

Abstract
Pancreas/duodenum homeobox protein 1 (PDX1) is an important transcription factor that regulates islet β-cell proliferation, differentiation, and function. Reduced expression of PDX1 is thought to contribute to β-cell loss and dysfunction in diabetes. Thus, promoting PDX1 expression can be an effective strategy to preserve β-cell mass and function. Previously, we established a PDX1 promoter-dependent luciferase system to screen agents that can promote PDX1 expression. Natural compound tectorigenin (TG) was identified as a promising candidate that could enhance the activity of the promoter for the PDX1 gene. In this study, we first demonstrated that TG could promote the expression of PDX1 in β-cells via activating extracellular signal-related kinase (ERK), as indicated by increased phosphorylation of ERK; this effect was observed under either normal or glucotoxic/lipotoxic conditions. We then found that TG could suppress induced apoptosis and improved the viability of β-cells under glucotoxicity and lipotoxicity by activation of ERK and reduction of reactive oxygen species and endoplasmic reticulum (ER) stress. These effects held true in vivo as well: prophylactic or therapeutic use of TG could obviously inhibit ER stress and decrease islet β-cell apoptosis in the pancreas of mice given a high-fat/high-sucrose diet (HFHSD), thus dramatically maintaining or restoring β-cell mass and islet size, respectively. Accordingly, both prophylactic and therapeutic use of TG improved HFHSD-impaired glucose metabolism in mice, as evidenced by ameliorating hyperglycemia and glucose intolerance. Taken together, TG, as an agent promoting PDX1 expression exhibits strong protective effects on islet β-cells both in vitro and in vivo.
Keywords: tectorigenin, PDX1, ER stress, β-cell protection, diabetes, drug discovery, pancreatic islet, type 2 diabetes, endoplasmic reticulum stress, lipotoxicity, apoptosis, glucotoxicity
Pancreatic islet β-cells, which account for about 80% of the total islet cells, are endocrine cells that secrete insulin and have a vital role in controlling the energy balance of the body (1). Loss or dysfunction of islet β-cells leads to the relative or absolute lack of insulin secretion, and in turn may generate hyperglycemia and diabetes (2, 3). Moreover, chronic hyperglycemia (glucotoxicity (4)), chronic dyslipidemia (lipotoxicity (5)), or a combination of these two conditions (glucolipotoxicity (6)) have been associated with the decreased number and impaired function of islet β-cells. Therefore, finding a way to protect islet β-cells could be crucial for treating metabolic disorders, such as type 2 diabetes mellitus (T2DM).
Pancreas/duodenum homeobox protein 1 (PDX1) is an important transcription factor that regulates islet β-cell proliferation, differentiation, and function (7, 8); PDX1 is highly conserved among different species (9). Complete deficiency of PDX1 has been associated with pancreatic agenesis, whereas partial deficiency leads to severe β-cell dysfunction, β-cell death, and diabetes (10). Under glucotoxicity and/or lipotoxicity, PDX1 expression in pancreatic islets is significantly reduced (11). Moreover, decreased expression of PDX1 has been closely related to islet β-cell apoptosis induced by oxidative stress and endoplasmic reticulum (ER) stress (12). Furthermore, studies have found that restoring PDX1 protein levels can normalize β-cell mass by modulating β-cell survival under the diabetic condition (13).
So far, several drugs, including DPP-4 inhibitors (14), GLP-1 analogs (15), and thiazolidinediones (16), have shown to be effective in protecting islet β-cells as well as in controlling blood glucose. However, novel, safe, and more efficient drugs aiming to protect and maintain islet β-cells are still in urgent need. To this end, in our previous study, we performed a small natural compound screening based on the PDX1 gene promoter-driven luciferase reporter screening system so as to identify new natural compounds that could promote PDX1 expression and protect islet β-cells (17). Tectorigenin (TG), a naturally occurring isoflavone compound isolated from the Pueraria thomsonii Benth (18), was identified as one of the promising candidate compounds that can significantly promote the activity of PDX1 gene promoter. TG has been reported to have hypoglycemic and hypolipidemic effects (19). TG could attenuate palmitate-induced endothelial insulin resistance (18) and ameliorate hyperglycemia by blocking preadipocyte differentiation and adipocytokines secretion (20). TG could also alleviate the progression of diabetic complications by improving vascular endothelium dysfunction (21) and inhibiting aldose reductase (22). Besides, TG has a protective effect on the liver (23), and exhibits anti-inflammation (24), antioxidation (25), anticancer (26), and other biological activities (27). Yet, the effects of TG on pancreatic β-cells have never been reported.
In this study, we further validated the effect of TG in promoting PDX1 expression and explored its protective effects on islet β-cells against glucotoxicity and lipotoxicity in vitro and in vivo. The molecular mechanisms underlying these beneficial effects of TG were also elucidated. This study provides a basis for the future development of TG as a novel drug targeting PDX1 to protect and repair pancreatic islet β-cells.
Results
TG increases PDX1 expression in the islet β-cells
TG, an O-methylated isoflavone, is a natural product (Fig. 1A). TG was selected by screening compounds that promote PDX1 expression using PDX1 promoter-dependent luciferase reporter gene vector (PGL3-PDX1-luc)3 (17). As shown in Fig. 1B, compared with the control group, TG could significantly increase the luciferase activity of PGL3-PDX1-luc–transfected cells (Fig. 1B), implying that TG may have potential to promote the PDX1 expression.
Figure 1.

TG increases PDX1 expression by activating ERK signaling and potentiates the insulin secretion of mice islets. A, the chemical structure of TG. B, luciferase activities of PGL3-PDX1-luc–transfected HEK293T cells treated with or without TG (40 µg/ml) for 24 h. Results are expressed as the fold-induction (over the activity of the negative control). C, RT-qPCR analysis of mRNA levels of PDX1 in INS-1 cells untreated or treated with 40 µg/ml of TG for 3 or 9 h. D, Western blotting analysis of PDX1 protein in INS-1 cells or islets untreated or treated with 40 µg/ml of TG for 24 h. E, Western blotting analysis of P-ERK levels in INS-1 cells or islets treated with 40 µg/ml of TG in the presence or absence of 10 μm U0126 for 10 min. F, luciferase activities of PGL3-PDX1-luc–transfected HEK293T cells treated with U0126, TG, or in combination for 24 h. Results are reported as a mean ± S.D. of fold-induction (the luciferase activity of TG, U0126, or combined treatment divided by the relative luciferase activity of control). G, Western blotting analysis of PDX1 levels in INS-1 cells or islets treated with TG, U0126, or a combination for 24 h. H–L, perifusion secretion assay of native mouse islets. Islets were perfused immediately after isolation with Ca5 buffer containing 3 mm glucose for 10 min, which were then changed to 3 mm glucose plus TG (H), 33 mm glucose Ca5 buffer or 33 mm glucose Ca5 buffer plus TG (I) for another 20 min. Otherwise, isolated islets were pretreated with TG (40 μg/ml) for 24 h before perifusion. Pretreated islets were perfused with 3 mm glucose for 10 min, which were then changed to 3 mm glucose plus TG (J), 33 mm glucose Ca5 buffer, or 33 mm glucose Ca5 buffer plus TG (K) for another 20 min. L, the area under the curve was calculated for the glucose-stimulated insulin secretion in H–L. Data are expressed as the mean ± S.D. of each experiment in triplicate. Image J was used for quantitative analysis. Error bars indicate S.D. (n = 3 biologically independent samples). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the control group.
To obtain direct evidence that TG can increase the expression of PDX1 in islet β-cells, we first tested the mRNA levels of PDX1 using RT-qPCR in TG-treated INS-1 cells. After treatment with TG for 3 and 9 h, mRNA transcription of PDX1 was increased in a time-dependent manner in INS-1 cells (Fig. 1C). We then examined the protein level of PDX1 by Western blotting. Consistently, TG effectively up-regulated the expression of PDX1 in INS-1 cells and RIN-m5F cells (Fig. 1D and Fig. S1, A and B). In addition, we further demonstrated that TG could also enhance PDX1 expression in native pancreatic islets isolated from mice (Fig. 1D). These results suggested that TG could promote the expression of PDX1 in pancreatic islet β-cells.
TG increases PDX1 expression requiring activated ERK signaling
ERK signaling has been reported to be involved in the regulation of PDX1 expression in islet β-cells (17, 28). Thus, in this study, we examined whether TG regulates the PDX1 expression also by activating ERK signaling. We first examined the levels of P-ERK by Western blotting and found that the P-ERK levels were markedly increased upon treatment with TG in INS-1 cells and native pancreatic islets isolated from mice (Fig. 1E). At the same time, the ERK inhibitor U0126 almost completely abolished the activation effect of TG on ERK (Fig. 1E).
Consequently, U0126 alone or in combination with TG was used to treat PGL3-PDX1 promoter- transfected HEK293T cells. We observed that U0126 alone could decrease the luciferase activity compared with control, and it could counteract TG-mediated increased luciferase activity as well (Fig. 1F). Consistently, U0126 treatment completely abolished TG-induced PDX1 expression at the protein level in islet β-cell lines or mouse islets (Fig. 1G and Fig. S1C). These results suggested that TG could increase PDX1 expression by activating ERK signaling.
TG improves insulin secretion of mouse islets
PDX1 is involved in the regulation of insulin transcription and pancreatic β-cell function (29, 30). To further investigate the effects of TG on β-cell function, we evaluated the influence of TG on the insulin secretion of β-cells through a perifusion secretion assay using isolated mouse islets. We observed that both immediate and 24-h treatment of 40 μg/ml of TG could enhance insulin secretion of islets (Fig. 1, H–L). Especially, TG-treated islets for 24 h showed a pronounced increase in the insulin peak after the 33 mm high glucose was loaded when compared with nontreated islets, which suggests that TG can markedly improve the insulin secretion under high-glucose conditions (Fig. 1, K and L). Therefore, these results indicated that TG could indeed potentiate β-cell function, which might be relevant to the increased expression of PDX1.
TG protects INS-1 cells from apoptosis induced by high-fat and high-glucose by activating the ERK pathway
Evidence has suggested that continuous exposure to high concentrations of free fatty acids and glucose may damage islet β-cells, activate apoptosis, and in turn, generate glucotoxicity and lipotoxicity (31–34). To test whether TG could have a protective role against glucotoxicity and lipotoxicity by increasing the expression of PDX1, we established glucotoxicity (35, 36) and lipotoxicity (32, 37) models in islet β-cell lines. We observed that 200 μm palmitic acid (PA) or 33 mm glucose (Glu) significantly decreased cell viability of INS-1 cells, which indicated that both lipotoxic and glucotoxic models were successfully established (Fig. 2, A and B). Contrary, we found that co-treatment with different concentrations of TG could significantly improve the cell viability of both lipotoxic (Fig. 2A) and glucotoxic INS-1 cells (Fig. 2B) in a dose-dependent manner. Moreover, we also demonstrated that TG dose-dependently improved cell viability of RIN-m5F cells under lipotoxicity (Fig. S1D). These data showed that TG indeed had protective effects on islet β-cells.
Figure 2.

TG has a protective role in islet cells against lipotoxicity and glucotoxocity-induced apoptosis by activating ERK. A and B, cell viability of INS-1 cells. The INS-1 cells were treated with 200 μm PA for 24 h (A) or 33 mm glucose for 72 h (B) with or without TG. Cell viability was evaluated using the MTT assay. Cell viability is expressed as fold-change of the control. C, representative scatter plots of the flow cytometry analysis of the fraction of apoptotic INS-1 cells treated with 200 μm PA for 24 h or 33 mm glucose for 72 h, with or without combined treatment with 40 μg/ml of TG. D–G, INS-1 cells were co-treated with the indicated combination for 24 h (D and E) or 72 h (F and G). Cell viability of lipotoxic (D) or glucotoxic (F) INS-1 cells was tested by MTT, or total cellular proteins (E and G) were obtained and subjected to Western blotting to detect the levels of PDX1, phosphorylation of ERK, and CC3. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the control group.
Next, we examined whether TG treatment may improve the cell viability of β-cells under lipotoxicity and glucotoxicity by inhibiting apoptosis. As shown in Fig. 2C, cells treated with 200 μm PA or 33 mm glucose increased early apoptosis in those cells compared with control cells (32.1 or 32.6 versus 12.0%). At the same time, 40 μg/ml of TG co-treatment decreased the proportion of 200 μm PA and 33 mm glucose-induced early apoptotic cells to 10.7 and 18.6%, respectively (Fig. 2C).
To further explore the mechanisms of action, the ERK inhibitor U0126 was applied to investigate whether ERK signaling was involved in this process (Fig. 1G). As shown in Fig. 2, D and F, as expected, 40 μg/ml of TG increased the viability of INS-1 cells regardless of high-fat or high-glucose conditions, whereas U0126 completely abolished this protective effect. Western blotting analysis showed that TG could reverse the inhibition of 200 μm PA or 33 mm glucose on ERK signaling and PDX1 expression in INS-1 cells, which, however, was blocked by U0126 (Fig. 2, E and G). Moreover, we also examined the levels of caspase 3 (C3) and its activation form, cleaved caspase-3 (CC3), the critical executor, and marker of apoptosis (38, 39). Consistent with previous reports (33) and our previous results (Fig. 2C), the levels of CC3 were obviously elevated in INS-1 cells following high-fat or high-glucose treatments (Fig. 2, E and G). Contrary, 40 μg/ml of TG co-treatment dramatically reduced the levels of CC3 in both PA (Fig. 2E) and glucose (Fig. 2G) damaged β-cells, which was in line with flow cytometry results (Fig. 2C). Nevertheless, U0126 abrogated the reduction effects of TG on CC3 levels in both lipotoxic and glucotoxic INS-1 cells (Fig. 2, E and G). Taken together, all of these results provided direct evidence showing that TG could prevent the down-regulation of PDX1 expression and islet β-cells apoptosis induced by lipotoxicity and glucotoxicity by activating ERK signaling.
TG prevents lipotoxicity- and glucotoxicity-induced oxidative stress and ER stress in INS-1 cells
It has been suggested that oxidative stress and ER stress contribute to islet β-cell apoptosis (12, 40). Herein, we examined whether TG has an antioxidant or anti-ER stress activity in INS-1 cells. DCFH-DA fluorescent probe was used to examine the ROS generation in INS-1 cells. As shown in Fig. 3, A and B, the fluorescence intensity of ROS was significantly increased in the INS-1 cells treated with either 200 μm PA or 33 mm glucose, whereas 40 μg/ml of TG decreased the fluorescence intensity of ROS in PA- or glucose-treated INS-1 cells (all p < 0.01). The above data suggested that TG might inhibit induced ROS production and oxidative stress.
Figure 3.
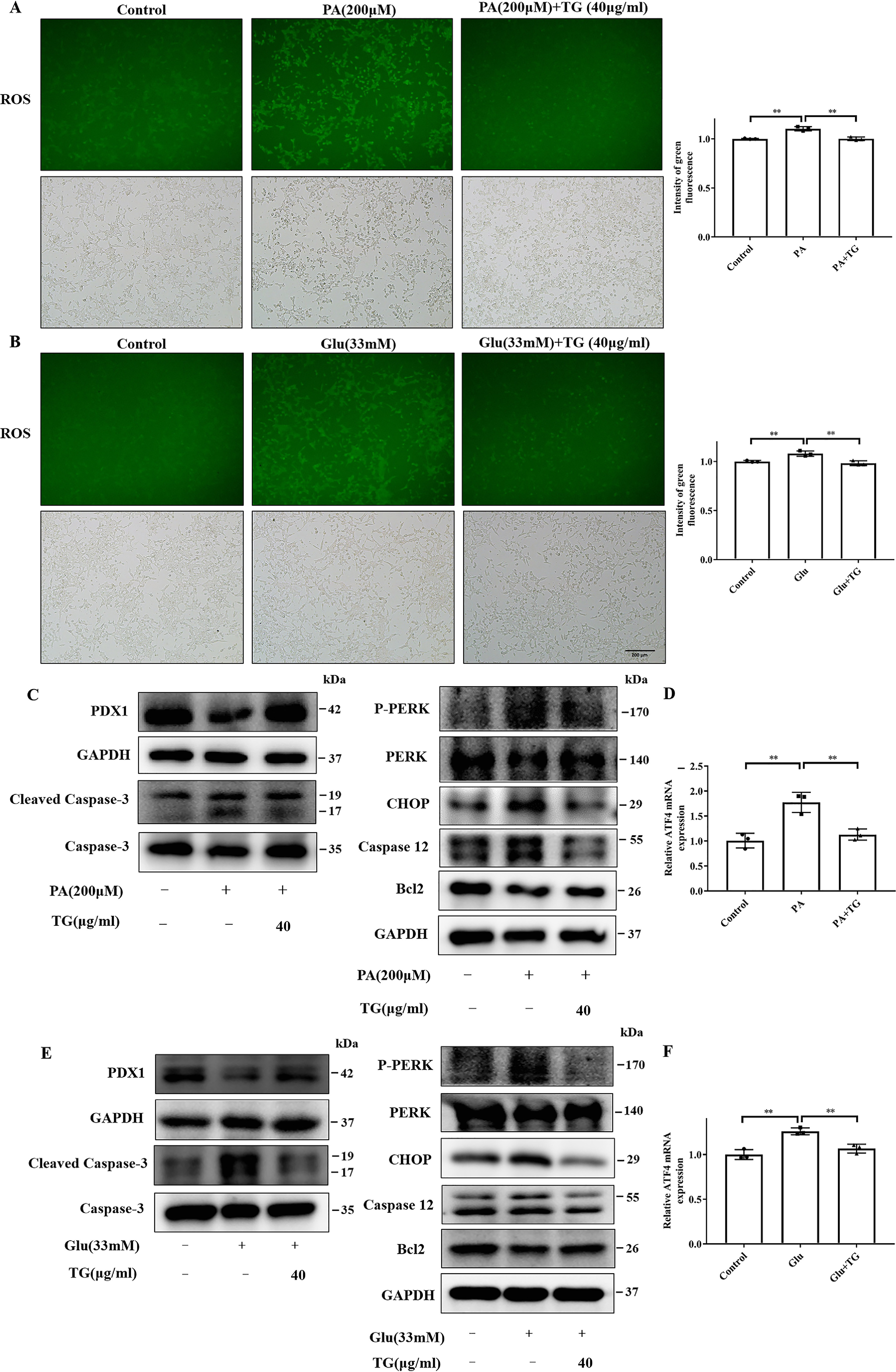
TG decreases ROS levels and prevents ER stress in INS-1 cells exposed to PA or glucose. A and B, effects of TG on the levels of ROS in INS-1 cells exposed to 200 μm PA for 24 h (A) or 33 mm glucose for 72 h (B). Intracellular ROS fluorescence images were analyzed using a fluorescence microscope; the intensity of green fluorescence was used to assess ROS production. C–F, effects of TG on the levels of ER stress markers in INS-1 cells exposed to 200 μm PA for 24 h (C and D) or 33 mm glucose for 72 h (E and F). P-PERK, CHOP, caspases-12, and anti-apoptotic signal Bcl2 were tested by Western blotting (C and E). The mRNA levels of ATF4 were evaluated with RT-qPCR (D and F). **, p < 0.01 versus the control group.
The proapoptotic transcription factor PERK, activating transcription factor 4 (ATF4), and C/EBP homologous protein (CHOP) mediate cell apoptosis induced by excess ER stress through activating caspases-12, which then activates caspases-3 (41, 42). It has been reported that suppressing CHOP expression could reduce apoptosis (12). We then analyzed these molecules associated with ER stress-related apoptosis in our experimental system to address the effect of TG on ER stress. As shown in Fig. 3, C and E, the expression levels of PDX1 and CC3 in INS-1 cells exhibited the same change upon 200 μm PA or 33 mm glucose treatment with or without TG as the previous results. Simultaneously, we also observed that treatment with PA (Fig. 3, C and D) or glucose (Fig. 3, E and F) significantly enhanced the protein levels of P-PERK, CHOP, and caspases-12 and mRNA levels of ATF4 in INS-1 cells. Contrary, TG co-treatment reversed the levels of ER stress molecules (Fig. 3, C–F). Moreover, compared with the control group, PA or the glucose group showed a decrease in the expression of Bcl-2, an antiapoptotic signal in INS-1 cells, which was also at least partially restored by TG (Fig. 3, C and E). Altogether, these data indicated that co-treatment with TG could markedly reduce the lipotoxicity- and glucotoxicity-induced oxidative stress and ER stress, which may contribute to its protective and anti-apoptotic effects on islet β-cells.
Prophylactic or therapeutic use of TG ameliorates hyperglycemia and glucose intolerance in the HFHSD-fed mice
Diet-induced obese C57BL/6J mice are commonly used as an animal model of T2DM and obesity with elevated blood glucose and impaired glucose tolerance (43). To validate the in vitro effects of TG on islet β-cells in vivo, we fed C57BL/6J mice with HFHSD to induce mouse islet β-cells dysfunction and diabetes; the model was established according to a previously reported method (44). TG was either prophylactically or therapeutically used in HFHSD-fed mice before or after the onset of diabetic phenotypes. During the prevention experiment, the control mice fed with HFHSD for 3 months gained weight and developed fasting hyperglycemia and fasting hyperinsulinism compared with normal diet (ND)-fed mice. Contrary, the prophylactic use of different doses of TG all significantly prevented weight gain and slowed down the increase of blood glucose and blood insulin levels of HFHSD-fed mice (Fig. 4, A–C). Besides, the level of homeostatic model assessment of β-cell (HOMA-β), an index for the secretory function of β-cells (45), was obviously decreased with 3-month-old HFHSD, which was also prevented in a dose-dependent manner by prophylactic use of TG (Fig. 4D), thus indicating that TG protects the function of β-cells from HFHSD impairment. To fully assess the pancreatic function, we performed intraperitoneal glucose tolerance test (IPGTT), which showed that prophylactic use of different doses of TG prevented glucose tolerance from decreasing in HFHSD-fed mice (Fig. 4E). These data suggested that TG had a certain preventive effect on the occurrence of diabetes.
Figure 4.
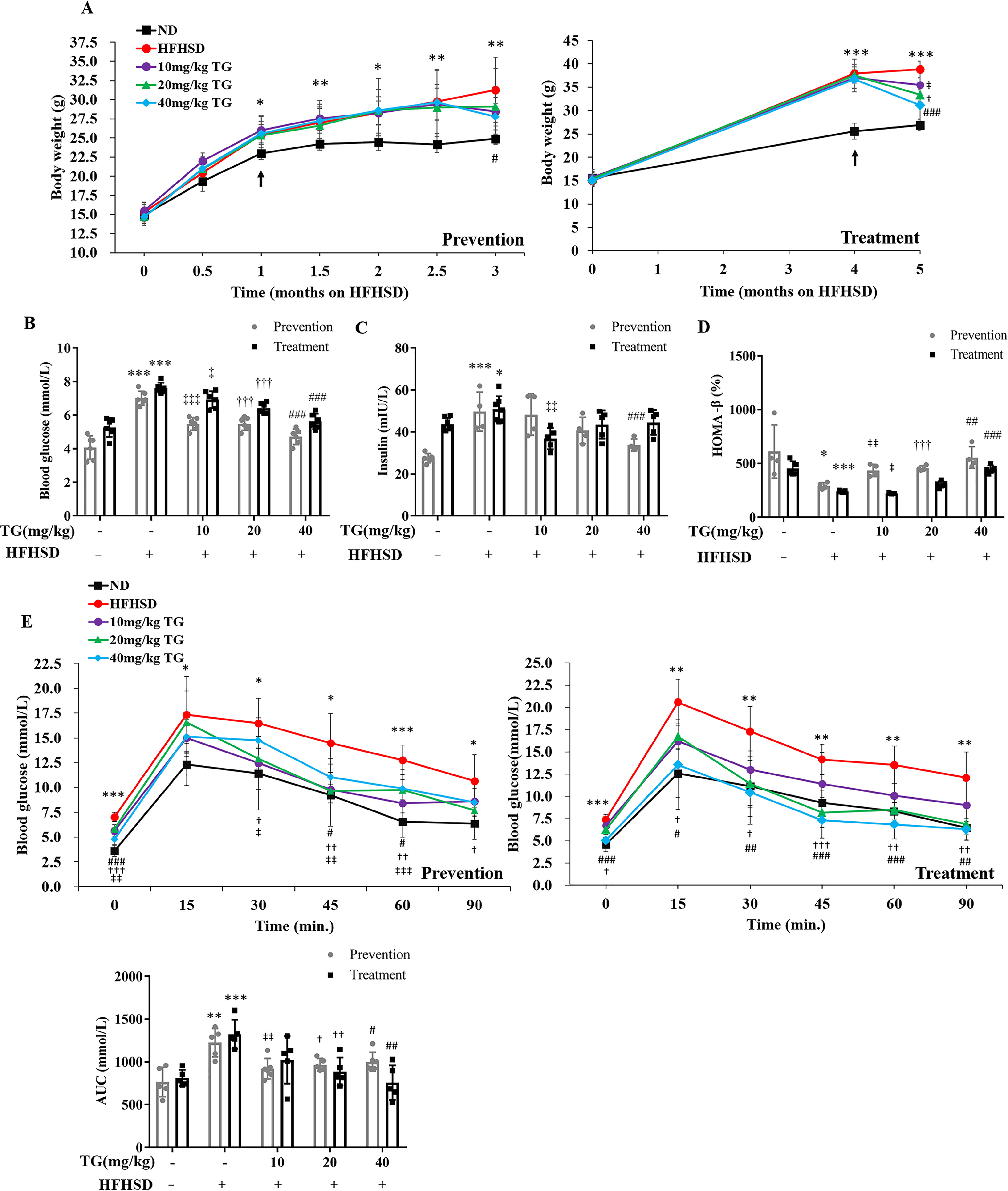
Prophylactic or therapeutic use of TG ameliorates hyperglycemia and glucose intolerance in the HFHSD-fed mice. A, body weight. B, fasting blood glucose levels. C, fasting blood insulin levels. D, HOMA-β of mice. E, IPGTT. IPGTT was evaluated after 8 h fasting. The area under the curve of IPGTT was analyzed. Values are expressed as mean ± S.D. *, p < 0.05 normal control versus the HFHSD group; ‡, p < 0.05, 10 mg/kg of TG versus the HFHSD group; †, p < 0.05, 20 mg/kg of TG versus the HFHSD group; #, p < 0.05, 40 mg/kg of TG versus the HFHSD group; n ≥ 6.
Furthermore, we analyzed the treatment effect of TG in HFHSD-induced diabetic mice. TG treatment for 1 month significantly reduced body weight, lowered fasting blood glucose levels (Fig. 4, A and B), and improved hyperinsulinemia (Fig. 4C) in HFHSD-induced diabetic mice. Besides, there was a significant increase in HOMA-β in 40 mg/kg of TG-treated mice compared with HFHSD control mice (Fig. 4D).
More importantly, different doses of TG treatment significantly enhanced glucose tolerance in diabetic mice compared with diabetic control mice (Fig. 4E). These results suggested that TG also had a therapeutic role in the development of diabetes.
Prophylactic use of TG prevents PDX1 expression decrease and protects islet β-cells from apoptosis by alleviating ER stress in the pancreas of HFHSD-fed mice
To elucidate whether the beneficial effects of TG on blood glucose homeostasis in HFHSD-fed mice were at least partly due to its protective effects on islet β-cells as revealed in vitro experiment, we further performed molecular and histological analyses of pancreas tissues on the HFHSD-fed mice described above. Under diabetic conditions, the expression level of PDX1 decreased, whereas its overexpression improved β-cell function (46). In this study, we observed that the expression of PDX1 in pancreatic tissues of HFHSD-fed mice (4-month HFHSD) was reduced compared with normal control (Fig. 5A). Albeit, prophylactic use of different doses of TG blocked the reduction and maintained PDX1 expression to different degrees where 40 mg/kg of TG had the strongest effect and reached statistical significance in quantity analysis (Fig. 5A).
Figure 5.
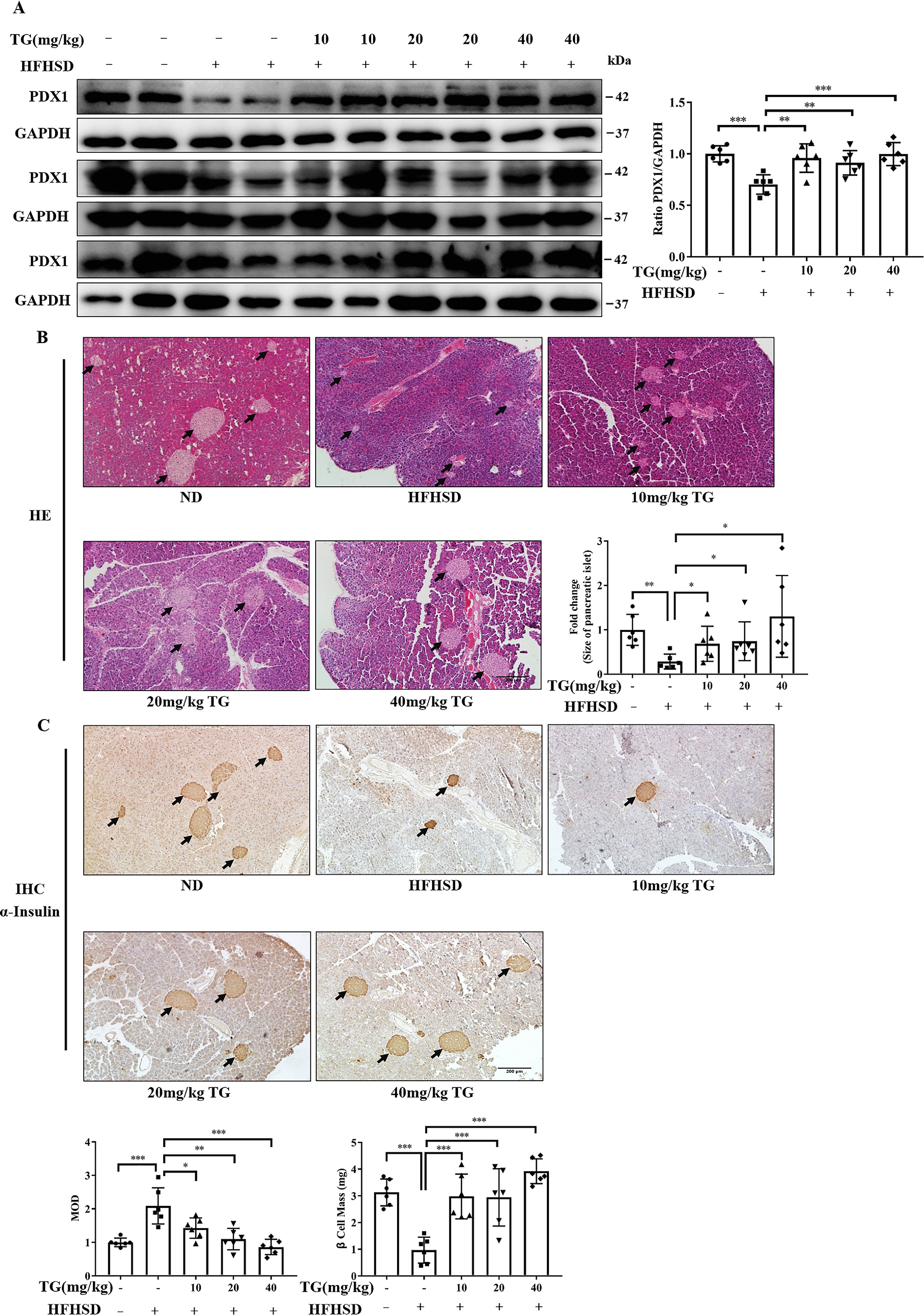
Prophylactic use of TG prevented PDX1 expression reduction and maintained islet and β-cell mass in the HFHSD mice. A, Western blotting analysis of PDX1 expression in pancreas tissue from mice. Image J software was used for quantitative analysis. B, representative images of H&E staining to show the islets histological changes (scale bar: 200 μm). IPP software was used for quantitative analysis of islet size. C, representative images of insulin IHC staining of pancreatic tissues (scale bar: 200 μm). MOD of insulin and β-cell mass are presented in the bar graph. IPP software was used for staining quantification. Values are expressed as mean ± S.D. *, p < 0.05 normal control versus the HFHSD group; ‡, p< 0.05, 10 mg/kg of TG versus the HFHSD group; †, p< 0.05, 20 mg/kg of TG versus the HFHSD group; #, p < 0.05, 40 mg/kg of TG versus the HFHSD group; n ≥ 6.
Next, we performed H&E staining on the pancreatic tissues. We found that the pancreatic islets of HFHSD-fed control mice were shrunken and smaller than those in normal mice, thus indicating that HFHSD conditions led to islet damage (Fig. 5B). In contrast, the preventive use of TG protected islets from HFHSD-induced damage. It prevented a decrease in islet size in a dose-dependent manner, where 40 mg/kg of TG maintained the islet size, which was the same as in normal islets (Fig. 5B).
We then performed insulin immunohistochemical (IHC) analysis. On the one hand, the insulin staining revealed the preventive effects of TG on HFHSD-induced islet impairment. On the other hand, these data showed that the number of insulin-positive β-cells was conspicuously lessened in pancreatic islets of HFHSD control mice, but relatively deeply stained (Fig. 5C), thus suggesting that a limited number of the islet β-cells had overloaded insulin secretion. However, this state was significantly prevented by prophylactic use of TG dose-dependently. As shown in Fig. 5C, the distribution area of the brown granules was larger, and the staining was lighter in different doses of the TG groups (Fig. 5C). Next, we quantified β-cell mass and found that the mass was more significant in the TG groups (Fig. 5C). Our results demonstrated that prophylactic use of TG could prevent pancreatic tissues from damage caused by long-term high-fat and high-sucrose diets.
Moreover, we analyzed the CC3 levels in the pancreas to evaluate apoptosis. Western blotting showed that the levels of CC3 were significantly elevated in pancreas tissues from HFHSD-fed control mice. At the same time, IHC analysis further exhibited deeply positive staining of CC3 in pancreatic islets, thus suggesting that apoptosis definitely occurred in islet β-cells of HFHSD-fed mice. Yet, prophylactic use of TG effectively blocked the HFHSD-induced occurrence of β-cell apoptosis in a dose-dependent manner as revealed by both Western blotting and IHC analysis (Fig. 6, A and B). To further delineate the involvement of ER stress in these processes, we analyzed CHOP expression in the pancreas of mice. As expected, a higher CHOP expression was observed in HFHSD-fed mice compared with ND-fed mice (Fig. 6C), whereas prophylactic use of distinct doses of TG blocked the up-regulation of CHOP expression in pancreas tissue (Fig. 6C). This result indicated that prophylactic use of TG could at least partly prevent islet β-cells from HFHSD-induced apoptosis by inhibiting ER stress, which was in line with in vitro data. Collectively, these data verified that TG might have preventive effects on the development of T2DM by protecting islet β-cells from glucotoxic and lipotoxic impairment.
Figure 6.
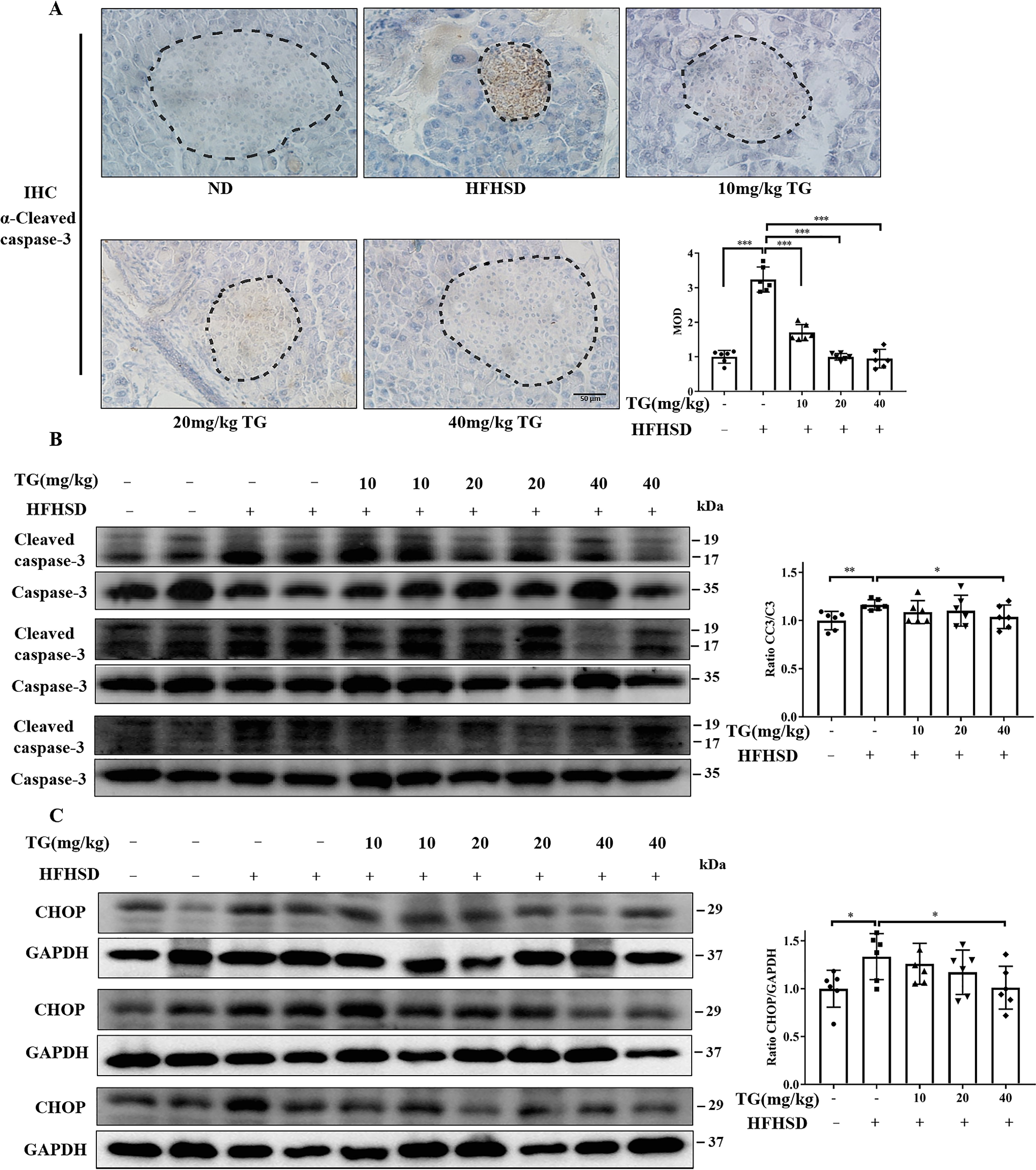
Prophylactic use of TG inhibits HFHSD-induced apoptosis by inhibiting ER stress in pancreatic tissues. A, representative images of CC3 immunohistochemical staining of pancreas tissues (scale bar: 50 μm). Islets were circled by dashed lines. MOD of CC3, which was quantified by IPP software is presented in the bar graph. B and C, the protein levels of CC3 (B) and CHOP (C) in pancreas tissues were analyzed by Western blotting. Image J software was used for quantitative analysis. Error bars represent ± S.D. *, p < 0.05 normal control versus the HFHSD group; ‡, p< 0.05, 10 mg/kg of TG versus the HFHSD group; †, p< 0.05, 20 mg/kg of TG versus the HFHSD group; #, p < 0.05, 40 mg/kg of TG versus the HFHSD group; n ≥ 6.
Therapeutic use of TG restores pancreatic PDX1 expression and islet mass and inhibits β-cell apoptosis as well as ER stress in HFHSD-induced diabetic mice
Next, we evaluated the therapeutic effect of TG on HFHSD damaged islet β-cells. Briefly, HFHSD-induced diabetic mice (5-month HFHSD) revealed a decrease in PDX1 expression, elevation of CC3 and CHOP levels, and shrunken and smaller islets with deep insulin staining in pancreas tissues; these were consistent with the prevention experiments mentioned above. Consequently, we demonstrated that the therapeutic use of TG, and especially a 40 mg/kg of dose, could obviously increase the PDX1 expression in the pancreas of diabetic mice (Fig. 7A). Concomitantly, H&E staining of pancreatic tissues showed that different doses of TG treatment improved the architecture of the pancreatic islets in diabetic mice, as revealed by relieved shrinking, enlarged islet size, and smooth border (Fig. 7B). Consistently, insulin IHC staining also showed that TG therapy improved islet structure and size, β-cell mass, and alleviated insulin expression burden on β-cells (Fig. 7C). These indicated that TG had therapeutic effects on damaged pancreatic islets in HFHSD diabetic mice.
Figure 7.
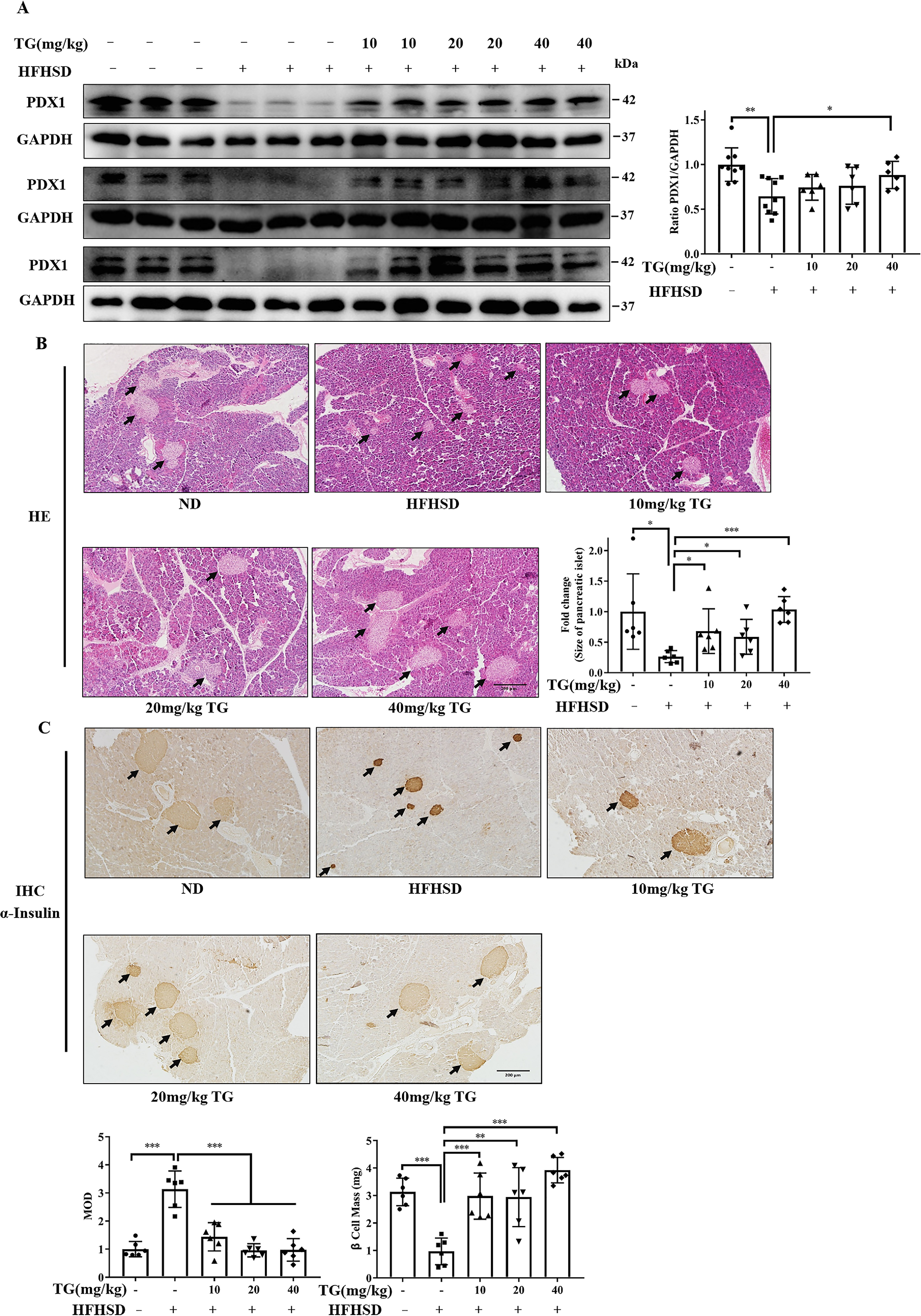
Therapeutic use of TG restores PDX1 expression and β-Cell mass in the HFHSD-induced diabetic mice. A, Western blotting analysis of PDX1 expression in pancreas tissues from HFHSD mice. Image J software was used for quantitative analysis. B, representative images of H&E staining to show the islet histological changes (scale bar: 200 μm). IPP software was used for quantitative analysis of islet size. C, representative images of insulin immunohistochemical staining of pancreatic tissues (scale bar: 200 μm). MOD of insulin and β-cell mass are presented in the bar graph. IPP software was used for staining quantification. Values are expressed as mean ± S.D. *, p < 0.05 normal control versus the HFHSD group; ‡, p < 0.05, 10 mg/kg TG versus the HFHSD group; †, p< 0.05, 20 mg/kg of TG versus the HFHSD group; #, p < 0.05 40 mg/kg of TG versus the HFHSD group; n ≥ 6.
As for the effects of TG on pancreatic apoptosis and ER stress in the treatment group, we observed that the levels of CC3 of HFHSD-induced diabetic mice was effectively reduced upon TG treatment, as indicated by both immunohistochemistry (Fig. 8A) and Western blotting analysis (Fig. 8B). Concomitantly, elevated CHOP expression in diabetic mice pancreas was reduced by TG treatment, and 40 mg/kg of TG treatment reached the statistical significance (Fig. 8C). Thus, these data suggested that the therapeutic use of TG could also attenuate apoptosis of islet β-cells induced through HFHSD by alleviating ER stress in vivo.
Figure 8.
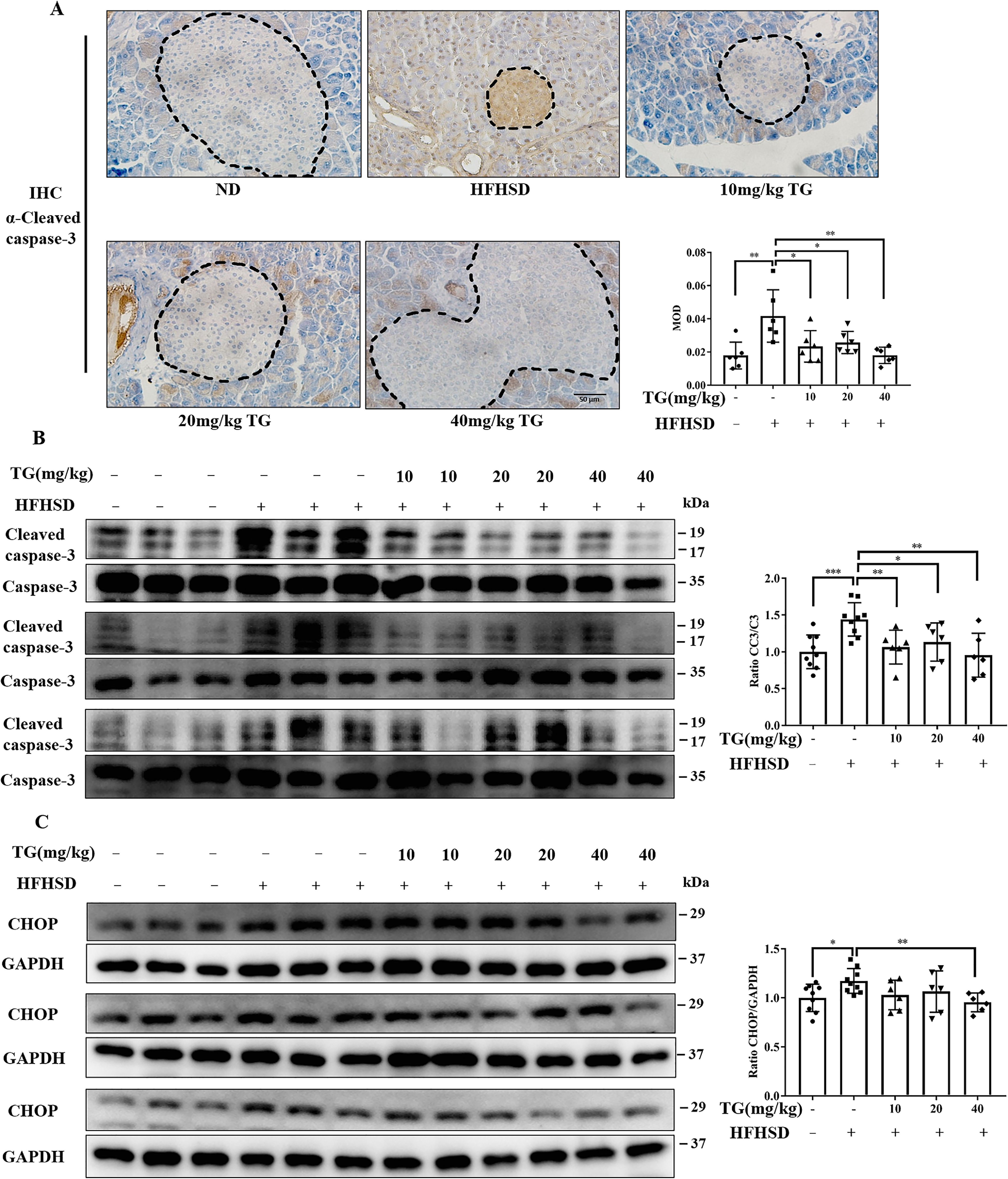
Therapeutic use of TG reduces apoptosis of islet β-cells by alleviating ER stress in HFHSD-induced diabetic mice. A, representative images of CC3 immunohistochemical staining of pancreas tissues (scale bar: 50 μm). Dashed lines and circled are islets. MOD of CC3, which was quantified by IPP software is presented in the bar graph. B and C, the protein levels of CC3 (B) and CHOP (C) were analyzed by Western blotting. Image J software was used for quantitative analysis. Error bars represent ±S.D. *, p < 0.05 normal control versus the HFHSD group; ‡, p < 0.05, 10 mg/kg of TG versus the HFHSD group; †, p < 0.05, 20 mg/kg of TG versus the HFHSD group; #, p < 0.05, 40 mg/kg of TG versus the HFHSD group; n ≥ 6.
Discussion
Currently, there are only limited drugs available that can protect islet β-cells and treat diabetes. PDX1 is an important anti-diabetes drug target (17, 47), which has a vital role in maintaining islet β-cell function and survival (48). Herein, we reported that TG, a natural compound, could promote the expression of PDX1 in islet β-cells, and effectively prevent and rescue β-cells from glucotoxicity and lipotoxicity in vitro and in vivo. These beneficial effects of TG on islet β-cells were related to its activation on the ERK pathway and alleviation of oxidative and ER stress. These findings could pave the way toward TG-targeted prevention and therapies of types 1 and 2 diabetes.
Glucotoxicity and lipotoxicity are among the main reasons leading to insufficient pancreatic β-cell mass and β-cell dysfunction in T2DM (33, 49). Besides, those conditions are associated with the down-regulation of PDX1 in β-cells (17, 50). Our analyses revealed that under both normal and toxic glycolipid conditions in vitro, TG might promote PDX1 expression and exert significant protective effects on islet β-cells against apoptosis by stimulating the ERK signaling. Although ERK is classically known as an important regulator of cell proliferation, differentiation, and survival (51, 52), its role in islet β-cell survival and function still remains controversial. Nonetheless, our previous studies (17, 47), as well as other reports (53, 54), suggest that activation of the ERK pathway has a prosurvival role at least for islet β-cells in resisting gluco- or lipotoxicity. As for the transcriptional regulation of the ERK pathway on PDX1, it is still unclear whether PDX1 is a direct downstream target gene of the ERK pathway or not.
Consistent with the in vitro results, we found that TG dramatically improved PDX1 expression and islet β-cell survival and well-maintained islet mass in HFHSD-fed mice. However, besides the protective effect we mentioned above, we could not exclude the possibility that TG may also trigger the mechanisms of islet β-cell regeneration and proliferation because therapeutic use of TG could restore the number of β-cells and the size of islets in diabetic mice. Thus, we plan to further investigate these mechanisms in our upcoming study. Our data indicate that TG could significantly prevent the onset or have a therapeutic effect on HFHS-induced diabetes. This result further consolidates the pharmacological activities of TG on protecting islet β-cells and implies its potential application as an anti-diabetic drug.
Current evidence suggests that oxidative stress and ER stress induced by glucotoxicity and lipotoxicity are associated with the pancreatic β-cell loss and impaired insulin secretion (55). It has been reported that TG has potent antioxidant activity in endothelial cells (18). Here, we found that TG ameliorated oxidative stress in islet β-cells under glucotoxicity and lipotoxicity conditions as well. Our data further confirm the antioxidant activity of TG, by which TG elicits protective effects on pancreatic β-cells to some extent. In addition, pancreatic β-cells, as professional secretory cells, are especially at risk of ER stress because β-cells are constantly under the demand from the body for insulin secretion (56). Manageable levels of ER stress could activate unfolded protein response (UPR), which would elicit adaptive changes to restore homeostasis and enable β-cells to maintain survival and functionality (57). However, UPR is “double-edged.” UPR also triggers degeneration and apoptosis in β-cells that experience continuous (chronic) or overwhelming ER stress, which is associated with the development and progression of diabetes (57, 58). There is increasing evidence to encourage the development of small molecules that facilitate protein folding and/or inhibit ER stress-induced cell death for therapeutic use in the treatment of diabetes. In our study, TG was demonstrated to reduce ER stress in both glucotoxic and lipotoxic islet β-cells in vitro or in the pancreas of HFHSD-fed mice in vivo. It was also reported that PDX1 expression was correlated with ER stress. For example, loss of PDX1 results in decreased expression of sarco/endoplasmic reticulum Ca(2+) pumps (SERCA2b), and in turn, leads to a reduction in ER Ca2+ concentration and subsequent induction of ER stress (59). Besides, previous studies have suggested that ERK activation is involved in apelin, caveolin-1 deficiency, or baicalein-mediated alleviation of diabetes-associated ER stress and protection of pancreatic β-cell against apoptosis (53, 54, 60). Based on these reports, we believe that TG partially elicits its protective effect on islet β-cells by preventing diabetes-related ER stress, which is also associated with ERK activation and subsequent improvement of PDX1 expression. According to our knowledge, this study first reported the effect of TG on ER stress. However, further studies are necessary to gain more insight into the molecular mechanisms underlying these effects. Moreover, chronic ER stress is associated with the initiation and progression of a variety of diseases (61), such as neurodegenerative diseases, metabolic syndromes, and cancer (61, 62). Whether TG restricts ER stress that occurs in other tissues or cells, and whether it has therapeutic potential for the treatment of other ER stress-related diseases need to be further investigated.
TG, also known as irisin, has been reported to possess various pharmacological functions. For example, its antioxidant activity (18), anti-inflammatory activity (20), and hypoglycemic and antilipemic effects have been demonstrated in streptozostin-induced diabetic rats (19). Yet, there are only a few studies on its working mechanisms. In this study, we elucidated the mechanism of TG in protecting and rescuing pancreatic islet β-cells, which may partially attribute to its hypoglycemic effects, whereas the direct molecular targets through which TG elicits its pharmacological effects still remain unclear. Future studies should focus on identifying TG targets. Taken together, our study delineates partial molecular mechanisms underlying TG pharmacological activities. It also implies that TG, as a β-cell protective agent, might be used as a promising preventive or therapeutic anti-diabetes drug in the future.
Experimental Procedures
Materials and reagents
TG (HPLC ≥ 98%) was purchased from Chengdu Biopurify Phytochemicals Ltd. (Chengdu, Sichuan, China). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and enhanced chemiluminescence (ECL) reagent were from Beyotime (Shanghai, China). U0126 monoethanolate (catalog number U120), d-(+)-glucose (catalog No. G7021), and PA (catalog number P5585) were acquired from Sigma-Aldrich. PDX1 promoter-dependent luciferase reporter plasmid (pGL3-PDX1-luc) was constructed in our laboratory. Anti-PDX1 (catalog number 20989-1-AP), anti-GAPDH (catalog number 60004-1-Ig), anti-PERK (catalog number 20582-1-AP), anti-CHOP (catalog number 15204-1-AP), and anti-insulin (catalog number 15848-1-AP) antibodies were from Proteintech (Wuhan, Hubei, China). Antibodies against ERK (catalog number 4695), phospho-(P-)ERK (catalog number 4370S), Caspase-3 (catalog number 9662), Cleaved Caspase-3 (catalog number 9661), P-PERK (catalog number 3179) and caspase 12 (catalog No. 2202) were from Cell Signaling Technology (Danvers, MA); anti-Bcl2 (catalog number sc-7382) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
Rat insulinoma cell lines, INS-1, and RIN-m5F cells were cultured in RPMI medium 1640 (R8758, Sigma-Aldrich) supplemented with 2 mm l-glutamine, 10 mm HEPES, 1 mm sodium pyruvate, 50 μm β-mercaptoethanol, and 10% (v/v) FBS (heat-inactivated FBS, GIBCO). HEK293T cells (human embryonic kidney) were maintained in Dulbecco's modified Eagle's medium (GIBCO) supplemented with 10% FBS. All of the cells were maintained under standard conditions (in a humidified atmosphere containing 5% CO2, 95% air at 37 °C) in the presence of 200 units/ml of gentamycin sulfate (Huangzhong Pharmaceutical Co., Ltd., Xiangyang, China).
Cell viability
The MTT assay assessed the INS-1 and RIN-m5F cells viability. Briefly, cells were seeded in 96-well–plates (5 × 104 cells/well) overnight. After incubating with TG in culture media containing 33 mm glucose for 72 h or 200 μm PA for 24 h, 20 μl of MTT (2.5 mg/ml) was added to each well, and cells were cultured for an additional 4 h (47). Subsequently, the medium was discarded, and blue formazan crystals were resolved with 100 μl of DMSO. Absorbance was measured at 490 nm using a microplate spectrophotometer (Thermo Electron Corporation, Shanghai, China). Cell viability was calculated relative to the control.
Luciferase assay
HEK293T cells were plated at a concentration of 1 × 105 cells/well in a 24-well–plate. After 24 h, cells were transfected with 1 µg of PDX1 promoter-dependent luciferase reporter plasmids (pGL3-PDX1-luc) or 1-1.5 µg of pGL3-basic vector plasmids per well using a Calcium Phosphate Cell Transfection kit (Beyotime, Shanghai, China) according to the manufacturer's instructions. β-Gal expression plasmid was co-transfected for normalizing transfection efficiency. The cells were incubated for 24 h and then treated with the 40 µg/ml of TG or 0.05% DMSO for 24 h. Subsequently, luciferase activity was measured using a FLUOstar OPTIMA system (BMG Labtech, Offenburg, Germany) as previously described (17).
Total RNA isolation and real-time quantitative PCR (RT-qPCR)
Total RNA was prepared from cultured cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using a real-time PCR kit (TransGen Biotech, Beijing, China). RT-qPCR was performed using 2× FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's protocol. Cycling conditions were 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 1 min, and 72 °C for 10 min. Primer sequences used for RT-qPCR are as follows: PDX1 (sense: 5′-CCGAATGGAACCGAGACTGG-3′ and antisense: 5′-GGGTCCTCTTATTCTCCTCC-3′), ATF4 (sense: 5′-ATGAGCCCTGAGTCCTACCT-3′ and antisense: 5′-GCTGTCTTGTTTTGCTCCAT-3′), β-actin (sense: 5′-CGTGCGTGACATTAAGGAGAAG-3′ and antisense: 5′-GGAAGGAAGGCTGGAAGAGTG-3′). Relative quantification of the expression of each gene was calculated using the △△CT method.
Western blotting analysis
Total proteins were extracted and Western blots were performed as previously described (63). Briefly, after different treatments, the total amount of proteins of cells or pancreatic tissues were extracted and electroblotted onto a polyvinylidene fluoride membrane following separation by 12% SDS-PAGE. The polyvinylidene fluoride membrane was incubated with 5% skim milk blocking solution for 1 h at room temperature, followed by incubation with a specific antibody overnight at 4 °C, and then with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, blots were visualized by ECL. The results were quantified using Image J.
Mouse pancreatic islets isolation
Pancreatic islets were isolated from male C57BL/6J mice as described previously (17). Briefly, C57BL/6 mice were sacrificed and the pancreas were removed from mice. Pancreas were shredded and digested with collagenase A (catalog number 11088793001, Roche Diagnostics, Mannheim, Germany) at 37 °C for 10 min. After rinsing three times with cold Ca5 buffer (125 mm NaCl, 5.9 mm KCl, 1.28 mm CaCl2, 1.2 mm MgCl2, 25 mm HEPES, 0.1% BSA), the intact islets were handpicked under a stereomicroscope (Szx7, Olympus Corp., Hamburg, Germany).
For perfusion secretion assays, some of the islets were immediately used for perifusion, and some of the islets were cultured in basic RPMI 1640 or in basic RPMI 1640 containing 40 μg/ml of TG in a sterile cell incubator at 37 °C for 24 h. For Western blotting analysis, matched islets were cultured in basic RPMI 1640 containing 40 μg/ml of TG for 10 min or in basic RPMI 1640 containing 40 μg/ml of TG for 24 h with or without 10 μm U0126.
Perifusion secretion assays
A total of 30 size-matched islets were placed in each column of the perifusion system. Then the column was gently closed with the top adaptor, immersed in a vertical position and controlled temperature in the incubator at 37 °C. The columns were perfused in parallel at a constant flow rate of 0.2 ml/min with Ca5 buffer containing 3 mm glucose at 37 °C for 30 min. They were further perifused with the same buffer for 10 min, and then the islets were stimulated with 40 μg/ml of TG, a high concentration (33 mm) of glucose or combination. Samples were collected every 1 min for 30 min, and the insulin concentration was measured using an insulin ELISA kit (Enzyme-linked Biotechnology, Shanghai, China) to delineate the curve of insulin secretion.
Flow cytometry analysis
The ratio of apoptotic cells was measured by annexin V-FITC Apoptosis Detection Kit (BD Biosciences, San Jose, CA) following the manufacturer's instructions. After exposure to 40 µg/ml of TG and 200 μm palmitic acid for 24 h or 33 mm glucose for 72 h, the cells were harvested, washed twice with PBS, stained according to the manufacturer's instructions, and then analyzed by flow cytometry. This method allowed us to discriminate viable cells (FITC negative, propidium iodide (PE) negative) from early apoptotic cells with intact cell membranes (FITC positive), late apoptotic and secondary necrotic cells (FITC positive, propidium iodide positive), and primary necrotic cells (FITC negative, propidium iodide positive) (64).
Intracellular ROS assay
The reactive oxygen species were determined with ROS-specific fluorescent dye DCFH-DA (number S0033; Beyotime, Shanghai, China). Briefly, INS-1 cells were treated with 40 µg/ml of TG and 200 μm of PA for 24 h or 33 mm glucose for 72 h. After medium removal, the INS-1 cells were incubated with serum-free medium containing 10 μm DCFH-DA for 20 min at 37 °C in an incubator with 5% CO2, after which INS-1 cells were washed with serum-free cell culture solution for three times. A fluorescent microscope was used to examine the intensity of fluorescence.
Animals and treatments
Male C57BL/6J mice (4 weeks old, 14-15 g) were purchased from Vital River Laboratory Animal Technology (Beijing, China). The mice were housed in a room at 25 ± 2 °C and 55 ± 5% relative humidity with controlled lighting (12:12 h light/dark cycle) and were allowed free access to food and water in the course of experiments. All animal studies (including the mouse euthanasia procedure) were approved by the Ethics Committee on the Care and Use of Laboratory Animals of Northeast Normal University and conducted according to the AAALAC and the IACUC guidelines.
After acclimatization, the mice were randomly divided into two groups (TG was injected at different times): prevention group and treatment group. The mice in each group were then randomly divided into five groups (n = 12/group): normal control group, fed with normal chow diet (ND)(4.5 kcal % fat diet, SPF growth feed, Beijing Keao Xieli Feed Co., Ltd.); diabetic control group, fed with 45% high-fat and 20% high-sucrose diet (HFHSD) (No. MD 45% fat; Mediscience Ltd., Yangzhou, China) (47, 65). The diets were replaced once every 2 days to prevent oxidization of the fat in diets; experimental groups, which were intraperitoneally injected with 10, 20, and 40 mg/kg of TG after 1 (prevention) or 4 months (treatment) of HFHSD. The TG solution was prepared in 0.9% NaCl (including 0.1% DMSO) and delivered by intraperitoneal injection once every 2 days. The control group was given a vehicle.
For the prevention research, mice were euthanized when the fasting blood glucose of the HFHSD diabetic control mice reached 7 mmol/liter, which occurred approximately 4 months after the administration of HFHSD (47). For the treatment research, mice were fed with HFHSD for 4 months until diabetic features, such as hyperglycemia, hyperinsulinemia, and impaired glucose tolerance, were observed. Also, the mice were euthanized 1 month after intraperitoneal injection of TG (47).
IPGTT
To perform the IPGTT, the mice were forced to fast for 8 h overnight and then intraperitoneally injected with glucose at a dose of 1 g of glucose/kg body weight. Blood glucose levels were measured before and 15, 30, 45, 60, and 90 min after glucose injection.
Biochemical analyses of serum
Blood glucose was measured by glucometer through tail-tip amputation. Serum insulin levels were measured by immunoassay using a mouse ELISA kit (Enzyme-linked Biotechnology, Shanghai, China) according to the manufacturer's protocols. HOMA-β (45) were calculated as HOMA-β = 20× fasting insulin (mIU/liter)/(fasting blood glucose (mmol/liter) − 3.5)(%).
Histopathology
Dissected mouse pancreases were fixed in Bouin's fixation and embedded in paraffin. The paraffin-embedded (5 μm) sections were stained with H&E and examined by light microscopy (47). IHC staining was performed using two steps. The sections were deparaffinized and rehydrated, and antigens were retrieved by autoclaving the slides in 10 mm citric acid buffer. The following antibodies were used: anti-insulin (catalog number 15848-1-AP, 1:200, Proteintech, Wuhan, China) and anti-cleaved caspase-3 (catalog number 9661, 1:100, Cell Signaling Technology, Danvers, MA). After washing the slides with PBS, they were treated with a Histostain-Streptavidin-Peroxidase kit (catalog number sp-0023, Bioss, Beijing, China), followed by visualization using DAB kit (catalog number C02-04001, Bioss, Beijing, China) according to the manufacturer's protocols. The area of each islet and the mean optical density (MOD) were analyzed using the Image-Pro Plus 5.0 software (Media Cybernetics, Rockville, MD). Consequently, two slides were selected from each animal, and five fields (×200 or ×400) in each slide were randomly selected for calculating the MOD. β-Cell mass (66) was obtained by the following formula: β-cell mass = (total β-cell area/total pancreatic area) × pancreas weight (mg).
Statistical analysis
All experiments were performed in triplicate, and the statistical significance of the experimental results was calculated by the Student's t test or tested by one-way and repeated measures analysis of variance (ANOVA). The statistical significance was set at p < 0.05, p < 0.01, p < 0.001. Error bars denote the mean ± S.D.
Data availability
All data are contained within the manuscript.
Supplementary Material
This article contains supporting information.
Author contributions—X. Y. and C. L. visualization; X. Y. writing-original draft; K. L., C. L., and Z. Z. methodology; K. L. project administration; C. L. software; L. S. and Y. L. conceptualization; L. S. supervision; L. S. and Y. L. writing-review and editing; Y. L. validation; X. Y. designed and performed the experiments, analyzed all data conducted and statistical analyses, and wrote the manuscript; K. L. performed a lot of cell and molecular biology experiments; C. L. contributed to the histological and morphometrical studies; Z. Z. contributed to cell culture and helped to complete supplementary experiments; J. W. performed the cell and molecular biology experiments; S. W. took care of mouse breedings and contributed to animal related experiments; L. L. contributed to the histological and morphometrical studies; C-L. Y. took care of mouse breedings and contributed to animal related experiments; Z-B. S. contributed to the analysis and interpretation of the data; Y-L. B. provided critical advice to the studies and valuable discussions; L-H. Z. took care of mouse breedings and contributed to animal related experiments; Y. S. contributed to cell culture; G. W. contributed to the histological and morphometrical studies; Y. H. contributed to the analysis and interpretation of the data; J. Y. performed the cell and molecular biology experiments; L. S. contributed to conception and design of the study, reviewed and approved the final version of all data; Y. L. contributed to conception and design of the study, reviewed and approved the final version of all data.
Funding and additional information—This work was supported by the National Natural Science Foundation of China Grants 81700709 and 81502284, the Fundamental Research Funds for the Central Universities Grant 111498001, Jilin Provincial Science & Technology Committee Grants 20170520031JH, 20170414028GH, 20180520105JH, and 20190304026YY), the Jilin Province Development and Reform Commission Grant 2018C051, Changchun Science and Technology Bureau Grant 17YJ003, and the National Engineering Laboratory for Druggable Gene and Protein Screening, Northeast Normal University Grant 130028901.
Conflict of interest—The authors declare no conflict of interest.
X. Yao et al., unpublished data.
- T2DM
- type 2 diabetes mellitus
- PDX1
- pancreas/duodenum homeobox protein 1
- ER
- endoplasmic reticulum
- ERK
- extracellular signal-regulated kinase
- TG
- tectorigenin
- PA
- palmitic acid
- ATF4
- activating transcription factor 4
- CHOP
- C/EBP homologous protein
- ND
- normal diet
- HOMA-β
- homeostatic model assessment of β-cell
- qPCR
- quantitative PCR
- ROS
- reactive oxygen species
- DCFH-DA
- 2′,7′-dichlorodihydrofluorescein diacetate
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IPGTT
- intraperitoneal glucose tolerance test
- UPR
- unfolded protein response
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- FBS
- fetal bovine serum
- HEK
- human embryonic kidney
- FITC
- fluorescein isothiocyanate
- IHC
- immunohistochemical
- MOD
- mean optical density
- CC3
- cleaved caspase-3.
References
- 1. Da Silva Xavier G. (2018) The cells of the islets of Langerhans. J. Clin. Med. 7, 54 10.3390/jcm7030054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brereton M. F., Rohm M., and Ashcroft F. M. (2016) β-Cell dysfunction in diabetes: a crisis of identity?. Diabetes Obes. Metab. 18, Suppl. 1, 102–109 10.1111/dom.12732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liston A., Todd J. A., and Lagou V. (2017) β-Cell fragility as a common underlying risk factor in Type 1 and Type 2 diabetes. Trends Mol. Med. 23, 181–194 10.1016/j.molmed.2016.12.005 [DOI] [PubMed] [Google Scholar]
- 4. Kaneto H. (2015) Pancreatic β-cell glucose toxicity in type 2 diabetes mellitus. Curr. Diabetes Rev. 11, 2–6 10.2174/1573399811666141216160217 [DOI] [PubMed] [Google Scholar]
- 5. Ertunc M. E., and Hotamisligil G. S. (2016) Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 57, 2099–2114 10.1194/jlr.R066514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansen J. B., Dos Santos L. R. B., Liu Y., Prentice K. J., Teudt F., Tonnesen M., Jonas J. C., Wheeler M. B., and Mandrup-Poulsen T. (2018) Glucolipotoxic conditions induce β-cell iron import, cytosolic ROS formation and apoptosis. J. Mol. Endocrinol. 61, 69–77 10.1530/JME-17-0262 [DOI] [PubMed] [Google Scholar]
- 7. Zhu Y., Liu Q., Zhou Z., and Ikeda Y. (2017) PDX1, Neurogenin-3, and MAFA: critical transcription regulators for β cell development and regeneration. Stem Cell Res. Ther. 8, 240 10.1186/s13287-017-0694-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao T., McKenna B., Li C., Reichert M., Nguyen J., Singh T., Yang C., Pannikar A., Doliba N., Zhang T., Stoffers D. A., Edlund H., Matschinsky F., Stein R., and Stanger B. Z. (2014) Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 19, 259–271 10.1016/j.cmet.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melloul D., Marshak S., and Cerasi E. (2002) Regulation of pdx-1 gene expression. Diabetes 51, Suppl. 3, S320–325 10.2337/diabetes.51.2007.s320 [DOI] [PubMed] [Google Scholar]
- 10. Fujimoto K., and Polonsky K. S. (2009) Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 11, Suppl. 4, 30–37 10.1111/j.1463-1326.2009.01121.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shimo N., Matsuoka T. A., Miyatsuka T., Takebe S., Tochino Y., Takahara M., Kaneto H., and Shimomura I. (2015) Short-term selective alleviation of glucotoxicity and lipotoxicity ameliorates the suppressed expression of key β-cell factors under diabetic conditions. Biochem. Biophys. Res. Commun. 467, 948–954 10.1016/j.bbrc.2015.10.038 [DOI] [PubMed] [Google Scholar]
- 12. Cnop M., Toivonen S., Igoillo-Esteve M., and Salpea P. (2017) Endoplasmic reticulum stress and eIF2α phosphorylation: the Achilles heel of pancreatic β cells. Mol. Metab. 6, 1024–1039 10.1016/j.molmet.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Claiborn K. C., Sachdeva M. M., Cannon C. E., Groff D. N., Singer J. D., and Stoffers D. A. (2010) Pcif1 modulates Pdx1 protein stability and pancreatic β cell function and survival in mice. J. Clin. Investig. 120, 3713–3721 10.1172/JCI40440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y., Xiao J., Tian H., Pei Y., Lu Y., Han X., Liu Y., Zhong W., Sun B., Fang F., and Shu H. (2013) The DPP-4 inhibitor MK0626 and exercise protect islet function in early pre-diabetic kkay mice. Peptides 49, 91–99 10.1016/j.peptides.2013.08.021 [DOI] [PubMed] [Google Scholar]
- 15. Jones B., Bloom S. R., Buenaventura T., Tomas A., and Rutter G. A. (2018) Control of insulin secretion by GLP-1. Peptides 100, 75–84 10.1016/j.peptides.2017.12.013 [DOI] [PubMed] [Google Scholar]
- 16. Kwon M. J., Lee Y. J., Jung H. S., Shin H. M., Kim T. N., Lee S. H., Rhee B. D., Kim M. K., and Park J. H. (2019) The direct effect of lobeglitazone, a new thiazolidinedione, on pancreatic β cells: a comparison with other thiazolidinediones. Diabetes Res. Clin. Pract. 151, 209–223 10.1016/j.diabres.2019.04.006 [DOI] [PubMed] [Google Scholar]
- 17. Liu L., Liang C., Mei P., Zhu H., Hou M., Yu C., Song Z., Bao Y., Huang Y., Yi J., Wang S., Wu Y., Zheng L., Sun Y., Wang G., et al. (2019) Dracorhodin perchlorate protects pancreatic β-cells against glucotoxicity- or lipotoxicity-induced dysfunction and apoptosis in vitro and in vivo. FEBS J. 286, 3718–3736 10.1111/febs.15020 [DOI] [PubMed] [Google Scholar]
- 18. Wang Q., Cheng X. L., Zhang D. Y., Gao X. J., Zhou L., Qin X. Y., Xie G. Y., Liu K., Qin Y., Liu B. L., and Qin M. J. (2013) Tectorigenin attenuates palmitate-induced endothelial insulin resistance via targeting ROS-associated inflammation and IRS-1 pathway. PLoS One 8, e66417 10.1371/journal.pone.0066417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee K. T., Sohn I. C., Kim D. H., Choi J. W., Kwon S. H., and Park H. J. (2000) Hypoglycemic and hypolipidemic effects of tectorigenin and kaikasaponin III in the streptozotocin-lnduced diabetic rat and their antioxidant activity in vitro. Arch. Pharmacal. Res. 23, 461–466 10.1007/BF02976573 [DOI] [PubMed] [Google Scholar]
- 20. Li Q. Y., Chen L., Yan M. M., Shi X. J., and Zhong M. K. (2015) Tectorigenin regulates adipogenic differentiation and adipocytokines secretion via PPARγ and IKK/NF-κB signaling. Pharm. Biol. 53, 1567–1575 10.3109/13880209.2014.993038 [DOI] [PubMed] [Google Scholar]
- 21. Yang S., Ma C., Wu H., Zhang H., Yuan F., Yang G., Yang Q., Jia L., Liang Z., and Kang L. (2020) Tectorigenin attenuates diabetic nephropathy by improving vascular endothelium dysfunction through activating AdipoR1/2 pathway. Pharmacol. Res. 153, 104678 10.1016/j.phrs.2020.104678 [DOI] [PubMed] [Google Scholar]
- 22. Moon H. I., Jung J. C., and Lee J. (2006) Aldose reductase inhibitory effect by tectorigenin derivatives from Viola hondoensis. Bioorg. Med. Chem. 14, 7592–7594 10.1016/j.bmc.2006.07.002 [DOI] [PubMed] [Google Scholar]
- 23. Zhang L., Zhao Y., Fan L., Xu K., Ji F., Xie Z., Ouyang X., Wu D., and Li L. (2019) Tectorigenin protects against experimental fulminant hepatic failure by regulating the TLR4/mitogen-activated protein kinase and TLR4/nuclear factor-κB pathways and autophagy. Phytother. Res. 33, 1055–1064 10.1002/ptr.6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ha Le M., Que do T. N., Huyen do T. T., Long P. Q., and Dat N. T. (2013) Toxicity, analgesic and anti-inflammatory activities of tectorigenin. Immunopharmacol. Immunotoxicol. 35, 336–340 10.3109/08923973.2013.770521 [DOI] [PubMed] [Google Scholar]
- 25. Park J. S., Jung J. S., Jeong Y. H., Hyun J. W., Le T. K., Kim D. H., Choi E. C., and Kim H. S. (2011) Antioxidant mechanism of isoflavone metabolites in hydrogen peroxide-stimulated rat primary astrocytes: critical role of hemeoxygenase-1 and NQO1 expression. J. Neurochem. 119, 909–919 10.1111/j.1471-4159.2011.07395.x [DOI] [PubMed] [Google Scholar]
- 26. Stettner M., Kaulfuss S., Burfeind P., Schweyer S., Strauss A., Ringert R. H., and Thelen P. (2007) The relevance of estrogen receptor-β expression to the antiproliferative effects observed with histone deacetylase inhibitors and phytoestrogens in prostate cancer treatment. Mol. Cancer Ther. 6, 2626–2633 10.1158/1535-7163.MCT-07-0197 [DOI] [PubMed] [Google Scholar]
- 27. Liu E. Y., Zheng Z. X., Zheng B. Z., Xia Y., Guo M. S., Dong T. T., and Tsim K. W. K. (2019) Tectorigenin, an isoflavone aglycone from the rhizome of Belamcanda chinensis, induces neuronal expression of erythropoietin via accumulation of hypoxia-inducible factor-1α. Phytother. Res. 34, 1329–1337 10.1002/ptr.6599 [DOI] [PubMed] [Google Scholar]
- 28. Zhang S. S., Hao E., Yu J., Liu W., Wang J., Levine F., and Feng G. S. (2009) Coordinated regulation by Shp2 tyrosine phosphatase of signaling events controlling insulin biosynthesis in pancreatic β-cells. Proc. Natl. Acad. Sci. U.S.A. 106, 7531–7536 10.1073/pnas.0811715106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brissova M., Shiota M., Nicholson W. E., Gannon M., Knobel S. M., Piston D. W., Wright C. V., and Powers A. C. (2002) Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 277, 11225–11232 10.1074/jbc.M111272200 [DOI] [PubMed] [Google Scholar]
- 30. Rutter G. A., Pullen T. J., Hodson D. J., and Martinez-Sanchez A. (2015) Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 466, 203–218 10.1042/BJ20141384 [DOI] [PubMed] [Google Scholar]
- 31. Ruan J. S., Lin J. K., Kuo Y. Y., Chen Y. W., and Chen P. C. (2018) Chronic palmitic acid-induced lipotoxicity correlates with defective trafficking of ATP sensitive potassium channels in pancreatic β cells. J. Nutr. Biochem. 59, 37–48 10.1016/j.jnutbio.2018.05.005 [DOI] [PubMed] [Google Scholar]
- 32. Bae G. D., Park E. Y., Baek D. J., Jun H. S., and Oh Y. S. (2018) Liquiritigenin prevents palmitate-induced β-cell apoptosis via estrogen receptor-mediated AKT activation. Biomed. Pharmacother. 101, 348–354 10.1016/j.biopha.2018.02.097 [DOI] [PubMed] [Google Scholar]
- 33. Lee S. J., Choi S. E., Jung I. R., Lee K. W., and Kang Y. (2013) Protective effect of nicotinamide on high glucose/palmitate-induced glucolipotoxicity to INS-1 β cells is attributed to its inhibitory activity to sirtuins. Arch. Biochem. Biophys. 535, 187–196 10.1016/j.abb.2013.03.011 [DOI] [PubMed] [Google Scholar]
- 34. Syeda K., Mohammed A. M., Arora D. K., and Kowluru A. (2013) Glucotoxic conditions induce endoplasmic reticulum stress to cause caspase 3 mediated lamin B degradation in pancreatic β-cells: protection by nifedipine. Biochem. Pharmacol. 86, 1338–1346 10.1016/j.bcp.2013.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kooptiwut S., Mahawong P., Hanchang W., Semprasert N., Kaewin S., Limjindaporn T., and Yenchitsomanus P. T. (2014) Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic β-cell death. J. Steroid Biochem. Mol. Biol. 139, 25–32 10.1016/j.jsbmb.2013.09.018 [DOI] [PubMed] [Google Scholar]
- 36. Guo L. X., Liu J. H., Zheng X. X., Yin Z. Y., Kosaraju J., and Tam K. Y. (2017) Geniposide improves insulin production and reduces apoptosis in high glucose-induced glucotoxic insulinoma cells. Eur. J. Pharmaceut. Sci. 110, 70–76 10.1016/j.ejps.2017.03.038 [DOI] [PubMed] [Google Scholar]
- 37. Cha S. H., Kim H. S., Hwang Y., Jeon Y. J., and Jun H. S. (2018) Polysiphonia japonica extract attenuates palmitate-induced toxicity and enhances insulin secretion in pancreatic β-cells. Oxid. Med. Cell. Longev. 2018, 4973851 10.1155/2018/4973851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vaux D. L., and Strasser A. (1996) The molecular biology of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 2239–2244 10.1073/pnas.93.6.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang C., Li L., Zhao B., Jiao A., Li X., Sun N., and Zhang J. (2016) Ghrelin protects against dexamethasone-induced INS-1 cell apoptosis via ERK and p38MAPK signaling. Int. J. Endocrinol. 2016, 4513051 10.1155/2016/4513051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y., Xue J., Li Y., Zhou X., Qiao S., and Han D. (2019) Telmisartan protects against high glucose/high lipid-induced apoptosis and insulin secretion by reducing the oxidative and ER stress. Cell Biochem. Funct. 37, 161–168 10.1002/cbf.3383 [DOI] [PubMed] [Google Scholar]
- 41. Fernandez A., Ordonez R., Reiter R. J., Gonzalez-Gallego J., and Mauriz J. L. (2015) Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J. Pineal Res. 59, 292–307 10.1111/jpi.12264 [DOI] [PubMed] [Google Scholar]
- 42. Cnop M., Welsh N., Jonas J. C., Jorns A., Lenzen S., and Eizirik D. L. (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54, Suppl. 2, S97–107 10.2337/diabetes.54.suppl_2.s97 [DOI] [PubMed] [Google Scholar]
- 43. Heydemann A. (2016) An overview of murine high fat diet as a model for type 2 diabetes mellitus. J. Diabetes Res. 2016, 2902351 10.1155/2016/2902351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cui L., Liu M., Chang X., and Sun K. (2016) The inhibiting effect of the Coptis chinensis polysaccharide on the type II diabetic mice. Biomed. Pharmacother. 81, 111–119 10.1016/j.biopha.2016.03.038 [DOI] [PubMed] [Google Scholar]
- 45. Wallace T. M., Levy J. C., and Matthews D. R. (2004) Use and abuse of HOMA modeling. Diabetes Care 27, 1487–1495 10.2337/diacare.27.6.1487 [DOI] [PubMed] [Google Scholar]
- 46. Yamamoto Y., Miyatsuka T., Sasaki S., Miyashita K., Kubo F., Shimo N., Takebe S., Watada H., Kaneto H., Matsuoka T. A., and Shimomura I. (2017) Preserving expression of Pdx1 improves β-cell failure in diabetic mice. Biochem. Biophys. Res. Commun. 483, 418–424 10.1016/j.bbrc.2016.12.128 [DOI] [PubMed] [Google Scholar]
- 47. Liang C., Hao F., Yao X., Qiu Y., Liu L., Wang S., Yu C., Song Z., Bao Y., Yi J., Huang Y., Wu Y., Zheng L., Sun Y., Wang G., et al. (2019) Hypericin maintians PDX1 expression via the Erk pathway and protects islet β-cells against glucotoxicity and lipotoxicity. Int. J. Biol. Sci. 15, 1472–1487 10.7150/ijbs.33817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Servitja J. M., and Ferrer J. (2004) Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia 47, 597–613 10.1007/s00125-004-1368-9 [DOI] [PubMed] [Google Scholar]
- 49. Robertson R. P., Harmon J., Tran P. O., Tanaka Y., and Takahashi H. (2003) Glucose toxicity in β-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 52, 581–587 10.2337/diabetes.52.3.581 [DOI] [PubMed] [Google Scholar]
- 50. Wang Y., Xie T., Zhang D., and Leung P. S. (2019) GPR120 protects lipotoxicity-induced pancreatic β-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin. Sci. 133, 101–116 10.1042/CS20180836 [DOI] [PubMed] [Google Scholar]
- 51. Tanimura S., and Takeda K. (2017) ERK signalling as a regulator of cell motility. J. Biochem. 162, 145–154 10.1093/jb/mvx048 [DOI] [PubMed] [Google Scholar]
- 52. Roux P. P., and Blenis J. (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68, 320–344 10.1128/MMBR.68.2.320-344.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kwak H. J., Yang D., Hwang Y., Jun H. S., and Cheon H. G. (2017) Baicalein protects rat insulinoma INS-1 cells from palmitate-induced lipotoxicity by inducing HO-1. PLoS One 12, e0176432 10.1371/journal.pone.0176432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zeng W., Tang J., Li H., Xu H., Lu H., Peng H., Lin C., Gao R., Lin S., Lin K., Liu K., Jiang Y., Weng J., and Zeng L. (2018) Caveolin-1 deficiency protects pancreatic β cells against palmitate-induced dysfunction and apoptosis. Cell. Signal. 47, 65–78 10.1016/j.cellsig.2018.03.013 [DOI] [PubMed] [Google Scholar]
- 55. Wang J., Yang X., and Zhang J. (2016) Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell. Signal. 28, 1099–1104 10.1016/j.cellsig.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 56. Back S. H., and Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 10.1146/annurev-biochem-072909-095555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ghosh R., Colon-Negron K., and Papa F. R. (2019) Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 27S, S60–S68 10.1016/j.molmet.2019.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scheuner D., and Kaufman R. J. (2008) The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 29, 317–333 10.1210/er.2007-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Johnson J. S., Kono T., Tong X., Yamamoto W. R., Zarain-Herzberg A., Merrins M. J., Satin L. S., Gilon P., and Evans-Molina C. (2014) Pancreatic and duodenal homeobox protein 1 (Pdx-1) maintains endoplasmic reticulum calcium levels through transcriptional regulation of sarco-endoplasmic reticulum calcium ATPase 2b (SERCA2b) in the islet β cell. J. Biol. Chem. 289, 32798–32810 10.1074/jbc.M114.575191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen H., Zheng C., Zhang X., Li J., Li J., Zheng L., and Huang K. (2011) Apelin alleviates diabetes-associated endoplasmic reticulum stress in the pancreas of Akita mice. Peptides 32, 1634–1639 10.1016/j.peptides.2011.06.025 [DOI] [PubMed] [Google Scholar]
- 61. Dandekar A., Mendez R., and Zhang K. (2015) Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 1292, 205–214 10.1007/978-1-4939-2522-3_15 [DOI] [PubMed] [Google Scholar]
- 62. Kaneko M., Imaizumi K., Saito A., Kanemoto S., Asada R., Matsuhisa K., and Ohtake Y. (2017) ER stress and disease: toward prevention and treatment. Biol. Pharm. Bull. 40, 1337–1343 10.1248/bpb.b17-00342 [DOI] [PubMed] [Google Scholar]
- 63. Zhang Y., Bao Y. L., Yang M. T., Wu Y., Yu C. L., Huang Y. X., Sun Y., Zheng L. H., and Li Y. X. (2010) Activin A induces SLC5A8 expression through the Smad3 signaling pathway in human colon cancer RKO cells. Int. J. Biochem. Cell Biol. 42, 1964–1972 10.1016/j.biocel.2010.08.007 [DOI] [PubMed] [Google Scholar]
- 64. Zhang W. J., Song Z. B., Bao Y. L., Li W. L., Yang X. G., Wang Q., Yu C. L., Sun L. G., Huang Y. X., and Li Y. X. (2016) Periplogenin induces necroptotic cell death through oxidative stress in HaCaT cells and ameliorates skin lesions in the TPA- and IMQ-induced psoriasis-like mouse models. Biochem. Pharmacol. 105, 66–79 10.1016/j.bcp.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 65. Wu J., Wang C., Li S., Li S., Wang W., Li J., Chi Y., Yang H., Kong X., Zhou Y., Dong C., Wang F., Xu G., Yang J., Gustafsson J. A., et al. (2013) Thyroid hormone-responsive SPOT 14 homolog promotes hepatic lipogenesis, and its expression is regulated by liver X receptor α through a sterol regulatory element-binding protein 1c-dependent mechanism in mice. Hepatology 58, 617–628 10.1002/hep.26272 [DOI] [PubMed] [Google Scholar]
- 66. Baena-Nieto G., Lomas-Romero I. M., Mateos R. M., Leal-Cosme N., Perez-Arana G., Aguilar-Diosdado M., Segundo C., and Lechuga-Sancho A. M. (2017) Ghrelin mitigates β-cell mass loss during insulitis in an animal model of autoimmune diabetes mellitus, the BioBreeding/Worcester rat. Diabetes Metab. Res. Rev. 33, e2813 10.1002/dmrr.2813 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.