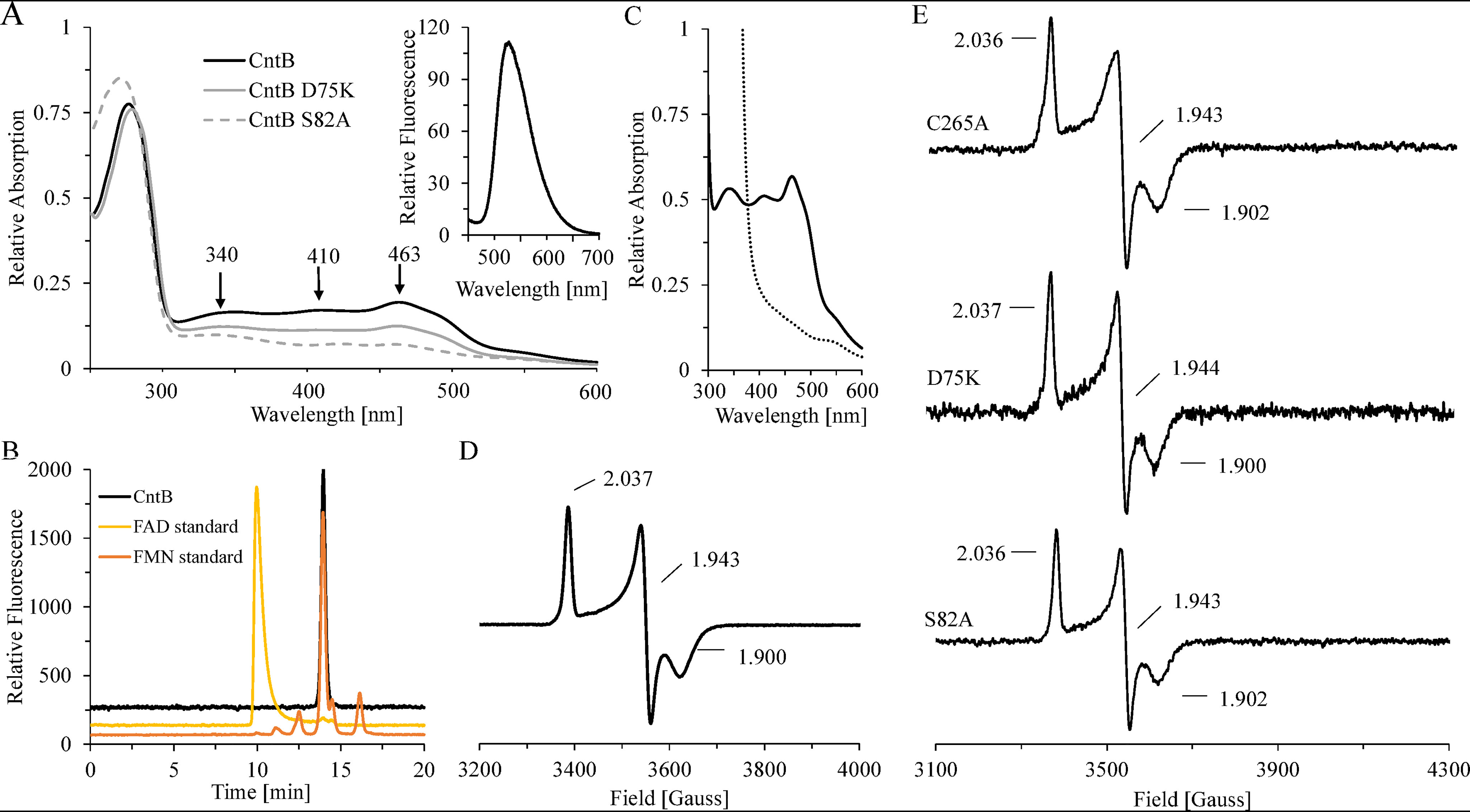
Spectroscopic characterization of CntB WT and variant proteins. A, room temperature UV-visible spectra of purified CntB (black line) and variants D75K (gray line) and S82A (gray dashed line). Inset, fluorescence spectrum of a CntB supernatant after heat denaturation and centrifugation (370 nm excitation). B, identification of the cofactor FMN by HPLC. The fluorescent sample from panel A was chromatographed on a Reprosil 100 C18 column using excitation/emission wavelengths of 370/526 nm (black). Authentic samples of FAD (yellow) or FMN (orange) were analyzed accordingly. C, room temperature UV-visible spectrum of purified CntB before (black continuous trace) and after (black dashed trace) treatment with 2 mm dithionite for 10 min. D and E, low-temperature (15 K) EPR spectra of ∼1 mm CntB samples (WT or C267A, D75K, and S82A mutants) after dithionite reduction (10 mm). All spectra were recorded at 9.653 GHz, 7.5-G modulation amplitude, 0.2-milliwatt microwave power, and 100-kHz modulation frequency.
Further experimental evidence for the presence of a flavin cofactor was obtained from fluorescence spectroscopy. In Fig. 4A, inset, the characteristic emission of the supernatant from a heat-denatured CntB sample at 526 nm (370 nm excitation) indicates the presence of a noncovalently bound flavin. Comparative HPLC analyses in the presence of authentic FAD/FMN samples clearly identified FMN as a cofactor (Fig. 4B). As judged from the supernatant of thermal denaturation experiments, an FMN content of 0.6 ± 0.1 mol per mol CntB was determined.
FMN binding was further explored using structure-guided mutagenesis. Functional relevance of amino acid residues Asp-75 and Ser-82 in the FMN binding pocket of CntB was proposed after assessment of the three-dimensional structure of the orthologous protein benzoate dioxygenase reductase (PDB entry 1KRH, 23% sequence identity). Purified CntB variant proteins D75K and S82A revealed a characteristic decrease of the absorption at 340 and 463 nm and the parallel reduction of the fluorescence signal at 526 nm (Fig. 4A and Table 1). In agreement with a significant decrease of the cofactor content (78% ± 5% and 15% ± 4% FMN), residual activities of 70% ± 8% and 35% ± 6% were observed compared with the WT protein (Table 1). From these data, we conclude that Asp-75 and Ser-82 contribute to the tight binding of FMN. NADH-dependent electron transfer of both variants was efficiently restored when the flavin cofactor was externally supplied to the assay.
Table 1.
Biochemical characterization of CntB wild-type and variant proteinsa
| Mutation | Function | Relative FMN content (%) | Relative l-carnitine depletion activity (%) | Relative TMA formation (%) | Relative NADH depletion activity (%) | Iron content (nmol protein−1) | Sulfur content (nmol protein−1) | EPR signal for [2Fe-2S] |
|---|---|---|---|---|---|---|---|---|
| D75K | [FMN] coordinating | 78 ± 5 | 70 ± 8 | 92 | 69 ± 3 | 1.85 ± 0.19 | 1.84 ± 0.16 | WT |
| S82A | [FMN] coordinating | 15 ± 4 | 35 ± 6 | 56 | 23 ± 2 | 2.12 ± 0.18 | 1.85 ± .019 | WT |
| C265A | No catalytic function | 96 ± 4 | 99 ± 11 | 70 | 90 ± 4 | 2.05 ± 0.17 | 1.95 ± 0.29 | WT |
| C267A | [2Fe-2S] coordinating | – | <5 | – | <3 | 0.10 ± 0.04 | 0.25 ± 0.17 | ND |
| C272A | [2Fe-2S] coordinating | – | <5 | – | <3 | 0.14 ± 0.05 | 0.05 ± 0.04 | ND |
| C275A | [2Fe-2S] coordinating | – | <5 | – | <3 | 0.23 ± 0.11 | 0.28 ± 0.09 | ND |
| C305A | [2Fe-2S] coordinating | – | <5 | – | <3 | 0.18 ± 0.09 | 0.24 ± 0.12 | ND |
a Table summarizes biochemical and spectroscopic properties of CntB variants. The FMN content (∼0.6 ± 0.1 mol per mol CntB), the specific activity of the l-carnitine depletion assay (771 ± 67 nmol min−1 mg−1), enzymatic TMA formation (determined by gas chromatography), and NADH depletion activity (407 ± 11 nmol min−1 mg−1) of the wild type were set as 100% (compared to experimental procedures). –, experiment not performed. <5 or <3, enzymatic activity below the detection limit of the respective assay. ND, an EPR signal was not detectable.
CntB carries a plant-type [2Fe-2S] cluster
EPR spectroscopy allows for the characterization of individual redox states of iron-sulfur cluster-containing proteins. The aerobically purified CntB protein (∼1 mm) was EPR silent. However, upon reduction with 2 mm sodium dithionite, a complete loss of color, a drastic reduction of the characteristic optical absorption maxima, and an S = 1/2 signal with g values of g1 = 2.037, g2 = 1.943, and g3 = 1.900, located in a region characteristic for a [2Fe-2S]+ cluster, were observed (Fig. 4C and D) (31). Concurrently, an iron content of 2.16 ± 0.18 mol Fe per mol CntB and a sulfur content of 2.07 ± 0.39 mol S per mol CntB was determined.
Theoretical sequence analysis indicated an overall five cysteine residues as potential ligands of the redox-active [2Fe-2S] cluster of CntB. Accordingly, the respective amino acids were individually mutagenized into alanine and the purified variants C265A, C267A, C272A, C275A, and C305A were characterized. Mutant C265A revealed WT-like enzymatic activity in the carnitine and NADH depletion assay. UV-visible experiments, flavin quantification, and EPR spectroscopy indicated the presence of FMN and the [2Fe-2S] cofactor by analogy to the WT protein (compare Fig. 4E and Table 1). Clear differences for variants C267A, C272A, C275A, and C305A were observed. Eluate fractions always contained substantially lower amounts of the target protein (∼1/10) contaminated with significant amounts of CntB degradation products. Attempts to further purify these variants were impeded because of the instability of the respective target proteins. Accordingly, freshly prepared protein samples were directly analyzed after affinity purification. Variants C267A, C272A, C275A, and C305A did not reveal detectable enzymatic activity, and EPR measurements of the related samples indicated the complete loss of the [2Fe-2S] center (∼200 μm sample concentration). From these data, we propose residues Cys-267, Cys-272, Cys-275, and Cys-305 as ligands of the plant-type [2Fe-2S] cluster of CntB. Apart from functioning as a redox-active component of carnitine monooxygenase, we conclude that this oxygen-insensitive [2Fe-2S] cluster is of central importance for the structural integrity of CntB.
EPR spectroscopy was also used to analyze the FMN-binding variants D75K and S82A (Fig. 4E). The dithionite-reduced samples indicated identical g values and approximately the same signal intensity as that observed for the WT protein. Obviously, these mutations in the FMN binding pocket do not impair the geometry or the maturation of the plant-type [2Fe-2S] cluster of CntB.
The redox midpoint potential of CntB was accessed by employing UV-visible spectro-electrochemistry in a custom-built cell described previously (32). Because of the limited stability of the protein during these experiments, only approximate values for the redox midpoint potential could be obtained (Fig. S1 and S2, left). For CntB, a midpoint potential of −162 ± 10 mV was established.
CntA is a functional trimer
Intersubunit electron transfer plays a central role for the redox catalysis of Rieske-type oxygenases. Accordingly, the oligomeric architecture of CntA was assessed by different experimental strategies. Analytical size exclusion chromatography revealed a native molecular weight of 139,000 Da, indicative of a functional CntA homotrimer (calculated monomer mass, 43,156 Da) (Fig. 5A).
Figure 5.

Relative molecular weight of CntA and CntB, small-angle X-ray scattering, and homology modeling of the functional CntA trimer. A, native molecular weight determination of CntA and CntB by analytical gel filtration. Purified proteins were analyzed on a Superdex 200 increase 5/150 GL column at a flow rate of 0.5 ml min−1, monitoring the absorption at 280 nm. Protein standards β-amylase (Mr = 200,000), alcohol dehydrogenase (ADH, Mr = 150,000), albumin (Mr = 66,000), carbonic anhydrase (Mr = 29,000), and cytochrome c (Mr = 12,400) were used for calibration. B, small-angle X-ray scattering (SAXS) analysis of CntA. Experimental SAXS data from a CntA solution (black dots) were related to the simulated SAXS curves of a potential monomeric (blue) or trimeric (red) structural model. The χ2 value reflects the fit quality with respect to the experimental data (best fit for CntA trimer). C, homology modeling of the CntA trimer. The Phyre server was used to predict a structural model of monomeric CntA. This model was superimposed onto the structure of the trimeric aromatic-ring hydroxylating dioxygenase (PDB code 3N0Q). The resulting CntA trimer (ABC) is depicted in two orientations as a surface/cartoon representation (left). Related interface regions I and II (dotted ovals) revealed a high degree of surface conservation (indicated as Shannon entropy H, ranging from 0 to 4.322 bit, top right). Both regions indicate an opposed electrostatic surface potential (φ, from −257 to +257 mV, middle right) and no increased hydropathy (−4.5 to 4.5, bottom right). The theoretical position of Glu-205 at the interface is indicated by an asterisk.
Subsequently, small-angle X-ray scattering (SAXS) experiments were performed to verify the proposed trimer. A dilute protein solution was used to determine the experimental CntA scattering curve. In Fig. 5B, the comparison of the experimental scattering curve (black dots) with the theoretical scattering curves for a hypothetical monomer (blue) or trimer (red) is shown. A significantly higher quality of fit was obtained for the trimer (χ2 = 4.95) compared with the monomer (χ2 = 99.27). Alternatively, processing of the SAXS raw data using the SAXSMoW 2.0 server (33) revealed a molecular weight of 123.7 kDa, in full agreement with the proposed trimer.
Trimeric Rieske-type oxygenases comprise two evolutionarily conserved contact regions that are centered by a bridging aspartate residue (34, 35) that might be replaced by a glutamate in CntA (Glu-205).
The Phyre2 server (36) was used for the three-dimensional modeling of the hypothetical structure of the CntA trimer (monomer templates 3VCA, 3N0Q, 2B1X, 1ULJ, and 1WQL, trimer superimposed to 3N0Q), and the sequence conservation among orthologous CntA sequences was indicated as a color code. As shown in Fig. 5C, the highest degree of surface conservation is found for the interface regions I and II of the depicted trimer interface (Shannon entropy H, shown from 0 to 4.322 bit, maximum conservation, top). These two regions further indicated an opposed electrostatic surface potential (φ, from −257 mV to +257 mV, middle), arguing for a tight monomer-monomer interaction that is not based on hydrophobic interactions (compare hydropathy from −4.5 to +4.5, bottom). The theoretical position of Glu-205 at the trimer interface is indicated by an asterisk. In summary, these experiments allowed us to clearly assign a functional CntA trimer that is tightly connected by polar protein interfaces.
CntA contains a [2Fe-2S] cluster and a mononuclear iron center
Aerobically purified CntA protein fractions showed a brownish color, and UV-visible spectroscopy revealed absorption maxima at 463 nm and 570 nm, indicative of an iron-sulfur protein. An iron content of 3.05 ± 0.21 mol Fe per mol CntA and a sulfur content of 2.11 ± 0.43 mol S per mol CntA was determined. A concentrated protein sample (∼1 mm) without reducing agent was subjected to EPR measurements (Fig. 6B). A strong S = 5/2 signal with a g value of 4.27 was observed, which is characteristic for the presence of a mononuclear iron (III) center. Conversion of such an [Fe] center into the EPR silent iron (II) state can be accomplished upon chemical reduction (37).
Figure 6.

Spectroscopic properties of CntA WT and variant proteins. A, room temperature UV-visible spectrum of purified CntA. Inset, spectral region from 400–600 nm for purified CntA before (black) and after (dashed) dithionite treatment (10 mm). B, low-temperature (15 K) EPR spectra of purified CntA (∼1 mm) before (a) and (b) after reduction (10 mm sodium dithionite).
Treatment of a related CntA sample with 10 mm sodium dithionite resulted in the bleaching of the brownish color, as indicated by drastically reduced absorption maxima at 463 nm and 570 nm, with the appearance of a new maximum at 520 nm, which is in agreement with previously reported spectroscopic data of related Rieske oxygenases (38) (Fig. 6A, inset). Subsequent EPR experiments indicated complete loss of the [Fe] center signal with the parallel appearance of an S = 1/2 signal with g values of g1 = 2.016, g2 = 1.921, and g3 = 1.759 (Fig. 6B). The obtained values are characteristic of a [2Fe-2S]+ center of the Rieske type (30).
These findings might be indicative for the artificial two-electron reduction of the CntA protein in the presence of dithionite. A first electron leads to the conversion of the mononuclear iron (III) center into the EPR silent iron (II) state. A subsequent electron is required to render the [2Fe-2S]2+ cluster into the EPR active [2Fe-2S]+ state. Within the overall EPR study, exclusive reduction of the [2Fe-2S]2+ cluster of CntA was not observed.
UV-visible spectro-electrochemical experiments of CntA were hampered, as a redox-dependent sample precipitation was observed. A lower limit for the midpoint potential of +300 mV could be estimated (Fig. S2, right).
Ligands of the Rieske [2Fe-2S] cluster and the mononuclear iron center of CntA
The protein sequence of CntA contains a motif that is typical for the ligation of a Rieske-type [2Fe-2S] cluster: CXHX17CX2H, with residues Cys-86, His-88, Cys-106, and His-109 as potential cluster ligands (in boldface). Mutant proteins C86A, H88A, C106A, and H109A were purified in close analogy to the WT protein. They revealed a substantial decrease of the iron and sulfur content (Fig. 3A, lanes 7–10, and Table 2). Related EPR experiments after dithionite reduction indicated complete loss of the respective [2Fe-2S] cluster signal (Table 2). No detectable l-carnitine monooxygenase activity was determined in related enzyme activity assays. These findings clearly indicate that the Rieske-type [2Fe-2S] cluster of CntA is coordinated by residues Cys-86, His-88, Cys-106, and His-109 (Table 2).
Table 2.
Biochemical characterization of CntA wild-type and variant proteinsa
| Mutation | Function | Relative l-carnitine depletion activity (%) | Relative TMA formation (%) | Iron content (nmol protein−1) | Sulfur content (nmol protein−1) | EPR signal for [Fe] | EPR signal for [2Fe-2S] |
|---|---|---|---|---|---|---|---|
| C86A | [2Fe-2S] coordinating | <5 | – | 0.12 ± 0.03 | 0.35 ± 0.11 | ND | ND |
| H88A | [2Fe-2S] coordinating | <5 | ND | 0.06 ± 0.03 | 0.64 ± 0.21 | Weak | ND |
| C106A | [2Fe-2S] coordinating | <5 | – | 0.03 ± 0.02 | 0.03 ± 0.02 | Weak | ND |
| H109A | [2Fe-2S] coordinating | <5 | – | 0.03 ± 0.02 | 0.67 ± 0.08 | ND | ND |
| E205Q | Intersubunit electron transfer | <5 | – | 2.78 ± 0.10 | 1.62 ± 0.14 | Weak | WT |
| E205D | Intersubunit electron transfer | <5 | ND | 2.53 ± 0.15 | 1.53 ± 0.07 | Weak | WT |
| H208A | [Fe] coordinating | <5 | – | 2.35 ± 0.11 | 2.08 ± 0.24 | ND | WT |
| H213A | [Fe] coordinating | <5 | ND | 2.00 ± 0.11 | 1.39 ± 0.15 | ND | WT |
| D323A | [Fe] coordinating | <5 | – | 2.71 ± 0.15 | 1.76 ± 0.28 | ND | WT |
aTable summarizes biochemical and spectroscopic properties of CntA variants. The specific activity of the l-carnitine depletion assay (771 ± 68 nmol min−1 mg−1) and of the enzymatic TMA formation (determined by gas chromatography) for the wild-type protein was set as 100% (compared to experimental procedures). ND, TMA formation not detectable. EPR signals for the [Fe] center (samples as purified) and the [2Fe-2S] cluster (samples after dithionite reduction) were classified. EPR signals comparable with the wild type are indicated as WT, substantially reduced EPR signals (<10% of wild-type signal) are indicated as weak, and the absence of a detectable EPR signal was indicated as not detectable (ND). <5 indicates enzymatic activity below the detection limit of the employed assay. –, not performed.
The four mutant proteins were also subjected to EPR spectroscopy in the nonreduced state to assess the integrity of the [Fe] center of CntA. Variants H88A and C106A indicated a barely detectable S = 5/2 signal, whereas proteins C86A and H109A were completely devoid of the [Fe] center (Fig. 7, left). These findings indicate that the presence of the Rieske [2Fe-2S] cluster is a prerequisite for the subsequent incorporation of the mononuclear iron.
Figure 7.

EPR spectra of CntA variants. Left, CntA variant protein C86A, H88A, C106A, and H109A as purified (∼1 mm). Right, variants H208A, H213A, and D323A (∼1 mm) after dithionite reduction (5 mm).
The related [Fe] center of Rieske proteins is typically ligated by two histidine residues and an aspartate or glutamate residue, forming a so-called 2-His-1-caboxylate triad (39). Residues His-208, His-213, and Asp-323 of CntA were proposed as candidate ligands of the [Fe] center of CntA (18). Compared with the WT, the purified variants H208A, H213A, and D323 indicated partial reduction of the iron content. The nonreduced EPR samples of these mutants revealed a complete loss of the [Fe] center. Proteins H208A, H213A, and D323 did not support any enzymatic activity (Table 2). These findings argue for a mononuclear iron center of CntA that is coordinated by protein ligands His-208, His-213, and Asp-323.
These three variants were also subjected to EPR experiments under reduced conditions. In all cases, a clear signal for the presence of the [2Fe-2S]+ cluster of CntA was determined (Fig. 7, right). Obviously, the presence of the mononuclear iron center is not a prerequisite for the maturation of the [2Fe-2S] cluster of CntA. The results of the overall mutagenesis study suggest a hierarchical metallocenter assembly of CntA: Rieske [2Fe-2S] cluster followed by the [Fe] center. However, intact Rieske cluster ligation might be a prerequisite for the spatial positioning of the [Fe] center ligands. According to this, the mutagenesis of residues Cys-86 and His-109 would result in the destabilization of the [Fe] center.
Mutagenesis of the bridging Glu-205
The importance of residue Glu-205 was recently demonstrated based on kinetic experiments of mutant CntA proteins (18). Involvement of this residue in the electron transfer from the Rieske-type [2Fe-2S] cluster to the [Fe] center of the adjacent subunit was proposed.
EPR spectroscopy of variants E205Q and E205D was used to further specify the functional role of this residue. Neither variant did revealed detectable enzymatic activity and a moderately reduced Fe and sulfur content (Table 2). Reduced EPR samples showed a clear signal of the Rieske-type [2Fe-2S] cluster. In contrast, the nonreduced samples showed almost complete loss of the [Fe] center signal (∼90% reduction) (Fig. 8). Obviously, mutagenesis of residue Glu-205 might hamper the maturation or the spatial ligation of the [Fe] center. In this context, variant E205Q indicates that an intact carboxylic group of Glu-205 is essential for the proposed intersubunit electron transfer of CntA. By analogy to related Rieske-type oxygenases, a bridging carboxylate between the [2Fe-2S] cluster and the [Fe] center of the adjacent CntA subunit was concluded. Variant E205D aims to “restore” the bridging aspartate that has been described for various Rieske proteins on the structural and functional level. Obviously, the slightly smaller aspartate residue is not capable of substituting for the bridging Glu-205 of CntA. We conclude that Glu-205 is important for the intersubunit electron transfer between the metallocenters of CntA (18) and/or the subsequent activation of molecular oxygen.
Figure 8.

EPR spectra of CntA variants of the bridging Glu-205. Purified CntA variants E205D and E205Q (∼1 mm) as purified (left) and dithionite-reduced samples (5 mm) (right).
Electron transfer pathway of carnitine monooxygenase
Components CntB and CntA catalyze carnitine monooxygenation (with subsequent TMA, maleic semialdehyde, and water formation, Fig. 2A) using one molecule of dioxygen and two electrons according to the following equation: carnitine + 2 e− + O2 → monooxygenated carnitine + H2O (substrate oxidized by 2 e−, O2 reduced by 4 e−, and 2 external reduction equivalents required).
Both electrons derived from NADH are transferred via the electron transport protein CntB onto the catalytic component CntA. This intercomponent electron transfer is not based on a tight interaction of CntB and CntA. Parallel overproduction of both components in E. coli followed by affinity chromatography of the GST-tagged component CntA did not result in the co-purification of detectable amounts of CntB. In vitro protein-protein interaction experiments using immobilized CntA (or CntB) did not allow for the trapping of CntB (or CntA). Accordingly, transient interaction of the monomeric CntB with the CntA trimer was concluded.
The presented EPR spectroscopic investigation of CntB and CntA already reflected individual redox states that are relevant for the understanding of the electron transfer pathway of carnitine monooxygenase. However, caution should be exercised for the interpretation of chemically reduced samples (inter alia because of the strong negative redox potential of dithionite [40]).
Accordingly, we made use of the natural two-electron donor NADH to exemplify intermediate states of carnitine monooxygenase catalysis. All subsequent experiments were performed as single-turnover experiments in the anaerobic workstation to strictly prevent O2-dependent reaction(s) prior to EPR measurements.
An immobilized CntB sample (∼0.5 mm on Co2+-loaded chelating Sepharose) was incubated in the presence of 1 mm NADH before the cofactor was removed by an excessive washing step. The resulting reduced elution fraction indicated a substantially differing EPR spectrum when related to the dithionite-reduced spectrum (Fig. 9A, gray trace, versus Fig. 4D). Compared with the dithionite reduced state, the characteristic signal of the plant-type [2Fe-2S]+ cluster remains almost unaltered (g1 = 2.037, g2 = 1.943, and g3 = 1.900), but a new dominant signal with g = 2.007 in the characteristic region for the semiquinone form of FMN was observed. The temperature-dependent spin relaxation behavior of both signals was further elucidated. Increasing the sample temperature (from 15 to 85 K) did not alter the semiquinone signal but led to an almost complete loss of the plant-type [2Fe-2S]+ signal (Fig. 9A, compare gray and black dashed traces). To further identify the slow relaxing g = 2 species, we performed a pulsed Q-band electron nuclear double resonance (ENDOR) experiment at 30 K (Fig. S3). The observed hyperfine splittings in the range of 1–24 MHz are consistent with a mostly protonated FMN radical (41). From these experiments, we conclude that the NADH-dependent two-electron reduction locates one electron to the FMN cofactor (in the form of a neutral FMNH* radical) and a second electron to the plant-type [2Fe-2S] cluster of CntB (42). The proposed CntB FMNH*/[2Fe-2S]+ species (I) is indicated in Fig. 10.
Figure 9.

EPR analysis of single-turnover experiments. A–C, intermediate states of the redox catalysis of CntB and CntA were spectroscopically characterized. All experiments performed under strict anaerobic conditions. A, an immobilized CntB sample was incubated in the presence of 2 mm NADH, the cofactor was removed by extensive washing, and the eluted protein was subjected to low-temperature EPR measurements at 15 K. A [2Fe-2S]+ cluster signal (g1 = 2.037, g2 = 1.945, and g3 = 1.900) and a semiquinone signal (g = 2.007) was observed (gray trace). Identical measurements at higher temperature (85 K) revealed the specific depletion of the g = 1.945 [2Fe-2S]+ cluster signal (black dashed line). A CntB FMNH*/[2Fe-2S]+ species (I) was proposed and was subsequently used in a single-turnover experiment. B, in the presence of CntA, a two-electron transfer from (I) resulted in the EPR-silent state CntB FMN [2Fe-2S]2+ (II) (after CntA removal and CntB elution). C, EPR spectroscopic characterization of the resulting CntA revealed the transfer of two electrons. A Rieske [2Fe-2S]+/[Fe]2+ state was concluded (III).
Figure 10.

Proposed redox catalytic cycle of carnitine monooxygenase. A redox catalytic cycle for carnitine monooxygenase was proposed based on the experimentally characterized redox states of the present study. Reduction of CntB by the two-electron donor NADH results in the FMNH*/[2Fe-2S]+ state (I). Subsequent two-electron transfer onto CntA reveals the electron donor in the CntB FMN/[2Fe-2S]2+ state (II) and activates the electron acceptor in the CntA [2Fe-2S]+/[Fe]2+ (III). Subsequent electron transfer onto O2 results in the CntA [2Fe-2S]2+/[Fe]2+ redox state, which resembles the as-purified state of this protein (IV) (compare to Fig. 7B). The reduction of O2 then leads to the formation of the monooxygenated l-carnitine product (not shown) (compare to Fig. 2A) and H2O. Cleavage of the C–N bond of the intermediate then results in the formation of malic semialdehyde and TMA.
Subsequently, we wanted to figure out if the NADH-reduced CntB species (I) facilitates the intercomponent electron transfer onto CntA. Therefore, a CntB sample was immobilized on Co2+-loaded chelating Sepharose, reduced by NADH, and the physiological reducing agent was removed by extensive washing. An equimolar amount of CntA then was incubated in the presence of the reduced electron donor protein, and the resulting CntA protein was subsequently eluted in the absence of CntB.
The immobilized CntB was specifically eluted and subjected to EPR measurements. An EPR-silent sample was observed (Fig. 9B) that might indicate electron translocation onto CntA, which in turn would leave the reductase CntB in the FMN/[2Fe-2S]2+ state (II).
Subsequent EPR measurements of the eluted CntA protein revealed the two-electron reduction of the Rieske protein, as indicated by g1 = 2.016, g2 = 1.921, and g3 = 1.762 (Fig. 9C). A Rieske [2Fe-2S]+/[Fe]2+ state was concluded (III) that might be linked to the subsequent activation of molecular oxygen on CntA.
Discussion
TMA formation in the gut microbiome came into focus in recent years, since the related metabolic processes were linked to the development of cardiovascular disease. Bacterial conversion of dietary l-carnitine into TMA and malic semialdehyde is catalyzed by an unusual Rieske-type monooxygenase, which is composed of the reductase component CntB and the catalytic unit CntA. The best-studied Rieske-type oxygenases play an important role in the degradation of aromatic compounds (23, 24), but the overall spectrum of reactions ranges from cis-hydroxylations, desaturations, and O- or N-dealkylations to the formation of chiral sulfoxides (22, 43). Protein crystallographic studies enabled the understanding of the substrate specificity of many Rieske-type proteins (44). These oxygenases share a conserved domain structure, but their sequences are highly divergent (45). From an evolutionary perspective, CntA was recently ascribed to a novel group of Rieske-type oxygenases (46, 47). The enzymes of this group V facilitate the degradation of quaternary ammonium compounds in N-dealkylation reactions. Glycine betaine monooxygenase is another member of this group and has been initially characterized with respect to the reductase BmoB and the oxygenase BmoA. BmoB is an NADH-dependent reductase that carries a noncovalently bound FAD cofactor. The Rieske-type unit BmoA enables the oxygen-dependent degradation of glycine betaine into dimethylglycine and formaldehyde (47).
In the present study, the multistep electron transfer of carnitine monooxygenase was elucidated based on biochemical and spectroscopic experiments. A chain of closely spaced redox relays enables the NAD(P)H-dependent activation of O2 with subsequent formation of TMA, malic semialdehyde, and water. Biochemical and spectroscopic approaches, in combination with site-directed mutagenesis and single-turnover experiments, allowed us to postulate a catalytic redox cycle of carnitine monooxygenase, which is schematically depicted in Fig. 10. All experimentally characterized redox states of this cycle were highlighted boldface (I–IV).
NADH facilitates the initial reduction of the flavin by two electrons. Subsequently, single-electron transfer from FMNH2 to the [2Fe-2S]2+ cluster is observed. The noncovalently linked FMN cofactor is held in place by residues Asp-75 and Ser-82, and the plant-type [2Fe-2S] cluster is ligated by Cys-267, Cys-272, Cys-275, and Cys-305. The resulting semiquinone form of CntB can be described as the FMNH*/[2Fe-2S]+ state (I). This reduced CntB species now facilitates the two-electron intersubunit electron transfer onto CntA by transient protein-protein interaction. Therefore, the reductase is regained in the CntB FMN/[2Fe-2S]2+ state (II). The Rieske-type protein CntA contains a typical [2Fe-2S] cluster, coordinated by residues Cys-86, His-88, Cys-106, and His-109 and a mononuclear [Fe] center with protein ligands His-208, His-213, and Asp-323. Upon reduction by CntB, a CntA [2Fe-2S]+/[Fe]2+ state (III) is observed that is capable of activating molecular oxygen in a two-electron transfer process with subsequent release of the oxygenase in the CntA [2Fe-2S]2+/[Fe]3+ redox state (IV). Glu-205 might play a fundamental role for the redox relay of CntA. This bridging glutamate might facilitate the intermolecular electron transfer between the subunits of the trimeric CntA protein (e.g. [2Fe-2S]subunit A → [Fe]subunit B; compare to Fig. 5C). Furthermore, we conclude a structural role of Glu-205 for the positioning of one of the histidinyl ligands of the mononuclear [Fe] center of the adjacent subunit. According to this, CntA is an unusual member of the Rieske-type protein family, because most other Rieske oxygenases make use of a fully conserved aspartate residue instead (29, 35). The reductive activation of O2 allows for the monooxygenation of l-carnitine paralleled by the formation of one molecule of H2O. Heterolytic cleavage of the C–N bond of this reaction intermediate then results in the formation of malic semialdehyde and TMA (Fig. 2A).
Future experiments will focus on the mechanism for the activation of molecular oxygen at the mononuclear [Fe] site of CntA and the related recognition of the l-carnitine substrate. The further understanding of this unusual Rieske-type monooxygenase will be of great interest for the targeted inhibition of carnitine monooxygenase as a future therapeutic strategy to combat cardiovascular disease.
Experimental procedures
Production and purification of CntA and CntB
Codon-optimized genes for the production of A. baumannii ATCC 19606 CntA and CntB were synthesized by GeneArt (Thermo Fisher). cntA and cntB genes were cloned into vectors pGEX-6P-1 and pACYCDuet-1 using the In-Fusion cloning system from Takara with the following primers: GGGCCCCTGGGATCCATGAGCGCAGTTGAAAAACTGC and GGCCGCTCGAGTCGACTTACTGGTGATACTGTGCAACC (for construct pGEX-6P-1-cntA) and ACCACAGCCAGATCCGATGGCCAGCCACTATGAAATG and CCGCAAGCTTGTCGACTTACAGATCCAGAACCAGTTTTTTGC (for construct pACYCDuet-1-cntB). The recombinant CntA fusion protein with an N-terminal GSH S-transferase tag or the N-terminally His-tagged CntB protein was produced in E. coli Tuner (DE3) cells. An overnight culture was used to inoculate 500 ml of LB medium containing 100 µg ml−1 ampicillin (pGEX-6P-1-cntA) or 34 µg ml−1 chloramphenicol (pACYCDuet-1-cntB). After ∼2 h at 37 °C and 200 rpm, an optical density at 578 nm of 0.4 was reached. The cell cultures were incubated for 15 min at 17 °C without agitation. Subsequently, the production of the target proteins was initiated by addition of 50 μm isopropyl-β-d-thiogalactopyranoside (IPTG), 1 mm Fe(III)-citrate, and 1 mm l-cysteine hydrochloride (to foster iron center maturation). After 16 h of cultivation at 17 °C and 180 rpm, the cells were harvested by centrifugation at 4,000 × g for 15 min at 4 °C, resuspended in 6 ml buffer 1 (100 mm HEPES-NaOH, pH 7.5, 150 mm NaCl), and disrupted by a single passage through a French press at 14,500 psi. Lysates were clarified by centrifugation at 112,000 × g for 60 min at 4 °C, and the supernatant was applied to 1 ml GSH-Sepharose (Macherey-Nagel) for the CntA fusion or 1 ml of a Co2+-loaded chelating Sepharose (Clontech) for His-tagged CntB. Affinity columns were subsequently washed with 18 ml buffer 1 to remove unbound proteins. Proteolytic on-column cleavage of the CntA fusion was performed with PreScission protease (400 units, 16 h at 4 °C) and the untagged CntA protein was eluted in 3× 1 ml buffer 1. CntB was eluted with 4× 1 ml buffer 1 containing 15 mm imidazole. Target protein fractions were identified by SDS-PAGE.
Purified protein samples were concentrated to ∼1 M using Amicon Ultra-0.5 centrifugal filter units (Millipore) equipped with a 30,000-Da cutoff membrane.
Determination of protein concentration
The concentration of purified proteins was determined using Bradford reagent (Sigma-Aldrich) according to the manufacturer's instructions with bovine serum albumin as a standard.
UV-visible light absorption spectroscopy
UV-visible light spectra of purified recombinant CntA and CntB were recorded at room temperature using a V-650 UV-Vis spectrophotometer (Jasco).
Iron determination method
Protein-bound iron was determined colorimetrically with o-phenanthroline after acid denaturation of purified CntA or CntB (48).
Sulfur determination method
Protein-bound sulfur was determined as described elsewhere (32).
UV-visible spectro-electrochemical measurements
Redox titrations were performed as described previously (49), using a 25-μl protein sample (0.5 mm) in 0.1 M HEPES-NaOH, pH 7.5, containing 150 mm NaCl. The potential was altered in steps of 10 mV with 10 min of equilibration before spectra were recorded. The redox reaction was followed by the absorbance change at 462 nm, and redox potentials were calculated by fitting the data points to the Nernst equation.
Standard l-carnitine depletion assay
A typical 100-µl standard l-carnitine depletion assay was performed in buffer 1 containing 5 μm purified CntA, 20 μm purified CntB, and 300 μm l-carnitine at 37 °C. Reactions were initiated by the addition of 2 mm NADH (or NADPH). At defined time points (0, 10, 30, 60, and 90 s), 100-µl samples were heat inactivated (99 °C, 10 min) and centrifuged for 10 min at 12,000 × g, and the l-carnitine concentration of the resulting supernatant was determined by a modified method according to reference 50. Samples of 50 µl were supplemented with 7.5 µl 2 mm 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (Sigma-Aldrich), 15 µl 2 mm acetyl-CoA, and 0.2 U carnitine acetyltransferase (Sigma-Aldrich) in a total volume of 300 µl. The carnitine acetyltransferase reaction is accompanied by the formation of CoA, which then leads to the cleavage of DTNB. The resulting 2-nitro-5-thiobenzoic acid was spectroscopically quantified at 412 nm using an extinction coefficient of ϵ412 = 14.15 mm−1 cm−1. A standard curve was determined using defined l-carnitine concentrations. The specific activity was determined by calculation of the slope in the linear range of the experiment. All experiments were performed in triplicate and completed by kinetic measurements in the absence of electron donor or CntA. The l-carnitine depletion assay was also performed under strict anaerobic conditions. Therefore, the purification of components CntB and CntA and the activity measurements were conducted in an anaerobic chamber (Coy Laboratories) using N2 saturated buffers. Sample manipulation procedures always resulted in an initial lack phase of 10 s. Relative l-carnitine depletion activities of CntB variant proteins were conducted at a reduced CntB concentration of 5 μm.
TMA quantification by GC
Enzymatic TMA formation was determined by GC according to established procedures (51, 52) (IBEN). Assay mixtures (1 ml) containing 5 μm CntA, 20 μm CntB, 2 mm l-carnitine, and 5 mm NADH were incubated for 2 min at 37 °C. Reactions were stopped by the addition of 1 M perchloric acid. Variant proteins of CntA or CntB or control experiments in the absence of CntB or NADH were processed accordingly.
NADH depletion assay
A colorimetric NADH depletion assay was performed at 37 °C in the presence of 2 μm CntA, 2 μm CntB, 300 μm l-carnitine, and 200 μm NADH using an extinction coefficient of ϵ340 = 6.2 mm−1 cm−1 for NADH (53). The specific activity was determined by calculation of the slope in the linear range of the assay. Control reactions in the absence of CntA or CntB or under strict anaerobic conditions (see above) were performed.
Flavin cofactor determination by HPLC
A purified CntB sample (50 μm) in a total volume of 300 µl was heat denatured at 99 °C for 10 min. The liberated cofactor was separated from the denatured protein by centrifugation (16,000 × g, 10 min at 22 °C). 10 µl of the supernatant was analyzed on a Reprosil 100 C18 column (Techlab) on an HPLC system equipped with a pump (PU-1580, Jasco), a column oven (X-LC, Jasco), a multiwavelength detector (MD-1515, Jasco), and a fluorescence detector (FP-1520, Jasco). Isocratic separation was performed at a flow rate of 0.5 ml min−1 at 30 °C using the following mobile phase: 14% (v/v) acetonitrile, 1.5% (v/v) TFA, and 0.09% (v/v) of a 75% (w/w) phosphoric acid in water. Authentic FAD or FMN samples (Sigma-Aldrich) were used as a standard, and the related elution profiles were normalized according to maximum signal of the CntB sample.
FMN cofactor quantification
The FMN content of purified CntB samples was quantified by UV-visible absorption spectroscopy using an extinction coefficient of ϵ450 = 12.2 mm−1 cm−1 for free FMN on a V-650 UV-Vis spectrophotometer (Jasco). FMN was liberated from the protein by heat denaturation as described above.
Analytical size exclusion chromatography
The oligomeric state of CntA and CntB was determined by analytical size exclusion chromatography using a Superdex 200 increase 5/150 GL column (GE Healthcare). The column was calibrated with protein standards (molecular weight marker kit MWGF 1000; Sigma) in the presence of buffer 1 at a flow rate of 0.45 ml min−1. Samples of CntB (20 µl) at a concentration of 350 μm or of CntA (20 µl) at a concentration of 280 μm were injected. Elution was monitored at 280 and 320 nm, respectively.
SAXS experiments
SAXS experiments were carried out on beamline BM29 (54) of the European Synchrotron Radiation Facility. A CntA sample with a concentration of 430 μm was exposed to an X-ray energy of 12,500 eV at a temperature of 293 K while steadily flowing through a quartz capillary. The scattering intensities were recorded with a Pilatus 1M hybrid photon counting detector within a momentum transfer range of 0.035–5 nm−1. A total of ten scattering images were collected with an exposure time of 0.5 s per image. The scattering data were processed with programs from the ATSAS suite (55). The scattering images were appraised for apparent effects of radiation damage and subsequently normalized, averaged, and baseline corrected through the subtraction of buffer scattering with the program PRIMUS (56). Theoretical scattering intensities for the trimer and the monomer of CntA were calculated and fitted to the experimental data with the program CRYSOL (57). CRYSOL reports χ2 values as an indicator for the general agreement of theoretical and experimental curves, which are calculated by
with N being the number of individual readout data points. For a more thorough assessment of the quality of fit throughout the resolution range, we calculated the residual
which was plotted as a function of momentum transfer/resolution. The best agreement is reflected by the smallest χ2 value and by residuals ΔI/σI only marginally different from zero throughout the whole resolution range.
Site-directed mutagenesis of cntA and cntB
Up to three nucleotides were exchanged using the QuikChange site-directed mutagenesis kit (Agilent Technologies) and the following oligonucleotides: C86A: CCTTTTATAACGTTGCGCCGCATCGTGG; H88A, CGTTTGTCCGGCGCGTGGTCATGAACTGC; C106A, GTTATTACCGCGCCGTATCACGCC; H109A, CCTGTCCGTATGCGGCCTGGACCTTTAAACTGG; E205Q, GGATAACTATATGCAGTGCTATCATTGTGG; E205D, GGATAACTATATGGATTGCTATCATTGTGG; H208A, GGAATGCTATGCGTGTGGTCCGGCAC; H213A, GTGGTCCGGCAGCGCCGGGTTTTGCCG; D323A, CCGTCCTGAGGCGCTGAATCTGGTTG (for the exchange of codons in cntA); D75K, GGTTTGTGTTCGTAAAAAAGTTGAA GGTAAAGG; S82A, GGTAAAGGTGGTGCGGTGTTTATGCATG; C265A, GAATATTGATGTTGAAGCGCTGTGTCGTGAAGG; C267A, GAATATTGATGTTGAATGTCTGGCGCGTGAAGG; C272A, CGTGAAGGTGTTGCGGGCACCTG TGAAACC; C275A, GGTGTTTGTGGCACCGCGGAAACCGCAATTCTGG; and C305A, CCAGAAAAGCATGATGATTGCGGTTAGCCGTGCC (for the exchange of codons in cntB). Exchanged nucleotides are indicated in boldface.
Preparation of EPR samples for WT and variant CntA and CntB proteins
Purified proteins were concentrated up to 1 mm using an Amicon Ultra-0.5 centrifugal filter unit (Millipore) equipped with a 30,000-Da cutoff membrane. Sodium dithionite was added as indicated. After 10 min of incubation, samples were transferred into 3-mm EPR tubes and frozen in liquid nitrogen.
Interaction of CntA and CntB
Theoretical interaction of CntA and CntB was explored using the existing pGEX-6P-1-cntA protein production vector in combination with the cntB gene with an additional ribosome binding site. Therefore, cntB was cloned into vector pGEX-6P-1-cntA using the primers CACCAGTAAGTCGACTTTCACACAGGAAACAGTATTCATGGCCAGCCAC (including ribosome binding site, indicated in boldface) and CAGTCACGATGCGGCCGCTTACAGATCCAGAACCAG. Parallel overproduction of CntA and CntB and the purification of the bait protein CntA was accomplished as described above. SDS-PAGE analysis was used to indicate potential copurification of CntA and CntB.
Alternatively, purified CntA or CntB was immobilized on the respective affinity material (0.3 ml resin). The bait proteins were incubated with the potential pray proteins (purified CntB or CntA) at a concentration of ∼50 μm for 5 min. After a subsequent washing step (5 ml buffer 1), the respective target proteins were eluted (as described above) and the related fractions were subjected to SDS-PAGE analysis.
EPR spectroscopy
X-Band EPR spectra were recorded on a Bruker Elexsys E-500 CW X-band spectrometer. The sample tubes of 3-mm outer diameter were placed in a standard TE102 resonator. Low-temperature measurements were obtained using an Oxford ESR 900 helium flow cryostat (3–300 K). Baseline corrections were performed by subtracting a background spectrum, obtained under the same experimental conditions, from an empty tube.
Q-band experiments were performed on a Bruker ELEXYS E580 spectrometer with a SuperQ-FT microwave bridge (5-W output power) using a home-built resonator described earlier (58). Cryogenic temperatures (10–30 K) were obtained by a closed-cycle helium flow cryostat (Cryogenic Ltd.). ENDOR experiments were performed using a 300-W ENI 300 L RF amplifier.
Single-turnover experiments
Single-turnover experiments were performed under strict anaerobic conditions in an anaerobic chamber (Coy Laboratories) with an oxygen partial pressure below 1 ppm (oxygen detector, Coy Laboratories). Buffers were N2 saturated prior to use. CntB was purified according to the standard procedure described above. The on-column immobilized protein (∼1 mm) was incubated in the presence of 2 mm NADH for 10 min which resulted in a clear bleaching of the immobilized sample. After an excessive washing step (6 ml buffer 1), the reduced protein was eluted in 2 ml buffer 1 containing 50 mm imidazole. The resulting protein fraction was concentrated (∼1 mm, 20–30 µl), transferred into 3 mm EPR tubes, and frozen in liquid nitrogen.
In related experiments, on-column immobilized CntB (after NADH reduction and extensive washing) was incubated in the presence of 1 mm CntA for 10 min. Subsequently, both proteins were specifically eluted. CntA was recovered in the presence of 2 ml buffer 1. Subsequently, CntB was liberated with 2 ml buffer 1 containing 50 mm imidazole. Both fractions were concentrated (∼1 mm, 20–30 µl), transferred into 3-mm EPR tubes, and frozen in liquid nitrogen.
Data availability
All data are contained within the present manuscript.
Supplementary Material
Acknowledgments
We thank Simone Virus for her excellent technical assistance and Anna Kopenhagen for supporting enzymatic measurements. We thank Dr. James Birrell for helpful discussions on the UV-visible spectro-electrochemistry experiments.
This article contains supporting information.
Author contributions—M. M., E. R., J. K., W. L., D. J., and J. M. conceptualization; M. M., E. R., J. K., C. L., W. L., D. J., and J. M. data curation; M. M., E. R., J. K., W. L., D. J., and J. M. formal analysis; M. M., E. R., J. K., W. L., D. J., and J. M. supervision; M. M., E. R., J. K., W. L., D. J., and J. M. validation; M. M., E. R., J. K., W. L., D. J., and J. M. investigation; M. M., E. R., J. K., W. L., D. J., and J. M. visualization; M. M., E. R., J. K., W. L., D. J., and J. M. writing-original draft; M. M., E. R., J. K., W. L., D. J., and J. M. project administration.
Funding and additional information—E.R. and W.L. thank the Max Planck Society for its continuous financial support.
Conflict of interest—The authors declare that they have no conflict of interest.
- TMA
- trimethylamine
- TMAO
- trimethylamine N-oxide
- DTNB
- 5,5'-dithiobis-(2-nitrobenzoic acid)
- TNB
- 2-thio-5-nitrobenzoic acid
- SAXS
- small angle X-ray scattering
- EPR
- electron paramagnetic resonance.
References
- 1. Gill S. R., Pop M., Deboy R. T., Eckburg P. B., Turnbaugh P. J., Samuel B. S., Gordon J. I., Relman D. A., Fraser-Liggett C. M., and Nelson K. E. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 10.1126/science.1124234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ley R. E., Turnbaugh P. J., Klein S., and Gordon J. I. (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- 3. Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., and Gordon J. I. (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- 4. Nicholson J. K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., and Pettersson S. (2012) Host-gut microbiota metabolic interactions. Science 336, 1262–1267 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- 5. Koren O., Spor A., Felin J., Fak F., Stombaugh J., Tremaroli V., Behre C. J., Knight R., Fagerberg B., Ley R. E., and Backhed F. (2011) Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl 1), 4592–4598 10.1073/pnas.1011383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cani P. D., Bibiloni R., Knauf C., Waget A., Neyrinck A. M., Delzenne N. M., and Burcelin R. (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 10.2337/db07-1403 [DOI] [PubMed] [Google Scholar]
- 7. Round J. L., and Mazmanian S. K. (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chistiakov D. A., Bobryshev Y. V., Kozarov E., Sobenin I. A., and Orekhov A. N. (2015) Role of gut microbiota in the modulation of atherosclerosis-associated immune response. Front. Microbiol. 6, 671 10.3389/fmicb.2015.00671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang W. H., and Hazen S. L. (2014) The contributory role of gut microbiota in cardiovascular disease. J. Clin. Invest. 124, 4204–4211 10.1172/JCI72331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miao J., Ling A. V., Manthena P. V., Gearing M. E., Graham M. J., Crooke R. M., Croce K. J., Esquejo R. M., Clish C. B., Vicent D., and Biddinger S. B, Morbid Obesity Study Group. (2015) Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 6, 6498 10.1038/ncomms7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koeth R. A., Wang Z., Levison B. S., Buffa J. A., Org E., Sheehy B. T., Britt E. B., Fu X., Wu Y., Li L., Smith J. D., DiDonato J. A., Chen J., Li H., Wu G. D., et al. (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 10.1038/nm.3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang W. H., Wang Z., Fan Y., Levison B., Hazen J. E., Donahue L. M., Wu Y., and Hazen S. L. (2014) Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J. Am. Coll. Cardiol. 64, 1908–1914 10.1016/j.jacc.2014.02.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Senthong V., Li X. S., Hudec T., Coughlin J., Wu Y., Levison B., Wang Z., Hazen S. L., and Tang W. H. (2016) Plasma trimethylamine N-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J. Am. Coll. Cardiol. 67, 2620–2628 10.1016/j.jacc.2016.03.546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Z., Roberts A. B., Buffa J. A., Levison B. S., Zhu W., Org E., Gu X., Huang Y., Zamanian-Daryoush M., Culley M. K., DiDonato A. J., Fu X., Hazen J. E., Krajcik D., DiDonato J. A., et al. (2015) Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595 10.1016/j.cell.2015.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleber H. P., Seim H., Aurich H., and Strack E. (1977) Utilization of trimethylammonium-compounds by Acinetobacter calcoaceticus. Arch. Microbiol. 112, 201–206 10.1007/BF00429336 [DOI] [PubMed] [Google Scholar]
- 16. Craciun S., and Balskus E. P. (2012) Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. U.S.A. 109, 21307–21312 10.1073/pnas.1215689109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falony G., Vieira-Silva S., and Raes J. (2015) Microbiology meets big data: the case of gut microbiota-derived trimethylamine. Annu. Rev. Microbiol. 69, 305–321 10.1146/annurev-micro-091014-104422 [DOI] [PubMed] [Google Scholar]
- 18. Zhu Y., Jameson E., Crosatti M., Schafer H., Rajakumar K., Bugg T. D., and Chen Y. (2014) Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl. Acad. Sci. U.S.A. 111, 4268–4273 10.1073/pnas.1316569111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barry S. M., and Challis G. L. (2013) Mechanism and catalytic diversity of rieske non-heme iron-dependent oxygenases. ACS Catal. 3, 2362–2370 10.1021/cs400087p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nam J. W., Nojiri H., Yoshida T., Habe H., Yamane H., and Omori T. (2001) New classification system for oxygenase components involved in ring-hydroxylating oxygenations. Biosci. Biotechnol. Biochem. 65, 254–263 10.1271/bbb.65.254 [DOI] [PubMed] [Google Scholar]
- 21. Yeh W. K., Gibson D. T., and Liu T. N. (1977) Toluene dioxygenase: a multicomponent enzyme system. Biochem. Biophys. Res. Commun. 78, 401–410 10.1016/0006-291X(77)91268-2 [DOI] [PubMed] [Google Scholar]
- 22. Resnick S. M., Lee K., and Gibson D. T. (1996) Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J. Ind. Microbiol. 17, 438–457 10.1007/BF01574775 [DOI] [Google Scholar]
- 23. Peng R. H., Xiong A. S., Xue Y., Fu X. Y., Gao F., Zhao W., Tian Y. S., and Yao Q. H. (2010) A profile of ring-hydroxylating oxygenases that degrade aromatic pollutants. Rev. Environ. Contam. Toxicol. 206, 65–94 10.1007/978-1-4419-6260-7_4 [DOI] [PubMed] [Google Scholar]
- 24. Gally C., Nestl B. M., and Hauer B. (2015) Engineering Rieske non-heme iron oxygenases for the asymmetric dihydroxylation of alkenes. Angew. Chem. Int. Ed. Engl. 54, 12952–12956 10.1002/anie.201506527 [DOI] [PubMed] [Google Scholar]
- 25. Van der Geize R., Yam K., Heuser T., Wilbrink M. H., Hara H., Anderton M. C., Sim E., Dijkhuizen L., Davies J. E., Mohn W. W., and Eltis L. D. (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 1947–1952 10.1073/pnas.0605728104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J., Simurdiak M., and Zhao H. (2005) Reconstitution and characterization of aminopyrrolnitrin oxygenase, a Rieske N-oxygenase that catalyzes unusual arylamine oxidation. J. Biol. Chem. 280, 36719–36727 10.1074/jbc.M505334200 [DOI] [PubMed] [Google Scholar]
- 27. Daughtry K. D., Xiao Y., Stoner-Ma D., Cho E., Orville A. M., Liu P., and Allen K. N. (2012) Quaternary ammonium oxidative demethylation: X-ray crystallographic, resonance Raman, and UV-visible spectroscopic analysis of a Rieske-type demethylase. J. Am. Chem. Soc. 134, 2823–2834 10.1021/ja2111898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Summers R. M., Louie T. M., Yu C. L., and Subramanian M. (2011) Characterization of a broad-specificity non-haem iron N-demethylase from Pseudomonas putida CBB5 capable of utilizing several purine alkaloids as sole carbon and nitrogen source. Microbiology 157, 583–592 10.1099/mic.0.043612-0 [DOI] [PubMed] [Google Scholar]
- 29. Parales R. E., Parales J. V., and Gibson D. T. (1999) Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity. J. Bacteriol. 181, 1831–1837 10.1128/JB.181.6.1831-1837.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mason J. R., and Cammack R. (1992) The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu. Rev. Microbiol. 46, 277–305 10.1146/annurev.mi.46.100192.001425 [DOI] [PubMed] [Google Scholar]
- 31. Voss I., Goss T., Murozuka E., Altmann B., McLean K. J., Rigby S. E., Munro A. W., Scheibe R., Hase T., and Hanke G. T. (2011) FdC1, a novel ferredoxin protein capable of alternative electron partitioning, increases in conditions of acceptor limitation at photosystem I. J. Biol. Chem. 286, 50–59 10.1074/jbc.M110.161562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beinert H. (1983) Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131, 373–378 10.1016/0003-2697(83)90186-0 [DOI] [PubMed] [Google Scholar]
- 33. Piiadov V., Ares de Araujo E., Oliveira Neto M., Craievich A. F., and Polikarpov I. (2019) SAXSMoW 2.0: online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein. Sci. 28, 454–463 10.1002/pro.3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kauppi B., Lee K., Carredano E., Parales R. E., Gibson D. T., Eklund H., and Ramaswamy S. (1998) Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6, 571–586 10.1016/S0969-2126(98)00059-8 [DOI] [PubMed] [Google Scholar]
- 35. Tarasev M., Pinto A., Kim D., Elliott S. J., and Ballou D. P. (2006) The bridging aspartate 178 in phthalate dioxygenase facilitates interactions between the Rieske center and the iron(II)–mononuclear center. Biochemistry 45, 10208–10216 10.1021/bi060219b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baratto M. C., Lipscomb D. A., Larkin M. J., Basosi R., Allen C. C. R., and Pogni R. (2019) Spectroscopic characterisation of the naphthalene dioxygenase from Rhodococcus sp. strain NCIMB12038. Int. J. Mol. Sci. 20, 3402 10.3390/ijms20143402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karlsson A., Parales J. V., Parales R. E., Gibson D. T., Eklund H., and Ramaswamy S. (2000) The reduction of the Rieske iron-sulfur cluster in naphthalene dioxygenase by X-rays. J. Inorg. Biochem. 78, 83–87 10.1016/S0162-0134(99)00213-5 [DOI] [PubMed] [Google Scholar]
- 39. Hegg E. L., and Que L. Jr. (1997) The 2-His-1-carboxylate facial triad–an emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 250, 625–629 10.1111/j.1432-1033.1997.t01-1-00625.x [DOI] [PubMed] [Google Scholar]
- 40. Mayhew S. G. (1978) The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 85, 535–547 10.1111/j.1432-1033.1978.tb12269.x [DOI] [PubMed] [Google Scholar]
- 41. Rostas A., Einholz C., Illarionov B., Heidinger L., Said T. A., Bauss A., Fischer M., Bacher A., Weber S., and Schleicher E. (2018) Long-lived hydrated FMN radicals: EPR characterization and implications for catalytic variability in flavoproteins. J. Am. Chem. Soc. 140, 16521–16527 10.1021/jacs.8b07544 [DOI] [PubMed] [Google Scholar]
- 42. Correll C. C., Batie C. J., Ballou D. P., and Ludwig M. L. (1992) Phthalate dioxygenase reductase: a modular structure for electron transfer from pyridine nucleotides to [2Fe-2S]. Science 258, 1604–1610 10.1126/science.1280857 [DOI] [PubMed] [Google Scholar]
- 43. Hudlicky T., Gonzalez D., and Gibson D. T. (1999) Enzymatic dihydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichimica Acta 33, 35–62 [Google Scholar]
- 44. Abu-Omar M. M., Loaiza A., and Hontzeas N. (2005) Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 105, 2227–2252 10.1021/cr040653o [DOI] [PubMed] [Google Scholar]
- 45. Capyk J. K., and Eltis L. D. (2012) Phylogenetic analysis reveals the surprising diversity of an oxygenase class. J. Biol. Inorg. Chem. 17, 425–436 10.1007/s00775-011-0865-9 [DOI] [PubMed] [Google Scholar]
- 46. Ertekin E., Konstantinidis K. T., and Tezel U. (2017) A Rieske-type oxygenase of Pseudomonas sp. BIOMIG1 converts benzalkonium chlorides to benzyldimethyl amine. Environ. Sci. Technol. 51, 175–181 10.1021/acs.est.6b03705 [DOI] [PubMed] [Google Scholar]
- 47. Shao Y. H., Guo L. Z., Zhang Y. Q., Yu H., Zhao B. S., Pang H. Q., and Lu W. D. (2018) Glycine betaine monooxygenase, an unusual Rieske-type oxygenase system, catalyzes the oxidative N-demethylation of glycine betaine in Chromohalobacter salexigens DSM 3043. Appl. Environ. Microbiol. 84, e00377–18 10.1128/AEM.00377-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lovenberg W., Buchanan B. B., and Rabinowitz J. C. (1963) Studies on the chemical nature of clostridial ferredoxin. J. Biol. Chem. 238, 3899–3913 [PubMed] [Google Scholar]
- 49. Birrell J. A., Laurich C., Reijerse E. J., Ogata H., and Lubitz W. (2016) Importance of hydrogen bonding in fine tuning the [2Fe-2S] cluster redox potential of HydC from Thermotoga maritima. Biochemistry 55, 4344–4355 10.1021/acs.biochem.6b00341 [DOI] [PubMed] [Google Scholar]
- 50. Marquis N. R., and Fritz I. B. (1964) Enzymological determination of free carnitine concentrations in rat tissues. J. Lipid Res. 5, 184–187 [PubMed] [Google Scholar]
- 51. Stockemer J. K. R. (1985) New methods for the determination of TVB-N (total volatile basic nitrogen) and TMA-N (trimethylamine nitrogen) in fish and fish products. Arch. Lebensmittelhyg. 36, 116–117 [Google Scholar]
- 52. Manthey M. (1988) Gaschrornatographische bestimmung von dimethyl-und trimethylamin in fisch und fischerzeugnissen. Informationen Fischwirtschaft 35, 131–135 [Google Scholar]
- 53. Hossain M. A., Nakano Y., and Asada K. (1984) Monodehydroascorbate reductase in spinach chloroplasts and its participation in regeneration of ascorbate for scavenging hydrogen peroxide. Plant Cell Physiol. 25, 385–395 10.1093/oxfordjournals.pcp.a076726 [DOI] [Google Scholar]
- 54. Pernot P., Round A., Barrett R., De Maria Antolinos A., Gobbo A., Gordon E., Huet J., Kieffer J., Lentini M., Mattenet M., Morawe C., Mueller-Dieckmann C., Ohlsson S., Schmid W., Surr J., et al. (2013) Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 10.1107/S0909049513010431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., and Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225 10.1107/S1600576717007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 57. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 58. Reijerse E., Lendzian F., Isaacson R., and Lubitz W. (2012) A tunable general purpose Q-band resonator for CW and pulse EPR/ENDOR experiments with large sample access and optical excitation. J. Magn. Reson. 214, 237–243 10.1016/j.jmr.2011.11.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the present manuscript.