

Abstract
As a model for genetic studies, Arabidopsis thaliana (Arabidopsis) offers great potential to unravel plant genome-related mechanisms that shape the root microbiome. However, the fugitive life history of this species might have evolved at the expense of investing in capacity to steer an extensive rhizosphere effect. To determine whether the rhizosphere effect of Arabidopsis is different from other plant species that have a less fugitive life history, we compared the root microbiome of Arabidopsis to eight other, later succession plant species from the same habitat. The study included molecular analysis of soil, rhizosphere, and endorhizosphere microbiome both from the field and from a laboratory experiment. Molecular analysis revealed that the rhizosphere effect (as quantified by the number of enriched and depleted bacterial taxa) was ~35% lower than the average of the other eight species. Nevertheless, there are numerous microbial taxa differentially abundant between soil and rhizosphere, and they represent for a large part the rhizosphere effects of the other plants. In the case of fungal taxa, the number of differentially abundant taxa in the Arabidopsis rhizosphere is 10% of the other species’ average. In the plant endorhizosphere, which is generally more selective, the rhizosphere effect of Arabidopsis is comparable to other species, both for bacterial and fungal taxa. Taken together, our data imply that the rhizosphere effect of the Arabidopsis is smaller in the rhizosphere, but equal in the endorhizosphere when compared to plant species with a less fugitive life history.
Subject terms: Microbial ecology, Plant ecology
Introduction
Over the past decade, the number of studies on plant-associated microbial communities has increased substantially aiming to understand how plant microbiomes support plant growth and health [1, 2]. The composition of microbial communities in the rhizosphere and to a greater extent in apoplastic spaces inside the root (here defined as the “endorhizosphere”) is markedly different from that in bulk soil. This phenomenon is referred to as the “rhizosphere effect” [3, 4]. Numerous studies have shown that the strength of the rhizosphere effect depends on a number of factors. One of these factors is the plant genotype, which suggests that the capacity of plants to shape the root microbiome has differentially evolved [5–11]. How this evolution works, and what the underlying mechanisms are is largely not known and studying this is challenging because the root microbiome comprises thousands of taxa among which are commensals, pathogens, and beneficial microorganisms. For example, whether plants can establish interactions with beneficial microbes in such complex settings, depends on their ability to promote these while not favoring pathogens in the root microbiome. Gaining insight into the molecular mechanisms underlying these processes requires deeper understanding of the plant genetic properties that can influence microbial preference, colonization, and performance.
Molecular plant-microbiome interaction studies can benefit greatly from working with a molecularly well-studied model plant species, such as Arabidopsis thaliana (Arabidopsis). For example, a study using hypo- and hyperimmune Arabidopsis mutant plants showed that salicylic acid plays a role in shaping the root microbiome [12], while another study on Arabidopsis revealed that the phosphate stress response alters the root-associated microbial community [10]. One of the major criteria to select Arabidopsis as a model for genetic studies, is its short life span, which is a general characteristic of fugitive plant species from early succession habitats [13]. However, this fugitive life history of Arabidopsis [14] might be a disadvantage when considering its rhizosphere effect as a blueprint to understand how other plant species may steer their rhizosphere microbiome composition. Ecological studies have shown that plant species with fugitive life histories invest in growth over defense, while slower growing species may invest more in defense mechanisms, as well as interactions with soil microbes [15–19]. This might imply that pioneer plants have a relatively small rhizosphere effect. However, very few, if any studies have compared the rhizosphere effect of Arabidopsis with that of plant species with a longer life history.
In the present study, we tested the hypothesis that Arabidopsis has a smaller rhizosphere effect than species with a longer life history. We measured the changes from bulk soil to the two plant “compartments”: rhizosphere and endorhizosphere—for both the bacterial and fungal community of Arabidopsis. We compared this with eight other plant species that occur in the same ecosystem, but that become more prominent in later stages of vegetation succession. We made the comparisons using plant and soil materials from both a field and a laboratory experiment under controlled conditions.
Results
Study site and experimental setup
We made use of an old field grassland ecosystem in the Veluwe area in The Netherlands, where Arabidopsis grows on spots that have opened up and it is surrounded by other plant species with distinct life histories. In this area, named “Mossel” (Figure S1A), ecological studies have been carried out following the initial transition from arable land to semi-natural grassland and the subsequent succession of the plant community [20, 21]. This involved intensive monitoring of the abundance of plant species over time (Fig. 1a). During this secondary successional trajectory, annuals are replaced by perennials and the various components of the soil community become increasingly correlated [18]. For a comparison with Arabidopsis, we selected eight plant species representing a diverse range of taxonomic clades (Figure S1) and of which the relative abundance was recorded over a 20-year time period (Fig. 1a). Immediately after stopping agricultural practices, the populations of the annual field forget-me-not (Myosotis arvensis) and the biannual smooth hawksbeard (Crepis capillaris) quickly increase and then decline with time since cessation of agriculture, while the perennials such as ox-eye daisy (Leucanthemum vulgare) and tansy (Tanacetum vulgare) gradually become more abundant during the first 20 years of succession. For four other species abundance patterns fluctuate over time (Fig. 1a). The cover of Arabidopsis has not been recorded in this long-term study, since the yearly measurements were done in July, almost 2 months after Arabidopsis completes its lifecycle and hence it is no longer detectable in the vegetation. Although Arabidopsis would be expected to disappear soon after land abandonment, the plant species can still be found mostly on bare soil patches created by burrowing animals like wild pigs, hares, and smaller organisms such as ants, which bring up subsoil to the soil surface (Figure S1A). In Spring 2018, we examined the relative area cover of Arabidopsis at plots in a fenced part of the Mossel area, where the experimental field is located. We found that there is a positive relationship between the cover of Arabidopsis in the vegetation and the percentage of bare soil (R2 = 0.72, p < 0.01; Fig. 1b). The fact that Arabidopsis stays in the grassland system even after more than 20 years of secondary succession, enables comparison of the rhizosphere effect of the pioneer Arabidopsis with that of perennial species such as T. vulgare and L. vulgare.
Fig. 1. Species relative abundances change over time.

a The vegetation cover of the plant species changes over time. The total cover of these eight plant species in (A) represent 59% of the total cover of all species in 2017. The annual M. arvensis and the biannual C. capillaris (solid lines) are declining after an presence immediately following land abandonment, while the perennials fluctuate or increase in percentage cover (dashed lines). b Cover of Arabidopsis (y-axis) measured in patches of a fixed size, in relation to the percentage bare soil present in those patches. Measurements were done in Spring 2018 and only plots where Arabidopsis was present were included in the analysis. When Arabidopsis is present, there is a positive relationship between the cover of Arabidopsis in the vegetation and the percentage bare soil. c The ratio of rhizosphere soil / fresh root weight is comparable for all plant species. Plant species are abbreviated as follows: AT Arabidopsis thaliana, CC Crepis capillaris, HP Hypericum perforatum, LC Lotus corniculatus, LV Leucanthemum vulgare, MA Myosotis arvensis, PL Plantago lanceolata, TO Taraxacum officinale, TV Tanacetum vulgare.
On an experimental plot in a recently fenced part of Mossel, where also the long-term field experiments are situated, the local Arabidopsis accession Msl [22] was grown alongside the other eight locally co-occurring plant species (Figure S1C). Bulk soil, rhizosphere, and the endorhizosphere were harvested 6 weeks after planting. The nine plant species yielded different quantities of rhizosphere soil; however, the ratio of fresh root weight and rhizosphere soil weight was comparable in all cases (Fig. 1c). We analysed the bacterial communities by 16 S rDNA amplicon sequencing using the HiSeq 2500 platform (Illumina). We were unable to isolate DNA from the Taraxacum officinale endorhizosphere samples, and therefore excluded these from further analysis. Clustering Operational Taxonomic Units (OTUs) by ≥97% sequence identity and removing low abundant OTUs, produced a dataset with over 25 million reads divided over 142 samples and 3187 OTUs.
Arabidopsis shows a relatively low yet significant bacterial rhizosphere effect
The rhizosphere effect is generally known to result in decreased evenness and richness (alpha diversity) of microbial species [23]. To test whether this is the case for Arabidopsis and the other species in our experimental setup, we calculated the Shannon diversity index of the soil samples and the plant rhizosphere and endorhizosphere samples. We did not find large decreases in alpha diversity in the rhizosphere samples (Fig. 2a), which is in contrast to most, but not all reports on alpha diversity of soil and rhizospheric soil studies [23]. The endorhizosphere of all the plant species reveals a substantial decrease in alpha diversity, indicating an increase in dominant bacterial species.
Fig. 2. Alpha diversity and phylum-level taxonomy indicate a rhizosphere effect in all plant species.

a The alpha diversity of the rhizosphere does not decrease considerably in the rhizosphere, while the endorhizosphere samples all show a significant decrease, indicating the presence of highly abundant species. b The relative abundance of the dominant (more than 1% of the total reads in at least one sample) phyla are shown for each plant species.
We further explored the shifts in the taxonomic composition due to the rhizosphere effect, by calculating the relative abundance of the major phyla in each individual sample. In line with published literature, we observed an increase of Proteobacteria in the rhizosphere of all the plant species [23–25]. In the endorhizosphere, Proteobacteria as well as Actinobacteria showed a considerable increase, while Acidobacteria were reduced. These shifts suggest that there are bacterial taxa that show relative enrichment and depletion in the rhizosphere and the endorhizosphere of all studied plant species (Fig. 2b).
To explore how the rhizosphere microbiome communities differ among plant species, we performed Principal Coordinate Analysis (PCoA). Using the Bray-Curtis dissimilarity measure on a rarefied OTU table, soil and rhizosphere samples were plotted along the first two principal coordinates (PCs). This revealed that the nine plant species tested established distinct bacterial rhizosphere communities in this field experiment (Fig. 3a, left panel). According to the results of pair-wise permutation analysis, the differences are significant (PERMANOVA, p-FDR < 0.01, see Table S1). Compared with the other plant species, the Arabidopsis rhizosphere samples cluster closest to the soil samples. This implies that the bacterial community in the Arabidopsis rhizosphere is less different from soil than that of the other eight plant species. In line with this, the soil-Arabidopsis rhizosphere comparison yielded the lowest R2-value (0.42 for Arabidopsis versus 0.55 for the average of the other plant species, see Fig. 3a). Although the separation on the first PCs and the corresponding R2-value of the Arabidopsis rhizosphere samples illustrate a lower rhizosphere effect, the separation between bulk soil and rhizosphere along the first PC axis was in the same direction for all plant species. This suggests that to a certain extent, the bacterial communities in the rhizosphere are established in a similar manner.
Fig. 3. The Arabidopsis bacterial rhizosphere is substantially changed.

a Principal coordinate analysis of soil and rhizosphere samples (left panel) or soil and endorhizosphere samples (right panel). The Arabidopsis rhizosphere samples are less dissimilar from bulk soil than other rhizosphere samples, while the endorhizospheres of all species are very distinct from bulk soil. Data were rarefied to 150,000 reads/sample (rhizosphere and soil data set) and 11,000 reads/sample (endorhizosphere and soil data set). Only samples with more than 11,000 reads were included. Colored numbers indicate the results of the pair-wise PERMANOVA results. Due to a technical issue, endophytic DNA of Taraxacum offcininale could not be isolated, and hence the results for the endorhizosphere are missing. b Number of affected OTUs. For log2-fold changes from up to six (x-axis), the number of differentially abundant OTUs are plotted as a dotted line (rhizosphere) or solid line (endorhizosphere). Significance was determined by the metagenomeSeq package in R. Enriched OTUs are on the right panel, depleted OTUs on the left. Arabidopsis has the lowest number of differentially detected OTUs in the rhizosphere, while the endorhizosphere values are relatively high. Results are shown for a p value cut-off of 0.01 and only OTUs are included which have a log2-normalized average read count of at least four per sample. c For log2-fold changes equal to or higher than 2 indicated by dotted lines in (b), absolute number of affected OTUs are given.
The PCoA analysis describes differences at the whole-community level. We next investigated whether these differences were also reflected in the number of differentially abundant (affected) OTUs between bulk soil and rhizosphere samples. In principle, the more OTUs that are significantly different from soil (enriched or depleted), the larger the rhizosphere effect. We counted the number of bacterial OTUs that were significantly different (according to the metagenomeSeq package in R) between rhizosphere and bulk soil for a range of log2-fold thresholds (−6 to 6, Fig. 3b). At a threshold of absolute log2-fold change of 2, we found that on average 350 out of 8782 tested OTUs were affected. A strong decline in affected OTUs is recorded when increasing the threshold to absolute log2-fold change of 3, indicating that a majority of the affected OTUs show a moderate change in abundancy (Fig. 3b, dotted lines).
In line with the multivariate analysis, the number of bacterial OTUs significantly affected in the Arabidopsis rhizosphere was lower than in any of the other species, along the whole gradient of fold-changes tested (Fig. 3b, dotted lines). This was the case for both depleted and enriched OTUs. However, at a log2-fold change of 2, it was still ~65% of the average of all other species, and 44% of bird’s-foot trefoil (Lotus corniculatus), which was the species with the highest number of affected bacterial OTUs (539 affected OTUs, see Fig. 2c and Table S2). Since L. corniculatus is the only species in our dataset with the ability to form nitrogen-fixing nodules, we checked the abundance of the taxonomic order Rhizobiales and particularly the genus Mesorhizobium as this is a widely accepted microsymbiont of Lotus species [26]. However, we did not find an overrepresentation of these taxa (See Table S3).
We confirmed our findings by using another statistical method (EdgeR [27]; as implemented by [7]) and by using p values ranging from 0.05 to 0.001 and different abundance filtering cut-offs. This revealed similar trends (Figure S2), indicating that the results were not dependent on the statistical method used. Based on these findings, we conclude that the effect of Arabidopsis on the composition of its rhizosphere bacterial microbiome is substantial, albeit ~ 35% lower when compared with the average value of the other eight plant species.
Arabidopsis is comparably selective to bacterial endophytes
We next examined the difference between the bulk soil and the endorhizosphere microbiomes. All species had established distinct bacterial endorhizosphere microbiomes and these were different from the bulk soil (Fig. 3a, right panel). Along the first PC axis (41% of variance), all endorhizosphere samples separated far from the bulk soil samples in the same direction, while individual species separated along the second PC axis (9% of variance). The R2 values resulting from the pair-wise PERMANOVA between bulk soil and all individual species only varied between 0.74 and 0.80 (p-FDR < 0.01) and the Arabidopsis R2 was 0.80 (Fig. 3a, right panel). The strong separation of soil and endorhizosphere samples was also reflected in the number of affected OTUs. At a log2-fold change threshold of 2, the number of affected OTUs ranged from 1116 (L. corniculatus) to 1293 (P. lanceolate; see Fig. 3c and Table S2). Furthermore, the number of affected OTUs dropped at high fold-changes relative to the rhizosphere-affected OTUs (Fig. 3b, solid lines). This shows that bacterial colonization of the endorhizosphere is far more restrictive than colonization of the rhizosphere. Arabidopsis exhibited a number of differentially abundant OTUs that were very similar to the other plant species (1244 at log2-fold change 2 Fig. 3c), indicating that the Arabidopsis root is comparably different from bulk soil in its endorhizosphere bacterial microbiome.
A majority of plant-enriched bacterial OTUs is shared with Arabidopsis
Plants can affect the composition of the microbiomes in a plant genotype-specific manner [24]. However, they also share affected OTUs [6, 9, 28]. We therefore studied to what extent the Arabidopsis-affected bacterial microbiome includes OTUs shared with the microbiomes of the other plant species. This is a crucial addition for evaluating Arabidopsis as a model, since it could be that the rhizosphere effect of the other plant species are not covered by that of Arabidopsis in terms of affected OTUs. We did this analysis in two different ways. First, we compared all OTUs and then we focused on the top 10 OTUs of the eight species. In both cases this was done for rhizo- and endorhizosphere. We determined for each OTU the log2-fold change between soil and (endo-)rhizosphere for Arabidopsis and each of the other plant species and used pairwise correlation analysis. We reasoned that a significant correlation and high correlation coefficient would show that, in general, OTUs that are affected in Arabidopsis are also affected in other species. Based on log2-fold changes, we found a strong correlation between Arabidopsis and other plant rhizospheres (on average r = 0.71, p < 0.01) and a very strong correlation between Arabidopsis and other plant endorhizospheres (on average r = 0.93, p < 0.01; Table S4).
The correlation of the fold changes gives similar weight to high and low abundant OTUs, regardless of whether they are affected by the rhizosphere effect. To examine whether Arabidopsis is also similar to the other plant species in the highly abundant and significantly changed OTUs, we chose the ten most abundant significantly enriched OTUs (“top 10”) of each plant species and tested whether they were enriched in Arabidopsis. The rhizosphere top 10 s of the eight (non-Arabidopsis) plant species together yielded 30 enriched OTUs, because a number of the OTUs were shared among species. For each plant species, at least 5 of the 10 enriched rhizosphere OTUs were also significantly enriched in Arabidopsis (Fig. 4, upper panel). Furthermore, 16 out of the total of 30 abundant OTUs are also enriched in the Arabidopsis rhizosphere. Of these 16, five bacterial OTUs were also present in the top ten of enriched OTUs in Arabidopsis.
Fig. 4. A majority of highly abundant plant-enriched bacterial OTUs is shared with Arabidopsis.

For each plant species (“y-axis”), the top 10 most abundant enriched bacterial OTUs from the rhizosphere (circles) and the endorhizosphere (squares) are plotted according to their mean relative abundance (x-axis). Colored filled shapes indicate OTUs also enriched in Arabidopsis, either in the rhizosphere (R), endorhizosphere (E), or both. Black open shapes represent OTUs not significantly enriched in Arabidopsis. The relative abundance of the shared OTUs in the soil is presented in the bottom row of each panel. Numbers inside each plot indicate how many OTUs in the top 10 are shared between the respective plant species and Arabidopsis. Arrowheads in the legend panel indicate OTUs that are also part of the Arabidopsis top 10 enriched OTUs. AT A. thaliana, CC C. capillaris, HP H. perforatum, LC L. corniculatus, LV L. vulgare, MA M. arvensis, PL P. lanceolata, TO T. officinale, TV T. vulgare.
In the endorhizosphere, a high number of OTUs are enriched in multiple species, and the top 10 s of most abundant enriched OTUs were encompassing 22 OTUs, of which 17 were shared with Arabidopsis. In line with this, the number of abundant enriched OTUs in the endorhizosphere that are shared with the Arabidopsis top 10 OTUs was higher than in the rhizosphere (Fig. 4, lower panel; on average 7.4 out of 10 in the endorhizosphere versus 5.4 out of 10 OTUs in the rhizosphere). Eight of these 17 shared OTUs were also present in the top 10 of enriched OTUs in Arabidopsis. Noteworthy, while most of the OTUs were indeed shared, the fold change was not the same in the different plant species and in several cases the top 10 enriched OTUs of the other plant species were less enriched in Arabidopsis.
We also used the top ten most abundant enriched bacterial OTUs in the Arabidopsis compartments and compared their abundance with that of the other plant species. (Figure S3). We found that in the rhizosphere, 9 of the 10 OTUs that were abundant and enriched in Arabidopsis were also enriched in at least one other plant species, but these OTUs were not necessarily present in their top 10. Five out of the 10 OTUs that were most enriched in Arabidopsis were also present in the top 10 enriched rhizosphere OTUs of at least one other plant species. Only OTU_1357 (associated with the genus Agrobacterium) was only enriched in Arabidopsis and not in any of the other plant species. In the endorhizosphere, all top 10 enriched OTUs in Arabidopsis were shared with at least one other plant species. One OTU (associated with the family order Actinomycetales) was not present in any of the other species’ top 10, but this OTU was nonetheless significantly enriched in all the plant endorhizospheres. The strong correlations of log2-fold changes, and the overlap in the 30 most abundant enriched OTUs suggests that the Arabidopsis rhizosphere effect to a large extent represents that of the other plant species tested.
Arabidopsis and the other plants also displayed plant species-specific enrichments in the rhizosphere and endorhizosphere. We next investigated whether and how the plant-specific rhizosphere effects differed between the plant species. In order to do so, we determined for each OTU in how many species (frequency) it was enriched and plotted the number of those OTUs per frequency. As shown in Fig. 5a, the number of enriched OTUs in the rhizosphere decreased with increasing frequency. The number of unique (plant specific) OTUs varies between the plant species, with the lowest number of enriched OTUs for P. lanceolata (4, representing <0.1% of the rhizosphere reads; see Fig. 5a and Table S3) and the highest number for L. corniculatus (77, representing 5.0% of the rhizosphere reads). The taxonomic profile of the L. corniculatus-specific enriched OTUs was again not dominated by taxa that can nodulate legumes, when we compared this to the taxonomic profile of the other plant species (Table S3). Arabidopsis had 15 plant-specific enriched OTUs, which represented 1.1% of the total rhizosphere reads. OTUs that were enriched in all species represented on average 1.7% of the rhizosphere community. In the endorhizosphere, the number of enriched OTUs per frequency increased after an initial decrease, and the highest number of enriched OTUs shared between all plant species (Fig. 5b). The lowest number of plant-specific enriched OTUs was found in T. vulgare (only one OTU with an endorhizosphere relative abundance of <0.1%; Fig. 5b and Table S3), whereas P. lanceolata showed the largest number and cumulative proportion of plant-specific enriched OTUs (37% and 6.0%, respectively). For Arabidopsis, there were 15 species specific enriched OTUs with a total relative abundance of 1.1%. The 156 OTUs that were enriched in all plant species represented on average 38% of the endorhizosphere communities (Table S3), indicating that a large group of bacteria is generally enriched in the plant endorhizosphere.
Fig. 5. OTUs can be affected in only one or in multiple plant species.

For each frequency (number of plant species an OTU is affected in), the number of OTUs is plotted for the rhizosphere (a) and endorhizosphere (b). For the specific enriched bacterial species, a division per plant species is given, the numbers and taxonomy of specific enriched taxa are given in Table S3.
The fungal communities in the rhizosphere and endorhizosphere
Using a similar sequencing approach, based on the ITS2 region, we then analysed the fungal communities of all the samples of the field experiment. As shown in Fig. 6a, the rhizosphere samples separated from the bulk soil, but in contrast to the bacterial dataset, not all in the same direction along the first two PC axes. According to a PERMANOVA, we found that there is a large spread in R2 values of the pair-wise comparisons between bulk soil and individual rhizosphere samples (ranging from 0.19 for Arabidopsis to 0.79 for L. corniculatus, p-FDR < 0.05). As reflected by the relatively low R2 value, the Arabidopsis fungal rhizosphere samples cluster closest to the soil in a PCoA.
Fig. 6. The rhizosphere effect on the fungal community.

Principal coordinate analysis of soil and rhizosphere samples (a) or soil and endorhizosphere samples (b). The separation between soil samples and Arabidopsis rhizosphere samples is the least pronounced when compared with the separation between soil and any of the other plants, which does not apply to the soil versus endorhizosphere separation. Colored numbers indicate the results of the pair-wise PERMANOVA results. c Arabidopsis has the lowest number of affected OTUs in the rhizosphere, but not in the endorhizosphere.
In line with this, the number of OTUs that differed between bulk soil and rhizosphere soil varied greatly between species (Fig. 6b). Arabidopsis and Crepis capillaris show a remarkable low number of affected fungal OTUs in the rhizosphere (for Arabidopsis, 11% of the average of the other plant species at log2-fold 2, and less than 1% of T. vulgare and T. officinale, the two species with the highest fungal rhizosphere effect; Table S2). The spread in both the PCoA and the number of enriched and depleted fungal OTUs indicates that plants are highly diverse in establishing the fungal microbiome.
A PCoA on the fungal microbiome in the soil and the endorhizosphere showed that at the community level, the endorhizosphere of individual species—including Arabidopsis—was comparably different from bulk soil (Fig. 6b). This was also evident from pairwise R2 values, which ranged from 0.37 to 0.46 (PERMANOVA, p-FDR < 0.01; Table S1) and the number of affected fungal OTUs (ranging from 143 to 211 at log2-fold 2; Table S2). When comparing the nine species, Arabidopsis ranked slightly above average, both in R2 value (0.43 versus 0.41 average) and in affected OTUs (172 versus 166 average) indicating that the Arabidopsis endorhizosphere is comparably different from bulk soil (see Fig. 6c). As a Brassicaceous species, Arabidopsis is not able to form symbiosis with arbuscular mycorrhizal fungi (AMF), while the other plant species are able to do so [29, 30]. Interestingly, we found for all endorhizosphere samples comparable relative abundances of the phylum Glomeromycota, which encompasses mainly AMF (on average 0.8%, see Table S3).
A lab experiment reconstitutes the findings of the field experiment
As most studies on mechanisms underlying the rhizosphere effect are performed under controlled conditions, we also performed a laboratory experiment. We grew the same plant species under conditions optimized for Arabidopsis and grew them in soil collected from the Mossel field one year earlier than the above described field experiment. First, we analysed the bacterial community in the rhizosphere and endorhizosphere samples as separate sample sets using PCoA. We found again that plant-specific microbiomes are established (65% of variance explained by constrained PCoA, p < 0.001, result not shown) and that the Arabidopsis rhizosphere samples cluster closest to the bulk soil (Figure S4A, left panel). The numbers of affected OTUs are in line with the PCoA: the species that cluster closer to bulk soil—namely Arabidopsis and M. arvensis—are also the species with a lower number of affected OTUs in the rhizosphere (Figure S4B). For example, at a log2-fold change of 2, Arabidopsis has ~50 affected OTUs, while the species with the highest number (L. vulgare) has ~150 (Figure S5). In contrast, analysing the endorhizosphere of Arabidopsis showed that both the clustering in a PCoA and the number of affected bacterial OTUs were similar to that of the other plant species (Figure S5A, B). This shows that also under laboratory conditions, the selectivity of the Arabidopsis endorhizosphere compartment is comparable to that of the other plant species tested.
To determine whether, and if so how, the lab experiment differs from the field experiment, we analysed the bacterial communities of all samples using PCoA (Fig. 7a). The first PC axis explained 36% of the variance and separated the samples according to compartment, but not according to field and lab experiment. Along the second PC axis, the two experiments were separated, but this separation explained a relatively low amount of variance (7%). This indicates that based on whole communities, the bacterial microbiomes of all compartments in the lab and the field experiment are comparable. Interestingly, the communities in the endorhizosphere and to a lesser extent in the rhizosphere differed more between the experiments than the starting soils did. This indicates that the plant-related bacterial communities established, to some extent, differently in the field and lab experiment and that the starting soils in these experiments are not the main determinant for this difference.
Fig. 7. The rhizosphere effect can be reconstituted in a laboratory experiment.

a Principal coordinate analysis of soil, rhizosphere and endorhizosphere samples of all the plant species (not per species indicated). The first PC axis separates the compartment. The second axis separates the lab and the field experiment with a relatively low amount of variance explained. b Each significantly affected OTU from the field experiment (see Fig. 3b, c) was tested for significance in the lab experiment. The absolute number of affected OTUs in the field experiment is indicated by the colored numbers at the bottom, and overlap with the laboratory experiment is plotted as a percentage of the total number of affected OTUs. The percentage of overlapping OTUs increases with log2-fold thresholds (1–8, x-axis), indicating that strongly affected OTUs are more likely to be affected in the lab experiment as well. Colors represent different plant rhizospheres (dotted lines) or endorhizospheres (solid lines). AT A thaliana, CC C. capillaris, HP H. perforatum, LC L. corniculatus, LV L. vulgare, MA M. arvensis, PL P. lanceolata, TO T. officinale, TV T. vulgare.
To obtain more quantitative insight in the differences between the field and lab study, we examined whether the OTUs that changed in the field experiment also changed in the lab experiment. For this, we compared each affected OTU in the field experiment with the same OTU in the lab experiment (Fig. 7b). For a range of log2-fold changes we counted the absolute number of OTUs that were affected in the field experiment (colored numbers in Fig. 7b) and plotted the fraction that is also affected in the lab experiment. Especially at higher log2-fold changes, the majority of affected OTUs are captured by the lab experiment. This is especially the case in the endorhizosphere, where at a log2-fold change of 6, almost all OTUs that changed in the field experiment also changed in the lab experiment. This indicates that – despite the subtle differences at the community level shown by the PCoA – the plants in the field and under lab conditions appeared to enrich largely the same microbial taxa.
Discussion
This study aimed to quantify the rhizosphere effect of Arabidopsis and to compare this effect with that of other plant species that occur in the same grassland ecosystem. Comparative microbiome studies on plant species originating from the same ecological site have been performed before [28, 31] but to the best of our knowledge, there has not yet been made an explicit comparison between the rhizosphere effect of Arabidopsis with that of plant species that have a less fugitive life history. Based on comparative analysis in the field as well as under controlled conditions, we conclude that the Arabidopsis rhizosphere microbiome significantly differed from soil, albeit to a lesser extent than that of plant species with a longer life history (as summarized in Fig. 8). Enriched bacterial taxa that are highly abundant in the rhizosphere of other plant species are for the larger part also enriched in Arabidopsis. The lower rhizosphere effect was not observed in the endorhizosphere, where a similar number and phylogenetic profile of bacterial taxa was enriched in all plant species, including Arabidopsis. The changes that were observed in the field experiment, broadly corresponded to what was observed in the laboratory experiment, especially for highly enriched bacterial taxa.
Fig. 8. Quantitative comparison of the Arabidopsis rhizosphere effect.
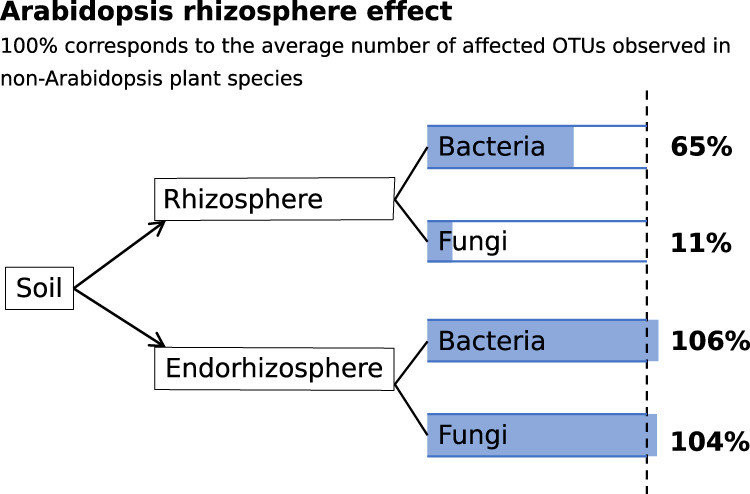
Arrows from soil to either rhizosphere or endorhizosphere indicate the change in the community as measured by affected OTUs, i.e., the rhizosphere effect. Percentages indicate the number of affected OTUs relative to the average of the other plant species.
The composition of the belowground microbiome of a plant is determined by a hierarchy of events. The first determinant is the initial microbial composition and heterogeneity of the soil. Then, plant species properties including root morphology, developmental stage, and rhizodeposition, determine which microbial species out this reservoir will be present in the root microbiomes [2]. Although part of the rhizosphere effect (as measured by enriched microbial taxa) seemed to be conserved for all the plant species investigated, we did in our study observe rhizosphere effects that varied between the plant species. This was especially the case for the fungal rhizosphere microbiome, where the magnitude of the rhizosphere effect had a broad range. Other studies have also shown that host properties have a stronger influence on plant-fungal communities than plant-bacterial communities [32, 33]. In the endorhizosphere, we did not observe differences in fungal reads affiliated to the phylum Glomeromycota (encompassing AMF) between Arabidopsis and the other species (Table S3). It could be that in general AMF are not highly abundant in the endorhizosphere of the tested plant species, due to relatively high phosphate levels [34] in the soil (926 mg/kg total phosphorus; based on a single measurement, results not shown), which are a remnant of the agricultural practices. In addition, it has been shown that AMF are capable of colonizing Arabidopsis roots [35], which could also in part explain the equal amounts of Glomeromycota in the roots of all plant species.
Of the bacterial and fungal rhizosphere communities, Arabidopsis was the closest to bulk soil and showed therefore by definition the smallest rhizosphere effect. Arabidopsis is a small plant and carbon release from the roots (e.g., low-molecular weight carbohydrates) is believed to be a largely passive process [36, 37] that depends on plant size. However, it is unlikely that only carbon release explains the differences between the strengths of the rhizosphere effect among the plant species. This is supported by a study showing that Pseudomonas cell numbers in rhizosphere soil of both small Arabidopsis plants and large Tobacco plants are 10- to 100-fold higher than those in bulk soil [38]. More likely, active secretion of phytochemicals is a factor by which the observed differences can be explained [39–41]. It has been shown that Arabidopsis secretes phytochemicals (triterpenes) that to a certain extend shape the rhizosphere composition [11]. Our results suggest that other plant species may invest more in the secretion of such phytochemicals, although this remains to be demonstrated.
Notably, Arabidopsis and the other eight plant species used in this study have different life histories and grow during different time periods of the year. For example, at the Mossel area, Arabidopsis is present during a period of ~6 weeks in the vegetation during early April and late September (personal observations), while the field experiment was conducted in July. Thus, the timing of the experiment is not “optimal” for all species, and the conditions were not optimised for any of the plant species, including Arabidopsis. Given the unpredictable conditions inherent to a field experiment, and the high probability of a changing soil community, we chose to grow them simultaneously under the same field conditions. However, due to their different life histories, not all species were at the same developmental stage at the time of harvesting. To alleviate some of those concerns, we decided to grow the plants also under conditions more optimal for Arabidopsis, i.e. in a lab. In addition, most studies on genetic mechanisms (e.g., mutant analyses) influencing the Arabidopsis (bacterial) root microbiome, are carried out under controlled conditions [10–12, 42–44]. Because we grew the plant species in both a field and laboratory setting, we were able to directly compare significantly affected bacterial taxa in the field setting, with the same OTUs in the laboratory setting. We found differences on the community level as well as differentially abundant OTUs (see Fig. 7a, b), indicating that lab studies cannot completely substitute field studies [45, 46]. However, the majority of OTUs in the bacterial data set showed similar trends in the field and the lab experiment, and this was the case for all the plant species. This suggests that our experimental setup did not play a major role in determining the strength of the rhizosphere effect. Furthermore, since the starting soil and growth conditions were different, our data indicate robust plant species-related rhizosphere effects for the plant species tested, including Arabidopsis. Interestingly, although the magnitude (number of enriched OTUs) in endorhizosphere is larger, the number of OTUs that were uniquely affected versus the number of OTUs affected in all species was higher in the rhizosphere (see Fig. 5). This is remarkable, because we would expect that plant-specific effects would be most pronounced in the endorhizosphere [28, 47]. This could be an indication that in our experimental system where plants are naturally occurring in the ecosystem, the rhizosphere effect is relatively specific. To investigate this further, growing the different plant species in varying soil types (with different “initial microbiomes”) would be a valuable future addition to our research.
In addition to shared plant-enriched OTUs we also found plant-specific effects, both in the rhizosphere and the endorhizosphere. L. corniculatus shows the highest number of plant-specific enriched OTUs, but we did not find known L. corniculatus root nodule symbionts that were specifically high in L. corniculatus. This is most likely due to the fact that we removed the nodules from the roots before we sequenced the endorhizosphere samples. A study by Zgadzaj et al. (2016) also showed a relatively high rhizosphere effect for Lotus species, and that study also excluded symbiotic structures [7]. The relatively high number of L. corniculatus specific OTUs could be indirectly related to its ability to fix nitrogen. Being better supplied with nitrogen, more nutrients or phytochemicals might be secreted by roots and this could drive niche occupation by more bacterial species.
An emerging follow up question is whether there is a phylogenetic signal which determines microbial composition, and whether Arabidopsis would follow this plant related signal. Previous work has shown a significant effect of plant lineage, especially on the composition of the endorhizosphere [28, 48] while other studies have shown the predictive value of specific plant traits such as root exudation profiles and root morphology [49]. In general, such studies require a broad, but congenic set of plant species and this goes beyond the scope of this study. Future work could include such a set of plants to explore the effect of plant lineage or life history. As an example, in Fig. 6, we saw a clear split along the first PC, separating the fungal communities of L. corniculatus and L. vulgare, but also such a split was observed between replicates of M. arvensis and H. perforatum. Because of the limited number of plant species, we were unable to link this separation to life history characteristics or other plant properties. For now, the underlying reason why samples from different plant species – and replicates of a single plant species—separate in such a rigorous manner, remains unclear.
In conclusion, our data show that there is a substantial rhizosphere effect in a diverse group of plant species grown in their native soil in a field setting. A laboratory setup could for the larger part repeat these findings, indicating a robust rhizosphere effect, and therefore lab experiments are a powerful setup to investigate the mechanisms by which microbiomes are established. According to our data, Arabidopsis has a relatively small though significant effect in the rhizosphere, whereas the effect on the endorhizosphere is similarly strong compared with the eight other species investigated. Therefore, we argue that many questions related to the rhizosphere effect can be addressed in Arabidopsis. Notably, Arabidopsis does not represent a common, shared fungal microbiome in the rhizosphere soil. The variation in composition of all the fungal rhizosphere communities suggest that it could be challenging to find an accurate model in general, and that it will be highly dependent on the research questions. Arabidopsis does have the advantage of being a well-studied model system with optimal molecular genetic tools available. In this respect, the relative low number of fungi in its rhizosphere might in some specific cases even be an advantage, as studies on molecular mechanisms underlying the interactions with the rhizosphere community can benefit from such relatively low complexity. Our data are a valuable addition to evaluate Arabidopsis as a model system, since it is widely used when (genetic) mechanisms are studied in relation to root microbiome establishment.
Materials and methods
Relative area cover measurements
We used an ex-arable field at the Veluwe area in the Netherlands (coordinates: “N52°03'35.5” “E5°45'06.4”). This area is a former agricultural site. Agricultural practices were ceased in 1996, and subsequently the area was restored as natural grassland. Part of this area is fenced. Annually, since 1997 the% cover of each species was recorded in 12 1 × 1 m permanent quadrates in each of five 10 × 10 m plots as described previously [50]. Only “NC” plots (plots that were naturally colonized, and not sown initially) were included. Arabidopsis cover was measured in mid-April 2018 as follows: in five NC replicate plots [50], ten vegetation recordings (25 × 25 cm) were taken along a diagonal line through each plot. Bare soil percentage as well as percentage Arabidopsis cover was scored.
Soil collection and preservation
Soil was collected at two time points, one for the lab experiment (April 2015) and one for the field experiment (May 2016) to pre-germinate seeds. Samples were taken from four spots within a radius of 100 m. When any vegetation was present, the top 5–10 cm was removed. Soil was sieved (2.5 mm), homogenized and stored at 4 °C until use.
Seed collection and treatment
Seeds of Arabidopsis Msl accession originate from wild plants at the Mossel location. Msl seeds used in this study originate from a single wild Arabidopsis thaliana plant after two rounds of propagation (F3). T. vulgare and L. vulgare seeds were collected at the Mossel field site. Seeds from the other species were obtained from De Cruydt Hoeck v.o.f in Abbendijk, the Netherlands, a provider of seeds of wild plants. All seeds were sterilized in 4x diluted household bleach for 10 min, washed seven times with sterile MQ water, transferred to plates with a wet filter paper, placed at 4 °C for 48 h and then moved to a 21 °C incubator in the dark for germination. In advance to the bleach treatment, the seeds of Arabidopsis Msl seeds got a short rinse with 70% ethanol and the seeds of L. corniculatus were treated 2 min with H2SO4. Seeds of T. officinale, T. vulgare, L. vulgare, and M. arvensis did not undergo the cold treatment and were placed directly in the 21 °C incubator.
Experimental set-up and plant growth conditions in the field
Field experiments were conducted in July 2016 at the Mossel area. Prior to growth in the field, plants were pre-germinated on collected Mossel soil under controlled conditions as follows: large particles such as dead roots and stones were removed from collected Mossel soil by hand. Soil was placed in pots (3 × 3 cm) within a tray (96 pots). To remove the endogenous seed population, the tray was watered and placed in the greenhouse for 2 days. After weeding, sterile 3-day-old seedlings on plates were transplanted into the tray. Seven days post transplantation, plants including the soil were planted into to the Mossel field. The experiment was set up as a semi-randomized design (Figure S1). In total, we planted 9 plant species, with 32 individual plants per species. The 32 plants per species were divided over 4 different blocks (8 per block), within an experimental plot of 2 × 5 meters as in Figure S1C. After 6 weeks of growth in the field, plants were harvested by digging 3–4 cm around the base of the plant and 10 cm deep. Plants (including attached soil) were transported to the laboratory for further processing. Four plant individuals were pooled into one sample.
Experimental set-up and plant growth conditions in the laboratory
All plants were grown in triplicate in a laboratory setting at the Wageningen University in May 2015. Pots (12 cm in diameter) were filled with Mossel soil, watered, and left in a growth chamber prior to transplanting of the seeds, to allow endogenous seed population to germinate. After weeding these seedlings, the 3-days-old seedlings from the sterile plates were transplanted, except for the Arabidopsis seeds, which were sown as seeds directly on the soil. The number of seedlings per pot varied from three for large species and five for small species. Pots were arranged a growth chamber (Temperature: 21.0 °C, humidity: 70.0%; 10.06 gr/kg, light intensity: 120 μMol/m2) and the position of the pots was regularly changed. Soil humidity was maintained at ~70% of the maximum water holding capacity by weighing the pots and watering three times a week. All plants were grown for 4–5 weeks until flowering, or until the moment that roots were seen through the holes in the bottom of the pot. Soil samples were collected from pots that contained unplanted Mossel soil and were harvested after 4 weeks.
Soil, rhizosphere, and endorhizosphere harvesting
Pots containing the plants were turned upside down which removed to plant including soil. Loose soil was removed from the roots by kneading and shaking by hand and by patting the roots on the back of a gloved hand. The soil that was sticking tightly to the roots was defined as rhizosphere soil. The harvesting protocol closely followed the procedures described previously [5, 6], with minor modifications, as described here. The entire root including the rhizosphere soil was put into a 50 mL Falcon tube containing 25 mL phosphate buffer (PB, per litre: 6.33 g NaH2PO4.H2O, 10.96 g Na2HPO4.2H2O and 200 μL Silwet L-77) and vortexed for 15 s. Next, the root was transferred to a new Falcon tube containing PB, and briefly vortexed. This procedure was repeated twice, until the PB stayed clear. Roots were sonicated for 10 cycles of 30 s bursts and 30 s pause. After vortexing briefly, roots were placed on filter paper and brown particles that remained within the root knot after the cleaning procedure was removed as much as possible. Meanwhile, rhizosphere from the first vortexing step was filtered through a 100 μm cell strainer (Falcon) and spun down for 10 min at 4000 × g. Supernatant was quickly poured off, and the pellet was transferred to a 2 mL tube. Endorhizosphere and rhizosphere samples were then weighed, frozen in liquid nitrogen and stored at −80 °C.
DNA isolation and 16 S rDNA amplicon sequencing
DNA from soil and rhizosphere samples was isolated using the Mo Bio PowerSoil kit (Qiagen) according to manufacturer’s instructions. According to the procedures described by Lundberg et al. (2012) DNA from the endorhizosphere samples was isolated using Fast DNA Spin Kit for Soil (MP Biomedicals). Quality and quantity of the DNA was checked by nanodrop and gel electrophoresis. Per sample, around 400 ng was sent for 16 S rDNA sequencing at Beijing Genomics Institute. Using primers 515 F and 806 R, the V4 region of the 16 S gene was sequenced. The laboratory experiment was sequenced using the MiSeq platform, while the field experiment samples from 1 year later were sequenced using the HiSeq sequencing platform.
Processing of the sequencing results (bacterial sequences)
Paired-end reads were merged into contigs using the RDP extension to PANDASeq [51], named Assembler [52] with a minimum overlap of 50 bp, Phred score of 25, and contig length of 100 bp. Contigs were converted to FASTA format using the fastx-toolkit and combined in a single file. Then, contigs were clustered into operational taxonomic units (OTUs) according to the UPARSE pipeline [53] implemented in VSEARCH 1.1.3 [54]. In short, the pipeline consisted of de-replication, sorting by abundance, and discarding singletons before clustering them into OTUs using the UPARSE algorithm [53], discarding chimeric sequences using the UCHIME algorithm [55] and mapping contigs to the OTUs using the usearch_global algorithm. The resulting OTU table was then converted into BIOM format using QIIME 1.9.1 [56]. Finally, we added taxonomic information for each OTU based on the GreenGenes database release 13_8 97 [57] using the RDP classifier 2.10.1 [58]. All processing steps were implemented in a SnakeMake workflow [59].
Processing of the sequencing results (fungal sequences)
Paired-end reads were quality trimmed to Q25 using the Phred algorithm with BBDuk [60]. Primers were removed with Cutadapt 1.18 [61] and reads without flanking primers or a length below 100 bp were discarded. The following steps were carried out with the software package PIPITS 2.4 [62]. First, reads were merged into contigs using VSEARCH 2.13.6 [54] with a minimum overlap length of 20 bp and contig length of 100 bp. Contigs were converted to FASTA format using the fastx-toolkit 0.0.14 and combined into a single file. Next, reads were dereplicated with VSEARCH [54]. The fungal ITS2 regions were extracted from the dereplicated sequences and reoriented with the tool ITSx 1.1b [63]. Singletons were removed and the extracted ITS2 regions were clustered at 97% identity. Chimeras were removed from the OTUs by screening against the UCHIME 28.06.2017 reference dataset [55]. The reinflated fungal ITS2 sequences were mapped to the non-chimeras. The preceding steps were all carried out with VSEARCH [54]. A custom PIPITS script was used to generate an OTU table. Taxonomy was assigned with the RDP classifier 2.0.2 [58] and the UNITE (02.02.2019) database [64]. All processing steps were implemented in a SnakeMake workflow [59].
Filtering of the OTU table
All analyses were executed in the R environment. The bacterial dataset (field and lab experiment) was processed as follows: first, OTUs related to mitochondrial and chloroplast sequences were removed, as were the OTUs that did not have 25 reads in at least 5 samples. This significantly reduced the read count of most endorhizosphere samples (due to plastid ribosomal RNA amplification). Therefore, we rarefied the OTU table to 11,000 reads for the field experiment and 2,480 for the lab experiment. This resulted in the exclusion of four M. arvensis endorhizosphere samples of the field experiment, and one T. officinale and one L. vulgare endorhizosphere sample of the lab experiment. The fungal dataset of the field experiment was processed in a similar way: the dataset was also rarefied to 10,000 reads per sample, resulting in the exclusion of two H. perforatum rhizosphere samples and two M. arvensis endorhizosphere samples. The number of discarded reads are given in Table S5.
Multivariate analysis and pairwise adonis
For each experiment, the OTU table was split into a “Rhizosphere” en “Endorhizosphere” dataset, both including the bulk soil samples. Using Bray-Curtis dissimilarity matrix, PCoA was executed, using a custom implementation of the publicly available scripts [7], largely based on the vegan package. Using the function adonis, multiple pairwise comparisons were done and corrected by the FDR method.
Differential abundance testing
Using a standard pipeline as implemented in the metagenomeSeq package in R [65], all OTUs were tested for differential abundance between soil and rhizosphere, and soil and endorhizosphere. We used the default settings to create the normalized measurable OTU table, resulting in 8782 OTUs that were tested for differential abundance using the fitZig function and default settings in the metagenomeSeq package. Next to that, we tested for differential abundance using the EdgeR package in R [27] based on custom scripts of a previously published pipeline (Zgadzaj et al., 2016). In this analysis, OTUs were included that were observed in 5 samples with at least 25 reads. (resulting in 3187 OTUs). Both analysis pipelines showed similar trends (Figure S2). Because of the low number of replicates, the metagenomeSeq package was not used for the lab experiment. Instead, we used the EdgeR implementation by Zgadzaj et al. (2016).
Supplementary information
Acknowledgements
We would like to thank Nicole van Dam for pointing out the Mossel area to study Arabidopsis in the most ecologically relevant setting. We would like to thank Victor Carrion for valuable help with the statistical tests using metagenomeSeq package in R. We would like to acknowledge Ruben Garrido-Oter for making his custom R-scripts publicly available. Thanks to Liesje Mommer for valuable comments on the fungal data. Furthermore, we would like to thank Elizabeth Prins and Aliesje Schneijderberg for helping with the harvesting of the field experiment.
Funding
This research was funded by ERC grant number 3100000843.
Author contributions
MS, XC, and CF executed the field and lab experiment. MdH, RvV, and LS constructed and implemented the bioinformatics pipelines. MS conducted in silico analyses. RH measured the relative area cover of Arabidopsis. MB and WvdP provided species abundance data and helped setting up this study. MS, MB, RG, and TB wrote the manuscript.
Data availability
The raw sequencing reads are available online under accession number PRJNA605923. The OTU table and custom R scripts are available upon request.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xu Cheng, Email: xu.cheng@wur.nl.
Ton Bisseling, Email: ton.bisseling@wur.nl.
Supplementary information
The online version of this article (10.1038/s41396-020-0695-2) contains supplementary material, which is available to authorized users.
References
- 1.Finkel OM, Castrillo G, Herrera Paredes S, Salas González I, Dangl JL. Understanding and exploiting plant beneficial microbes. Curr Opin Plant Biol. 2017;38:155–63. doi: 10.1016/j.pbi.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH. Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol. 2013;11:789–99. doi: 10.1038/nrmicro3109. [DOI] [PubMed] [Google Scholar]
- 3.Hartmann A, Schmid M, van Tuinen D, Berg G. Plant-driven selection of microbes. Plant Soil. 2009;321:235–57. [Google Scholar]
- 4.PAHM Bakker, Berendsen RL, Doornbos RF, PCA Wintermans, CMJ. Pieterse. The rhizosphere revisited: root microbiomics. Front Plant Sci. 2013;4:165. doi: 10.3389/fpls.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- 6.Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zgadzaj R, Garrido-Oter R, Jensen DB, Koprivova A, Schulze-Lefert P, Radutoiu S. Root nodule symbiosis in Lotus japonicus drives the establishment of distinctive rhizosphere, root, and nodule bacterial communities. Proc Natl Acad Sci. 2016;113:E7996–E8005. doi: 10.1073/pnas.1616564113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haney CH, Samuel BS, Bush J, Ausubel FM. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat Plants. 2015;1:15051. doi: 10.1038/nplants.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-Jaramillo JE, Carrión VJ, Bosse M, Ferrão LFV, De Hollander M, Garcia AAF, et al. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017;11:2244–57. doi: 10.1038/ismej.2017.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castrillo G, PJPL Teixeira, Paredes SH, Law TF, De Lorenzo L, Feltcher ME, et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature. 2017;543:513–8. doi: 10.1038/nature21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang AC, Jiang T, Liu YX, Bai YC, Reed J, Qu B, et al. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science. 2019;364:eaau6389. doi: 10.1126/science.aau6389. [DOI] [PubMed] [Google Scholar]
- 12.Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–4. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- 13.Kardol P, De Deyn GB, Laliberté E, Mariotte P, Hawkes CV. Biotic plant-soil feedbacks across temporal scales. J Ecol. 2013;101:309–15. [Google Scholar]
- 14.Pigliucci M. Ecology and evolutionary biology of Arabidopsis. Arabidopsis Book. 2002;1:e0003. doi: 10.1199/tab.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warembourg FR. The’ rhizosphere effect’: a plant strategy for plants to exploit and colonize nutrient-limited habitats. Bocconea. 1997;7:187–94. [Google Scholar]
- 16.Deyn GBD. Plant life history and above–belowground interactions: missing links. Oikos. 2017;126:497–507. [Google Scholar]
- 17.Hakes AS, Cronin JT. Successional changes in plant resistance and tolerance to herbivory. Ecology. 2012;93:1059–70. doi: 10.1890/10-1637.1. [DOI] [PubMed] [Google Scholar]
- 18.Morriën E, Hannula SE, Snoek LB, Helmsing NR, Zweers H, De Hollander M, et al. Soil networks become more connected and take up more carbon as nature restoration progresses. Nat Commun. 2017;8:14349. doi: 10.1038/ncomms14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheres B, Van Der Putten WH. The plant perceptron connects environment to development. Nature. 2017;543:337–45. doi: 10.1038/nature22010. [DOI] [PubMed] [Google Scholar]
- 20.Van der Putten WH, Mortimer SR, Hedlund K, Van Dijk C, Brown VK, Lepä J, et al. Plant species diversity as a driver of early succession in abandoned fields: a multi-site approach. Oecologia. 2000;124:91–99. doi: 10.1007/s004420050028. [DOI] [PubMed] [Google Scholar]
- 21.Bezemer TM, Van der Putten WH. Diversity and stability in plant communities. Nature. 2007;446:E6–7. doi: 10.1038/nature05749. [DOI] [PubMed] [Google Scholar]
- 22.Schneijderberg M, Schmitz L, Cheng X, Polman S, Franken C, Geurts R, et al. A genetically and functionally diverse group of non-diazotrophic Bradyrhizobium spp. colonizes the root endophytic compartment of Arabidopsis thaliana. BMC Plant Biol. 2018;18:61. doi: 10.1186/s12870-018-1272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vieira S, Sikorski J, Dietz S, Herz K, Schrumpf M, Bruelheide H, et al. Drivers of the composition of active rhizosphere bacterial communities in temperate grasslands. ISME J. 2019;14:463–75. doi: 10.1038/s41396-019-0543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinhold-Hurek B, Bünger W, Burbano CS, Sabale M, Hurek T. Roots shaping their microbiome: global hotspots for microbial activity. Annu Rev Phytopathol. 2015;53:403–24. doi: 10.1146/annurev-phyto-082712-102342. [DOI] [PubMed] [Google Scholar]
- 25.Hacquard S. Disentangling the factors shaping microbiota composition across the plant holobiont. N. Phytol. 2016;209:454–7. doi: 10.1111/nph.13760. [DOI] [PubMed] [Google Scholar]
- 26.Lorite MJ, Estrella MJ, Escaray FJ, Sannazzaro A, Videira E Castro IM, et al. The Rhizobia-Lotus symbioses: deeply specific and widely diverse. Front Microbiol. 2018;9:2055. Frontiers Media S.A., 9 [DOI] [PMC free article] [PubMed]
- 27.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson MTJ, et al. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc Natl Acad Sci. 2018;28:E1157 LP–E1165. doi: 10.1073/pnas.1717617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosme M, Fernández I, Van der Heijden MGA, Pieterse CMJ. Non-Mycorrhizal Plants: The Exceptions that Prove the Rule. Trends Plant Sci. 2018;23:577–87. doi: 10.1016/j.tplants.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Demars BG, Boerner REJ. Vesicular arbuscular mycorrhizal development in the Brassicaceae in relation to plant life span. Flora. 1996;191:179–89. [Google Scholar]
- 31.Dawson W, Hör J, Egert M, van Kleunen M, Peste M. A small number of low-abundance bacteria dominate plant species-specific responses during rhizosphere colonization. Front Microbiol. 2017;8:975. doi: 10.3389/fmicb.2017.00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergelson J, Mittelstrass J, Horton MW. Characterizing both bacteria and fungi improves understanding of the Arabidopsis root microbiome. Sci Rep. 2019;9:24. doi: 10.1038/s41598-018-37208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hannula SE, Kielak AM, Steinauer K, Huberty M, Jongen R, De Long JR, et al. Time after time: temporal variation in the effects of grass and forb species on soil bacterial and fungal communities. MBio. 2019;10:e02635–19. doi: 10.1128/mBio.02635-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Der Wal A, De Boer W, Lubbers IM, Van Veen JA. Concentration and vertical distribution of total soil phosphorus in relation to time of abandonment of arable fields. Nutr Cycl Agroecosystems. 2007;79:73–79. [Google Scholar]
- 35.Veiga RSL, Faccio A, Genre A, Pieterse CMJ, Bonfante P, van der Heijden MGA. Arbuscular mycorrhizal fungi reduce growth and infect roots of the non-host plant Arabidopsis thaliana. Plant Cell Environ. 2013;36:1926–37. doi: 10.1111/pce.12102. [DOI] [PubMed] [Google Scholar]
- 36.Jones DL, Nguyen C, Finlay RD. Carbon flow in the rhizosphere: carbon trading at the soil-root interface. Plant Soil. 2009;321:5–33. [Google Scholar]
- 37.Hirsch PR, Miller AJ, Dennis PG. Do root exudates exert more influence on rhizosphere bacterial community structure than other rhizodeposits? Mol Micro Ecol Rhizosph. 2013;1:229–42. [Google Scholar]
- 38.Micallef SA, Shiaris MP, Colón-Carmona A. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J Exp Bot. 2009;60:1729–42. doi: 10.1093/jxb/erp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.PAHM Bakker, Berendsen RL, Doornbos RF, PCA Wintermans, CMJ Pieterse. The rhizosphere revisited: Root microbiomics. Front Plant Sci. 2013;4:165. doi: 10.3389/fpls.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan J, Zhao J, Wen T, Zhao M, Li R, Goossens P, et al. Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome. 2018;6:156. doi: 10.1186/s40168-018-0537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berendsen RL, Vismans G, Yu K, Song Y, De Jonge R, Burgman WP, et al. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018;12:1496–507. doi: 10.1038/s41396-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Badri DV, Quintana N, El Kassis EG, Kim HK, Choi YH, Sugiyama A, et al. An ABC transporter mutation alters root exudation of phytochemicals that provoke an overhaul of natural soil microbiota. Plant Physiol. 2009;151:2006–17. doi: 10.1104/pp.109.147462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stringlis IA, Yu K, Feussner K, De Jonge R, Van Bentum S, Van Verk MC, et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc Natl Acad Sci USA. 2018;115:E5213–E5222. doi: 10.1073/pnas.1722335115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MJEEE Voges, Bai Y, Schulze-Lefert P, Sattely ES. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc Natl Acad Sci USA. 2019;116:12558–65. doi: 10.1073/pnas.1820691116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sergaki C, Lagunas B, Lidbury I, Gifford ML, Schäfer P. Challenges and approaches in microbiome research: from fundamental to applied. Front Plant Sci. 2018;9:1205. doi: 10.3389/fpls.2018.01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sessitsch A, Pfaffenbichler N, Mitter B. Microbiome applications from lab to field: facing complexity. Trends Plant Sci. 2019;24:194–8. doi: 10.1016/j.tplants.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Bulgarelli D, Schlaeppi K, Spaepen S, van Themaat EVL, Schulze-Lefert P. Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol. 2013;64:807–38. doi: 10.1146/annurev-arplant-050312-120106. [DOI] [PubMed] [Google Scholar]
- 48.Yeoh YK, Dennis PG, Paungfoo-Lonhienne C, Weber L, Brackin R, Ragan MA, et al. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nat Commun. 2017;8:215. doi: 10.1038/s41467-017-00262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilschut RA, van der Putten WH, Garbeva P, Harkes P, Konings W, Kulkarni P, et al. Root traits and belowground herbivores relate to plant–soil feedback variation among congeners. Nat Commun. 2019;10:1–9. doi: 10.1038/s41467-019-09615-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukami T, Bezemer TM, Mortimer SR, Van Der Putten WH. Species divergence and trait convergence in experimental plant community assembly. Ecol Lett. 2005;8:1283–90. [Google Scholar]
- 51.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinforma. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Sun Y, Cole JR. A scalable and accurate targeted gene assembly tool (SAT-Assembler) for next-generation sequencing data. PLoS Comput Biol. 2014;10:e1003737. doi: 10.1371/journal.pcbi.1003737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 54.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koster J, Rahmann S. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 60.Bushnell B, Rood J, Singer E. BBMerge – Accurate paired shotgun read merging via overlap. PLoS One. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- 62.Gweon HS, Oliver A, Taylor J, Booth T, Gibbs M, Read DS, et al. PIPITS: an automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol Evol. 2015;6:973–80. doi: 10.1111/2041-210X.12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. 2013;4:n/a–n/a. [Google Scholar]
- 64.Nilsson RH, Larsson K-H, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47:D259–D264. doi: 10.1093/nar/gky1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paulson JN, Colin Stine O, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–2. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing reads are available online under accession number PRJNA605923. The OTU table and custom R scripts are available upon request.