

Abstract
Beta2-glycoprotein I (β2-GPI) is an abundant plasma glycoprotein with unknown physiological function and is currently recognized as the main target of anti-phospholipid antibodies responsible for complement activation and vascular thrombosis in patients with anti-phospholipid syndrome (APS). Here, we provide evidence that Mannose Binding Lectin (MBL) binds to β2-GPI in Ca++ and dose-dependent manner and that this interaction activates complement and promotes complement-dependent thrombin generation. Surprisingly, a significant binding was observed between MBL and isolated domains II and IV of β2-GPI while the carbohydrates chains, domain I and domain V were not involved in the interaction documenting a non-canonical binding mode between MBL and β2-GPI. Importantly, this interaction may occur on endothelial cells since binding of MBL to β2-GPI was detected on the surface of human umbilical vein endothelial cells and co-localization of MBL with β2-GPI was observed on the endothelium of a biopsy specimen of femoral artery from an APS patient. Since β2-GPI-mediated MBL-dependent thrombin generation was increased after priming the endothelium with TNF-α, our data suggests that this mechanism could play an important yet unrecognized role under physiological conditions and may be upregulated in pathological situations. Moreover, the complement activation and the pro-coagulant effects of β2-GPI/ MBL complex may contribute to amplify similar activities of anti-β2-GPI antibodies in APS and possibly act independently of antibodies raising the issue of developing appropriate therapies to avoid recurrences and disability in patients at risk for these clinical conditions.
Introduction
Beta2-glycoprotein I (β2-GPI) is a 50-kDa protein synthesized in the liver that circulates in the blood at approximately 200 μg/ml.(1) Although the function of this protein is largely unknown, the finding that individuals and mice with β2-GPI deficiency lead an apparently healthy life suggests that its role is not critical under physiologic conditions.(2),(3),(4) The interest in β2-GPI has greatly increased over the last two decades after the discovery that it serves as a preferential target for antibodies present in patients with anti-phospholipid syndrome (APS).(5) Animal models of APS have clearly shown that these antibodies mediate the onset of thrombosis and adverse pregnancy outcomes that represent the two main clinical manifestations of the syndrome.(6–8) In vitro studies have revealed that β2-GPI binds to several cell types including platelets, monocytes, endothelial cells and trophoblast interacting with various receptors differently expressed on the cell targets.(9),(10),(11),(12) However, analysis of the in vivo distribution of the purified protein have shown that β2-GPI localizes on decidual endothelial cells and extravillous trophoblast while its deposition on the endothelium of the other vascular districts required priming with LPS.(13) β2-GPI is structurally organized into five domains, four of which are composed of 60 aminoacids also found in complement (C) control proteins and the fifth domain characterized by an extra stretch of positively charged aminoacids that allows binding of the protein to anionic phospholipids.(14) Clinical and animal studies have provided convincing evidence that the pathogenic antibodies are directed against the DI domain that is exposed after the deposition of β2-GPI on the cell surface.(15),(16)
The C system plays a critical role in mediating anti-β2-GPI-induced clot formation and pregnancy loss as revealed by the failure of these antibodies to cause vascular thrombi and pregnancy loss in C deficient animals.(8) This conclusion is also supported by the finding that C activation products are deposited on the vascular endothelium at site of thrombus formation(17) and in the placentae collected from APS patients and animal models of APS.(8) Antibody-mediated C activation through the classical pathway in this pathological condition leads to the release of C5a and the assembly of the terminal C complex involved in the onset of pregnancy loss and thrombus formation.(6, 7)
In this work, we have investigated the ability of β2-GPI to trigger C activation independently of antibody and to promote biological effects associated with C activation products. Our working hypothesis was that the protein may bind Mannose Binding Lectin (MBL) due to the high carbohydrate content that accounts for approximately 20% w/w of the molecular mass(18),(19) and activates the lectin pathway. MBL has long been known to contribute to host defense, particularly in early childhood, targeting mannose and N-acetylglucosamine oligosaccharides expressed on infectious agents.(20) In addition, MBL-mediated activation of the lectin pathway has been implicated in the development of various pathological conditions including ischemia/reperfusion,(21) autoimmune diseases(22),(23) and IgA nephropaties.(24) More recently, activation of the lectin pathway has been shown to promote coagulation through MASPs-mediated activation of prothrombin and other coagulation factors.(25–28) Since antibody/β2-GPI interaction activates the C classical pathway, which in turn stimulates coagulation leading to thrombus formation, we hypothesized that MBL/β2-GPI complex can have a similar effect. Data will be presented showing that MBL binds to β2-GPI in Ca++ dependent manner and triggers the C lectin pathway and prothrombin cleavage to generate thrombin.
Material and Methods
Sera
Sera were obtained from APS patients containing antibodies either to DI or to DV domains of β2-GPI after obtaining informed consent and the IgG was purified by affinity chromatography using Protein G column (HiTrap Protein G HP, GE Healthcare, Milan, Italy).(16) Serum from a C1q deficient patient(29) was used to activate the C lectin pathway.
Purification of serum β2-GPI
Human β2-GPI was isolated from pooled human sera obtained from blood donors by perchloric acid treatment, and further purified by affinity chromatography on Heparin column (Hi-Trap Heparin HP; GE Healthcare) and ion-exchange chromatography (Resource-S; GE Healthcare). The purity of the preparation was checked by SDS-PAGE.(13, 30) DI, DII and DIV domains of β2-GPI were generated as previously reported.(31)
Generation of human recombinant MBL
Recombinant human MBL was produced in the Chinese ovary hamster cell line DG44 as described(32) and purified on mannan coated beads. This procedure results in pure highly oligomerized MBL assessed by SDS-PAGE followed by Coomassie Blue staining.
Generation of human recombinant β2-GPI
The cDNA of human recombinant β2GPI was cloned in a pDEST-40 vector. After sequence verification, the protein was expressed in HEK293 cells as described before.(33) Briefly, stable cell lines were generated by G418 selection (0.5–1.0 mg/ml). The secreted protein was purified under native conditions by immunoaffinity using an N-terminal engineered HPC-4 tag and further cleaned up by step-gradient elution from heparin-sepharose (GE Healthcare). The N-terminal tag was quantitatively removed by enterokinase before subjecting the protein to size exclusion chromatography (SEC), the final purification step before biochemical and structural studies. The purity of each preparation was >98%, as judged by SDS-PAGE and RP-HPLC. The chemical identify was verified by N-terminal sequencing and X-ray crystallography(33)
Preparation of deglycosylated β2-GPI
Enzymatic digestion of serum β2-GPI to remove glycosylations was obtained under native conditions using enzyme mix II from New England Biolabs (Ipswich, MA, USA), as previously described(33). Briefly, a solution of wild-type β2-GPI (0.6 mg/ml, 500 μl) was incubated with 12 μl of enzyme mix and 50 of buffer 1 for 30 min at RT and then transferred to 37C for 48 hours or until completion. The reaction was monitored by taking samples at 0, 1, 2, 3, 8, 24 and 48 hours and analyzed by SDS-PAGE. Lack of sugars was verified by Pierce™ Glycoprotein Staining Kit. The mixture was then purified by SEC to recover the deglycosylated protein that typically elutes ~1 mL after the glycosylated protein due to the smaller hydrodynamic radius.
Recombinant deglycosylated human β2-GPI was obtained blocking O- and N-glycosylations. Residues T130, N143, N164, N174 and N234 were mutated to serine (S) or glutamine (Q) by site-directed mutagenesis to generate the β2-GPI mutant T130S/N143Q/N164Q/N174Q/N234Q. After sequence verification, deglycosylated β2GPI was expressed in HEK293 cells and purified from the media following the protocol developed for the wild-type protein. The purity of each preparation was >98%, as judged by SDS-PAGE and RP-HPLC. Lack of sugars was verified by Pierce™ Glycoprotein Staining Kit (Suppl. Figure 1).
Binding of MBL to β2-GPI
The interaction of MBL with β2-GPI was measured by ELISA using γ-irradiated polystyrene plates (Thermo Fisher Scientific, Milan, Italy) coated overnight with serum purified or recombinant β2-GPI (10 μg/mL) in 100 mM Na2CO3/NaHCO3 pH 8.8. After washing with phosphate-buffered saline (PBS) and blocking the residual binding sites with PBS containing 2% BSA (Sigma-Aldrich, Milan, Italy), the immobilized β2-GPI was incubated with either 50% pooled normal human sera (NHS) as source of MBL or recombinant MBL (1μg/ml) in Veronal-buffered saline (VBS) containing 5mM barbital buffer (Sigma-Aldrich), 142 mM NaCl, 0.15 mM CaCl2, 0.5 mM MgCl2 and 0.05% gelatin to a final volume of 100 μl for 2 hr at 37°C. Bound MBL was revealed by a monoclonal antibody anti-human MBL (Bioporto, Copenhagen, Denmark) followed by alkaline phosphatase (AP)-conjugated secondary antibodies (Sigma-Aldrich). After addition of pNPP (1 mg/ml) (Sigma-Aldrich) as a substrate, the plates were read at 405 nm using a Titertek Multiskan ELISA reader (Flow Labs, Milano, Italy).
Binding of MBL to cell surface β2-GPI was also investigated using human umbilical vein endothelial cells (HUVECs) grown to confluence in 96-well tissue culture plates (GE Healthcare) in Human Endothelial serum-free medium (GE Healthcare) supplemented with 10% fetal calf serum (GE Healthcare). After overnight culture in serum-free medium (GE Healthcare), the cells were exposed either to human β2-GPI (10 μg/mL) or to 10% NHS for 60 minutes at 37°C followed by MBL (1μg/ml) for 2 hours at 37°C. The bound antibodies were detected with AP-conjugated goat anti-human IgG (Sigma-Aldrich). In some experiments the cells were treated overnight with TNF-α (100 ng/ml, GE Healthcare) prior to addition of β2-GPI. The results are expressed as mean +/− SD of at least two different experiments performed in triplicate.
Immunofluorescence analysis
The biopsy specimen of popliteal artery obtained from an APS patient undergoing femoral-popliteal bypass previously reported(17) was examined by immunofluorescence for deposits of β2-GPI and MBL. The tissue was stained for β2-GPI with biotin-labeled recombinant antibody CH2-deleted MBB2(17) followed by FITC-labeled streptavidin (Sigma-Aldrich) and for MBL with primary rabbit IgG anti MBL (Sigma-Aldrich) revealed by sheep anti-rabbit CY3-labeled F(Ab’)2 (Sigma-Aldrich). The sections were examined under a Leica DM2000 fluorescence microscope (Leica) equipped with a digital camera (DFC 490; Leica). Images were acquired by using Leica Application Suite Software. Original magnification 200x
Evaluation of C activation
C activation by MBL bound to HUVEC-adsorbed β2-GPI prepared as detailed above was evaluated by incubating the cells carrying the MBL/β2-GPI complex with 1/50 human C1q deficient serum for 60 min at 37°C. C4 deposition was detected by sheep anti-C4 IgG (The Binding Site, UK) followed by incubation with AP-conjugated secondary antibodies (Sigma-Aldrich).
Prothrombin activation assay
Cleavage of prothrombin by C activated through the lectin pathway was assessed by coating the plates with 200 μg/ml mannan (Sigma-Aldrich) in 0.1 M bicarbonate buffer pH 8.8 overnight at +4°C. The free binding sites were blocked with PBS containing 2% BSA and the bound mannan was then incubated with 1/50 C1q-deficient serum in VBS for 2 hr at 37°C. After washing with VBS, a mixture of 694 nM prothrombin (Haematologic Technologies, VT, USA) and the substrate S-2238 (Chromogenix) in 0,5 mM Tris buffered saline pH 8,5 containing 20 mM CaCl2 was added and incubated at 37°C. The reaction was read at 405nm. A similar approach was used to evaluate thrombin generation by C activated through the lectin pathway by the MBL/β2-GPI complex assembled on HUVEC and incubated with 1/10 C1q-deficient serum, with or without MBL (1μg/ml). Thrombin formation was evaluated by adding prothrombin and the S-2238 substrate in Human Endothelial serum-free medium (GE Healthcare) at pH 7.4.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 for Windows. The data were expressed as mean +/− SD and analyzed with the paired Student’s t test. Probabilities of <0.05 were considered statistically significant.
Results
MBL binds to β2-GPI
Hydrophobic plates currently employed to analyze the sera of APS patients for the presence of antibodies to β2-GPI were used to investigate MBL/β2-GPI interaction. Binding of MBL to β2-GPI was dose and calcium-dependent and was already detected at MBL concentration of 125ng/ml in VBS containing 0.15 mM CaCl2 increasing progressively with the addition of higher amount of the protein (Figure 1A). Interaction of MBL with β2-GPI was negligible in calcium-free buffer and in VBS containing 0.5 mM MgCl2, suggesting that Ca++ ions are required and cannot be replaced by Mg2+ ions. Further addition of CaCl2 up to 5 mM in the assay system resulted in a slight increase in MBL binding (Figure 1B). To assess if MBL also recognizes soluble β2-GPI, the interaction of MBL with solid-phase bound β2-GPI was evaluated in the presence of serum purified β2-GPI (10 μg/mL). The results presented in Fig 1C clearly show that soluble β2-GPI failed to exert an inhibitory effect at ten-fold higher concentration than MBL.
Figure 1. Binding of MBL to immobilized β2-GPI.
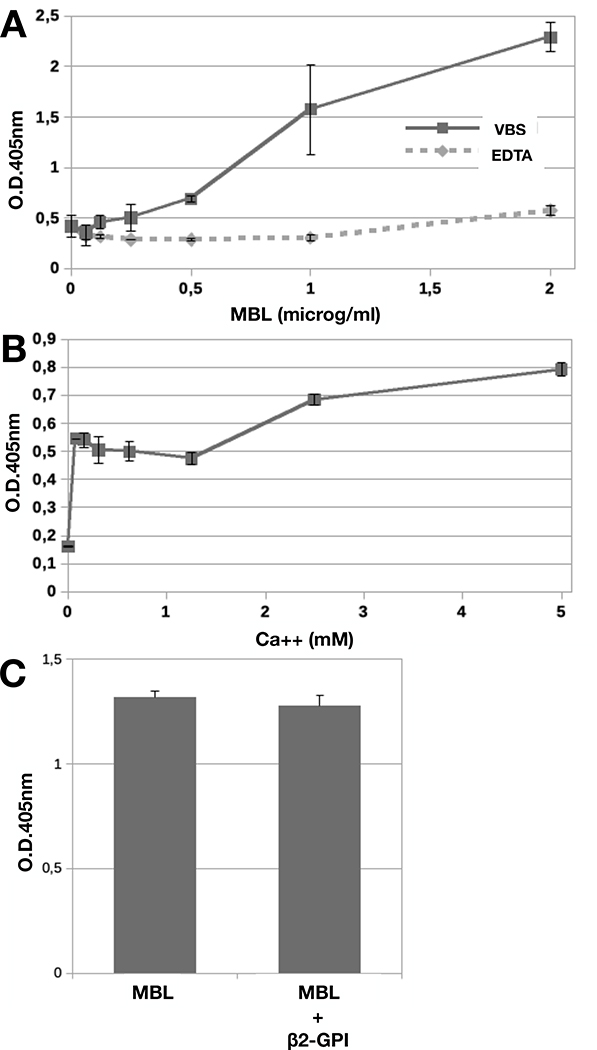
A) β2-GPI (10 μg/mL) immobilized on ELISA plate was incubated with different concentration of MBL for 2 hours at 37°C. B) β2-GPI (10 μg/mL) immobilized on ELISA plate was incubated with MBL (1μg/ml) in VBS containing 0.5 mM Mg++ and increasing Ca++ concentrations for 2 hours at 37°C. C) β2-GPI (10 μg/mL) immobilized on ELISA plate was incubated with MBL (1μg/ml) in the presence or in the absence of soluble β2-GPI (10 μg/mL) for 2 hours at 37°C. Bound MBL was revealed using anti-MBL mAb and alkaline phosphatase-labeled anti-mouse secondary Ab. The results are expressed as mean +/− SD and derived from at least 3 independent experiments (3 or 4 replicates per groups).
Analysis of sugar recognition by MBL
The finding that calcium was required for MBL/β2-GPI interaction led us to investigate whether the sugars, that constitute approximately 20% of beta2-GPI, represent a preferential target of MBL. To this end, the binding of MBL (1μg/mL) was evaluated in the presence of N-acetylglucosamine and D-mannose. Surprisingly, neither of the two sugars exhibited an inhibitory effect at the concentration of 100 mM (Figure 2A). The role of sugars as ligands of MBL was further investigated coating the wells of the microtiter plates with two sugar-free preparations of β2-GPI (10 μg/mL) obtained either by enzymatic digestion of the oligosaccharide chains of the glycoprotein purified from serum or by site-directed mutagenesis of recombinant β2-GPI to generate a mutant protein lacking the polysaccharides. Both protein variants were recognized by MBL equally well as serum-derived and glycosylated recombinant β2-GPI (Figure 2B).
Figure 2. Binding of MBL to β2-GPI is sugar independent.

A) β2-GPI (10 μg/mL) was immobilized on ELISA plate and incubated with MBL (1μg/ml) in the absence or in the presence of GlcNAc or Mannose (100 mM) for 2 hours at 37°C. B) Serum-derived β2-GPI, glycosylated recombinant β2-GPI, and recombinant β2-GPI deglycosylated either by enzymatic treatment or by site-directed mutagenesis (10 μg/mL) were immobilized on ELISA plate and incubated with MBL (1μg/ml) for 2 hours at 37°C. Bound MBL was revealed using anti-MBL mAb and alkaline phosphatase-labeled anti-mouse secondary Ab. The results are expressed as mean +/− SD and derived from at least 3 independent experiments (3 replicates per groups).
Examination of β2-GPI domains reacting with MBL
Two different approaches were employed to identify the domains recognized by MBL. First, bound-β2-GPI was incubated with saturating dose of IgG (100 μg/ml) reacting either with DI or DV domains prior to the addition of MBL. The serum IgG from APS patients were selected for their ability to activate C-mediated coagulation in the case of anti-DI Abs or, alternatively, to inhibit β2-GPI binding to endothelial cells for anti-DV Abs.(16) As shown in Figure 3A, none of these antibodies prevented MBL binding to β2-GPI suggesting that both DI and DV are unlikely to provide a binding site for MBL. To obtain more direct information on the domains of β2-GPI recognized by MBL, we tested the binding of MBL to immobilized β2-GPI domains. Only three domains comprising DI, DII and DIV were available for the study. The results presented in Figure 3B show a modest, but significant, binding of MBL to DII and DIV domains and a significantly higher value of MBL/β2-GPI interaction while the binding to DI was undetectable.
Figure 3. Interaction between β2-GPI domains and MBL.

A) β2-GPI (10 μg/mL) was immobilized on ELISA plate and incubated with MBL (1μg/ml) in the presence or in the absence of IgG (100 μg/ml) against domain I or domain V of β2-GPI for 2 hours at 37°C. B) β2-GPI, and the domains I, II or IV of β2-GPI (10 μg/mL) were immobilized on ELISA plate and incubated with MBL (1μg/ml) for 2 hours at 37°C. Bound MBL was revealed using anti-MBL mAb and alkaline phosphatase-labeled anti-mouse secondary Ab. The data are expressed as mean +/− SD and derived from at least 3 independent experiments. * = p<0,05; ** = p<0,001.
Endothelial cells as target of β2-GPI/MBL complex
To ascertain if the interaction between MBL and β2-GPI may also occur on the cell surface, we analyzed HUVEC as cell target of β2-GPI. The cells were kept overnight in serum-free medium to favor the release of cell-bound β2-GPI and then incubated with either NHS or the two purified proteins in succession. As expected, both β2-GPI and MBL were detected on HUVEC exposed to NHS containing the two proteins (Figure 4A). Purified β2-GPI was also found to bind to HUVEC and subsequent addition of MBL resulted in MBL deposition that was undetectable on cells incubated with this protein in the absence of β2-GPI (Figure 4B).
Figure 4. β2-GPI and MBL deposition on endothelial cells.
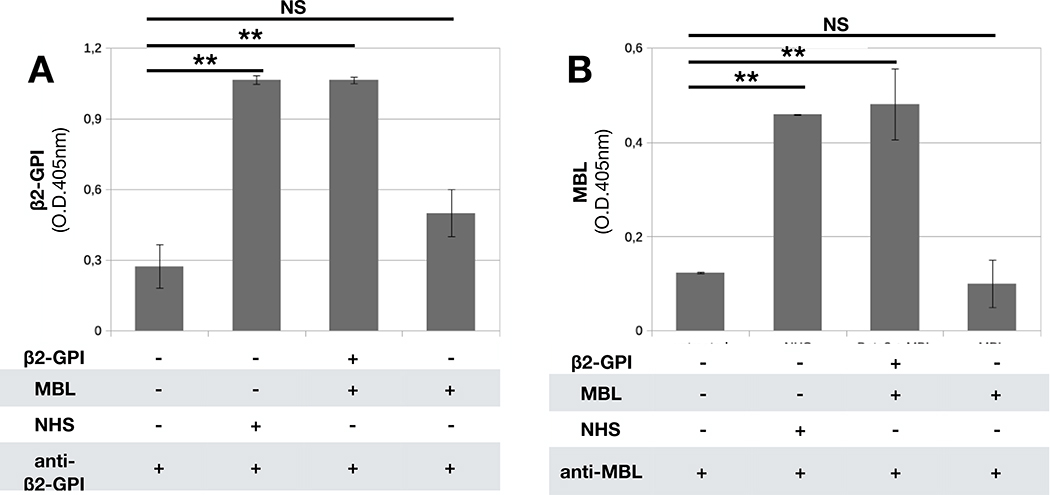
HUVEC were maintained in serum free medium and then incubated with purified β2-GPI (10 μg/mL) and MBL (1μg/mL), NHS (1/50) or only MBL as a control. A) Bound β2-GPI was revealed using MBB2 recombinant Ab and alkaline phosphatase-labeled anti-human secondary Ab. B) Bound MBL was revealed using anti-MBL mAb and alkaline phosphatase-labeled anti-mouse secondary Ab. The data are expressed as mean +/− SD and derived from at least 3 independent experiments (3 or 4 replicates per groups). * = p<0,05; ** = p<0,01; NS: Not Significant
Immunofluorescence analysis of the popliteal artery biopsy obtained from an APS patient revealed linear deposition of β2-GPI on the endothelial cells as opposed to the uneven distribution of MBL. Double staining of the arterial wall for the two proteins showed that MBL was deposited at sites of vascular endothelium where it co-localized with β2-GPI (Figure 5). No staining was observed using unrelated rabbit IgG antibodies as negative control.
Figure 5. β2-GPI and MBL deposition on the wall of occluded femoral artery.

Immunofluorescence analysis of the wall of the occluded femoral artery for the deposition of β2-GPI and MBL. The tissue was stained for β2-GPI using the recombinant antibody MBB2 followed by FITC-labeled anti-human IgG. MBL was stained using rabbit IgG anti MBL and sheep anti rabbit CY3-labeled F(Ab’)2. Two different sections are shown. Original magnification ×200.
Functional effect of MBL bound to β2-GPI
Since activation of the lectin C pathway is a well-known function of target-bound MBL, we measured C4 deposition on HUVEC bearing MBL bound to β2-GPI. To focus the analysis on the lectin pathway-dependent activation of C4, we used a C1q-deficient serum to prevent a potential activation of the classical pathway. The data presented in Figure 6 show that incubation of β2-GPI-coated cells with MBL-containing serum resulted in deposition of C4 that increased with further addition of MBL. C4 binding was undetected when the cells were exposed to C1q-deficient serum in the absence of β2-GPI and MBL. To mimic a pro-inflammatory scenario, the β2-GPI-mediated binding of MBL to HUVEC was further studied after TNF-α priming. Under these experimental conditions, C4 deposition on cell surface was found to be significantly higher than that of untreated cells (Figure 6) thus indicating that priming of the endothelium promotes binding of β2-GPI and stimulates MBL-dependent C activation.
Figure 6. C4 deposition on endothelial cells.

HUVECs were maintained in serum free medium in the presence or in the absence of TNF-α (100 ng/ml) and then incubated with purified β2-GPI (10 μg/mL) and MBL (1μg/mL). C1q deficient serum (1/50) was used as a source of C (see Materials and Methods for experimental details). C4 deposition was revealed using anti-C4 antibody and phosphatase-labeled secondary antibody. The results are expressed as mean +/− SD and derived from at least 3 independent experiments (3 replicates per groups). * = p<0,05; ** = p<0,01.
Given that C and clotting cascades crosstalk, we next sought to determine if C activated by β2-GPI/MBL complex may also have a procoagulant effect. To obtain this information, we used an experimental approach similar to that employed to investigate C4 activation, with the only difference that prothrombin and the chromogenic thrombin substrate were added after incubation with the serum. Thrombin generation was first tested in the classical assay of C activation through the lectin pathway incubating C1q-deficient serum in mannan-coated wells. Thrombin was produced under these conditions as assessed by the amount of cleaved chromogenic substrate (Suppl. Figure 2). C1q-deficient serum incubated with β2-GPI-coated HUVEC induced thrombin generation which was more evident after addition of MBL, suggesting that the concentration of this protein in C1q deficient serum was a limiting factor in the stimulation of substantial thrombin formation (Figure 7). In keeping with our results in Figure 6, priming of HUVEC with TNF-α led to enhanced thrombin generation, as indicated by the increase in the cleavage of the chromogenic substrate (Figure 7).
Figure 7. Prothrombin activation on endothelial cells.

HUVEC were maintained in serum free medium in the absence or in the presence of TNF-α (100 ng/ml) and then incubated with purified β2-GPI (10 μg/mL) and MBL (1μg/mL). C1q deficient serum (1/10) was used as a source of C. Prothrombin and the prothrombin substrate were then added and cleavage of the substrate was followed as detailed in Materials and Methods. Data were expressed as mean +/− SD and derived from at least 3 independent experiments (2 or 3 replicates per groups). * = p<0,05.
Discussion
Beta2-GPI/antibody complexes are known to activate the C system and to stimulate blood coagulation. In this work, we have identified MBL as a novel ligand for β2-GPI and report data showing that the MBL complexed with surface-bound β2-GPI can elicit effects analogous to those induced by the anti-β2-GPI /β2-GPI complex. Hence, the MBL/β2-GPI system may be an important yet unappreciated pathway that contributes to thrombus formation.
Ca++ was required for the binding of MBL to β2-GPI in line with similar results reported on the interaction of MBL with a variety of infectious and non-infectious targets.(20) We reckoned that the most likely explanation for this finding was that the carbohydrate chains of β2-GPI offer binding sites for the carbohydrate-recognition domain (CRD) of MBL. However, contrary to what was expected, both mannose and GlcNac, two major carbohydrate components of β2-GPI, failed to inhibit MBL binding. These results were further supported by the finding that enzymatically deglycosylated β2-GPI and a sugar-free recombinant molecule were recognized by MBL equally well as fully glycosylated β2-GPI. Taken together, these data suggest that the carbohydrates of β2-GPI are not involved in the interaction with MBL.
While the molecular mechanisms responsible for the interaction between MBL and β2-GPI could not be fully elucidated from these studies, it is tempting to speculate that Ca++ ions may act as a bridge between MBL and regions of β2-GPI that are rich in negatively charged amino acids. Consistent with this premise, MBL interacted with domains II and IV, which are negatively charged, yet failed to react with domain 1, which is positively charged.(33) Additional support to this hypothesis came from the experiments with anti-DI and anti-DV antibodies, which did not prevent MBL binding to β2-GPI. Unfortunately, recombinant domain III was not available and therefore its interaction with MBL could not be investigated. However, it is unlikely that this domain bears a recognition site for MBL because it is shielded by the polysaccharides that are mostly localized in this part of the molecule.(19),(33)
An interesting observation of this study is that soluble β2-GPI did not inhibit binding of MBL to immobilized β2-GPI suggesting that, similar to other ligands,(33) the preferential interaction of MBL with bound β2-GPI may arise from the ability of β2-GPI to form oligomeric structures upon binding to negatively charged phospholipids.
The type of molecular interaction between MBL and β2-GPI does not meet the classical criteria of Ca++ dependent and sugar binding of MBL to infectious and non-infectious targets(20), suggesting that the CRD is probably not involved in this process. Another possible candidate for an alternative recognition site for β2-GPI is the collagen domain of MBL. This portion of the molecule binds MASPs in a Ca++ dependent manner(34) and may interact with β2-GPI in a similar way, as it does with the bone morphogenetic protein-1 (BMP-1)/tolloid-like proteinases (BTPs).(35) BMP-1/ BTPs and β2-GPI share a similar behavior in their interaction with MBL, which recognizes both proteins only if immobilized on solid supports in the presence of calcium and not as soluble molecules. One major difference is that the formation of BMP-1/ BTPs -MBL complex is inhibited by MASP2, indicating that BMP-1/ BTPs and MASP2 compete for the same binding site on MBL. Conversely, the β2-GPI/MBL complex is able to activate C through the lectin pathway, suggesting that the collagen-like domain is still free to bind MASPs after the assembly of the complex. Larvie and colleagues(36) have identified a binding site for amyloid β peptides in the cysteine-rich domain at the N-terminus of MBL, but this site does not seem to be involved in the binding of β2-GPI since the interaction between the amyloid peptides and MBL, unlike that of β2-GPI and MBL, is calcium-independent. Overall, our data tend to exclude the binding of β2-GPI to the N-terminal cysteine-rich and collagenous domains of MBL and rather support the localization of the binding site at the C terminus of the molecule, though the exact site needs to be identified.
Binding of MBL was not restricted to β2-GPI immobilized on the wells of ELISA plates but was also extended to the protein deposited on the surface of HUVEC. These endothelial cells were selected for this study because in vitro experiments(37) and an in vivo model(13) have shown that they constitutively bind β2-GPI in the absence of priming factors such as LPS or pro-inflammatory cytokines required to favor the protein deposition on the endothelium of all the other vascular districts.(13) The in vivo relevance of these observations was indicated by the finding that MBL and β2-GPI were found to co-localize on the endothelium of a vessel occluded by a blood clot in an APS patient with recurrent thrombosis undergoing vascular anastomosis. The linear distribution of β2-GPI and the irregular deposition of MBL at sites of the endothelial surface, where it co-localized with β2-GPI, reflect the relative amounts of the two proteins circulating in blood with a large preponderance of β2-GPI. MBL bound to β2-GPI share with the β2GPI/anti-β2GPI complex formed in APS patients the ability to induce C activation and to trigger the coagulation cascade. These two processes are closely linked since the induction of coagulation by anti-β2GPI antibodies is dependent on C activated through the classical pathway(7, 8) and that induced by MBL via the lectin pathway as documented by C4 deposition in the presence of C1q deficient serum. Our finding that thrombin is generated by MBL bound to mannan and also to β2GPI is in line with previous reports showing that activation of the lectin C pathway leads to MASPs-mediated cleavage of prothrombin resulting in thrombin generation and clot formation.(25–28)
In conclusion, we have presented evidence that MBL binds to solid phase- and cell surface-bound β2-GPI in a calcium-dependent manner. We have also shown that the CRD of MBL is not involved in the recognition of β2-GPI and that the β2-GPI/MBL complex assembled on endothelial cells activates C and in turn induce prothrombin cleavage leading to the generation of thrombin. This system may be physiologically relevant and could be upregulated during inflammatory conditions thus contributing to thrombus formation.
Supplementary Material
Key points.
Mannose-Binding Lectin interacts with bound-Beta2-GPI
MBL/Beta2-GPI complex activates the lectin pathway of the complement system
Complement activated by the MBL/Beta2-GPI complex promotes thrombin generation
Acknowledgments
The authors thank Prof. Marina Botto of the Imperial college, London (UK) for the generous supply of human C1q deficient serum and Luca De Maso for his valuable technical contribution.
Grant support
This work was supported by grants 20C003_2010 Ricerca Corrente from Italian Ministry of Health (PL.M.), the National Institutes of Health Research Grant HL150146 (N.P.), the Danish Research Council for Independent Research DFF-6110-00489 (P.G.).
Footnotes
Conflict-of-interest: The authors declare no competing financial interests.
References
- 1.de Groot PG, and Meijers JCM 2011. β(2) -Glycoprotein I: evolution, structure and function. J. Thromb. Haemost 9: 1275–1284. [DOI] [PubMed] [Google Scholar]
- 2.Miyakis S, Robertson SA, and Krilis SA 2004. Beta-2 glycoprotein I and its role in antiphospholipid syndrome-lessons from knockout mice. Clin. Immunol 112: 136–143. [DOI] [PubMed] [Google Scholar]
- 3.Yasuda S, Tsutsumi A, Chiba H, Yanai H, Miyoshi Y, Takeuchi R, Horita T, Atsumi T, Ichikawa K, Matsuura E, and Koike T 2000. beta(2)-glycoprotein I deficiency: prevalence, genetic background and effects on plasma lipoprotein metabolism and hemostasis. Atherosclerosis 152: 337–346. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi R, Atsumi T, Ieko M, Takeya H, Yasuda S, Ichikawa K, Tsutsumi A, Suzuki K, and Koike T 2000. Coagulation and fibrinolytic activities in 2 siblings with beta(2)-glycoprotein I deficiency. Blood 96: 1594–1595. [PubMed] [Google Scholar]
- 5.Giannakopoulos B, and Krilis SA 2013. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med 368: 1033–1044. [DOI] [PubMed] [Google Scholar]
- 6.Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, and Salmon JE 2003. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Invest 112: 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, Bossi F, Ziller F, Sblattero D, Meroni P, and Tedesco F 2005. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 106: 2340–2346. [DOI] [PubMed] [Google Scholar]
- 8.Tedesco F, Borghi MO, Gerosa M, Chighizola CB, Macor P, Lonati PA, Gulino A, Belmonte B, and Meroni PL 2018. Pathogenic Role of Complement in Antiphospholipid Syndrome and Therapeutic Implications. Front Immunol 9: 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meroni PL, Borghi MO, Raschi E, and Tedesco F 2011. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol 7: 330–339. [DOI] [PubMed] [Google Scholar]
- 10.Urbanus RT, Derksen RHWM, and de Groot PG 2008. Platelets and the antiphospholipid syndrome. Lupus 17: 888–894. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H, Wang H, Li N, Yu Y, Huang H, Yan Y, and Wang T 2009. Annexin A2 mediates anti-beta 2 GPI/beta 2 GPI-induced tissue factor expression on monocytes. Int. J. Mol. Med 24: 557–562. [DOI] [PubMed] [Google Scholar]
- 12.Di Simone N, Luigi MP, Marco D, Fiorella DN, Silvia D, Clara DM, and Alessandro C 2007. Pregnancies complicated with antiphospholipid syndrome: the pathogenic mechanism of antiphospholipid antibodies: a review of the literature. Ann. N. Y. Acad. Sci 1108: 505–514. [DOI] [PubMed] [Google Scholar]
- 13.Agostinis C, Biffi S, Garrovo C, Durigutto P, Lorenzon A, Bek A, Bulla R, Grossi C, Borghi MO, Meroni P, and Tedesco F 2011. In vivo distribution of β2 glycoprotein I under various pathophysiologic conditions. Blood 118: 4231–4238. [DOI] [PubMed] [Google Scholar]
- 14.de Laat B, Mertens K, and de Groot PG 2008. Mechanisms of disease: antiphospholipid antibodies-from clinical association to pathologic mechanism. Nat Clin Pract Rheumatol 4: 192–199. [DOI] [PubMed] [Google Scholar]
- 15.de Laat B, Derksen RHWM, van Lummel M, Pennings MTT, and de Groot PG 2006. Pathogenic anti-beta2-glycoprotein I antibodies recognize domain I of beta2-glycoprotein I only after a conformational change. Blood 107: 1916–1924. [DOI] [PubMed] [Google Scholar]
- 16.Durigutto P, Grossi C, Borghi MO, Macor P, Pregnolato F, Raschi E, Myers MP, de Groot PG, Meroni PL, and Tedesco F 2019. New insight into antiphospholipid syndrome: antibodies to β2glycoprotein I-domain 5 fail to induce thrombi in rats. Haematologica 104: 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meroni PL, Macor P, Durigutto P, De Maso L, Gerosa M, Ferraresso M, Borghi MO, Mollnes TE, and Tedesco F 2016. Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood 127: 365–367. [DOI] [PubMed] [Google Scholar]
- 18.Kristensen T, Schousboe I, Boel E, Mulvihill EM, Hansen RR, Møller KB, Møller NP, and Sottrup-Jensen L 1991. Molecular cloning and mammalian expression of human beta 2-glycoprotein I cDNA. FEBS Lett. 289: 183–186. [DOI] [PubMed] [Google Scholar]
- 19.Lozier J, Takahashi N, and Putnam FW 1984. Complete amino acid sequence of human plasma beta 2-glycoprotein I. Proc. Natl. Acad. Sci. U.S.A 81: 3640–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, and Skjoedt M-O 2016. A journey through the lectin pathway of complement-MBL and beyond. Immunol. Rev 274: 74–97. [DOI] [PubMed] [Google Scholar]
- 21.Farrar CA, Asgari E, Schwaeble WJ, and Sacks SH 2012. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol 3: 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garred P, Voss A, Madsen HO, and Junker P 2001. Association of mannose-binding lectin gene variation with disease severity and infections in a population-based cohort of systemic lupus erythematosus patients. Genes Immun. 2: 442–450. [DOI] [PubMed] [Google Scholar]
- 23.Tsutsumi A, Takahashi R, and Sumida T 2005. Mannose binding lectin: genetics and autoimmune disease. Autoimmun Rev 4: 364–372. [DOI] [PubMed] [Google Scholar]
- 24.Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, and Fujita T 1998. Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol. Dial. Transplant 13: 1984–1990. [DOI] [PubMed] [Google Scholar]
- 25.Krarup A, Wallis R, Presanis JS, Gál P, and Sim RB 2007. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS ONE 2: e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess K, Ajjan R, Phoenix F, Dobó J, Gál P, and Schroeder V 2012. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE 7: e35690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenny L, Dobó J, Gál P, and Schroeder V 2015. MASP-1 of the complement system promotes clotting via prothrombin activation. Mol. Immunol 65: 398–405. [DOI] [PubMed] [Google Scholar]
- 28.Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, Skjoedt M-O, Huber-Lang M, Garred P, Ekdahl KN, and Nilsson B 2016. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J. Thromb. Haemost 14: 531–545. [DOI] [PubMed] [Google Scholar]
- 29.Mehta P, Norsworthy PJ, Hall AE, Kelly SJ, Walport MJ, Botto M, and Pickering MC 2010. SLE with C1q deficiency treated with fresh frozen plasma: a 10-year experience. Rheumatology (Oxford) 49: 823–824. [DOI] [PubMed] [Google Scholar]
- 30.Cavazzana A, Pengo V, Tonello M, Noventa F, Grossi C, Borghi MO, de Moerloose P, Reber G, and Ruffatti A 2009. Anti-beta(2)-glycoprotein I ELISA assay: the influence of different antigen preparations. Thromb. Haemost 101: 789–791. [PubMed] [Google Scholar]
- 31.van Os GMA, Meijers JCM, Agar Ç, Seron MV, Marquart JA, Åkesson P, Urbanus RT, Derksen RHWM, Herwald H, Mörgelin M, and D E Groot PG 2011. Induction of anti-β2 -glycoprotein I autoantibodies in mice by protein H of Streptococcus pyogenes. J. Thromb. Haemost 9: 2447–2456. [DOI] [PubMed] [Google Scholar]
- 32.Larsen F, Madsen HO, Sim RB, Koch C, and Garred P 2004. Disease-associated mutations in human mannose-binding lectin compromise oligomerization and activity of the final protein. J. Biol. Chem 279: 21302–21311. [DOI] [PubMed] [Google Scholar]
- 33.Ruben E, Planer W, Chinnaraj M, Chen Z, Zuo X, Pengo V, Filippis VD, Alluri RK, McCrae KR, Macor P, Tedesco F, and Pozzi N 2020. X-ray and solution structures of human beta-2 glycoprotein I reveal a new mechanism of autoantibody recognition,. Biochemistry. [Google Scholar]
- 34.Thielens NM, Cseh S, Thiel S, Vorup-Jensen T, Rossi V, Jensenius JC, and Arlaud GJ 2001. Interaction properties of human mannan-binding lectin (MBL)-associated serine proteases-1 and −2, MBL-associated protein 19, and MBL. J. Immunol 166: 5068–5077. [DOI] [PubMed] [Google Scholar]
- 35.Lacroix M, Tessier A, Dumestre-Pérard C, Vadon-Le Goff S, Gout E, Bruckner-Tuderman L, Kiritsi D, Nyström A, Ricard-Blum S, Moali C, Hulmes DJS, and Thielens NM 2017. Interaction of Complement Defence Collagens C1q and Mannose-Binding Lectin with BMP-1/Tolloid-like Proteinases. Sci Rep 7: 16958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larvie M, Shoup T, Chang W-C, Chigweshe L, Hartshorn K, White MR, Stahl GL, Elmaleh DR, and Takahashi K 2012. Mannose-binding lectin binds to amyloid β protein and modulates inflammation. J. Biomed. Biotechnol 2012: 929803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Papa N, Guidali L, Sala A, Buccellati C, Khamashta MA, Ichikawa K, Koike T, Balestrieri G, Tincani A, Hughes GR, and Meroni PL 1997. Endothelial cells as target for antiphospholipid antibodies. Human polyclonal and monoclonal anti-beta 2-glycoprotein I antibodies react in vitro with endothelial cells through adherent beta 2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 40: 551–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.