

Abstract
Otitis media (OM), a very common disease in young children, can result in hearing loss. In order to potentially replicate previously reported associations between OM and PLG, exome and Sanger sequencing, RNA-sequencing of saliva and middle ear samples, 16S rRNA sequencing, molecular modeling, and statistical analyses including transmission disequilibrium tests (TDT) were performed in a multi-ethnic cohort of 718 families and simplex cases with OM. We identified four rare PLG variants c.112A > G (p.Lys38Glu), c.782G > A (p.Arg261His), c.1481C > T (p.Ala494Val) and c.2045 T > A (p.Ile682Asn), and one common variant c.1414G > A (p.Asp472Asn). However TDT analyses for these PLG variants did not demonstrate association with OM in 314 families. Additionally PLG expression is very low or absent in normal or diseased middle ear in mouse and human, and salivary expression and microbial α-diversity were non-significant in c.1414G > A (p.Asp472Asn) carriers. Based on molecular modeling, the novel rare variants particularly c.782G > A (p.Arg261His) and c.2045 T > A (p.Ile682Asn) were predicted to affect protein structure. Exploration of other potential disease mechanisms will help elucidate how PLG contributes to OM susceptibility in humans. Our results underline the importance of following up findings from genome-wide association through replication studies, preferably using multi-omic datasets.
Subject terms: Medical genetics, Genetic predisposition to disease, Genetic association study, DNA sequencing, Next-generation sequencing, RNA sequencing, Sequence annotation, Targeted resequencing, Microbial genetics, Bacterial genetics, Genetics, Gene expression
Introduction
Otitis media (OM) is one of the most common diseases in young children, with 80% of children under the age of 3 years having at least one episode of acute OM1. OM with effusion can result in hearing loss2, and can occasionally lead to an auditory processing disorder3. The frequent occurrence of this disease continues to be a burden on the healthcare system. From 2009 to 2011, 8.6 million emergency department visits resulted in otologic diagnoses in the United States, with OM as the most common diagnosis (60.6%) for ear cases4. This healthcare burden results in great costs. Management of OM, including office visits, antibiotics, and surgeries from 1998–2008 was estimated to have cost over $5.3 billion annually in the United States5.
There are multiple known risk factors for OM, such as allergies, viral upper respiratory infection, previous history of OM, family history, lack of breastfeeding, multiple siblings, day care attendance, second-hand smoke and low social status6. Despite the public health measures taken to lower these risk factors and reduce the incidence of OM, prevalence remains high, suggesting additional factors such as genetic predisposition. Previous studies have shown a strong correlation between genetic risk factors and both recurrent acute otitis media (RAOM) and chronic otitis media with effusion (COME), with heritability estimated from 22–74% based on type of OM and cohort7,8.
In a genome-wide association study (GWAS) involving > 120,000 European-descent individuals, a heterozygous PLG variant c.112A > G (p.Lys38Glu) was associated with childhood ear infections [OR = 1.43; 95%CI = 1.26, 1.63; p = 3.8 × 10−8] 9. Additionally PLG variants c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) were reported to be co-inherited in families with type I plasminogen deficiency10.
PLG (MIM 173,350) encodes plasminogen which is a secreted blood zymogen that, when activated by proteolysis, is converted to plasmin. The product plasmin is a serine protease that degrades fibrin clots and promotes degradation of the extracellular matrix11. Plasminogen has been shown to play a crucial role in other cellular processes such as wound healing, immunity, tissue remodeling, inflammation, and cell migration12. Interestingly, there is evidence that some bacteria possess plasminogen-binding adhesins on their cell surface to exploit the fibrinolytic system by degrading host junction proteins and initiating signaling events that facilitate bacterial uptake and invasion11,13. In previous studies, mice deficient in plasminogen spontaneously developed chronic OM by 18 weeks of age, and local injection of plasminogen led to healing of eardrum perforations14,15. For these mouse studies, only homozygous Plg-deficient mice were examined.
In this study, we want to determine whether individuals with heterozygous PLG variants also have increased susceptibility to OM. We aim to identify coding PLG variants co-segregating in families with OM. For the identified PLG variants, we followed up our findings with phenotypic description, tissue expression and microbiome analyses to determine the role that these variants potentially play in OM pathogenesis. Identification of associations between pathogenic variants and OM will increase the overall understanding of OM susceptibility and the mechanisms behind the disease.
Results
PLG variants identified from exome data in multi-ethnic families with OM
We submitted DNA samples from 259 multi-ethnic families for exome sequencing. In nine families, we identified four rare and damaging PLG variants c.112A > G (p.Lys38Glu), c.782G > A (p.Arg261His), c.1481C > T (p.Ala494Val) and c.2045 T > A (p.Ile682Asn), and one common and benign variant c.1414G > A (p.Asp472Asn; Table 1; Figs. 1; 2). The variants c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) were both found in two Minnesota families and one Finnish trio with an autosomal dominant pattern of inheritance with reduced penetrance (Fig. 1A–C). For each identified exome variant, Sanger sequencing of the same variant(s) was performed in the rest of the relatives of the proband to check for co-segregation. Family UMN48 co-segregates c.112A > G (p.Lys38Glu) with OM, however only two out of three affected siblings are heterozygous for c.1414G > A (p.Asp472Asn) due to a recombination event between these two variants in the third sibling (Fig. 1A). The second Minnesota family UMN469 has incomplete co-segregation for both c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn); only the affected individuals in the second generation are heterozygous for both variants, while the affected children in the third generation are wildtype, which may suggest intra-familial genetic heterogeneity (Fig. 1B) 16,17. Of the seven family members of UMN469 that carry the c.1414G > A (p.Asp472Asn) variant, four are unaffected with OM (Fig. 1B). The third family is Finnish trio UHF18: the proband has both c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn), but the mother who also has both c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) was normal at examination and has no written record of being affected with OM in childhood (Fig. 1C).
Table 1.
PLG variants identified in nine multi-ethnic families with otitis media.
| ID | hg19 Position1 | cDNA variant | Amino acid variant | rsID | Damaging prediction2 | CADD | gnomAD MAF |
|---|---|---|---|---|---|---|---|
| UMN483, UMN4693, UHF183 | 161,127,501 | c.112A > G | p.Lys38Glu | rs73015965 | FA,MA,mLR,mSVM, MT,PP2,SI | 19.0 | NFE = 0.005, FIN = 0.0005 |
| UHF48, UHF68, UHF116 | 161,137,790 | c.782G > A | p.Arg261His | rs4252187 | MA,MT,PP2,PR,SI | 27.6 | NFE = 0.004, FIN = 0.005 |
| UMN483, UMN4693, PKOM18, UHF183, UMN5014a4 | 161,152,240 | c.1414G > A | p.Asp472Asn | rs4252125 | – | 1.4 | NFE = 0.29, SAS = 0.10, FIN = 0.26 |
| UHF520 | 161,152,819 | c.1481C > T | p.Ala494Val | rs4252128 | MA, SI, PP2 | 26.8 |
NFE = 0.004, FIN = 0.00004 |
| UMN5014a4 | 161,162,369 | c.2045 T > A | p.Ile682Asn | rs147175166 | FA,MT,PP2,PR | 23.3 | NFE = 0.001 |
CADD scaled combined annotation-dependent depletion score, FA FATHMM, FIN gnomAD finnish, gnomAD genome aggregation database, MA mutation assessor, MAF population-matched minor allele frequency, mLR MetaLR, mSVM MetaSVM, MT MutationTaster, NFE gnomAD non-Finnish European, PKOM Pakistani family; PP2 PolyPhen2 HVAR, PR PROVEAN, SAS gnomAD South Asian, SI SIFT, UHF Finnish family, UMN Minnesota family.
1mRNA accession number = NM_00301.
2Damaging prediction from bioinformatics tools in dbNSFP v.3.3a.
3Families UMN48, UMN469, and UHF18 have both the c.112A > G (p.Lys38Glu) and the c.1414G > A (p.Asp472Asn) variant. In two families UMN469 and UHF48, not all affected individuals carry the PLG variant. In one trio UHF18, one unaffected individual carries both variants.
4Family UMN5014a has both the c.1414G > A (p.Asp472Asn) and the c.2045 T > A (p.Ile682Asn) variant. Two unaffected individuals carry the c.1414G > A (p.Asp472Asn) variant.
Figure 1.

Four multi-ethnic families with the PLG c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) variants. Two Minnesota families (A) UMN48 and (B) UMN469 and (C) one Finnish trio UHF18 carry both variants with an autosomal dominant pattern of inheritance with reduced penetrance. (D) Despite Pakistani family PKOM18 being highly consanguineous, PLG c.1414G > A (p.Asp472Asn) co-segregated with OM in an autosomal dominant pattern.
Figure 2.
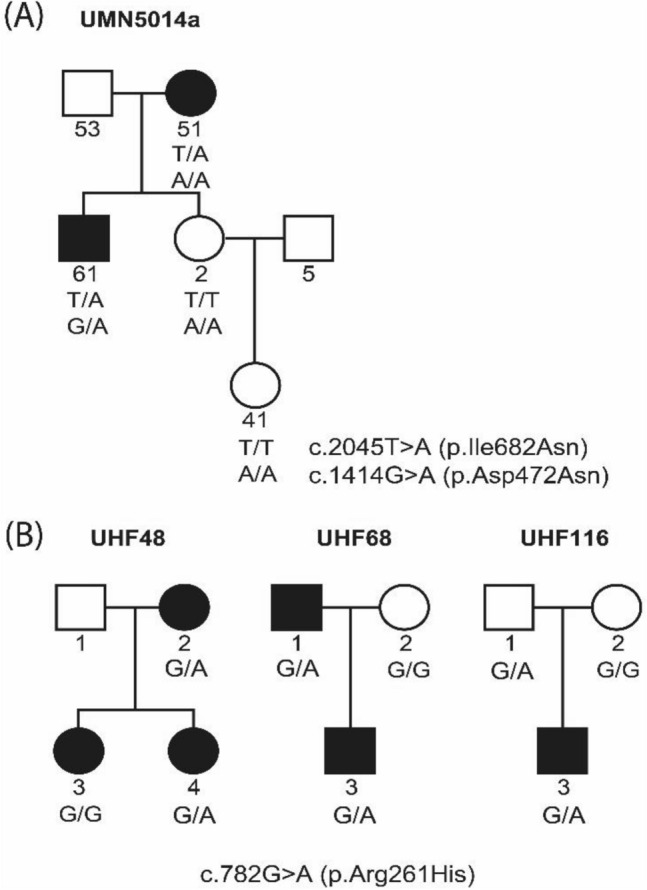
Four multi-ethnic families with the PLG c.782G > A (p.Arg261His), c.2045 T > A (p.Ile682Asn) and c.1414G > A (p.Asp472Asn) variants. (A) In Minnesota family UMN5014a, the c.2045 T > A (p.Ile682Asn) variant co-segregates with OM, however two unaffected individuals carry the c.1414G > A (p.Asp472Asn) variant. (B) Three Finnish families have the c.782G > A (p.Arg261His) variant.
The exome data from an affected individual from Pakistani family PKOM18 is heterozygous for c.1414G > A (p.Asp472Asn) but this individual does not carry the c.112A > G (p.Lys38Glu) variant. Despite PKOM18 being highly consanguineous, Sanger sequencing of the DNA samples from the rest of the PKOM18 family showed that the c.1414G > A (p.Asp472Asn) variant co-segregated with OM in an autosomal dominant pattern (Fig. 1D).
Additional rare, predicted-to-be-damaging PLG variants co-segregated with OM in four families (Fig. 2; Table 1). In one Minnesota family UMN5014a, the c.2045 T > A (p.Ile682Asn) variant co-segregates with OM. However, in this family all relatives with DNA samples including two affected and two unaffected individuals carry the c.1414G > A (p.Asp472Asn) variant (Fig. 2A). Three Finnish families have the c.782G > A (p.Arg261His) variant (Fig. 2B). Two out of three affected individuals in family UHF48 carry the c.782G > A (p.Arg261His) variant. In two Finnish trios, UHF68 and UHF116, the proband is heterozygous for the c.782G > A (p.Arg261His) variant. In both trios the variant was inherited from the father, but the father was considered affected in only one of these two trios (Fig. 2B). We identified c.1481C > T (p.Ala494Val) in the exome data for one Finnish individual with OM, however DNA was not available for both parents, and therefore we could not verify co-segregation or inheritance within that family (pedigree not shown).
In summary, three out of five families with multiple affected relatives co-segregate at least one PLG variant with OM. In three trios the PLG variant was inherited, but in only one trio was a parent who transmitted the minor allele for the PLG variant labeled as affected. One affected proband without family data carried the minor allele for a rare PLG variant.
In the five Finnish families with PLG variants and exome data, five out of the eight affected individuals have both RAOM and COME. We also checked an additional 25 Minnesota and 213 Finnish families with exome data for additional PLG variants, and only found the common c.1414G > A (p.Asp472Asn) variant in 16 (64%) Minnesota and 95 (45%) Finnish families. However the allele counts in these families are not significantly different from counts for the c.1414G > A (p.Asp472Asn) variant in non-Finnish European and Finnish gnomAD alleles. In Finnish families, no significant association was found between the PLG c.1414G > A (p.Asp472Asn) variant and OM type. Additionally, in OM patients from Colorado and Texas, carriage of either c.112A > G (p.Lys38Glu) or c.1414G > A (p.Asp472Asn) was not associated with either RAOM or COME. These data suggest that PLG variants are not associated with a specific OM type.
Additional non-OM traits in families with PLG variants
In three Minnesota families with heterozygous PLG variants, two of which have [c.112A > G(p.Lys38Glu);c.1414G > A(p.Asp472Asn)] and one with [c.2045 T > A(p.Ile682Asn);c.1414G > A(p.Asp472Asn)], a higher proportion of affected individuals have hay fever, asthma or allergy, with allergy being significant (Fisher exact p = 0.047). On the other hand, out of five Finnish families with PLG variants, in family UHF48 only one of two affected individuals had asthma, as well as hypertension and psoriasis. A second family UHF18 had other potentially autoimmune phenotypes such as diabetes, nephritis and uveitis. In Pakistani family PKOM18, only one affected relative had eczema. In three Texan trios with transmission of the minor allele for the c.112A > G (p.Lys38Glu) variant (Table 2), only one out of six variant carriers have allergic rhinitis history. Taken together, these findings do not show a consistent pattern of association across cohorts between heterozygous PLG variants and additional traits.
Table 2.
Transmission disequilibrium test for PLG variants in 314 US trios.
| Variant | White | Hispanic | Black | Mixed/Other | Transmitted | Non-transmitted | TDT p |
|---|---|---|---|---|---|---|---|
| p.Lys38Glu | 232 | 44 | 21 | 17 | 3 | 4 | 0.71 |
| p.Arg261His | 30 | 3 | 0 | 7 | 0 | 0 | – |
| p.Ala494Val | 49 | 7 | 2 | 9 | 0 | 1 | 0.31 |
| p.Ile682Asn | 190 | 41 | 22 | 10 | 1 | 0 | 0.32 |
| p.Asp472Asn | 221 | 44 | 23 | 17 | 101 | 116 | 0.31 |
| All variants1 | 215 | 43 | 21 | 17 | 104 | 119 | 0.32 |
1For UTMB59, the father transmitted the minor allele for both c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) variants to the child. On the other hand, for each of two trios UTMB132 and UTMB206 one parent carried both c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) variants but did not transmit the minor alleles to the child.
TDT analysis for PLG variants in US trios
In order to potentially replicate OM GWAS findings for PLG, the c.112A > G (p.Lys38Glu), c.782G > A (p.Arg261His), c.1481C > T (p.Ala494Val), c.2045 T > A (p.Ile682Asn), and c.1414G > A (p.Asp472Asn) variants were Sanger-sequenced in 314 US trios with OM. To determine genetic linkage and association, transmission of the minor alleles for these PLG variants were assessed using the transmission disequilibrium test (TDT)18. The minor allele for variant c.782G > A (p.Arg261His) was not present in Colorado trios. The minor allele for c.1481C > T (p.Ala494Val) was non-transmitted in one trio and transmitted in no trios (Table 2). For c.112A > G (p.Lys38Glu), three alleles were transmitted and four were non-transmitted. The variant c.1414G > A (p.Asp472Asn) also had a higher number of non-transmitted alleles, with 101 transmitted and 116 non-transmitted alleles. Lastly, the variant c.2045 T > A (p.Ile682Asn) had one transmitted allele and zero non-transmitted. Based on the TDT p-values for all variants, and their combined p value (p = 0.32), these variants were not significant in family-based association tests. Based on posthoc power analysis19, and an odds ratio of 1.43 for the PLG variant c.112A > G (p.Lys38Glu) from GWAS9,our trio study is underpowered to detect this small effect size and will require at least 16,200 trios to obtain sufficient power. On the other hand, in a previous study we were able to obtain a significant result for a rare FUT2 c.412C > T (p.Arg138Cys) variant using the same trio set16.
Low or absent PLG expression in saliva and middle ear
Plg expression was checked in microarray data of wildtype mouse middle ears that were inoculated with non-typeable Haemophilus influenzae (NTHi) vs. control20. Plg expression was low in mouse middle ear. In general, regulation of Plg in the mouse middle ear following infection with NTHi was minimal, although significant down-regulation was observed at a single time point at 48 h after inoculation.
From human data from GTEx, PLG is most highly expressed in the liver, with lower expression in the kidney and spleen. However no GTEx data is available on middle ear expression and the four rare PLG variants we identified. On the other hand, in GTEx, the common c.1414G > A (p.Asp472Asn) variant is a significant expression quantitative trait locus (eQTL) in esophageal mucosa. We previously performed RNA-sequencing of 19 saliva samples from OM patients in the US21. In these saliva samples, PLG was expressed at a very low level (DESeq2 normalized reads: range 0 to 8.5). A Wilcoxon rank sum test was then used to compare the salivary PLG expression for individuals who are wildtype vs. carriers of c.1414G > A (p.Asp472Asn) variant alleles. There was no significant difference in PLG expression in saliva in the two groups (data not shown). Likewise PLG was expressed at a very low level in the cholesteatoma and mucosa samples collected during OM surgery (DESeq2 normalized reads: range 0 to 8.3, average cholesteatoma 1.96, average mucosa 2.79). Thus in our available samples from human saliva and middle ear, PLG expression was very low or absent.
Network analysis
The PLG variant c.112A > G (p.Lys38Glu) was identified along with 31 other significant loci in a previous GWAS of OM9,22. In order to identify common biological processes between PLG and other loci associated with OM, this list of 32 genes was uploaded into NetworkAnalyst 3.0 and mapped (Reactome; Supplementary Figure S3A)23–25. In the resulting network, SP3 and PLG were the two largest nodes and were joined by 4 different nodes: SERPINE1, FN1, IGFBP3, and EP300. A secondary network was then mapped from the gene list: SP3, PLG, SERPINE1, FN1, IGFBP3, and EP300 (GO:BP; Supplementary Figure S3B). The most significant pathways identified within these networks from the Reactome database were “dissolution of fibrin clot”, “platelet degranulation” and “response to elevated platelet cytosolic Ca2+” (Supplementary Table S3), which are related to the known functions of plasminogen in fibrinolysis.
16S rRNA sequencing and analyses of middle ear and nasopharyngeal samples from OM patients in the US according to carriage of the c.1414G > A (p.Asp472Asn) variant
We also performed microbiome sequencing and analyses of the middle ears and nasopharynges of carriers of the c.1414G > A (p.Asp472Asn) variant compared to wildtype individuals, however we found no significant differences in overall microbiome composition based on α-diversity indices (Supplementary Table 1). Only Bifidobacterium had an increased relative abundance in the nasopharynx of variant carriers (nominal p = 0.0024), which was non-significant after correction for multiple testing (false discovery rate/FDR-adjusted p = 0.13; Supplementary Fig. 1). Unfortunately, we do not have saliva, middle ear or microbial samples for probands with the rare PLG variants.
Predicted effect of three rare variants on PLG protein
All three novel variants [c.782G > A (p.Arg261His), c.1481C > T (p.Ala494Val) and c.2045 T > A (p.Ile682Asn)] are located near disulfide bonds between cysteine residues (Cys166-Cys243, Cys483-Cys524, and Cys548-Cys666 respectively) that are potentially important for protein stabilization26. More specifically, these disulfide bonds are important for the structure and interface between domains. If these bonds are impacted by these variants, the stability and enzymatic activity of the whole protein could be affected27,28.
The c.2045 T > A (p.Ile682Asn) variant is within the β-chain of the trypsin domain near the “94-shunt” surface loop which is an important component in the accessibility of the catalytic triad residues that are necessary for trypsin-like protease activity29. The 6-residue difference between the typical “99-loop” of serine proteases and the “94-shunt” in PLG is predicted to be an important factor in its increased range of specificity30. In the native form, the isoleucine at this position forms an H-bond with Pro593 (Supplementary Fig. 2A) whereas the asparagine variant is predicted to form two extra H-bonds with Cys567 and Pro683 (Supplementary Fig. 1B). Due to this variant, there is a potential decrease in the accessibility of the catalytic triad that mediates the main enzymatic function of the protein.
The c.782G > A (p.Arg261His) variant occurs within the kringle 2 (KR2) domain of the PLG α-chain and could disrupt the single disulfide bond that serves as the interface between the KR2 and KR3 domains. The closed conformation of the native PLG, where fibrin is bound and less active, is in part stabilized by a chloride ion bond located near Arg26126,31. The histidine variant is predicted to interfere with this chloride ion bond and destabilize the protein (Supplementary Fig. 2C–D). Additionally other previously identified variants within this region have been predicted to interfere with the interface between the serine protease domain and KR232.
The c.1481C > T (p.Ala494Val) variant occurs within the KR5 domain of the β-chain26. The KR5 domain is crucial for the change in conformation from closed to open via fibrin binding to the lysine binding sites present in this domain31. There were no changes in the H-bonds observed for this variant compared to the native protein and based on the location of the valine variant, the effect of the extra side chain on the valine is predicted to be minimal (Supplementary Fig. 2E–F).
Discussion
We identified four rare variants and one common variant within PLG in probands with OM. The PLG variants c.112A > G (p.Lys38Glu) or c.1414G > A (p.Asp472Asn) co-segregate with OM in two out of five families with multiple affected relatives (Fig. 1; Fig. 2A), but we do not have additional evidence from trio or expression studies to support association between these variants and OM. In a previous GWAS, the heterozygous genotype for c.112A > G (p.Lys38Glu) was a risk factor for OM. Additionally this variant was often found to be co-inherited in cis with c.1414G > A (p.Asp472Asn), which was previously associated with increased susceptibility to fungal infection in mice33. However we did not see an association between any of the five identified PLG variants and OM in our trios (Table 2). It might be that these variants have weak effects that are only detectable in very large cohorts, e.g. with an OR of 1.43 for the p.Lys38Glu variant, we would need > 16,000 trios to detect a risk effect19. Furthermore, our families with OM may have other common or rare variants in other genes that have stronger phenotypic effects, whether risk or protective variants, and are concealing the effect of these PLG variants.
Hereditary plasminogen deficiency with reduced protein activity is referred to as Type 1 Plasminogen Deficiency (MIM 217090). This disease is inherited in an autosomal recessive manner, and most commonly manifests as ligneous conjunctivitis, in which the wood-like pseudomembranous lesions develop on the mucous membranes in the eyes34,35. Furthermore, similar lesions have been observed on the skin and other mucous membranes including the middle ear, gingiva, respiratory tract, and gastrointestinal or female genital tract, with OM occurring in 14% of patients with plasminogen deficiency. Notably none of the PLG carriers among our families have these lesions. PLG c.112A > G (p.Lys38Glu) is the most commonly identified variant in 34% of patients with type 1 plasminogen deficiency, whether as homozygous or compound heterozygous12. However individuals with either homozygous or compound heterozygous genotypes for c.112A > G (p.Lys38Glu) may have decreased serum plasminogen activity and may also be healthy10. We also considered the possibility that the known PLG variants are only causal of susceptibility when inherited in an autosomal recessive manner. However in the few individuals in the Colorado cohort that are homozygous for c.1414G > A (p.Asp472Asn), we did not see a difference in salivary PLG expression or α-diversity in microbial samples compared to wildtype or heterozygous (Supplementary Table 1). Likewise, none of the affected individuals in our families appear to be compound heterozygous for PLG variants.
Plasminogen has a potential role in pathogenic invasion by bacteria that bind it and thus can activate fibrin degradation to aid entry via cell junctions11. It is not uncommon for pathogens to use the host plasminogen activation system as a means of invasion and many bacteria with PLG receptors have been identified, including Haemophilus influenzae and Streptococcus pneumoniae36,37. Bifidobacterium is a common probiotic for use in preventing recurrent acute OM though its efficacy is unproven38. Additionally, a previous study did not detect Bifidobacterium as having a significant role in acute OM, suggesting it may be a commensal colonizer of the nasopharynx39. Thus the increased relative abundance of Bifidobacterium in the c.1414G > A (p.Asp472Asn) variant as compared to the wild-type does not corroborate previous findings that this common variant is associated with reduced risk of OM.
In human and mouse middle ear tissue PLG expression was low. Additionally, plasminogen activator activity could not be detected in middle ear effusion samples from adult patients40. However, in the mouse middle ear study reported here, expression of the plasminogen activator gene (Plat) is upregulated in the middle ear six hours after NTHi inoculation, suggesting some Plg activity within middle ear. Plasminogen is a blood protein that is essential for wound healing, including healing of tympanic membrane perforations41. This is further supported by our network analyses (Supplementary Table 3), wherein the most significant pathways linking PLG to other OM susceptibility genes involve processes for coagulation and cell adhesion which are both important for healing of injured tissues. In plasminogen-deficient patients, ligneous lesions primarily develop on the tympanic membrane, rather than the middle ear mucosa or cavity42. Unfortunately, we do not have data from the tympanic membrane. There could be other proteins such as activators or binding partners involved that result in PLG having an indirect effect on OM susceptibility. For example, in the GTEx database the common c.1414G > A (p.Asp472Asn) variant is an eQTL for a lincRNA in esophageal mucosa, suggesting other potential disease mechanisms for PLG variants such as epigenetic regulation. These are however outside the scope of this study. Because PLG is secreted by the cell, it is possible that the protein is active in circulation and in the middle ear space, as shown by previous studies involving mice and secretion assays for other rare variants12,14.
Interestingly, in COS-7 cells transfected with PLG mutants, secretion kinetics of both the c.112A > G (p.Lys38Glu) and c.1414G > A (p.Asp472Asn) variants were similar to the wild-type control12. This further supports the finding that the effects of these two variants on OM susceptibility are very weak to none. On the other hand, in vitro transfection of COS-7 cells from other variants including missense substitutions of arginine resulted in decreased cell secretion of PLG, suggesting that other novel variants are potentially functional12.
Three rare, heterozygous PLG variants, c.782G > A (p.Arg261His), c.1481C > T (p.Ala494Val) and c.2045T > A (p.Ile682Asn), were identified in US and Finnish families with OM. Two of these variants are predicted to change conformation of the protein and either affect enzymatic function or ligand binding (Supplementary Fig. 2). A previously reported variant c.782G > A (p.Arg261His) was identified in both cases and controls with multiple sclerosis43, but is novel for OM. A variant at the same amino acid position p.Arg261Cys was identified in a family with all heterozygous individuals having reduced PLG levels to 64–68%44. This might suggest that c.782G > A (p.Arg261His), and other rare variants in PLG, are likely to be functional or pathogenic. Additionally, previously reported PLG variants that affect secretion kinetics in COS-7 cells fall within the same domains as the novel PLG variants reported here12: variant c.2045 T > A (p.Ile682Asn) occurs within the β-chain of the trypsin domain, c.1481C > T (p.Ala494Val) within the KR5 domain of the β-chain, and c.782G > A (p.Arg261His) fall within the kringle 2 (KR2) domain of the α-chain45. All three domains in which these identified variants occur harbor previously reported variants that affect secretion of PLG. Due to the rarity of these three novel variants, their effects on tissue expression or the microbiome cannot be assessed at present.
To conclude, we identified three novel rare, missense variants and two known PLG variants which co-segregate with OM in some, but not all of our families with these variants. We hypothesized that these variants may play a role in OM susceptibility, however there was no association between these PLG variants and OM in our trios. Although the novel rare variants we identified might have potential effects on protein structure based on molecular modeling, PLG expression is very low or absent in normal or diseased middle ear tissues in both mouse and human. Our previous studies support a common mechanism by which the OM susceptibility variant affects expression of the encoded protein and other genes downstream, which in turn shifts the middle ear microbiome by changing the overall biodiversity or relative abundance of specific pathogens or commensal taxa16,21,46,47. The mostly negative results of our multiple studies showed that this disease mechanism does not apply to PLG variants in relation to OM susceptibility in humans. Based on the Plg-knockout mouse model with chronic OM and non-healing eardrum perforations, PLG variants may act on the healing process within the tympanic membrane or the secretion of plasminogen into the middle ear cavity through other disease mechanisms, including epigenetic, that are not detectable by the methods used in this study. However such effects are easily concealed by other stronger factors contributing to OM susceptibility within the middle ear mucosa, including other risk or protective variants with larger phenotypic effects in our families. Our results underline the importance of following up findings from genome-wide association through replication studies, preferably using multi-omic datasets.
Materials and methods
Ethics approval
Guidelines from the Declaration of Helsinki were followed throughout the conduct of this study. Prior to start of the study, ethical approval was obtained from the following institutional review boards (IRB): Bahauddin Zakariya University; Colorado Multiple IRB; Helsinki University Hospital; University of Maryland Baltimore; University of Minnesota; University of Texas Medical Branch (UTMB) Galveston; University of Virginia; and University of Washington. Informed consent was obtained from all adult participants and parents of children enrolled in the study. In addition, approval was obtained from the Institutional Animal Care and Use Committee of the Veterans Affairs Medical Center, San Diego, California. All experiments were performed in accordance with the Animal Welfare Act and the Health Research Extension Act of 1985.
Subject ascertainment
Except in Pakistan, OM-affected families from different cohorts were first identified upon referral of a child or proband for OM surgery (Supplementary Table 2). Samples were obtained from previously established family cohorts for OM, including 257 trios from Texas, 85 trios from Colorado, 140 families from Minnesota, 217 families from Finland, and 19 families from Pakistan. For Texan and Coloradan trios, clinical data from the proband including age, sex, breastfeeding history, allergic history, OM status and surgical technique, and saliva samples from both proband and parents were obtained. All saliva samples were collected using Oragene DNA collection kits and DNA was isolated using the manufacturer’s protocol.
In the three cohorts from Minnesota, Finland and Pakistan, DNA was extracted from blood samples provided by family members who participated in the study. These families were characterized as previously described16,48. In Minnesota, all family members were examined by an otolaryngologist and tested by tympanometry for middle ear function, and family members were considered affected if ≥ 2 data sources, whether otoscopy, tympanometry, medical records, or personal history, were positive for OM. For Finnish families, clinical data, including history of OM, risk factors for OM, and otolaryngologic surgery, were obtained. Finnish individuals were considered positive for OM if they had insertion of tympanostomy tubes, effusive OM for > 2 months, or recurrent OM (i.e., > 3 episodes in 6 months or > 4 episodes in 12 months). Families from Pakistan were identified through referral for familial occurrence of OM. For Pakistani families, age at onset and recurrence of OM episodes were determined from medical history and OM status by otoscopy. For all cohorts, individuals with known genetic, craniofacial, and immunodeficiency syndromes were excluded.
DNA sequencing
DNA samples from individuals with OM from 28 Minnesota, 217 Finnish, and 14 Pakistani families were submitted for exome sequencing using an Illumina HiSeq instrument, at an average 40–60 × coverage. For the Minnesota and Finnish families, sequence capture was performed at the University of Washington using the Roche NimbleGen SeqCap EZ Human Exome Library v.2.0. For the Pakistani families, exome sequencing was performed at the University of Maryland and genomic libraries were recovered for exome enrichment using the Agilent SureSelect Human Expanded All Exon V5 (62 Mb) kit. For all exome data, alignment and variant calling were performed using Burrows-Wheeler Aligner49and Genome Analysis Toolkit50, respectively.
Sanger sequencing was used to confirm co-segregation of five PLG variants (NM_00301.3) identified in exome data from nine multi-ethnic families (Table 1). An additional 70 Coloradan trios, 246 Texan trios, 1 Finnish family, 2 Minnesota families, and 1 Pakistani family without exome data were Sanger-sequenced for the PLG variants.
Bioinformatic and statistical analyses
PLG variants found in exome data from OM-affected individuals were selected based on whether they are predicted to be damaging according to bioinformatic tools, including the scaled Combined Annotation-Dependent Depletion (CADD) score ≥ 15.051, FATHMM52, MutationAssessor53, MutationTaster54, PolyPhen255, PROVEAN56, SIFT57, MetaLR and MetaSVM from dbNSFP58,59. Variants predicted to be damaging were further filtered for rare minor allele frequency (MAF < 1%), with the exception of common variant c.1414G > A (p.Asp472Asn), which was previously found to be co-inherited with c.112A > G (p.Lys38Glu). A Transmission Disequilibrium Test (TDT) was performed using genotypes for PLG variants c.782G > A (p.Arg261His) and c.1481C > T (p.Ala494Val) for Coloradan trios, and c.112A > G (p.Lys38Glu), c.1414G > A (p.Asp472Asn) and c.2045 T > A (p.Ile682Asn) for Coloradan and Texan trios18.
For 314 families with PLG variants, non-genetic variables such as age, sex, ethnicity, breastfeeding history, previous diagnoses of allergy or asthma, and type of OM were investigated. Logistic regression analyses were performed with OM type (RAOM, COME) as outcome variable, carriage of PLG variants as independent variable and other non-genetic variables as covariates. For Finnish probands, Fisher exact test was used to compare occurrence of RAOM vs. COME according to the PLG c.1414G > A (p.Asp472Asn) genotype. All variants tested were in Hardy–Weinberg equilibrium in the Texan, Coloradan and Finnish probands.
RNA expression in infected mouse middle ear
PLG expression in the mouse middle ear was derived from microarray analysis of mRNA from wild-type mice with acute OM after inoculation with non-typeable H. influenzae (NTHi) compared to healthy control mice, as described previously20. In order to discover genes that are regulated during acute OM, transbullar inoculation of the middle ears were performed on 320 WBxB6 F1 hybrid mice with PBS (sham control) or NTHi. Two independent samples were generated for each time point after infection or PBS injection: 0 h (0 h, uninfected controls), 3 h, 6 h, 1 day (1d), 2d, 3d, 5d, and 7d, from initiation of OM to resolution. RNA was profiled on Affymetrix mouse 430 2.0 whole-genome microarrays. A total of 3,605 genes, approximately 14.4% of the mouse genome, defined the signature of acute, NTHi-induced OM across time.
RNA expression in human saliva and middle ear
Salivary RNA samples were collected from 19 pediatric patients undergoing OM surgery at the Children’s Hospital Colorado (CHCO) and submitted for RNA sequencing, as previously described21. In brief, RNA samples were processed with the NuGen Trio RNA-Seq Kit (Tecan, Redwood City, CA, USA) at the University of Colorado Denver Genomics and Microarray Core and sequenced on an Illumina HiSeq 4,000 with average ~ 31 million reads per sample. Reads were trimmed using the FASTX-Toolkit (v0.0.13) and aligned using STAR v2.5.3a60. Counts were normalized, summed and analyzed according to carriage of PLG variant c.1414G > A (p.Asp472Asn) using the Wilcoxon test.
Three cholesteatoma samples and four mucosa samples were collected from patients undergoing OM surgery at the University of Colorado Hospital or CHCO. Samples were immediately isolated using the QIAGEN RNeasy Micro Kit. The tissue was initially homogenized with glass beads and QIAGEN buffer RLT, using a bead vortexer for 4 min. All four mucosa and three cholesteatoma samples passed QC and were submitted for RNA-sequencing. Libraries were constructed using the NuGEN Trio RNA-Seq kit. Sequencing was completed on the Illumina NovaSeq, with paired-end 2 × 151 bp reads.
For human tissue samples, reads were trimmed with BBDuk v.38.5061. Salmon v0.13.1 was used to quantify the transcripts from the RNA-sequence data62. Salmon was run in the mapping-based mode, which includes indexing and quantification. The Salmon index was created using the Ensembl release 96 GRCh38 human reference genome with a k-mer setting of 31. Transcript quantification was performed using the ‘–validateMappings’ flag. R was used to extract gene-summarized counts from the salmon quantification output, using the tximport package63. Counts were filtered to have an average of more than 3 reads in either the cases or controls. Principal components (PC) plots were generated for each dataset. One sample (CHCO86) was removed from further analyses due to an insufficient mapping rate and not clustering with the other samples in the PC analysis. Counts were normalized using DESeq264.
Network analysis
Network analysis of PLG was performed with NetworkAnalyst 3.023–25. A list of 32 genes previously found to be associated with OM22 and the IMEx Interactome generic protein–protein interaction database were used as input. Significant pathways were identified using the databases from the Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, Gene Ontology (GO), and PANTHER via the Function Explorer tool in NetworkAnalyst. Functions associated with the network that had an FDR-adjusted p-value less than 0.05 were considered significant.
Microbiome sequencing and analysis
16S rRNA sequencing and microbiome analysis was performed for 42 middle ear and 77 nasopharyngeal samples from Coloradan individuals with OM. To construct the 16S amplicon library, broad-range amplification and sequence analysis of 16S rRNA genes to determine bacterial profiles were used with previously described methods16. Analysis of the alpha-diversity indices of species richness, diversity, abundance and evenness (Chao1, Goods coverage, observed species index, Shannon Diversity Index) were tested for significance in association with PLG c.1414G > A genotype via t-test. Individual OTU associations were determined using Wilcoxon test for taxa that passed a prevalence threshold of 10% and relative abundance greater than 1%. R software was used for data analyses and generating figures.
Molecular modeling for rare PLG variants p.Arg261His, p.Ala494Val and p.Ile682Asn
For the three rare variants that we identified, protein models of PLG (PDB ID 4DUU) and the rare variants p.Arg261His, p.Ala494Val and p.Ile682Asn were constructed in Phyre2 and homology was visually analyzed and compared in DeepView/Swiss-PdbViewer65,66.
Data availability
Novel variants were deposited in ClinVar (accession numbers SCV000996504.1, SCV000996506.1, SCV000996507.1, VCV000013583.6 and VCV000692202.1). RNA-seq data from cholesteatoma and middle ear mucosal tissues are available in dbGaP phs001941.v1.p1. Demultiplexed paired-end 16S rRNA sequence data are accessible in the NCBI Short Read Archive under Accession Number PRJNA439435.
Supplementary information
Acknowledgements
We thank all study participants. We also thank E Valdivia for data collection efforts at UTMB, E Leinonen for collection and handling of Finnish samples, and the Office of Population Studies Foundation research and data collection teams. This manuscript is dedicated to Dr. Michele Sale, who passed away during the conduct of this research. The full list of current members of University of Washington Center for Mendelian Genomics is available at http://uwcmg.org/docs/Crediting_UW-CMG/UW_CMG_Banner.pdf.
Author contributions
All authors read and approved the manuscript. RSC conceptualized the study. TC, HP and WS provided clinical data and DNA samples from Texan families. MSc, NF, SS, SC, PY, KC, TW, HJ, JP and SG provided clinical data and DNA samples from Coloradan families. KD, MSa and AM provided clinical data and DNA samples from Minnesota families. LH, EE, JK and PM provided clinical data and DNA samples from Finnish families. ZA, SR, RSh, RI and AY provided clinical data and DNA samples and performed exome and Sanger sequencing for Pakistani families. UWCMG, MB, DN and SL performed exome sequencing for 12 Minnesota families. TB performed DNA isolations from saliva and performed Sanger sequencing for Minnesota, Texan, Coloradan and Finnish families. TB and EB performed bioinformatics analyses. EL and EB performed RNA-sequencing analyses. AR provided data from mouse middle ear expression studies. DF and CE performed microbiome analyses. CE performed molecular modeling. TB, CE and RSC performed statistical analyses and wrote the manuscript.
Competing interests
Dr. Allen Ryan is a co-founder of, shareholder in and uncompensated consultant to Otonomy, Inc., a relationship that was approved by the University of California San Diego. All other authors declare no competing interests.
Footnotes
A list of authors and their affiliations appears at the end of the paper.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Regie Lyn P. Santos-Cortez, Email: regie.santos-cortez@cuanschutz.edu
University of Washington Center for Mendelian Genomics (UW-CMG):
Michael J. Bamshad, Deborah A. Nickerson, and Suzanne M. Leal
Supplementary information
is available for this paper at 10.1038/s41598-020-70498-w.
References
- 1.Vergison A, et al. Otitis media and its consequences: beyond the earache. Lancet Infect. Dis. 2010;10:195–203. doi: 10.1016/S1473-3099(10)70012-8. [DOI] [PubMed] [Google Scholar]
- 2.Cai T, McPherson B, Li C, Yang F. Pure tone hearing profiles in children with otitis media with effusion. Disabil. Rehabil. 2018;40:1166–1175. doi: 10.1080/09638288.2017.1290698. [DOI] [PubMed] [Google Scholar]
- 3.Khavarghazalani B, Farahani F, Emadi M, Hosseni Dastgerdi Z. Auditory processing abilities in children with chronic otitis media with effusion. Acta Otolaryngol. 2016;136:456–459. doi: 10.3109/00016489.2015.1129552. [DOI] [PubMed] [Google Scholar]
- 4.Kozin ED, et al. Epidemiology of otologic diagnoses in United States emergency departments. Laryngoscope. 2015;125:1926–1933. doi: 10.1002/lary.25197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casey JR, Pichichero ME. Payment analysis of two diagnosis and management approaches of acute otitis media. Clin. Pediatr. (Phila) 2014;53:865–873. doi: 10.1177/0009922814533592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, et al. Risk factors for chronic and recurrent otitis media: a meta-analysis. PLoS ONE. 2014;9:e86397. doi: 10.1371/journal.pone.0086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hafren L, et al. Genetic background and the risk of otitis media. Int. J. Pediatr. Otorhinolaryngol. 2012;76:41–44. doi: 10.1016/j.ijporl.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 8.Casselbrant ML, et al. The heritability of otitis media: a twin and triplet study. JAMA. 1999;282:2125–2130. doi: 10.1001/jama.282.22.2125. [DOI] [PubMed] [Google Scholar]
- 9.Pickrell JK, et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 2016;48:709–717. doi: 10.1038/ng.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donmez-Demir B, et al. Novel plasminogen gene mutations in Turkish patients with type I plasminogen deficiency. Blood Coagul. Fibrinolysis. 2016;27:637–644. doi: 10.1097/MBC.0000000000000383. [DOI] [PubMed] [Google Scholar]
- 11.Ayon-Nunez DA, Fragoso G, Bobes RJ, Laclette JP. Plasminogen-binding proteins as an evasion mechanism of the host's innate immunity in infectious diseases. Biosci. Rep. 2018 doi: 10.1042/BSR20180705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tefs K, et al. Molecular and clinical spectrum of type I plasminogen deficiency: a series of 50 patients. Blood. 2006;108:3021–3026. doi: 10.1182/blood-2006-04-017350. [DOI] [PubMed] [Google Scholar]
- 13.Raymond BB, Djordjevic S. Exploitation of plasmin(ogen) by bacterial pathogens of veterinary significance. Vet. Microbiol. 2015;178:1–13. doi: 10.1016/j.vetmic.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Eriksson PO, Li J, Ny T, Hellstrom S. Spontaneous development of otitis media in plasminogen-deficient mice. Int. J. Med. Microbiol. 2006;296:501–509. doi: 10.1016/j.ijmm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Prestwich AH, et al. Lack of plasminogen does not alter the early inflammatory response following a tympanic membrane perforation: a study in plasminogen-deficient mice. Acta Otolaryngol. 2008;128:1294–1302. doi: 10.1080/00016480701361996. [DOI] [PubMed] [Google Scholar]
- 16.Santos-Cortez RLP, et al. FUT2 variants confer susceptibility to familial otitis media. Am. J. Hum. Genet. 2018;103:679–690. doi: 10.1016/j.ajhg.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehman AU, et al. Challenges and solutions for gene identification in the presence of familial locus heterogeneity. Eur. J. Hum. Genet. 2015;23:1207–1215. doi: 10.1038/ejhg.2014.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am. J. Hum. Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez M, et al. The transcriptome of a complete episode of acute otitis media. BMC Genomics. 2015;16:259. doi: 10.1186/s12864-015-1475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larson ED, et al. A2ML1 and otitis media: novel variants, differential expression and relevant pathways. Hum. Mutat. 2019 doi: 10.1002/humu.23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos-Cortez RLP, et al. Panel 3: genomics, precision medicine and targeted therapies. Int. J. Pediatr. Otorhinolaryngol. 2020;130(Suppl 1):109835. doi: 10.1016/j.ijporl.2019.109835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou G, et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia J, Gill EE, Hancock RE. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015;10:823–844. doi: 10.1038/nprot.2015.052. [DOI] [PubMed] [Google Scholar]
- 25.Xia J, Benner MJ, Hancock RE. NetworkAnalyst–integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014;42:W167–174. doi: 10.1093/nar/gku443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen TE, Martzen MR, Ichinose A, Davie EW. Characterization of the gene for human plasminogen, a key proenzyme in the fibrinolytic system. J. Biol. Chem. 1990;265:6104–6111. [PubMed] [Google Scholar]
- 27.Urano T, Castellino FJ, Suzuki Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018 doi: 10.1111/jth.14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005;93:647–654. doi: 10.1160/TH04-12-0842. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, et al. Human plasminogen catalytic domain undergoes an unusual conformational change upon activation. J. Mol. Biol. 2000;295:903–914. doi: 10.1006/jmbi.1999.3397. [DOI] [PubMed] [Google Scholar]
- 30.Wu G, et al. Structural studies of plasmin inhibition. Biochem. Soc. Trans. 2019;47:541–557. doi: 10.1042/BST20180211. [DOI] [PubMed] [Google Scholar]
- 31.Law RH, et al. The X-ray crystal structure of full-length human plasminogen. Cell. Rep. 2012;1:185–190. doi: 10.1016/j.celrep.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 32.Schuster V, Hugle B, Tefs K. Plasminogen deficiency. J. Thromb. Haemost. 2007;5:2315–2322. doi: 10.1111/j.1538-7836.2007.02776.x. [DOI] [PubMed] [Google Scholar]
- 33.Zaas AK, et al. Plasminogen alleles influence susceptibility to invasive aspergillosis. PLoS Genet. 2008;4:e1000101. doi: 10.1371/journal.pgen.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bateman JB, Pettit TH, Isenberg SJ, Simons KB. Ligneous conjunctivitis: an autosomal recessive disorder. J. Pediatr. Ophthalmol. Strabismus. 1986;23:137–140. doi: 10.3928/0191-3913-19860501-09. [DOI] [PubMed] [Google Scholar]
- 35.Schuster V, Seregard S. Ligneous conjunctivitis. Surv. Ophthalmol. 2003;48:369–388. doi: 10.1016/S0039-6257(03)00056-0. [DOI] [PubMed] [Google Scholar]
- 36.Attali C, Durmort C, Vernet T, Di Guilmi AM. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect. Immun. 2008;76:5350–5356. doi: 10.1128/IAI.00184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanderson-Smith ML, De Oliveira DM, Ranson M, McArthur JD. Bacterial plasminogen receptors: mediators of a multifaceted relationship. J. Biomed. Biotechnol. 2012;2012:272148. doi: 10.1155/2012/272148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laursen RP, et al. Risks for upper respiratory infections in infants during their first months in day care included environmental and child-related factors. Acta Paediatr. 2018 doi: 10.1111/apa.14320. [DOI] [PubMed] [Google Scholar]
- 39.Chonmaitree T, et al. Nasopharyngeal microbiota in infants and changes during viral upper respiratory tract infection and acute otitis media. PLoS ONE. 2017;12:e0180630. doi: 10.1371/journal.pone.0180630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamaguchi Y, Ohi M, Sakakura Y, Miyoshi Y. Activities of antiplasmin and antiplasminogen activator in serous middle ear effusions. Ann. Otol. Rhinol. Laryngol. 1985;94:293–296. [PubMed] [Google Scholar]
- 41.Li J, Eriksson PO, Hansson A, Hellstrom S, Ny T. Plasmin/plasminogen is essential for the healing of tympanic membrane perforations. Thromb. Haemost. 2006;96:512–519. doi: 10.1160/TH06-03-0168. [DOI] [PubMed] [Google Scholar]
- 42.Marcus DM, et al. Ligneous conjunctivitis with ear involvement. Arch. Ophthalmol. 1990;108:514–519. doi: 10.1001/archopht.1990.01070060062050. [DOI] [PubMed] [Google Scholar]
- 43.Sadovnick AD, et al. Analysis of plasminogen genetic variants in multiple sclerosis patients. G3 (Bethesda) 2016;6:2073–2079. doi: 10.1534/g3.116.030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin-Fernandez L, et al. The unravelling of the genetic architecture of plasminogen deficiency and its relation to thrombotic disease. Sci. Rep. 2016;6:39255. doi: 10.1038/srep39255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell AL, et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019;47:D351–D360. doi: 10.1093/nar/gky1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santos-Cortez RL, et al. Middle ear microbiome differences in indigenous Filipinos with chronic otitis media due to a duplication in the A2ML1 gene. Infect. Dis. Poverty. 2016;5:97. doi: 10.1186/s40249-016-0189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank, D. N. et al. Otitis media susceptibility and shifts in the head and neck microbiome due to SPINK5 variants. J. Med. Genet. 10.1136/jmedgenet-2020-106844 (2020). [DOI] [PMC free article] [PubMed]
- 48.Santos-Cortez RL, et al. Rare A2ML1 variants confer susceptibility to otitis media. Nat. Genet. 2015;47:917–920. doi: 10.1038/ng.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKenna A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 55.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013 doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sim NL, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bushnell B, Rood J, Singer E. BBMerge: accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Novel variants were deposited in ClinVar (accession numbers SCV000996504.1, SCV000996506.1, SCV000996507.1, VCV000013583.6 and VCV000692202.1). RNA-seq data from cholesteatoma and middle ear mucosal tissues are available in dbGaP phs001941.v1.p1. Demultiplexed paired-end 16S rRNA sequence data are accessible in the NCBI Short Read Archive under Accession Number PRJNA439435.