

Abstract
Hepatic fibrosis is an inflammatory response that leads to liver cirrhosis in the most advanced condition. Liver cirrhosis is a leading cause of deaths associated with liver diseases; hence, understanding the underlying mechanisms of hepatic fibrosis is critical to develop effective therapies. Tripartite motif (TRIM) family proteins have been shown to be involved in liver fibrosis; however, the exact role of several TRIM proteins in this process remained unexplored. In this study, we investigated the role of TRIM37 in hepatitis B virus (HBV)-associated hepatic fibrosis. We analyzed TRIM37 expression in hepatic fibrosis patients and performed functional and mechanistic studies in tissue culture and mouse models to identify the role of TRIM37 in hepatic fibrosis. We found an increased expression of TRIM37 in hepatic fibrosis patients. Mechanistically, we showed that TRIM37 physically interacts with SMAD7 and promotes ubiquitination-mediated degradation of SMAD7, and that SMAD7 is a key mediator of TRM37-induced hepatic fibrosis. Furthermore, we showed nuclear factor κB (NF-κB) activation mediated by reactive oxygen species (ROS) is necessary for the transcriptional induction of TRIM37 during HBV infection. Our study shows TRIM37 as an important promoter of HBV-associated hepatic fibrosis.
Keywords: TRIM37, SMAD7, NF-κB, hepatic fibrosis, hepatitis B virus, reactive oxygen species
Graphical Abstract
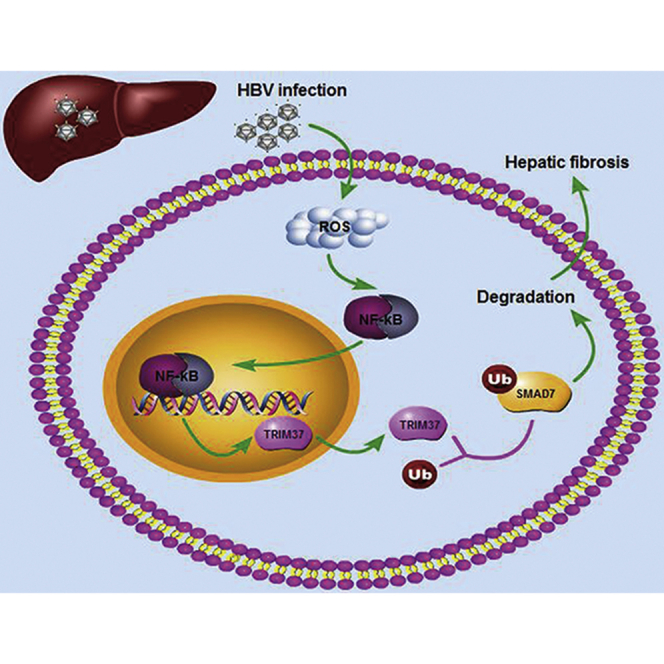
TRIM37 is increased in hepatic fibrosis patients and induced hepatic fibrosis by physically interacting with SMAD7 and promoting ubiquitination-mediated degradation of SMAD7. Meanwhile, NF-κB activation mediated by reactive oxygen species (ROS) is necessary for the transcriptional induction of TRIM37 during HBV infection.
Introduction
Hepatic cirrhosis is a common cause of death associated with liver disease. Hepatic fibrosis is a condition that can often lead to hepatic cirrhosis.1 A number of factors, such as autoimmune hepatitis, bile obstruction, iron overload, alcoholic liver disease, and hepatitis virus infection, can cause hepatic fibrosis.2, 3, 4 It is a complex fibrogenic and inflammatory response with continuous deposition of extracellular matrix (ECM) proteins, such as collagen, in perisinusoidal space. This leads to scarring of liver, and, when left untreated, it causes cirrhosis, portal hypertension, hepatic encephalopathy, hepatocellular carcinoma, liver failure, and ultimately death.5 Hepatitis B virus (HBV) infection is known to be a common cause of hepatic fibrosis and the abovementioned liver diseases. HBV infects and activates hepatic stellate cells (HSCs). The activation of HSCs is characterized by an increase in cell proliferation and secretion of ECM proteins, including collagen, matrix metalloproteinase (MMP), α-smooth muscle actin (α-SMA), and transforming growth factor β (TGF-β).6 Furthermore, HBV infection elicits innate as well as adaptive immune responses, and the type of immune response against the virus has been suggested to determine the severity of liver diseases.7,8 Although it is clear that HBV infection leads to hepatic fibrosis and related liver diseases, our understanding of the factors that are critical in mediating hepatic fibrosis during HBV infection remains incomplete.
Tripartite motif (TRIM) family proteins have more than 100 proteins that contain a highly conserved TRIM. They contain RING domain, which has E3 ubiquitin ligase activity. TRIM proteins are ubiquitously expressed, and they have important functions in the immune response as well as many other biological processes.9, 10, 11 Several members of TRIM proteins are important for antiviral activity against murine leukemia virus,12,13 HIV-114 and HBV.15 In a screening performed to identify specific TRIM proteins that affect HBV infection, eight TRIM proteins were shown to inhibit HBV replication.15 In the same screening, some of the TRIM proteins were found to increase the expression of HBV antigens;15 however, their precise role in replication of HBV and any of the HBV-associated liver diseases remains unexplored.
TRIM37 is widely known as the protein that is responsible for Mulibrey nanism syndrome. The syndrome is characterized by defects in growth of multiple organs, and it is caused by mutation in TRIM37.16,17 In recent years, TRIM37 has been suggested to be involved in the progression of several cancers. It has been shown to increase proliferation in lung cancer cells,18,19 glioma cells,20,21 colorectal cancer cells22 and pancreatic cancer cells.23 In addition, it has been reported to promote epithelial-to-mesenchymal transition (EMT)24 and metastasis.24,25 The role of TRIM37 in other diseases, including liver diseases, remains unknown. TRIM37 is one of the TRIM proteins that was found to increase expression of some of the HBV antigens,15 indicating a potential role of this protein in promoting HBV infection. However, it is unclear whether the correlation has any functional implications in HBV infection and associated liver diseases. In this study, we investigated the role of TRIM37 in HBV-mediated hepatic fibrosis and identified the underlying mechanism.
Results
HBV Induces TRIM37 Expression
To test the potential role of TRIM37 in HBV infection, the expression of TRIM37 in patients with hepatic fibrosis with or without HBV infection was measured by reverse transcriptase polymerase chain reaction (RT-PCR). We found an increase in the expression of TRIM37 in hepatic fibrosis patients with HBV compared to the normal controls as well as hepatic fibrosis patients who were negative for HBV (Figure 1A). Other tested TRIM genes did not show the similar pattern (Figure 1A). To verify the specific role of HBV in TRIM37 induction, we isolated serum from HBV-infected hepatic fibrosis patients and treated LX-2 cells, a human HSC line, with increasing doses of HBV virus. The cells showed dose-dependent induction of TRIM37 in both mRNA and protein levels (Figures 1B and 1C). The other tested TRIM genes did not show an induction in response to the virus (Figure 1B). Taken together, these results suggest that the TRIM37 induced by HBV is involved in hepatic fibrosis.
Figure 1.

HBV Induces TRIM37 Expression
(A) The mRNA levels of the indicated TRIM proteins in liver tissues of normal controls and hepatic fibrosis patients with or without HBV infection were measured by RT-PCR (n = 15). (B and C) LX-2 cells were treated with serum containing HBV DNA (104, 105, and 106 IU/mL) from patients with hepatic fibrosis, and, after 48 h, (B) mRNA and (C) protein levels of the indicated TRIM proteins were measured by RT-PCR and western blot, respectively.
TRIM37 Promotes HBV-Induced Hepatic Fibrosis
To test the role of TRIM37 in HBV-induced hepatic fibrosis, we first confirmed that HBV infection produced HBV viral proteins, hepatitis B e antigen (HBeAg) and hepatitis B surface antigen (HBsAg), in LX-2 cells (Figure 2A). We also verified an increase in HBV viral DNA upon HBV infection (Figure 2B). Then, we examined the role of TRIM37 in an HBV-mediated increase in cell proliferation by using short hairpin RNA (shRNA) that efficiently knocks down TRIM37 (Figure 2C). HBV infection increased the proliferation of LX-2 cells transfected with control shRNA, and knock down of TRIM37 blunted the increase in proliferation (Figure 2D), suggesting the role of the TRIM37 in promoting HBV infection-mediated cell proliferation.
Figure 2.

TRIM37 Promotes HBV-Induced Hepatic Fibrosis
(A and B) LX-2 cells were transfected with HBV replicon or control vector, and (A) the concentrations of hepatitis B e antigen (HBeAg) and hepatitis B surface antigen (HBsAg) in cell culture supernatant were assessed by ELISA, and (B) the HBV DNA copy number was determined using qPCR. (C) LX-2 cells were transfected with shTRIM37 (−1, −2, and −3), and mRNA and protein levels of TRIM37 were measured. (D–H) LX-2 cells were transfected with control vector or HBV along with control shRNA or shTRIM37 (shTRIM37-2 and shTRIM37-3), and the effect on (D) cell viability was measured by a CCK-8 kit, (E) LX-2 morphology was observed, (F) Hyp secretion was measured by an A030 Hyp assay kit, (G) collagen I/III in cell culture supernatant was measured by ELISA, and (H) protein levels of TRIM37, α-SMA, and MMP-2 were assessed by western blot. Scale bar, 50 μm. ∗∗∗p < 0.01 compared with control or shNC; ##p < 0.01, ###p < 0.001 compared with HBV + shNC.
Normally, LX-2 cells without HBV infection show the characteristics of unactivated primary HSCs, as seen in Figure 2E. In contrast, the cells strongly exhibited the activated form of HSCs (myofibroblast-like phenotype) after HBV infection. Interestingly, TRIM37 knockdown blunted the myofibroblast-like phenotype induced by HBV infection (Figure 2E). Meanwhile, hepatic fibrosis involves secretion of ECM proteins, such as collagen, and hydroxyproline (Hyp).26 Hence, we examined the levels of Hyp and collagen I/III in LX-2 cells infected with HBV. We found a significant increase in the level of Hyp and collagen I/III secreted by cells infected with HBV, and the levels of Hyp and collagen I/III were significantly reduced in TRIM37 knockdown cells (Figures 2F and 2G). The expression of other proteins involved in fibrosis, such as α-SMA and MMP-2, were also reduced upon TRIM37 knockdown in LX-2 cells infected with HBV (Figure 2H). Collectively, these results show the role of TRIM37 in promoting HBV-mediated hepatic fibrosis.
TRIM37 Regulates SMAD7 Ubiquitination
To uncover the underlying mechanism associated with TRIM37 mediated hepatic fibrosis, we applied mass spectrometry (MS) to identify proteins that interact with TRIM37 in LX-2 cells (Figure 3A). As expected, TRIM37 was the protein with the most peptides (Figure 3B). The second most peptides were from SMAD7 (Figure 3B). The interaction between TRIM37 and SMAD7 was further validated with co-immunoprecipitation (coIP) followed by immunoblotting (IB) (Figure 3C). To test whether the interaction between TRIM37 and SMAD7 regulates the expression of SMAD7, we examined the expression of SMAD7 in LX-2 cells overexpressing TRIM37 (oeTRIM37) (Figure 3D). TRIM37 overexpression caused a reduction in SMAD7 protein level (Figure 3E). This regulation was not observed in the SMAD7 mRNA level (Figure 3E), suggesting that TRIM37 regulates expression of SMAD7 in a post-transcriptional level. Polyubiquitination-mediated protein degradation by proteasome is a common form of post-transcriptional regulation, and since TRIM37 is an E3 ubiquitin ligase, we tested whether TRIM37 regulates the ubquitination of SMAD7. We treated oeTRIM37 cells with MG132, an inhibitor of proteasome, to prevent the ubiquitination-mediated protein degradation, and checked the level of SMAD7. Indeed, the treatment increased the level of SMAD7 in the oeTRIM37 cells (Figure 3E). Furthermore, oeTRIM37 increased the level of ubiquitinated SMAD7 (Figure 3F). Collectively, these results demonstrate that TRIM37 ubiquitinates SMAD7 and targets it for degradation.
Figure 3.

TRIM37 Regulates SMAD7 Ubiquitination
(A) TRIM37-associated proteins in LX-2 cells were identified by mass spectrometric analysis and are listed in (B). (C) LX-2 cell lysates were immunoprecipitated with either control IgG or TRIM37 antibody (top panel) and control IgG or SMAD7 antibody (bottom panel), and immunoblotted with indicated antibodies. (D) LX-2 cells were transduced with TRIM37 overexpression (oeTRIM37) or empty vector, and TRIM37 expression was measured by RT-PCR and western blot. (E) LX-2 cells transduced with oeTRIM37) or empty vector were treated with vehicle or MG132, and western blot was performed with the indicated antibodies. (F) Cell lysates from oeTRIM37 or empty vector-transfected LX-2 cells were immunoprecipitated with anti-SMAD7 or IgG and then immunoblotted with anti-ubiquitin. ∗∗∗p < 0.01 compared with vector.
TRIM37 Promotes Hepatic Fibrosis by Targeting SMAD7 for Degradation
Next, we tested whether SMAD7 is a critical mediator of hepatic fibrosis caused by TRIM37. We overexpressed SMAD7 in LX-2 cells (oeSMAD7) (Figure 4A) and checked the effect on hepatic fibrosis markers. TRIM37 overexpression significantly increased the cell proliferation in LX-2 cells (Figure 4B). However, the overexpression of SMAD7 in the context of oeTRIM37 blunted the effect of TRIM37 overexpression in cell proliferation (Figure 4B). LX-2 cells with TRIM37 overexpression also exhibited the myofibroblast-like phenotype, and the overexpression of SMAD7 in the context of oeTRIM37 blunted the effect of TRIM37 overexpression (Figure 4C). Likewise, oeSMAD7 reduced Hyp (Figure 4D) and collagen I/III secretion (Figure 4E), suggesting an inhibitory effect of SMAD7 on hepatic fibrosis. A similar effect was observed in the expression of α-SMA and MMP-2 (Figure 4F). Furthermore, overexpression of SMAD7 in oeTRIM37 cells completely reversed the effect of TRIM37 overexpression on the secretion of Hyp (Figure 4D) and collagen I/ III (Figure 4E), as well as the expression of α-SMA and MMP-2 (Figure 4F). These results suggest that TRIM37 promotes hepatic fibrosis by promoting SMAD7 degradation.
Figure 4.

TRIM37 Promotes Hepatic Fibrosis by Targeting SMAD7 for Degradation
(A) Generation of the SMAD7 overexpression system. LX-2 cells were transduced with SMAD7 overexpression (oeSMAD7) or empty vector, and expression of SMAD7 was measured in mRNA levels by RT-PCR and in protein levels by western blotting. (B–F) LX-2 cells were transduced with empty vector, SMAD7 (oeSMAD7), TRIM37 (oeTRIM37), and oeSMAD7 + oeTRIM37, and the effect on (B) cell viability was measured by a CCK-8 kit, (C) LX-2 morphology was observed, (D) Hyp secretion was measured by an A030 Hyp assay kit, (E) collagen I/III in cell culture supernatant was measured by ELISA, and (F) protein levels of α-SMA and MMP-2 were assessed by western blot. Scale bar, 50 μm. ∗∗∗p < 0.01 compared with vector; ##p < 0.01, ###p < 0.001 compared with oeTRIM37.
TRIM37 Promotes Hepatic Fibrosis In Vivo
To test the role of TRIM37 in hepatic fibrosis in vivo, we examined the effect of Trim37 knockdown in mice injected with AAV8-HBV 1.2 vector. Masson stain demonstrated an increase in collagen expression in the liver of mice injected with the AAV8-HBV 1.2 vector, and the expression was suppressed upon Trim37 knockdown (Figure 5A). HBV infection was verified by staining for the expression of HBsAg and HBV core antigens (HBcAgs) (Figure 5A). As expected, the AAV8-HBV 1.2 vector injection led to an increase in Hyp secretion, and knockdown of Trim37 significantly reduced the level of Hyp (Figure 5B). Likewise, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), markers of fibrosis,27 also showed an increase in AAV8-HBV 1.2 vector-injected liver, and the levels were reduced significantly upon Trim37 knockdown (Figure 5C). We also observed a decrease in SMAD7 protein level upon AAV8-HBV 1.2 vector injection, and knockdown of Trim37 reversed it (Figure 5D). All other previously described ECM proteins and markers of fibrosis showed an increase in the protein level upon AAV8-HBV 1.2 vector injection, and the effects were reversed upon Trim37 knockdown (Figure 5D). Collectively, these results establish an important role of TRIM37 in hepatic fibrosis in vivo.
Figure 5.

TRIM37 Promotes Hepatic Fibrosis In Vivo
(A–D) AAV8-HBV 1.2 vector was injected in mice through the tail vein along with control shRNA (ad-shNC) or Trim37 shRNA (ad-shTRIM37), and the effect on (A) collagen secretion was measured by Masson’s trichrome staining in liver tissue, and HBV infection was verified by HBsAg and hepatitis B core antigen (HBcAg) staining; (B) the level of Hyp secretion was measured by an A030 Hyp assay kit; (C) the amounts of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were quantified by commercial kits; and (D) protein levels of TRIM37, SMAD7, collagen I/III, α-SMA, and MMP-2 were measured by western blotting using the indicated antibodies. Scale bar, 100 μm. ∗∗∗p < 0.01 compared with control; ##p < 0.01, ###p < 0.001 compared with AAV8-HBV 1.2 + ad-shNC.
HBV-Mediated Nuclear Factor κB (NF-κB) Activation Transcriptionally Regulates TRIM37 Expression
Finally, we investigated the mechanism through which HBV regulates TRIM37 activity. Previous studies have shown that HBV induces the generation of reactive oxygen species (ROS).28 We validated the increase in intracellular ROS upon HBV infection in LX-2 cells (Figure 6A). We observed an increase in the level of mRNA and protein of TRIM37 in HBV-infected cells (Figure 6B and 6C), and N-acetylcysteine (NAC), an antioxidant, treatment reduced the level of both mRNA and protein level of TRIM37 (Figures 6B and 6C), suggesting that ROS is critical for the induction of TRIM37. Previous studies have shown that ROS can activate NF-κB,29 so we tested whether HBV infection and NAC treatment affect the activation of NF-κB. Indeed, HBV infection increased the nuclear translocation of the p65 subunit of NF-κB (NF-κB p65), and the nuclear translocation decreased upon NAC treatment (Figure 6D).
Figure 6.

HBV-Mediated NF-κB Activation Transcriptionally Regulates the Expression of TRIM37
(A–D) LX-2 cells were transfected with HBV replicon or control vector, cells were treated with NAC (10 mM) for 12 h, and (A) ROS production was measured by dichlorofluorescin diacetate (DCF-DA) staining followed by fluorescence-activated cell sorting (FACS), and the fluorescence intensity was quantified; (B–D) expression levels of TRIM37 and NF-κB p65 were measured by RT-PCR and/or western blotting. (E and F) LX-2 cells were transduced with either control vector or HBV replicon, treated with PDTC (10 μM) or vehicle for 24 h, and (E) luciferease reporter activity of TRIM37 promoter was measured, and (F) mRNA and protein levels of TRIM37 were measured by RT-PCR and western blotting, respectively. (G) The NF-κB binding site in the the TRIM37 promoter was predicted by the JASPAR algorithm. (Left) The predicted sequence, binding site (BS), and schematic diagram of TRIM37 gene, promoter region, and 3′ UTR are shown. (Right) Chromatin from LX-2 cells was immunoprecipitated using control IgG or NF-κB p65 antibody, and binding of the antibody in the BS region of the promoter or negative control 3′ UTR region of TRIM37 (NC) was measured by qPCR. ∗∗∗p < 0.001 compared with control or IgG; ###p < 0.001 compared with HBV.
Next, we tested whether NF-κB directly activates the transcription of TRIM37. The promoter region of TRIM37 was cloned to drive luciferase expression, and the transcriptional role of NF-κB in HBV-infected cells was evaluated by treating cells with pyrrolidine dithiocarbamate (PDTC), an NF-κB inhibitor. We observed higher luciferase activity in the cells infected with HBV, and the activity was reduced in cells treated with PDTC (Figure 6E). In addition, we observed an increase in the mRNA as well as protein level of TRIM37 in the cells infected with HBV, and they were reduced in the cells treated with PDTC (Figure 6F), further supporting the transcriptional role of NF-κB in inducing TRIM37 expression.
We applied JASPAR,30 a promoter analysis algorithm, to identify potential NF-κB binding sites in the promoter region of TRIM37. We found an NF-κB binding motif upstream of the transcription start site (TSS) of TRIM37 (Figure 6G). Then, we performed chromatin immunoprecipitation (ChIP) to test whether the predicted site is bound by NF-κB. The ChIP analysis showed a specific enrichment of NF-κB binding in the predicted region of the TRIM37 promoter, whereas the enrichment was absent in the 3′ UTR region of TRIM37 (Figure 6G). Collectively, these results demonstrate that HBV infection-dependent ROS activates NF-κB, which in turn induces TRIM37 expression.
Discussion
In this study, we showed that TRIM37 is important for HBV-induced hepatic fibrosis. We further showed that SMAD7 is the downstream target of TRIM37, and ROS and NF-κB are the upstream regulators of TRIM37. Identification of these key players involved in the regulation of hepatic fibrosis will have a positive impact in identifying some of the key therapeutic targets for HBV-induced hepatic fibrosis treatments, as it remains a major liver disease with unmet clinical needs.
TRIM family members have been previously reported to be important regulators of HBV infection and fibrosis. Eight members of the TRIM proteins have been shown to suppress HBV infection by inhibiting transcription of the HBV mRNA.15 In addition, TRIM52 has been recently shown to promote HBV-mediated hepatic fibrosis by promoting ubiquitination of PPM1A that has an inhibitory role in TGF-β signaling.28 In this study, we showed that TRIM37 is also an important promoter of hepatic fibrosis. However, the mechanism through which TRIM37 promotes hepatic fibrosis is distinct from that of TRIM52. This suggests that different members of TRIM family proteins participate in hepatic fibrosis, both negatively and positively, through diverse mechanisms. Other members of the TRIM family proteins could also participate in this process through their unique mechanisms. Future studies might reveal more of the TRIM family proteins’ role in fibrosis with different mechanisms.
We identified SMAD7 as a key substrate of TRIM37 that is targeted for degradation through ubiquitination. This targeted degradation is critical for the role of TRIM37 in hepatic fibrosis. SMAD7 is a transcriptional target of TGF-β signaling that functions as an important negative regulator of TGF-β signaling by suppressing the activity of SMAD2/4, SMAD3/4, and SMAD1/4, which are the effector transcription factors activated by TGF-β.31 Furthermore, TGF-β is a potent fibrogenic cytokine that regulates cell proliferation, ECM production, and EMT, all of which play an important role in fibrosis.32 SMAD7 has been previously shown to prevent the activation of HSCs and hepatic fibrosis by counteracting TGF-β signaling.33 Hence, our findings support a model where TRIM37 further enhances the activity of TGF-β by suppressing the SMAD7-mediated negative feedback loop of TGF-β signaling.
In this study, we showed that ROS is an upstream regulator of TRIM37 expression that in turn promotes hepatic fibrosis. This is consistent with previous studies that have shown that ROS is involved in fibrosis.32,34,35 Furthermore, HBV infection has been shown to generate ROS through HBx protein that is targeted to host mitochondria,34 and ROS has been shown to promote transcriptional activity of NF-κB.29 Thus, our study provides a direct link between HBV infection and hepatic fibrosis that is mediated through ROS generation, NF-κB activation, TRIM37 expression, and SMAD7 degradation. Future studies will be focused on investigating whether any of the components in the TRIM37 axis that we described in this study can be targeted for therapeutic purposes.
Materials and Methods
Tissue Specimens
Liver tissue specimens from control patients without liver disease and HBV infection, and from hepatic fibrosis patients with or without HBV infection, were obtained from Shanghai Guanghua Hospital of Integrative Medicine. The tissues were first snap-frozen in liquid nitrogen before storing at −80°C for future use. Approval of this study was obtained from the Ethics Committee of Shanghai Guanghua Hospital of Integrative Medicine and all patients gave their written informed consent.
Cell Culture
The blood serum samples from hepatic fibrosis patients infected with HBV (HBV DNA = 6.75 × 106 IU/mL) were diluted and added into LX-2 cells (American Type Culture Collection). The final concentrations of HBV DNA were 104, 105, and 106 IU/mL, respectively.
HBV Transfection
LX-2 cells (1 × 105 cells/well in six-well plates) were plated in RPMI 1640 medium. The next day, the cells were transfected with HBV 1.3-mer P null replicon plasmid (Addgene, 65462). After 48 h, HBV DNA was quantified using TaqMan RT-PCR. Primers used in this study are described in Zhou et al.36
Gene Knockdown and Overexpression
Genes of interest were knocked down using shRNAs. Three independent shRNAs against human TRIM37 (shTRIM37-1, shTRIM37-2, and shTRIM37-3) were used. The sequences encoding shRNAs for TRIM37 are as follows: shTRIM37-1, 5ʹ-CCGGTGGAGAAGATTCAGAATGAACTCGAGTTCATTCTGAATCTTCTCCTTTTTG-3ʹ; shTRIM37-2, 5ʹ-CCGGTCCAGTAGTTTACTAGACATCTCGAGATGTCTAGTAAACTACTGGTTTTTG-3ʹ; and shTRIM37-3, 5ʹ-CCGGTGCCTTGATACATGGCAGTACTCGAGTACTGCCATGTATCAAGGCTTTTTG-3ʹ. shRNAs were cloned into pLKO.1 plasmid (Addgen, USA). To overexpress TRIM37 and SMAD7, coding DNA sequences of the genes were cloned in pLVX-Puro plasmid (Clontech). To generate virus particles, the shRNA plasmids or the overexpression plasmids were co-transfected with pMD2G and psPAX2 plasmids into 293T cells. Lipofectamine 2000 (Invitrogen, USA) was used for transfection. The virus particles were collected 48 h after transfection. pLKO.1-scramble shRNA (shNC) was used as the negative control for shRNA experiments.
Cell Proliferation Assay
Cell Counting Kit-8 (CCK-8) (Signalway Antibody) was used to measure cell proliferation, following the manufacturer’s instructions. Briefly, 3 × 103 cells/well were seeded into 96-well plates. At the the indicated time points, 10 μL of CCK-8 solution was added in each well and incubated for 1 h. The viable cells were quantified with a microplate reader at optical density 450 (OD450).
Hyp Assay
Hyp in the cell culture medium was measured using an A030 Hyp assay kit (Nanjing Jiancheng Bioengineering Institute, A030-2).
Enzyme-Linked Immunosorbent Assay (ELISA)
The amounts of HBeAg and HBsAg were quantified using a human HBeAg ELISA kit (Cusabio Technology, CSB-E13557h) and a human HBsAg ELISA kit (Cusabio Technology, CSB-E10089h), respectively. Collagen I and III concentrations were measured using a human collagen type I (Col I) ELISA kit (BioVision Technologies, E4617-100) and a human Col III ELISA kit (United States Biological, 354899) following the manufacturers’ protocols.
Analysis of Intracellular ROS Levels
The intracellular ROS level was measured as described in Lim et al.37
RT-PCR
RNAs from LX-2 cells and liver tissues were extracted using TRIzol reagent (Invitrogen), and cDNA was synthesized using a cDNA synthesis kit (Fermentas). The mRNA levels were measured by a SYBR Green PCR kit (Thermo Fisher Scientific). The following primers were used for RT-PCR: TRIM37, 5ʹ-TGGACTTACTCGCAAATG-3ʹ (forward) and 5ʹ-ATCTGGTGGTGACAAATC-3ʹ (reverse); TRIM16L, 5ʹ-GCCGAGATGGAGAAGAGTAAG-3ʹ (forward) and 5ʹ-GCCGAGATGGAGAAGAGTAAG-3ʹ (reverse); TRIM56, 5ʹ-GAAACGCTTCTCCCTCAAC-3ʹ (forward) and 5ʹ-GAAACGCTTCTCCCTCAAC-3ʹ (reverse); TRIM40, 5ʹ-ATGCCCTCAGCCACTACAAG-3ʹ (forward) and 5ʹ-TCCCGTGGTCTACCTGAAAC-3ʹ (reverse); TRIM59, 5ʹ-TGCCTTACCATAGGTCAAC-3ʹ (forward) and 5ʹ-GATTGCCAACATCACAGAG-3ʹ (reverse); SMAD7, 5ʹ-CGGACTCCAGGACGCTGTT-3ʹ (forward) and 5ʹ-CTGCAGGCTGTACGCCTTCT-3ʹ (reverse). GAPDH primers used in this study are described in Guo et al.38
Western Blot Analysis
Total protein was extracted from LX-2 cells using radioimmunoprecipitation assay (RIPA) lysis buffer (Sigma-Aldrich). Nuclear and cytoplasm proteins were fractionated using a NE-PER kit (Thermo Fisher Scientific). The protein concentration was quantified by a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). 25 μg of protein was loaded on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel, transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore), and the protein of interest was probed with primary antibodies at 4°C overnight, followed by secondary antibodies (Beyotime Biotechnology, A0208 and A0216) for 1 h at 25°C. The following antibodies were used: anti-TRIM37 (Cell Signaling Technology, 96167), anti-MMP-2 (Abcam; ab2462), anti-α-SMA (Abcam; ab5694), anti-SMAD7 (Abcam, ab90086), anti-NF-κB p65 (Abcam, ab16502), anti-H3 (Abcam, ab1791), and anti-GAPDH (Cell Signaling Technology, 5174). The binding of the secondary antibodies was visualized by an enhanced chemiluminescence (ECL) system (GE Healthcare/Amersham Biosciences).
IP and Liquid Chromatography (LC)/MS
Tag2-TIRM37 or empty vector was transfected into 293T cells, and cells were harvested in RIPA buffer after 48 h. The lysate was pre-cleared for 2 h with protein A/G beads (Santa Cruz Biotechnology, sc-2003) at 4°C and immunoprecipitated with anti-FLAG beads (Sigma-Aldrich, M8823) overnight at 4°C. The bound proteins were eluted with FLAG peptide (Sigma-Aldrich). The proteins were resolved in SDS-PAGE, and the gel was stained with Coomassie brilliant blue. The bands that were differentially present were excised, digested with trypsin, and the proteins were identified by LC/MS.
CoIP and Ubiquitination Assay
For coIP, cells were harvested in RIPA buffer, and 2 mg of protein lysate was pre-cleared as described above, immunoprecipitated with anti-TRIM37 (Abcam, ab264190), anti-SMAD7 (Santa Cruz Biotechnology, sc-365846), or control immunoglobulin G (IgG) (Santa Cruz Biotechnology, sc-2027) overnight at 4°C and immunoblotted as described above.
For the ubiquitination assay, SMAD7 from control or TRIM37 overexpressing LX-2 cells was immunoprecipitated as described above with 1 μg of IgG or anti-SMAD7. The ubiquitination in SMAD7 was probed using anti-ubiquitin antibody (Abcam, ab7780) by western blotting as described above.
ChIP Assay
Cells were transfected with HBV replicon or control vector for 24 h and were fixed with 1% formaldehyde at room temperature. The fixed cells were harvested and sonicated for chromatin fragmentation. The chromatin solutions were then incubated with anti-NF-κB p65 (Abcam, ab16502) or control IgG antibody overnight at 4°C. NF-κB p65 binding in the TRIM37 promoter region was quantified by PCR using the following TRIM37 promoter primers: 5′-CACGATGCTGGGTGAAAAC-3′ (forward) and 5′-TTGGGGCTCTGCTACGC-3′ (reverse).
Luciferase Reporter Assay
The full-length TRIM37 promoter was cloned into pGL3 vector (Promega). To measure TRIM37 promoter activity, cells were transfected with the pGL3-TRIM37 promoter, and the luciferase activity was measured with a luciferase assay kit (Promega). The data were normalized to the control sample transfected with pGL3.
Adenovirus Production
The mouse Trim37 shRNA (ad-shTrim37) (Sangon Biotech, China) or non-specific scramble shRNA (adNC) was cloned in pShuttle-H1 adenovirus vector. The plasmids and the packaging plasmids were co-transfected into HEK293T cells to generate high-titer adenovirus.
Animal Experiments
Wild-type (WT) male C57BL/6 mice (6–8 weeks old; Vitalriver, Beijing, China) were used in the experiments. Mice were bred and maintained at the Laboratory Animal Facility of the Shanghai Guanghua Hospital of Integrative Medicine. All procedures conducted in this study followed the Guide for the Care and Use of Laboratory Animals, and they were approved by the Institutional Animal Care and Use Committee at the Shanghai Guanghua Hospital of Integrative Medicine. For the virus experiments, 200 μL of AAV8-HBV 1.2 vector (2 × 1011 vector genome equivalents [vg]) resuspended in phosphate-buffered saline (PBS) was injected into the tail vein of mice (n = 6). The ad-shNC and ad-shTrim37 vectors were also injected into the tail vein. In the control group, PBS was injected. After 4 weeks, the mice were euthanized, serum was collected, and liver was harvested. The samples were frozen in liquid nitrogen for biochemical, hematoxylin and eosin (H&E), Masson’s trichrome (Beijing Leagene Biotechnology), and immunohistochemistry (IHC) staining and western blotting. AST, ALT, and Hyp levels were measured with commercial assay kits (Nanjing Jiancheng Bioengineering Institute).
IHC
Paraffin-embedded tissue sections of liver tissues were used for immunohistochemical studies. IHC staining was performed on liver specimens following a standard protocol using anti-HBsAg (Novus Biologicals, NB100-62652) and anti-HBcAg (Abcam, ab8638), followed by incubation with secondary antibody (Shanghai Long Island Biotec, D-3004). Two independent pathologists without prior knowledge evaluated the IHC slides.
Statistics and Data Analysis
All experiments had three biological triplicates. The data were quantified and expressed as mean ± SD. All statistics were conducted by GraphPad Prism 7.0 (GraphPad, USA). Comparisons between different experimental groups were performed with a Tukey’s post hoc multiple test using ANOVA. p <0.05 was used as a cutoff for statistical difference.
Author Contributions
H.X., D.X., and J.Z. designed the experiments; W.J., Y.L., and J.Y. conducted the experiments; and Z.P. and D.X. analyzed data and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81774252).
Contributor Information
Zhiqiang Pan, Email: pzq527@163.com.
Donghao Xie, Email: xdhdh2012@126.com.
References
- 1.Lim Y.S., Kim W.R. The global impact of hepatic fibrosis and end-stage liver disease. Clin. Liver Dis. 2008;12:733–746, vii. doi: 10.1016/j.cld.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Sebastiani G., Gkouvatsos K., Pantopoulos K. Chronic hepatitis C and liver fibrosis. World J. Gastroenterol. 2014;20:11033–11053. doi: 10.3748/wjg.v20.i32.11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trautwein C., Friedman S.L., Schuppan D., Pinzani M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015;62(1, Suppl):S15–S24. doi: 10.1016/j.jhep.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 4.Hernandez-Gea V., Friedman S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C.Y., Yuan W.G., He P., Lei J.H., Wang C.X. Liver fibrosis and hepatic stellate cells: etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016;22:10512–10522. doi: 10.3748/wjg.v22.i48.10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai Q., An J., Wu X., You H., Ma H., Liu T., Gao N., Jia J. HBV promotes the proliferation of hepatic stellate cells via the PDGF-B/PDGFR-β signaling pathway in vitro. Int. J. Mol. Med. 2012;30:1443–1450. doi: 10.3892/ijmm.2012.1148. [DOI] [PubMed] [Google Scholar]
- 7.Chisari F.V., Ferrari C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 8.Guidotti L.G., Chisari F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 9.Ozato K., Shin D.M., Chang T.H., Morse H.C., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munir M. TRIM proteins: another class of viral victims. Sci. Signal. 2010;3:jc2. doi: 10.1126/scisignal.3118jc2. [DOI] [PubMed] [Google Scholar]
- 11.McNab F.W., Rajsbaum R., Stoye J.P., O’Garra A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011;23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Wolf D., Cammas F., Losson R., Goff S.P. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J. Virol. 2008;82:4675–4679. doi: 10.1128/JVI.02445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perron M.J., Stremlau M., Song B., Ulm W., Mulligan R.C., Sodroski J. TRIM5α mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. USA. 2004;101:11827–11832. doi: 10.1073/pnas.0403364101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stremlau M., Owens C.M., Perron M.J., Kiessling M., Autissier P., Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S., Guo J.T., Wu J.Z., Yang G. Identification and characterization of multiple TRIM proteins that inhibit hepatitis B virus transcription. PLoS ONE. 2013;8:e70001. doi: 10.1371/journal.pone.0070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlberg N., Jalanko H., Perheentupa J., Lipsanen-Nyman M. Mulibrey nanism: clinical features and diagnostic criteria. J. Med. Genet. 2004;41:92–98. doi: 10.1136/jmg.2003.014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hämäläinen R.H., Avela K., Lambert J.A., Kallijärvi J., Eyaid W., Gronau J., Ignaszewski A.P., McFadden D., Sorge G., Lipsanen-Nyman M., Lehesjoki A.E. Novel mutations in the TRIM37 gene in Mulibrey nanism. Hum. Mutat. 2004;23:522. doi: 10.1002/humu.9233. [DOI] [PubMed] [Google Scholar]
- 18.Dong S., Pang X., Sun H., Yuan C., Mu C., Zheng S. TRIM37 targets AKT in the growth of lung cancer cells. OncoTargets Ther. 2018;11:7935–7945. doi: 10.2147/OTT.S183303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y., Deng L., Zhao X., Li B., Ren D., Yu L., Pan H., Gong Q., Song L., Zhou X., Dai T. Tripartite motif-containing 37 (TRIM37) promotes the aggressiveness of non-small-cell lung cancer cells by activating the NF-κB pathway. J. Pathol. 2018;246:366–378. doi: 10.1002/path.5144. [DOI] [PubMed] [Google Scholar]
- 20.Chang C., Zhu S., Xu Z., Liu Y. Tripartite motif-containing protein 37 is overexpressed in human glioma and its downregulation inhibits human glioma cell growth in vitro. Int. J. Clin. Exp. Pathol. 2018;11:502–511. [PMC free article] [PubMed] [Google Scholar]
- 21.Tang S.L., Gao Y.L., Wen-Zhong H. Knockdown of TRIM37 suppresses the proliferation, migration and invasion of glioma cells through the inactivation of PI3K/Akt signaling pathway. Biomed. Pharmacother. 2018;99:59–64. doi: 10.1016/j.biopha.2018.01.054. [DOI] [PubMed] [Google Scholar]
- 22.Zhao P., Guan H.T., Dai Z.J., Ma Y.G., Liu X.X., Wang X.J. Knockdown of tripartite motif-containing protein 37 (TRIM37) inhibits the proliferation and tumorigenesis in colorectal cancer cells. Oncol. Res. 2017;25:115–122. doi: 10.3727/096504016X14732772150181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J., Tian S., Yu C., Chen M., Sun C. TRIM37 promoted the growth and migration of the pancreatic cancer cells. Tumour Biol. 2016;37:2629–2634. doi: 10.1007/s13277-015-4078-7. [DOI] [PubMed] [Google Scholar]
- 24.Chen D., You X., Pan Y., Liu Q., Cao G. TRIM37 promotes cell invasion and metastasis by regulating SIP1-mediated epithelial-mesenchymal transition in gastric cancer. OncoTargets Ther. 2018;11:8803–8813. doi: 10.2147/OTT.S178446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J., Yu C., Chen M., Tian S., Sun C. Over-expression of TRIM37 promotes cell migration and metastasis in hepatocellular carcinoma by activating Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2015;464:1120–1127. doi: 10.1016/j.bbrc.2015.07.089. [DOI] [PubMed] [Google Scholar]
- 26.Toyoki Y., Sasaki M., Narumi S., Yoshihara S., Morita T., Konn M. Semiquantitative evaluation of hepatic fibrosis by measuring tissue hydroxyproline. Hepatogastroenterology. 1998;45:2261–2264. [PubMed] [Google Scholar]
- 27.Kim W.R., Flamm S.L., Di Bisceglie A.M., Bodenheimer H.C., Public Policy Committee of the American Association for the Study of Liver Disease Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology. 2008;47:1363–1370. doi: 10.1002/hep.22109. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov A.V., Valuev-Elliston V.T., Tyurina D.A., Ivanova O.N., Kochetkov S.N., Bartosch B., Isaguliants M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget. 2017;8:3895–3932. doi: 10.18632/oncotarget.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen A.C., Arany P.R., Huang Y.Y., Tomkinson E.M., Sharma S.K., Kharkwal G.B., Saleem T., Mooney D., Yull F.E., Blackwell T.S., Hamblin M.R. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS ONE. 2011;6:e22453. doi: 10.1371/journal.pone.0022453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bryne J.C., Valen E., Tang M.H., Marstrand T., Winther O., da Piedade I., Krogh A., Lenhard B., Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–D106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S., Fei T., Zhang L., Zhang R., Chen F., Ning Y., Han Y., Feng X.H., Meng A., Chen Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell. Biol. 2007;27:4488–4499. doi: 10.1128/MCB.01636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morry J., Ngamcherdtrakul W., Yantasee W. Oxidative stress in cancer and fibrosis: opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017;11:240–253. doi: 10.1016/j.redox.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H.M., Lorenzen J., Ten Dijke P., Gressner A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- 34.Richter K., Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res. 2016;365:591–605. doi: 10.1007/s00441-016-2445-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonzalez-Gonzalez F.J., Chandel N.S., Jain M., Budinger G.R.S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 2017;190:61–68. doi: 10.1016/j.trsl.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J., Lan Q., Li W., Yang L., You J., Zhang Y.M., Ni W. Tripartite motif protein 52 (TRIM52) promoted fibrosis in LX-2 cells through PPM1A-mediated Smad2/3 pathway. Cell Biol. Int. 2020;44:108–116. doi: 10.1002/cbin.11206. [DOI] [PubMed] [Google Scholar]
- 37.Lim W., Kwon S.H., Cho H., Kim S., Lee S., Ryu W.S., Cho H. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J. Mol. Med. (Berl.) 2010;88:359–369. doi: 10.1007/s00109-009-0563-z. [DOI] [PubMed] [Google Scholar]
- 38.Guo Q., Xiong Y., Song Y., Hua K., Gao S. ARHGAP17 suppresses tumor progression and up-regulates P21 and P27 expression via inhibiting PI3K/AKT signaling pathway in cervical cancer. Gene. 2019;692:9–16. doi: 10.1016/j.gene.2019.01.004. [DOI] [PubMed] [Google Scholar]