

Abstract
Long non-coding RNAs (lncRNAs) play a crucial role in the growth of vascular smooth muscle cells (VSMCs), the dysfunction of which is closely associated with the initiation and progression of cardiovascular diseases (CVDs). Abnormal phenotypic switching and proliferation of VSMCs constitute a significant event in the progression of atherosclerosis. The present study identified a novel lncRNA, PEBP1P2, which serves as a valuable regulator of VSMCs in phenotypic transformation and proliferation. The expression of PEBP1P2 was remarkably decreased in proliferating VSMCs and pathological arteries when using a balloon injury model of rats. Furthermore, we found that PEBP1P2 represses proliferation, migration, and dedifferentiation during phenotype switching in VSMCs induced by platelet-derived growth factor BB (PDGF-BB). Mechanistically, cyclin-dependent kinase 9 (CDK9) was confirmed to be the direct target of PEBP1P2, which was proven to mediate phenotypic switching and proliferation of VSMCs and was rescued by PEBP1P2. Then, we explored the clinical significance, as we observed the decreased expression of PEBP1P2 in the serum of coronary heart disease (CHD) patients and human advanced carotid atherosclerotic plaques. Finally, PEBP1P2 overexpression distinctly suppressed neointima formation and VSMC phenotypic switching in vivo. Taken together, PEBP1P2 inhibits proliferation and migration in VSMCs by directly binding to CDK9, implying that it may be a promising therapeutic target for the treatment of proliferative vascular diseases.
Keywords: cardiovascular diseases, vascular smooth muscle cells, long non-coding RNAs, phenotypic switching, CDK9
Graphical Abstract
He et al. addressed the suppressive role of lncRNA PEBP1P2 on phenotypic switching in VSMCs and neointima formation. These findings provide important insights into the underlying mechanism of atherosclerosis, and the restoration of lncRNA PEBP1P2 provides a potential therapeutic strategy against proliferative vascular diseases.
Introduction
Cardiovascular diseases (CVDs), at the top of the 10 most common causes of death, remain the leading causes of mortality in the world.1,2 According to the latest report, the prevalence of CVDs continues to rise, causing at least 40% of deaths in China.3 Vascular smooth muscle cells (VSMCs), one of the main cellular constituents of the blood vessel wall, are vital determinant for CVDs, which exist in two states, including differentiation (contractile phenotype) and dedifferentiation (synthetic phenotype) during vascular development.4,5 Generally, VSMCs transform from a differentiated phenotype to a dedifferentiation phenotype in response to the stimulation of vascular wall injury or some growth factors, such as platelet-derived growth factor BB (PDGF-BB) and tumor necrosis factor alpha (TNF-α). The differentiated phenotype is characterized by low proliferation and migration and high expression of contractile genes such as α-smooth muscle actin (α-SMA), calponin 1 (CNN1), and smooth muscle myosin heavy chain (SMHC). Conversely, the synthetic phenotype presents high proliferation and migration with low expression of contractile markers.4,6, 7, 8 Increasingly, studies have proved that VSMC phenotypic switching contributes to the initiation and development of most CVDs such as atherosclerosis, restenosis after coronary artery angioplasty or bypass, diabetic vascular complications, and hypertension.9,10 Thus, more studies to investigate the mediators and mechanisms of the pathogenesis of VSMCs involved in CVDs remain necessary.
Long non-coding RNAs (lncRNAs) are the critical epigenetic regulators of genetic expression, which are defined as a cluster of RNA molecules characterized by more than 200 nt in length and the inability to encode proteins.11,12 To date, there is increasing evidence suggesting that lncRNA abnormalities are directly related to CVDs, including atherosclerosis, pathological hypertrophy and the development of cardiocytes, and dyslipidemia.13 For example, lncRNAs H19, ANRIL, SENCR, GAS5, MYOSLID, and MEG3 are associated with atherosclerosis through regulating phenotypic switching, proliferation, and migration in VSMCs.14 Therefore, understanding the intimate regulatory mechanisms of lncRNAs is critical to investigate novel strategies for CVD diagnosis and therapy.
Cyclin-dependent kinase 9 (CDK9), a transcriptional regulator that has few effects on cell cycle control, is activated by binding with cyclin T1 to form positive transcription elongation factor b (P-TEFb), which enhances transcription activity through RNA polymerase II to facilitate RNA synthesis for cell growth and differentiation.15, 16, 17 In addition to its crucial role in cancer cell metabolism and cardiomyocyte proliferation, recent studies have shown that it may also play an important role in CVDs, as it was detected to be upregulated in artery plaque, serum, and monocyte samples of coronary heart disease (CHD) patients.16,18,19 However, the regulatory function and mechanism of CDK9 in VSMC phenotypic conversion are mostly unknown.
Although several studies have explored the role of lncRNAs in VSMC phenotypic switching, its role and the molecular mechanism are largely unknown. In this study, we identified lncRNA PBRP1P2 (also known as lncRNA5) as a novel modulator in phenotypic transformation, proliferation, and migration of VSMCs and proliferative vascular diseases, which could provide a potential therapeutic target for intervention in CVDs.
Results
Identification and Characterization of PEBP1P2
PEBP1P2 is a 409-bp lncRNA derived from pseudogene PEBP1P2 via the UCSC Genome Browser (http://genome.ucsc.edu/), which was previously reported and named as lncRNA5. A previous study reported that PEBP1P2 was enriched in human vessel cell lines (human coronary artery smooth muscle cells [HCASMCs] and human umbilical vein endothelial cells [HUVECs]) and tissues.20 Therefore, we speculate that it could play a potential role in vascular diseases. First, we used two common tools, CPAT (coding potential assessment tool) and CPC (coding potential calculator), to evaluate the coding possibility of PEBP1P2 in the following study. For CPAT, the coding probability was 0.0029, indicating that PEBP1P2 was a non-coding transcript (Figure S1A). For CPC, the coding potential score was −1.0995, also indicating that it had little coding possibility (Figure S1B).
Next, to explore the function of PEBP1P2 in CVDs, we evaluated its expression in the serum of patients with CHD and in human advanced carotid atherosclerotic plaques by quantitative real-time PCR. The results demonstrated that PEBP1P2 expression was markedly decreased in CHD samples compared with the healthy individuals (Figure 1A), and the baseline demography data and biochemical test results are shown in Table S1. Similarly, the expression of PEBP1P2 was significantly reduced in plaque compared with normal carotid intima (Figure 1B). Then, the liftOver tool21 was used to detect whether transcripts with a similar genomic organization on rat (rn4) reference genomes would assess the conservation of PEBP1P2. We found that the potential rat PEBP1P2 ortholog was located on chromosome 4, similar to the human genome (Figure 1C), whereas there was no homology with the mouse genome (Figure S1C). We also used NCBI BLAST22 for sequence comparison, which showed that human PEBP1P2 shares 82.35% homology with the predicted rat gene (Figure S1D). Subsequently, the quantitative real-time PCR analysis revealed that PEBP1P2 was enriched in rat aorta compared to other muscle tissues such as cardiac and skeletal muscle (Figure 1D). Meanwhile, we observed that PEBP1P2 was highly expressed in VSMCs compared to other cell lines associated with atherosclerosis (Figure 1E). We next tried to investigate the expression of PEBP1P2 in a balloon injury model of the rat carotid artery that simulates human angioplasty, in which VSMCs switch from the differentiation to dedifferentiation phenotype and promote the neointimal formation.23 The results showed that PEBP1P2 expression was significantly downregulated in the arteries at 3, 7, and 21 days after injury compared with the sham group by quantitative real-time PCR (Figure 1F).
Figure 1.

Identification and Characterization of PEBP1P2
(A) The PEBP1P2 expression in CHD patients (n = 27) and healthy controls (n = 29) analyzed using quantitative real-time PCR. (B) Quantitative real-time PCR analysis of PEBP1P2 expression in human advanced carotid atherosclerotic plaque and normal carotid intima tissues. (C) Genomic location of PEBP1P2 on rat assembly (rn4), predicted using the UCSC liftOver tool. (D and E) Quantitative real-time PCR analysis for PEBP1P2 expression in rat muscle tissues (D) and atherosclerosis-related cell lines (E). (F) Quantitative real-time PCR was used to analyze the expression of PEBP1P2 in rat carotid arteries after sham operation or balloon injury at different points in time as indicated. Data are presented as mean ± SD. n = 9 for (B), n = 3 for (E), n = 5 for (D) and (F). ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl, ∗∗∗p < 0.001 versus Ctl.
In short, these results demonstrated that PEBP1P2 was enriched in VSMCs and associated with CVDs and arterial injury, which indicated it might play an essential role in regulating VSMC function and CVDs.
Knockdown of PEBP1P2 Enhanced VSMC Proliferation, Migration, and Dedifferentiation during Phenotype Switching
We performed loss-of-function studies to evaluate the potential role of PEBP1P2 on proliferation, migration, and phenotypic transformation. Two small interfering RNAs (siRNAs), especially targeting PEBP1P2, were designed and synthesized to decrease PEBP1P2 expression in VSMCs. PEBP1P2 expression was reduced more than 70% or 50% using transfecting siRNAs (Figure 2A). PEBP1P2 knockdown significantly boosted proliferation of VSMCs by 5-ethynyl-2′-deoxyuridine (EdU) (Figure 2B) and Cell Counting Kit-8 (CCK-8) (Figure 2C) assays. Meanwhile, wound healing (Figure 2D) and transwell (Figure 2E) assays indicated that cell migration was remarkably increased. Because the phenotype switching was closely related to the proliferation and migration of VSMCs,4 we further detected the role of PEBP1P2 knockdown in this process. After PEBP1P2 knockdown, mRNA (Figure 2F) and protein (Figure 2G) levels of contractile markers were downregulated, including α-SMA, CNN1, and SMHC. Interestingly, there was no significant change in cyclin D1 (CCDN1) mRNA (Figure 2F) and protein (Figure 2G) levels. In brief, these results suggested that PEBP1P2 knockdown most likely promoted VSMC proliferation and migration through accelerating phenotype switching rather than regulating the cell cycle.
Figure 2.

Knockdown of PEBP1P2 Enhanced VSMC Proliferation, Migration, and Dedifferentiation during Phenotype Switching
(A) siRNAs were used to knock down PEBP1P2 in VSMCs. The expression of PEBP1P2 was determined using quantitative real-time PCR. (B and C) Cell-Light EdU staining (B) and Cell Counting Kit-8 (CCK-8) (C) analysis for cell proliferation of VSMCs with PEBP1P2 knockdown. Hoechst and EdU staining show blue and green, respectively. (D and E) The cell migration after PEBP1P2 knockdown in VSMCs was analyzed by wound-healing (D) and transwell (E) analysis. (F and G) Quantitative real-time PCR (F) and western blotting (G) analyses were conducted to measure the mRNA and protein expression level of contractile markers and CCND1 after PEBP1P2 knockdown in VSMCs. Results were quantified using ImageJ software. Data are presented as mean ± SD. Scale bars, 100 μm. n = 3. ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl, ∗∗∗p < 0.001 versus Ctl.
Overexpression of PEBP1P2 Suppressed VSMC Proliferation, Migration, and Dedifferentiation during Phenotype Switching and Pathophysiological Functions Induced by PDGF-BB
Gain-of-function analysis was conducted to study the role of PEBP1P2 in VSMCs further. Cells with stable overexpression of PEBP1P2 were established and validated. After transfection of the overexpression plasmids for 24 h, PEBP1P2 expression increased remarkably compared to the control group (Figure 3A). Contrary to PEBP1P2 knockdown, we found that overexpression of PEBP1P2 reduced both proliferation (Figures 3B and 3C) and migration (Figures 3D and 3E). Moreover, mRNA (Figure 3F) and protein (Figure 3G) levels of contractile markers were upregulated. However, there was no observable difference in the expression of CCDN1 (Figures 3F and 3G), which was consistent with PEBP1P2 knockdown.
Figure 3.

Overexpression of PEBP1P2 Suppressed VSMC Proliferation, Migration, and Dedifferentiation during Phenotype Switching and Pathophysiological Functions Induced by PDGF-BB
(A) The expression levels of PEBP1P2 were measured in VSMC-transfected pcDNA3.0-PEBP1P2 by quantitative real-time PCR. (B and C) Cell-Light EdU staining (B) and CCK-8 (C) analysis confirmed the cell proliferation after PEBP1P2 overexpression in VSMCs. (D and E) Wound-healing (D) and transwell (E) analysis for cell migration after PEBP1P2 overexpression in VSMCs. (F and G) Quantitative real-time PCR (F) and western blotting (G) analysis was performed to determine the mRNA and protein levels of contractile genes and CCND1 after PEBP1P2 overexpression in VSMCs. (H) Quantitative real-time PCR analysis of PEBP1P2 expression in VSMCs induced by PDGF-BB (20 ng/mL) at different points in time. (I and J) Cell-Light EdU staining (I) and CCK-8 (J) analysis detection of the cell proliferation of VSMCs. (K and L) Cell migration was detected by wound-healing (K) and transwell (L) analyses. (M) Western blotting analysis for the expression level of contractile markers. The plasmid-mediated overexpression of PEBP1P2 was performed in VSMCs, then treated with 20 ng/mL PDGF-BB as indicated (for I–M). Results were quantified by ImageJ software. Data are presented as mean ± SD. Scale bars, 100 μm. n = 3. ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl; #p < 0.01 versus Ctl + PDGF-BB, ##p < 0.001 versus Ctl + PDGF-BB.
Next, we explored the effect of PEBP1P2 in proliferating VSMCs. We found that PEBP1P2 expression levels in VSMCs treated with PDGF-BB (20 ng/mL) were reduced in a time-dependent manner. In particular, 24 h of treatment exhibited the most significant decrease (Figure 3H). Additionally, overexpression of PEBP1P2 attenuated PDGF-BB-caused proliferation (Figures 3I and 3J), migration (Figures 3K and 3L), and phenotypic transformation (Figure 3M) in VSMCs.
In summary, PEBP1P2 overexpression blocked proliferation, migration, and phenotypic transformation of VSMCs and inhibited pathological functions induced by PDGF-BB.
PEBP1P2 Directly Binds to CDK9
There have been increasing numbers of studies proving that lncRNAs regulate gene expression either in cis or in trans.12 Through quantitative real-time PCR, we found that the expression of PEBP1P2 in neighboring genes at chromosome 2, including RETSAT, CAPG, ELMOD3, TCF7L1 and TGOLN2, had no significant change after PEBP1P2 knockdown or overexpression (Figure S2A), which suggested that PEBP1P2 may work in trans. Previous studies have demonstrated that CDK9 played a crucial role in cardiomyocyte proliferation and atherosclerosis.16,19 Therefore, we assessed the possible interaction between PEBP1P2 and CDK9 by bioinformatics analyses. Using RNA-protein interaction prediction (RPISeq), the potential PEBP1P2-binding protein was predicted, and CDK9 was shown to interact with PEBP1P2 by the random forest (RF) classifier (=0.80) and support vector machine (SVM) classifier (=0.58) (Figure 4A). Moreover, the potential PEBP1P2/CDK9 interaction was tested by the prediction of lncRNA-protein interactions, which showed a high score of 63.73 (Figure S2B). These findings were also further confirmed by catRAPID express, a prediction tool for identification of co-expressed protein and RNA pairs, and the nucleotides between 1 and 220 of PEBP1P2 were predicted to the central residues directly bind to CDK9. This prediction yielded the interaction propensity of 103 and the discriminative power of 99% (Figure 4B). Additionally, the results from the catRAPID signature module, a prediction tool for RNA-binding propensity, showed the overall prediction score of CDK9 was 0.65 (Figure 4C), suggesting that CDK9 may directly bind to RNAs.24 Then, we testified the direct interaction between PEBP1P2 and CDK9 by RNA immunoprecipitation (RIP) assays (Figure 4D), which confirmed the accuracy of our prediction.
Figure 4.

PEBP1P2 Directly Binds to CDK9
(A) The RPISeq (http://pridb.gdcb.iastate.edu/RPISeq/) results showed that the CDK9 protein was predicted to interact with PEBP1P2. (B) Predicted interaction of PEBP1P2 and CDK9 protein by catRAPID analysis (http://service.tartaglialab.com/update_submission/247977/d4d773c37b). (C) catRAPID signature module (http://service.tartaglialab.com/update_submission/247978/970c69ee6e) for prediction of the RNA-binding propensity of CDK9 protein. Overall prediction scores above 0.5 indicate a propensity to bind. (D) RNA immunoprecipitation (RIP) detection of the interaction between CDK9 and PEBP1P2 in VSMCs by agarose gel electrophoresis. (E and F) The protein expression levels of CDK9 in VSMCs after PEBP1P2 knockdown or overexpression were assessed using western blotting (E) and immunofluorescence (F) analysis. Quantified using ImageJ software. Data are presented as mean ± SD. Scale bar, 25 μm. n = 3 for (D)–(F). ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl.
To further explore whether PEBP1P2 was involved in the regulation of CDK9 expression, we knocked down PEBP1P2 and proved that the expression levels of CDK9 increased by using western blot and immunofluorescence. Conversely, the expression of CDK9 was downregulated after PEBP1P2 overexpression (Figures 4E and 4F). Additionally, we sought to investigate the special function of PEBP1P2 in the regulation of the classic molecular pathways associated with atherosclerosis. The levels of the relevant proteins, including nuclear factor κB (NF-κB) p65, phosphorylated (p-)NF-κB p65, p38 mitogen-activated protein kinase (MAPK), p-p38MAPK, c-Jun, p-c-Jun, Akt, and p-Akt, were detected. The results showed that PEBP1P2 knockdown suppressed the expression of p-p38 and p-c-Jun, whereas PEBP1P2 overexpression exhibited the opposite effect. There were no significant differences in the expression of other proteins after PEBP1P2 knockdown or overexpression (Figure S2C).
Briefly, PEBP1P2 could directly bind to CDK9 and negatively regulated its expression, and the p-p38/p-c-Jun pathway may also be its potential regulatory pathway.
Knockdown of CDK9 Inhibited VSMC Proliferation, Migration, and Phenotype Switching, Which Were Involved in the PEBP1P2 Regulatory Pathway
CDK9-knockdown VSMCs were established through transfection of siRNAs and confirmed that CDK9 expression was successfully downregulated by quantitative real-time PCR and western blot, the expression of which could be partially recovered by knockdown of PEBP1P2 (Figures 5A and 5B). Remarkably slower proliferation (Figure 5C) and migration (Figures 5D and 5E) rates were observed in the CDK9 knockdown of VSMCs compared with control, which could be rescued by knockdown of PEBP1P2. Also, CDK9 knockdown upregulated the expression of contractile genes and offset after PEBP1P2-knockdown, whereas there was no effect on CCND1 expression (Figure 5F).
Figure 5.

Knockdown of CDK9 Inhibited VSMC Proliferation, Migration, and Phenotype Switching, Which Were Involved in the PEBP1P2 Regulatory Pathway
(A and B) siRNAs were conducted to knock down CDK9 in VSMCs. In the other group, both CDK9 and PEBP1P2 were knocked down. The mRNA and protein expression levels of CDK9 were measured by quantitative real-time PCR (A) and western blotting (B) analysis. (C) CCK-8 analysis was used to analyze the cell proliferation after CDK9 knockdown or both CDK9 and PEBP1P2 knockdown. (D and E) Wound-healing (D) and transwell (E) analysis for cell migration. (F) Western blotting analysis for the expression of contractile markers and CCND1. (G) The protein levels of CDK9 in VSMCs induced by PDGF-BB (20 ng/mL) were determined by western blotting analysis. (H and I) The siRNA-mediated knockdown of CDK9 was performed in VSMCs, then treated with 20 ng/mL PDGF-BB as indicated. Cell proliferation and migration were detected by CCK-8 (H) and transwell (I) analyses, respectively. Results were uantified using ImageJ software. Data are shown as mean ± SD. Scale bars, 100 μm. n = 3. ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl; #p < 0.01 versus si-CDK9#1 + si-PEBP1P2, ##p < 0.001 versus si-CDK9#1 + si-PEBP1P2 or si-CDK9 + PDGF-BB.
Concomitantly, the expression of CDK9 in VSMCs induced by 20 ng/mL PDGF-BB was increased in a time-dependent manner (Figure 5G), which exhibited the opposite results of PEBP1P2. Likewise, the proliferation and migration in VSMCs under PDGF-BB treatment were offset by CDK9 knockdown (Figures 5H and 5I). Collectively, the proliferation, migration, and phenotypic alternation of VSMCs were depressed after CDK9 knockdown, and which could be recovered by PEBP1P2 knockdown.
Overexpression of CDK9 Promoted VSMC Proliferation, Migration, and Phenotype Switching, Which Were Negatively Regulated by PEBP1P2
To further verify the critical role of CDK9, pAsRed2-CDK9 was constructed. After transfection in VSMCs, we observed that the CDK9 expression was dramatically upregulated using quantitative real-time PCR and western blot analyses, while this upregulation was restored by PEBP1P2 overexpression (Figures 6A and 6B). In contrast to CDK9 knockdown, overexpression of CDK9 accelerated both proliferation (Figure 6C) and migration (Figures 6D and 6E), which could be recovered by PEBP1P2 overexpression. Afterward, we found that α-SMA, CNN1, and SMHC were significantly downregulated after transfection of CDK9 constructs, whereas there was no noticeable effect on CCND1 expression. Concomitantly, the protein levels of these markers were rescued by PEBP1P2 overexpression (Figure 6F). In conclusion, PEBP1P2 regulated the proliferation, migration, and phenotypic transformation through the targeting of CDK9 in VSMCs.
Figure 6.

Overexpression of CDK9 Promoted VSMC Proliferation, Migration, and Phenotype Switching, Which Were Negatively Regulated by PEBP1P2
(A and B) pAsRed2-CDK9 was conducted to overexpress CDK9 in VSMCs. Both CDK9 and PEBP1P2 were overexpressed in the other group. The mRNA and protein levels of CDK9 were measured by quantitative real-time PCR (A) and western blotting (B) analysis. (C) The cell proliferation after CDK9 overexpression or both CDK9 and PEBP1P2 overexpression in VSMCs was determined by CCK-8 analysis. (D and E) Wound-healing (D) and transwell (E) analysis for cell migration in each group. (F) Western blotting analysis for the expression of contractile genes and CCND1 after CDK9 overexpression or both CDK9 and PEBP1P2 overexpression in VSMCs. Results were quantified using ImageJ software. Data are shown as mean ± SD. Scale bars, 100 μm. n = 3. ∗p < 0.05 versus Ctl, ∗∗p < 0.01 versus Ctl, ∗∗∗p < 0.001 versus Ctl; #p < 0.01 versus over-CDK9 + si-PEBP1P2, ##p < 0.001 versus over-CDK9 + si-PEBP1P2.
Overexpression of PEBP1P2 Attenuated Neointima Formation and VSMC Phenotype Transformation Induced in a Balloon-Injured Carotid Artery Model
To determine the role of PEBP1P2 in neointimal formation and VSMC phenotype transformation in vivo, a highly characteristic carotid balloon injury model was executed in rats. Using a plasmid/polyethylenimine (PEI)/polyethylene glycol (PEG) cocktail to perform in vivo transfection,25 the pcDNA3.0-PEBP1P2/PEI/PEG or pcDNA3.0/PEI/PEG cocktail was generated and then injected via the tail vein after the balloon injury every 7 days for a total of 21 days (Figure 7A). Additionally, pcDNA3.0-PEBP1P2/PEI/PEG complex labeling with the fluorescence dye Cy5.5 was conducted intravenously after the balloon injury to investigate whether the injection was successful. The result showed that epifluorescence in vivo was mainly distributed in the injury area 6 h after injection using the in vivo imaging system (IVIS) (Figure 7B). We also detected the expression of PEBP1P2 in injured arteries after 21 days. We found that PEBP1P2 expression was significantly increased in injury arteries infected with the pcDNA3.0-PEBP1P2/PEI/PEG cocktail (Figure 7C).
Figure 7.

PEBP1P2 Impeded Neointima Formation and VSMC Phenotype Transformation In Vivo
(A) Systematic administration of cocktail in rat carotid balloon injury model. (B) The in vivo bioluminescence images of the biodistribution of Cy5.5-labeled pcDNA3.0-PEBP1P2/PEI/PEG complexes in the carotid balloon injury model. (C) Quantitative real-time PCR was performed to detect the expression of PEBP1P2 in carotid arteries from pcDNA3.0/PEI/PEG or pcDNA3.0-PEBP1P2/PEI/PEG cocktail-infected rat carotid arteries at 21 days after balloon injury. (D) Representative H&E-stained arterial sections from pcDNA3.0/PEI/PEG or pcDNA3.0-PEBP1P2/PEI/PEG cocktail-infected rat carotid arteries after balloon injury. (E) Morphometric analysis of the ratio of intima to media. (F) Western blot analysis of the level of contractile markers and CDK9 in carotid arteries in each group on day 21 after balloon injury. Results were quantified using ImageJ software. Data are shown as mean ± SD. Scale bar, 100 μm. n = 5 for (B)–(D). ∗p < 0.05 versus Injury + pcDNA3.0/PEI/PEG cocktail.
Importantly, balloon-injured arteries infected with the pcDNA3.0-PEBP1P2/PEI/PEG cocktail demonstrated a marked decrease in neointima formation compared with arteries transduced with the pcDNA3.0/PEI/PEG cocktail (Figure 7D). Then, we calculated the intima/media ratio, with the results showing that the ratio was lower in the PEBP1P2-treated group than in the control group (Figure 7E), suggesting a protective role for PEBP1P2 in limiting intimal hyperplasia. Furthermore, western blot analysis showed that the expression of VSMC transformation markers was upregulated in pcDNA3.0-PEBP1P2/PEI/PEG complex-infected, balloon-injured arteries compared with the control group, and this increase was accompanied by a marked decrease in CDK9 expression (Figure 7F). These data suggested that PEBP1P2 plays a critical role in VSMC phenotypic switching and neointimal hyperplasia in vivo.
Discussion
It is well known that the abnormal proliferation and migration of VSMCs are closely associated with CVDs, such as atherosclerosis, hypertension, pulmonary hypertension, and aortic aneurysm, especially in-stent restenosis (ISR).26,27 To date, restenosis after stent implantation continues to present several knotty clinical problems. Significantly, the development of restenosis was due to neointima hyperplasia chiefly caused by aberrant VSMC proliferation and migration after vascular wall injury.28 Although the improvement of drug-eluting stents (DESs) significantly reduced the in-stent stenosis incidence rate by releasing antiproliferative drugs to inhibit VSMC proliferation and migration, it also posed a new problem: late stent thrombosis due to the side effect of antiproliferative drug inevitably suppressing re-endothelialization.27 Therefore, it is necessary to explore the regulators and molecular mechanisms in VSMC proliferation and migration. As a kind of considerable epigenetic regulator, an increasing amount of evidence has shown that dysregulation of lncRNA expression and function were firmly related to vascular cell functions and pathologies, working through a variety of mechanisms.27,29 Among them, some have been considered as critical regulators for VSMC phenotypic switching during vascular development. For example, Ahmed et al.23 reported that lncRNA NEAT1 promoted VSMC proliferation and migration by binding WDR5 to restrain differentiation during phenotypic switching. Jin et al.30 revealed a novel VSMC enriched lncRNA, AK098656, which was upregulated in hypertension patient plasma and accelerated VSMC phenotypic switching via direct interacting with MYH11/FN1 occured in VSMCs. Wang et al.31 demonstrated that GAS5 repressed VSMCs proliferation, migration, and phenotypic conversion via β-catenin signaling. Therefore, it is promising to develop non-coding RNAs as significant targets for the regulation of phenotype transformation in VSMCs.
In this study, we identified and explored the functional roles and regulatory mechanisms of a novel pseudogene-derived lncRNA, PEBP1P2. We found the PEBP1P2 was enriched in human VSMCs and rat aortic tissue. Additionally, it was downregulated in human VSMCs stimulated by PDGF-BB and in rat carotid artery tissues injured by a balloon. These results indicated that PEBP1P2 could be involved in the proliferation and migration of VSMCs, and it may play a significant role in regulating vascular function. In a knockdown experiment, we further found that PEBP1P2 remarkably increased VSMC proliferation, migration, and phenotypic transformation. Alternatively, these processes were observably suppressed due to PEBP1P2 overexpression. Meanwhile, we observed that this evidence occurred under PDGF-BB-induced VSMCs. Interestingly, PEBP1P2 knockdown or overexpression had no significant effect on CCND1 expression. However, above all, an in vivo experiment showed that PEBP1P2 could play an essential part in inhibiting neointimal formation and VSMC phenotypic switching. These results suggested the vital contribution of PEBP1P2 in regulating VSMC proliferation, migration, and phenotype alternation, and it may act as a promising therapeutic target in vascular diseases, such as in-stent restenosis after angioplasty.
The regulatory mechanisms of lncRNAs are complex. Among them, regulation of target gene expression in trans, depending on the direct interaction with its protein partner, RNA-binding protein, is one of the important pathways.12,32 For example, Atianand et al.33 reported that lncRNA EPS, a negative regulator of inflammatory responses, inhibited the expression of immune genes by directly binding to hnRNPL to maintain the nucleosome position in macrophages. We found that PEBP1P2 may be directly bound to CDK9 by bioinformatics analysis, which was further confirmed using RIP analysis. Next, we observed that PEBP1P2 knockdown distinctly increased CDK9 expression, while its levels declined after PEBP1P2 overexpression. Interestingly, in contrast to PEBP1P2, CDK9 was upregulated in PDGF-BB-treated human VSMCs and injured rat carotid arteries, which suggested that CDK9 was probably capable of regulating VSMC proliferation and migration, which has been proven in other diseases. Sano et al.34 demonstrated that CDK9 was increased and activated in cardiac hypertrophy, and its chronic activation predisposed mice to heart failure. Tarhriz et al.35 showed that CDK9 was a crucial factor in cardiac differentiation by inducing miR-1/206 expression and inhibiting miR-133. However, the function of CDK9 in VSMCs was virtually unknown. Consequently, we investigated the role of CDK9 in regulating VSMCs. The results showed that the expression of CDK9 could be increased by PEBP1P2 knockdown. Conversely, CDK9 expression also could be offset by PEBP1P2 upregulation. In addition, we have also proven that CDK9 regulated proliferation, migration, and the expression of differentiation markers in both resting and proliferating VSMCs, which have already been shown to be closely associated with PEBP1P2. Collectively, our results suggested that CDK9 serves as the target of PEBP1P2, which was strictly related to VSMC proliferation, migration, and phenotypic transition.
Clinically, the cost of CVDs in human suffering and economic burden is almost impossible to calculate because of its high incidence and chronic course.36,37 Due to these characteristics of CVDs, they face many difficulties in prevention and treatment. Recently, increasing discoveries have shown that ncRNAs play a critical regulatory role in atherosclerosis, mal-remodeling in cardiac hypertrophy, myocardial ischemic reperfusion injury and infarction, and diabetic cardiomyopathy.38, 39, 40, 41 Additionally, several lncRNAs have been successfully demonstrated as biomarkers or therapeutic targets for some CVDs.14 This information on lncRNAs has provided novel concepts on CVD pathogenesis, and in some conditions promoted the opportunities for early diagnosis and therapeutic intervention.42, 43, 44 In our study, we found that PEBP1P2 was differentially expressed in CHD and healthy people. In future studies, we plan to increase the sample size to further study the crucial involvement of PEBP1P2 in different periods of CHD, which may provide more convincing evidence in the diagnosis of CHD in the future.
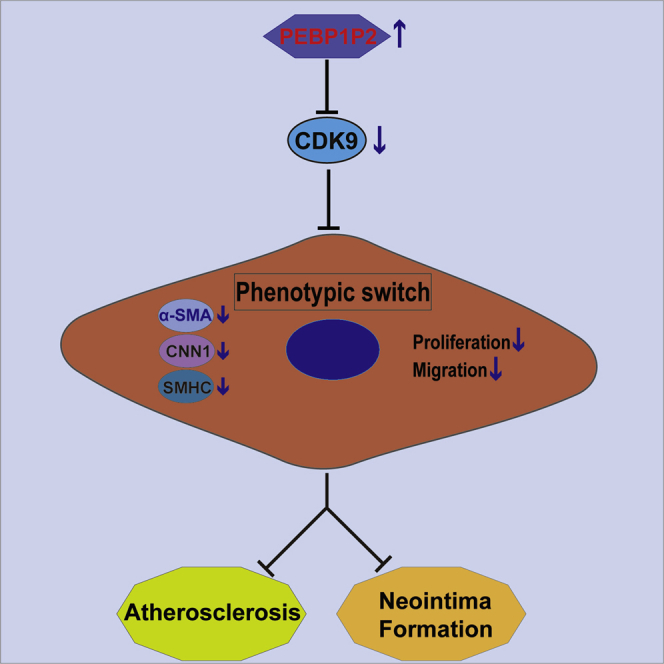
In conclusion, our data revealed that PEBP1P2 presented beneficial protective effects against abnormal proliferation, migration, and phenotypic switching of VSMCs and atherosclerosis mainly via targeting the CDK9 pathway. In these pathological processes, PEBP1P2 works to restrain CDK9 expression in VSMCs through binding directly to it (Figure 8). These findings implied that PEBP1P2 might be a promising therapeutic target for atherosclerosis and CVDs with dyshomeostasis of VSMCs.
Figure 8.

Schematic Diagram Showing the Regulatory Roles of PEBP1P2 in Atherosclerosis
PEBP1P2 might directly bind to CDK9 to restrain its expression and inhibit p-p38 and p-c-Jun expression. This process was connected with VSMC proliferation, migration, and phenotype switching. As a consequence, PEBP1P2 suppressed atherosclerosis by regulating CDK9 expression.
Materials and Methods
Clinical Specimens
The peripheral blood samples were obtained from 56 individuals at the affiliated hospital of Qingdao University (Qingdao, China) from March 2018 to February 2019. Among them, there were 27 CHD patients without any stent implantation-confirmed lesion stenosis ≥50% in any of the main epicardial coronary arteries (i.e., left main artery, left anterior descending artery, left circumflex artery, or right coronary artery) by angiography and 29 control samples from healthy individuals without any diseases. These blood samples were collected before taking any medicine. Sera were extracted from the samples and stored at –80°C for RNA isolation and quantitative real-time PCR analysis. Advanced carotid atherosclerotic plaque tissues were harvested from patients undergoing carotid endarterectomy for carotid stenosis. These tissues were usually discarded after routine surgery, so these collections for the present work did not have any adverse effects on the patients. The normal carotid intima tissues were collected from voluntary body donors. The adventitial layers were removed, and the tissues were stored in liquid nitrogen for RNA isolation and quantitative real-time PCR analysis. Exclusion criteria were diabetes, cancer, immunological diseases, hematological disorder, or infection. Informed consent was required for all people. The Research Ethics Committees of The Affiliated Hospital of Qingdao University approved this study, and all experiments were conducted following the principles of the Declaration of Helsinki.
Cell Culture, Transfection, and Treatment
VSMCs (a human aortic smooth muscle cell line) (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS) (ExCell Bio, Shanghai, China), penicillin (50 U/mL), and streptomycin (50 μg/mL) in a humidified atmosphere at 37°C and 5% CO2. According to the manual of the manufacturer, cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
For PDGF-BB induction experiments, VSMCs were treated with PDGF-BB (20 ng/mL, Calbiochem, Burlington, MA, USA) for 12 or 24 h.
Preparation of siRNA, Plasmid Expression Vector, and In Vivo Transfection Cocktail
The siRNA sequence specially targeting PEBP1P2 or CDK9 was designed and synthesized (GenePharma, Shanghai, China). VSMCs with knockdown of PEBP1P2 or CDK9 were achieved by stably transfecting si-PEBP1P2 or si-CDK9 and transfected with control non-silencing siRNA as control following the RNAi manual of the manufacturer. The sequences of the siRNA oligonucleotides are presented in Table S2.
The PEBP1P2 sequence was synthesized and subcloned into the pcDNA3.0 vector (BGI, Shenzhen, China), and the cDNA that encoded the CDS of CDK9 was multiplied via PCR and subcloned into the SalI and EcoRI sites of the pAsRed2 vector. VSMCs with ectopic expression of lncRNA PEBP1P2 or CDK9 were achieved by stably transfecting pcDNA3.0-PEBP1P2 or pAsRed2-CDK9, and VSMCs were transfected with empty pcDNA3.0 or pAsRed2 vector as control, respectively.
The plasmid/PEI/PEG cocktail was prepared by mixing the solution of plasmids and aldehyde-PEG (molecular weight [MW] of 20,000 Da, Solarbio, Beijing, China), followed by the addition of dendritic PEI (MW of 20,000, Polysciences, Warrington, PA, USA) solution. The pH of the final solution was adjusted to 8.0, and the molar ratio of plasmid/PEI/PEG was 1:10:100. The detailed steps were performed as described previously.25
Rat Carotid Artery Injury Model, In Vivo Transfection, and Imaging
Male Sprague-Dawley rats (300–350 g) were purchased from Vital River Laboratory Animal Technology (Beijing, China) for this study. The rat carotid artery injury models were conducted according to previous reports.23,45 Briefly, rats were anesthetized by an intraperitoneal injection of 10% chloral hydrate solution (300 mg/kg) and then injected with heparin (200 U/kg) by tail vein to prevent thrombosis. The 8 × 1.2-mm coronary dilatation catheter (Mini TREK, Abbott Laboratories, Chicago, IL, USA) was placed into the common carotid artery via the left external carotid artery. The balloon was inflated (8 atmospheres [atm], 5 s) and partially withdrawn and reinserted three times to ensure endothelial injury completely. The catheter was removed, the external carotid artery was then permanently ligated, and the blood flow in the common carotid artery and its inner branch was restored. The common carotid artery was only isolated without dilatation injury as the sham group. Rats were sacrificed after 3, 7, and 21 days, and aortic tissues were collected for RNA extraction for subsequent PEBP1P2 detection.
In vivo transfection was performed using a plasmid/PEI/PEG cocktail. The suspension of the cocktail of plasmids coding the PEBP1P2 gene (3.5 mg/kg) was injected into the tail vein after the balloon injury every 7 days for a total of 21 days, and saline or cocktail of empty plasmids were injected respectively as controls. After 21 days, the left uninjured common carotid artery and the injured artery were respectively taken out either for total RNA and protein extraction or OCT (optimum cutting temperature) embedding.
In order to investigate the targeting capability of the cocktail in injured arteries, the complexes labeled with the fluorescence dye CY5.5 were injected via tail vein after the balloon injury. The rats were anesthetized for a whole-body IVIS (Caliper Life Sciences, Hopkinton, MA, USA) 6 h later. Then, the rats were euthanized, and carotid arteries were dissected for fluorescence imaging immediately. The right uninjured carotid artery served as control.
RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
Total RNA was abstracted from the blood, tissue, and cell samples with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Then, the cDNA synthesis was achieved with a PrimeScript reverse transcription (RT) reagent kit (Takara, Kyoto, Japan). Quantitative real-time PCR was implemented using Hieff UNICON Power qPCR SYBR Green master mix (Yeasen, Shanghai, China). GAPDH was used as an endogenous control. The ΔΔCt method was performed for analysis of relative gene expression data. All experimental procedures were conducted according to the instructions of the manufacturers. PCR primers are shown in Table S3.
Western Blot Analysis
Cells were washed twice in PBS solution and lysed with radioimmunoprecipitation assay (RIPA) buffer (Solarbio, Beijing, China) according to the instructions of the manufacturer. Protein concentration was measured using the bicinchoninic acid (BCA) (Solarbio, Beijing, China) method. Then, the protein was heated in SDS-PAGE loading buffer (Solarbio, Beijing, China) at 95°C for 5 min, equal quantities of protein were isolated using 10% SDS-PAGE, and gels were imprinted onto the 0.45-μm polyvinylidene fluoride (PVDF) membrane. After blocking in Tris-buffered saline with Tween 20 (TBS-T) and 5% fat-free milk for 1 h, primary antibodies (a-SAM, ab5694, 1:2,000, Abcam; SMHC, ab53219, 1:2,000, Abcam, Cambridge, MA, USA; CNN1, 24855-1-AP, 1:3,000, Proteintech; CCND1, 60186-1-Ig, 1:5,000, Proteintech, Chicago, IL, USA; CDK9, #2316, 1:1,000, Cell Signaling Technology, Boston, MA, USA; Akt, #4691, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p-Akt (Ser473), #4060, 1:1,000, Cell Signaling Technology, Boston, MA, USA; c-Jun, #9165, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p-c-Jun, #91952, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p38, #9212, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p-p38, #4511, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p65, #8242, 1:1,000, Cell Signaling Technology, Boston, MA, USA; p-p65, bs-17502R, 1:1,000, Bioss Antibodies, Beijing, China; and GAPDH, CAB932MI22, 1:5,000, Cloud-Clone, Wuhan, China) were applied. The secondary antibodies were purchased from Abcam. Images were detected by the Fusion SoloS system (Paris, France), and band densities were quantified using ImageJ 1.5.1.
Cell Proliferation Analysis
Cells were sowed in 96-well plates, and then cell proliferation was tested by the Cell-Light EdU Apollo 567 in vitro kit (RiboBio, Guangzhou, China) and CCK-8 (Yeasen, Shanghai, China). The experimental procedures were conducted according to the instructions of the manufacturer. Optical density was measured by ImageJ 1.5.1.
Cell Migration Analysis
Detection of cell migration was accomplished by wound healing and transwell assays, respectively.
Wound Healing Assay
VSMCs were seeded in six-well plates for the wound healing assay. Wounds through the cell monolayer were made by 1,000-μL plastic tips after cells were treated for 12 h, and images were taken using a Nikon Ti-S inverted phase-contrast microscope (Nikon, Tokyo, Japan) at different times.
Transwell Assay
VSMCs were cultured in DMEM medium with 10% FBS for 24 h after treatment. Then, the treated cells (1 × 105 cells) were trypsinized and seeded into chambers (8-μm pores, 24-well format, Corning Life Sciences, Corning, NY, USA) in FBS-free DMEM medium (200 μL). Chambers were then dipped into lower chambers with 10% FBS medium (500 μL) and/or containing PDGF-BB (20 ng/mL, PeproTech, NJ, USA) for 24 h. Next, cells in lower membranes were fixed with 4% paraformaldehyde after removal of medium and stained with 0.1% gentian violet for 0.5 h, separately. Images with five stochastic fields per membrane were obtained with a Nikon Ti-S inverted phase-contrast microscope.
Wound healing areas and migrated cell numbers were measured by ImageJ 1.5.1.
RIP
Approximately 1 × 107 VSMCs were used for each sample. Cells were cleaned twice by PBS and lysed with RIPA buffer (Solarbio, Beijing, China) with ribonuclease inhibitor (Solarbio, Beijing, China) for 5 min. Lysates were centrifuged at 12,000 rpm at 4°C for 4 min and supernatants were collected to determine protein concentration by a BCA assay. Subsequently, protein (500 μg) and magnetic beads (30 μL) were incubated with CDK9 antibody (5 μg) or control mouse immunoglobulin G (IgG) (5 μg) overnight at 4°C, respectively. The protein, beads, and RNA complexes were immunoprecipitated, and then RNA was abstracted using TRIzol followed by reverse transcription, RT-PCR, and agarose gel electrophoresis.
Fluorescence Confocal Microscopy
VSMCs were seeded on coverslips for 24 h. Then, cells were washed twice in PBS solution and fixed with 4% paraformaldehyde for 15 min. Next, cells were incubated with anti-CDK9 antibody (Cell Signaling Technology, #2316, rabbit, 1:100) and subsequently with fluorescein isothiocyanate (FITC)-conjugated AffiniPure goat anti-rabbit IgG (#111-095-003, 1:100, Jackson ImmunoResearch, West Grove, PA, USA). DAPI was used to stain nuclei. Images were taken by a Leica TCS SP8 confocal laser scanning microscope. Optical density was measured using ImageJ 1.5.1.
Sectioning and Hematoxylin and Eosin (H&E) Staining
Serial cross-sections of carotid arteries (8 μm) were obtained at the injured area, and six sections from each vessel were collected for morphometric analysis. H&E staining was performed following the instructions of the H&E staining kit (Meilunbio, Dalian, China), and images were captured by a Nikon Ti-S inverted phase-contrast microscope. For the quantitative analysis, areas of intima and media were measured using ImageJ software.
Statistical Analysis
Continuous and categorical variables are represented as mean ± SD and number (percentages), respectively. Statistical analysis was performed via GraphPad Prism 8.0 and SPSS 25.0. Statistical differences were measured through unpaired Student’s t tests, one-way ANOVA with a Tukey’s post hoc test, or a Mann-Whitney test depending on the distribution of variables. p < 0.05 was considered statistically significant. Each experiment was repeated at least three times.
Author Contributions
X.H. and Y.L. carried out the cell and protein experiments; Z.W., X.H., and Z.L. conducted clinical analysis; Y.Y. and J.T. carried out plasmid constructs and molecular experiments; T.Y., Y.Y., and Z.W. participated in the data analysis, performed the statistical analysis, and drafted the manuscript; and T.Y. and H.X. conceived and designed the study, participated in the data analysis and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant nos. 31701208 and 81870331), the Natural Science Foundation of Shandong Province (grant no. ZR2017MC067), and by The People’s Livelihood Science and Technology Project of Qingdao (grant nos. 18-2-2-65-jch and 19-6-1-7-nsh).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.08.013.
Contributor Information
Tao Yu, Email: yutao0112@qdu.edu.cn.
Hui Xin, Email: xinhuiqy@163.com.
Supplemental Information
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Roth G.A., Johnson C.O., Abate K.H., Abd-Allah F., Ahmed M., Alam K., Alam T., Alvis-Guzman N., Ansari H., Ärnlöv J., Global Burden of Cardiovascular Diseases Collaboration The burden of cardiovascular diseases among US States, 1990–2016. JAMA Cardiol. 2018;3:375–389. doi: 10.1001/jamacardio.2018.0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu S., Li Y., Zeng X., Wang H., Yin P., Wang L., Liu Y., Liu J., Qi J., Ran S. Burden of cardiovascular diseases in China, 1990–2016: findings from the 2016 Global Burden of Disease Study. JAMA Cardiol. 2019;4:342–352. doi: 10.1001/jamacardio.2019.0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li F.J., Zhang C.L., Luo X.J., Peng J., Yang T.L. Involvement of the miR-181b-5p/HMGB1 pathway in Ang II-induced phenotypic transformation of smooth muscle cells in hypertension. Aging Dis. 2019;10:231–248. doi: 10.14336/AD.2018.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 6.Basatemur G.L., Jørgensen H.F., Clarke M.C.H., Bennett M.R., Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019;16:727–744. doi: 10.1038/s41569-019-0227-9. [DOI] [PubMed] [Google Scholar]
- 7.Lacolley P., Regnault V., Segers P., Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol. Rev. 2017;97:1555–1617. doi: 10.1152/physrev.00003.2017. [DOI] [PubMed] [Google Scholar]
- 8.Jun M.Y., Karki R., Paudel K.R., Sharma B.R., Adhikari D., Kim D.W. Alkaloid rich fraction from Nelumbo nucifera targets VSMC proliferation and migration to suppress restenosis in balloon-injured rat carotid artery. Atherosclerosis. 2016;248:179–189. doi: 10.1016/j.atherosclerosis.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 9.Shi N., Chen S.Y. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J. Biomed. Res. 2014;28:40–46. doi: 10.7555/JBR.28.20130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett M.R., Sinha S., Owens G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devaux Y., Zangrando J., Schroen B., Creemers E.E., Pedrazzini T., Chang C.P., Dorn G.W., 2nd, Thum T., Heymans S., Cardiolinc network Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015;12:415–425. doi: 10.1038/nrcardio.2015.55. [DOI] [PubMed] [Google Scholar]
- 12.Kopp F., Mendell J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sallam T., Sandhu J., Tontonoz P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function. Circ. Res. 2018;122:155–166. doi: 10.1161/CIRCRESAHA.117.311802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leeper N.J., Maegdefessel L. Non-coding RNAs: key regulators of smooth muscle cell fate in vascular disease. Cardiovasc. Res. 2018;114:611–621. doi: 10.1093/cvr/cvx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H., Pandey S., Travers M., Sun H., Morton G., Madzo J., Chung W., Khowsathit J., Perez-Leal O., Barrero C.A. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. 2018;175:1244–1258.e26. doi: 10.1016/j.cell.2018.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matrone G., Wilson K.S., Maqsood S., Mullins J.J., Tucker C.S., Denvir M.A. CDK9 and its repressor LARP7 modulate cardiomyocyte proliferation and response to injury in the zebrafish heart. J. Cell Sci. 2015;128:4560–4571. doi: 10.1242/jcs.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Y., Zhan Y., Hou G., Li L. Cyclin-dependent kinase 9 may as a novel target in downregulating the atherosclerosis inflammation (Review) Biomed. Rep. 2014;2:775–779. doi: 10.3892/br.2014.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itkonen H.M., Poulose N., Walker S., Mills I.G. CDK9 inhibition induces a metabolic switch that renders prostate cancer cells dependent on fatty acid oxidation. Neoplasia. 2019;21:713–720. doi: 10.1016/j.neo.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Y., Zhao S., Gong Y., Hou G., Li X., Li L. Serum cyclin-dependent kinase 9 is a potential biomarker of atherosclerotic inflammation. Oncotarget. 2016;7:1854–1862. doi: 10.18632/oncotarget.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell R.D., Long X., Lin M., Bergmann J.H., Nanda V., Cowan S.L., Zhou Q., Han Y., Spector D.L., Zheng D., Miano J.M. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler. Thromb. Vasc. Biol. 2014;34:1249–1259. doi: 10.1161/ATVBAHA.114.303240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haeussler M., Zweig A.S., Tyner C., Speir M.L., Rosenbloom K.R., Raney B.J., Lee C.M., Lee B.T., Hinrichs A.S., Gonzalez J.N. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019;47:D853–D858. doi: 10.1093/nar/gky1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed A.S.I., Dong K., Liu J., Wen T., Yu L., Xu F., Kang X., Osman I., Hu G., Bunting K.M. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA. 2018;115:E8660–E8667. doi: 10.1073/pnas.1803725115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang B., Armaos A., Tartaglia G.G. RNAct: protein-RNA interaction predictions for model organisms with supporting experimental data. Nucleic Acids Res. 2019;47:D601–D606. doi: 10.1093/nar/gky967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakae M., Ito T., Yoshihara C., Iida-Tanaka N., Yanagie H., Eriguchi M., Koyama Y. Highly efficient in vivo gene transfection by plasmid/PEI complexes coated by anionic PEG derivatives bearing carboxyl groups and RGD peptide. Biomed. Pharmacother. 2008;62:448–453. doi: 10.1016/j.biopha.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Brozovich F.V., Nicholson C.J., Degen C.V., Gao Y.Z., Aggarwal M., Morgan K.G. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol. Rev. 2016;68:476–532. doi: 10.1124/pr.115.010652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Indolfi C., Iaconetti C., Gareri C., Polimeni A., De Rosa S. Non-coding RNAs in vascular remodeling and restenosis. Vascul. Pharmacol. 2019;114:49–63. doi: 10.1016/j.vph.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y., He X., Liu R., Qin Y., Wang S., Yao X., Li C., Hu Z. lncRNA CRNDE regulates the proliferation and migration of vascular smooth muscle cells. J. Cell. Physiol. 2019;234:16205–16214. doi: 10.1002/jcp.28284. [DOI] [PubMed] [Google Scholar]
- 29.Das S., Zhang E., Senapati P., Amaram V., Reddy M.A., Stapleton K., Leung A., Lanting L., Wang M., Chen Z. A novel angiotensin II-induced long noncoding RNA giver regulates oxidative stress, inflammation, and proliferation in vascular smooth muscle cells. Circ. Res. 2018;123:1298–1312. doi: 10.1161/CIRCRESAHA.118.313207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin L., Lin X., Yang L., Fan X., Wang W., Li S., Li J., Liu X., Bao M., Cui X. AK098656, a novel vascular smooth muscle cell-dominant long noncoding RNA, promotes hypertension. Hypertension. 2018;71:262–272. doi: 10.1161/HYPERTENSIONAHA.117.09651. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y.N., Shan K., Yao M.D., Yao J., Wang J.J., Li X., Liu B., Zhang Y.Y., Ji Y., Jiang Q., Yan B. Long noncoding RNA-GAS5: a novel regulator of hypertension-induced vascular remodeling. Hypertension. 2016;68:736–748. doi: 10.1161/HYPERTENSIONAHA.116.07259. [DOI] [PubMed] [Google Scholar]
- 32.Wapinski O., Chang H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. [Google Scholar]
- 33.Atianand M.K., Hu W., Satpathy A.T., Shen Y., Ricci E.P., Alvarez-Dominguez J.R., Bhatta A., Schattgen S.A., McGowan J.D., Blin J. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell. 2016;165:1672–1685. doi: 10.1016/j.cell.2016.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sano M., Abdellatif M., Oh H., Xie M., Bagella L., Giordano A., Michael L.H., DeMayo F.J., Schneider M.D. Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat. Med. 2002;8:1310–1317. doi: 10.1038/nm778. [DOI] [PubMed] [Google Scholar]
- 35.Tarhriz V., Eyvazi S., Musavi M., Abasi M., Sharifi K., Ghanbarian H., Hejazi M.S. Transient induction of Cdk9 in the early stage of differentiation is critical for myogenesis. J. Cell. Biochem. 2019;120:18854–18861. doi: 10.1002/jcb.29204. [DOI] [PubMed] [Google Scholar]
- 36.Celermajer D.S., Chow C.K., Marijon E., Anstey N.M., Woo K.S. Cardiovascular disease in the developing world: prevalences, patterns, and the potential of early disease detection. J. Am. Coll. Cardiol. 2012;60:1207–1216. doi: 10.1016/j.jacc.2012.03.074. [DOI] [PubMed] [Google Scholar]
- 37.Bansilal S., Castellano J.M., Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int. J. Cardiol. 2015;201(Suppl 1):S1–S7. doi: 10.1016/S0167-5273(15)31026-3. [DOI] [PubMed] [Google Scholar]
- 38.Das A., Samidurai A., Salloum F.N. Deciphering non-coding RNAs in cardiovascular health and disease. Front. Cardiovasc. Med. 2018;5:73. doi: 10.3389/fcvm.2018.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang N., Jiang S., Yang Y., Liu S., Ponnusamy M., Xin H., Yu T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018;36:e12436. doi: 10.1111/1755-5922.12436. [DOI] [PubMed] [Google Scholar]
- 40.Liu S., Yang Y., Jiang S., Tang N., Tian J., Ponnusamy M., Tariq M.A., Lian Z., Xin H., Yu T. Understanding the role of non-coding RNA (ncRNA) in stent restenosis. Atherosclerosis. 2018;272:153–161. doi: 10.1016/j.atherosclerosis.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 41.Liu S., Yang Y., Jiang S., Xu H., Tang N., Lobo A., Zhang R., Liu S., Yu T., Xin H. miR-378a-5p regulates proliferation and migration in vascular smooth muscle cell by targeting CDK1. Front. Genet. 2019;10:22. doi: 10.3389/fgene.2019.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J., Rossi J.J. Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol. Ther. Nucleic Acids. 2014;3:e169. doi: 10.1038/mtna.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon S., Rossi J.J. Aptamers: uptake mechanisms and intracellular applications. Adv. Drug Deliv. Rev. 2018;134:22–35. doi: 10.1016/j.addr.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W., Halligan K.E., Zhang X., Bisaillon J.M., Gonzalez-Cobos J.C., Motiani R.K., Hu G., Vincent P.A., Zhou J., Barroso M. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ. Res. 2011;109:534–542. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.