

Summary
Dysregulation of inositol-requiring enzyme 1 (IRE1), the primary transducer of Unfolded Protein Response (UPR), has been observed in tumor initiation and progression, but the underlying mechanism remains to be further elucidated. In this study, we identified that the IRE1 gene is frequently amplified and over-expressed in aggressive luminal B breast cancer cells and that IRE1 upregulation is significantly associated with worse overall survival of patients with breast cancer. IRE1 processes and mediates degradation of a subset of tumor suppressor microRNAs (miRNAs), including miR-3607, miR-374a, and miR-96, via a mechanism called Regulated IRE1-Dependent Decay (RIDD). IRE1-dependent degradation of tumor suppressor miR-3607 leads to elevation of RAS oncogene GTPase RAB3B in breast cancer cells. Inhibition of IRE1 endoribonuclease activity with the pharmacological compound 4μ8C or genetic approaches effectively suppresses luminal breast cancer cell proliferation and aggressive cancer phenotypes. Our work revealed the IRE1-RIDD-miRNAs pathway that promotes malignancy of luminal breast cancer.
Subject Areas: Bioinformatics, Cancer, Molecular Mechanism of Gene Regulation
Graphical Abstract
Highlights
-
•
IRE1 gene is amplified and over-expressed in Luminal B breast cancer cells
-
•
IRE1 processes and mediates degradation of tumor suppressor miRNAs
-
•
IRE1-dependent degradation of tumor suppressor miRNAs elevates expression of RAB3B
-
•
Inhibition of IRE1 suppresses Luminal breast cancer cell proliferation
Bioinformatics; Cancer; Molecular Mechanism of Gene Regulation
Introduction
Inositol-requiring enzyme 1 (IRE1, also known as IRE1α and ENR1), an endoplasmic reticulum (ER)-resident, type I transmembrane protein, was originally identified as a primary transducer of Unfolded Protein Response (UPR) (Tirasophon et al., 1998). IRE1 possesses two distinct catalytic domains: an endoribonuclease (RNase) and a serine/threonine kinase. Upon a variety of cell-intrinsic or extrinsic stress conditions that trigger accumulation of unfolded or misfolded proteins in the ER (collectively known as ER stress), such as hypoxia, nutrient deprivation, and protein over-expression, IRE1 oligomerizes and autophosphorylates to render its RNase activity (Zhang and Kaufman, 2008). IRE1 executes unconventional splicing of the mRNA that encodes a potent transcription factor, XBP1, which activates the expression of genes involved in protein folding, secretion, and degradation of misfolded proteins. In addition to processing the XBP1 mRNA, IRE1 can process select mRNAs or precursor microRNAs (pre-miRNAs), leading to their degradation, a process known as regulated IRE1-dependent decay (RIDD) (Hollien and Weissman, 2006; Upton et al., 2012; Wang et al., 2017). Recent studies suggested that IRE1 undergoes dynamic conformational changes and switches its functions depending on the duration of ER stress (Li et al., 2010; Lin et al., 2007; Tam et al., 2014). It has been proposed that IRE1 quickly forms oligomeric clusters to process XBP1 mRNA splicing (XBPs) in response to acute ER stress. However, under chronic or physiological ER stress, or when IRE1 is exogenously over-expressed in mammal cells, IRE1's functional specificity as an RNase shifts to cleaving primarily ER-targeted mRNAs or miRNAs, instead of XBP1 mRNA, via the RIDD pathway (Han et al., 2009; Hollien et al., 2009; So et al., 2012; Wang et al., 2017, 2018). Importantly, the RIDD targets may depend on the tissue context and biological processes (Dejeans et al., 2014; Dufey et al., 2020; Lhomond et al., 2018). Previous studies on the role of IRE1 in human cancer, particularly breast cancer, have focused on the IRE1/XBP1 pathway (Chen et al., 2014; McGrath et al., 2018; Zhao et al., 2018). However, whether the IRE1-RIDD pathway plays a role in breast cancer malignancy remains largely unknown.
Dysregulation of miRNAs was found to be involved in the initiation of tumorigenesis, as well as the progression, invasion, and metastasis of cancers (Dawson and Kouzarides, 2012; Farazi et al., 2011; Jacobsen et al., 2013; Stecklein et al., 2012; Wang and Wang, 2012; You and Jones, 2012). Increasing evidence indicates that miRNAs are less abundant in tumors, including breast cancer, than in their normal tissue counterparts, leading to the notion that miRNAs are predominantly tumor suppressors rather than tumor promoters (Jacobsen et al., 2013; Liu, 2012; Zhang et al., 2013). Canonical miRNA biogenesis requires a series of sequential enzymatic processing reactions, and the key enzyme among these is the DICER RNase, which cleaves the pre-miRNA to generate a mature miRNA (Ha and Kim, 2014). miRNAs regulate gene expression by inducing degradation of target mRNAs and/or by interfering with protein translational machinery (Ameres and Zamore, 2013; Lin and Gregory, 2015). A search for general regulators of cancer metastasis identified a set of miRNAs whose expression is repressed in metastatic human breast cancer cells, suggesting crucial roles of miRNAs in repressing breast cancer progression (Tavazoie et al., 2008).
In this study, we revealed that the UPR transducer IRE1 acts as an oncogenic factor to repress a subset of tumor suppressor miRNAs via the RIDD mechanism and subsequently control expression of a major oncogene, RAB3B, in aggressive luminal breast cancer cells. Our work suggests that the IRE1-RIDD-miRNAs axis has a functional role during tumor development and progression and therefore provides a foundation for treating aggressive breast cancers by specifically targeting oncogenic RNase activity of IRE1.
Results
IRE1 Is Frequently Gained/Amplified in Luminal B Breast Cancers and Associated with Patient Survival
As copy number alteration (CNA) is an important mechanism that activates oncogenes or inactivates tumor suppressors in human cancers (Albertson, 2006; Albertson et al., 2003; Beroukhim et al., 2010), we first queried CNAs of the IRE1 gene in more than 10,000 tumor samples across 32 cancer types from the Pan-Cancer Atlas of The Cancer Genome Atlas (TCGA) via cBioPortal (Cerami et al., 2012; Gao et al., 2013). Although IRE1 is altered across many types of human tumors, the highest frequency (6.26%) of the IRE1 gene amplification was observed in breast cancers (Figure 1A). When we incorporated low-level gain of IRE1, 39.35% TCGA breast cancer samples had an increased IRE1 copy number. Based on the expression of 50 select genes (PAM50), breast cancer is categorized into five molecular subtypes: estrogen receptor-positive luminal A and B, epidermal growth factor receptor 2-enriched (HER2+), basal-like (commonly known as triple-negative), and normal-like breast cancers (Cancer Genome Atlas, 2012; Perou et al., 2000; Riaz et al., 2013). To determine the breast cancer subtypes that possess IRE1 CNAs, we performed the CNA analysis of IRE1 in different subtypes of breast cancer. We found that luminal B breast cancer had the highest frequency (68.53%) of the IRE1 gene gain/amplification, whereas the normal-like breast cancer subtype had the lowest frequency (8.57%) (Figure 1B). Luminal B breast cancer has been reported to have lower expression of estrogen receptors, higher expression of proliferation markers, and higher histologic grade than luminal A (Ades et al., 2014; Tran and Bedard, 2011). In addition, luminal B breast cancer exhibits worse prognosis and has a distinct response profile to hormone therapy and chemotherapy (Ades et al., 2014; Tran and Bedard, 2011).
Figure 1.

The IRE1 Gene is Amplified and Over-expressed in Luminal Breast Cancer
(A) Frequencies of high-level amplification of the IRE1 gene in more than 10,000 TCGA tumor samples across 32 tumor types.
(B) Frequencies of Gain/Amp of the IRE1 gene in five subtypes of TCGA breast cancer samples. The frequencies were based on the TCGA dataset of 1,084 human patients with breast cancer.
(C) Kaplan-Meier plots of overall survival associated with gain/amplification of IRE1 in METABRIC breast cancers. The plot was based on the METABRIC dataset, which includes 1,974 human patients with breast cancer.
(D) Expression levels of the IRE1 mRNA in a panel of normal-like, luminal, HER2-positive, and basal-like breast cancer cell lines, determined by qPCR (n = 3 biological repeats). The mRNA expression levels in MCF10A cells were arbitrarily set as 1. Relative expression levels are shown as fold changes compared with those of MCF10A cells. (Blue bar, luminal subtype; pink bar, Her2+ subtype; red bar, basal-like subtype). ∗p < 0.05.
(E) Levels of IRE1, estrogen receptor 1 (ESR1), and HER2 proteins in a panel of normal-like, luminal, HER2-positive, and basal-like breast cancer cell lines, determined by western blot analysis.
To validate our findings from the TCGA breast cancer dataset regarding the IRE1 gene amplification, we conducted an independent analysis using the METABRIC dataset that contains approximately 2,000 breast cancer samples with long-term clinical follow-up data. Again, we found that IRE1 had high-level amplification in 8.47% of METABRIC breast cancer samples. We also found that luminal B METABRIC breast cancer had the highest frequency (40.57%) of IRE1 gene gain/amplification, compared with other subtypes (Figure S1A). We then examined the correlation between IRE1 copy number and the probability of overall survival in METABRIC breast cancer patients. Amplification or gain of IRE1 was significantly associated with shorter survival in METABRIC breast cancer patients (p = 0.0017) (Figure 1C).
Breast cancer cell lines retain many of the molecular characteristics of the tumors from which they were derived and represent excellent models for investigating breast cancer pathobiology (Daemen et al., 2013; Liu et al., 2009, 2016). We examined the genetic alterations and expression levels of IRE1 in a panel of breast cancer cell lines. Based on our data and published array-based comparative genomic hybridization data, we identified that, among 19 breast cancer cell lines, 11 lines had IRE1 amplification/gain (Barretina et al., 2012; Liu et al., 2009; Neve et al., 2006; Ray et al., 2004). Notably, the luminal B breast cancer cell line SUM52 has a focal amplification at the IRE1 loci (17q23.3) (Figure S1B). Furthermore, luminal B (e.g., SUM52) and luminal-like HER2+ breast cancer cell lines (e.g., SUM225) exhibited higher expression of IRE1 at both mRNA and protein levels, compared with the other breast cancer cell lines (Figures 1D and 1E). Together, these data indicated that IRE1 is frequently upregulated in aggressive luminal B breast cancers, and its gain/amplification is significantly associated with survival of patients with breast cancer.
IRE1 Promotes Expression of the RAS Oncogene Family Member RAB3B in Luminal Breast Cancer Cells
To understand the mechanistic basis by which IRE1 is involved in luminal breast cancer malignancy, we analyzed the transcriptomic signature that IRE1 regulates in breast cancer. Under ER stress, IRE1 is activated through trans-autophosphorylation at Ser/Thr residues to elicit its RNase activity (Zhang and Kaufman, 2008). Previously, we and others demonstrated that Lys to Ala mutations that destroy IRE1 kinase activity (K599A) or RNase activity (K907A) decrease the Xbp1 mRNA splicing and RIDD activity (Tirasophon et al., 1998; Wang et al., 2017) (Figure 2A). We suppressed the activity of IRE1 RNase in SUM52 line by adenoviral-based over-expression of the IRE1 dominant-negative K599A or K907A and then performed RNA sequencing (RNA-seq) analysis with IRE1 dominant-negative and control SUM52 cells. Through the RNA-seq analysis, we identified 98 genes that were commonly upregulated (65 genes) or downregulated (33 genes) in K599A-expressing SUM52 (SUM52-K599A) or K907A-expressing SUM52 (SUM52-K907A) cells (Figure 2B). SUM52-K907A cells had significantly more altered genes than SUM52-K599A cells. This was consistent with the previous observation that over-expression of K907A was more efficient in suppressing IRE1 RNase activity than that of K599A (Tirasophon et al., 1998; Zhang et al., 2011). Based on the RNA-seq result and gene ontology and pathway analysis, most of the upregulated mRNAs upon IRE1 inhibition were those encoding enzymes or regulators involved in ER protein folding homeostasis or ER stress response, such as PDIA4, DERL2, SEC61B, HSPA1A, HSPB1, ERO1LB, EDEM2, SEL1L, DNAJB11, and MANF (Figures 2B and S2). Notably, PDIA4 mRNA has been identified as an IRE1 RIDD target (Maurel et al., 2014). It is possible that the mRNAs upregulated by IRE1 inhibition are the targets of IRE1-RIDD activity even in the absence of ER stress in breast cancer, an interesting question to be elucidated in the future. Furthermore, gene ontology enrichment analysis revealed that most of the downregulated genes upon IRE1 inhibition, including RAS oncogene family members (RAB3B) and RET proto-oncogene, were involved in cell growth, invasion, and cancer pathways (Figures 2B and S3). To confirm the results of RNA-seq analysis, we performed qPCR and western blot assays of the major oncogenic factors downregulated in SUM52 cells expressing IRE1 dominant-negative or treated with a specific IRE1 RNase inhibitor, 4μ8C (Cross et al., 2012; Qiu et al., 2013). Among others, we confirmed that expression of the classic oncogene RAB3B in IRE1 dominant-negative-expressing or 4μ8C-treated SUM52 cells was significantly decreased at both mRNA and protein levels, compared with that in the control SUM52 cells (Figures 2C and 2D). These results suggest that RAB3B is a downstream target of IRE1 RNase activity in breast cancer. In addition, we examined expression of RET proto-oncogene in SUM52 cells in which IRE1 RNase activity is suppressed by IRE1 dominant-negative over-expression. Consistent with the RNA-seq analyses, expression levels of the RET mRNA or protein in SUM52-K599A or SUM52-K907A cells were reduced, compared with that of the control SUM52 cells (Figures S4A and S4B).
Figure 2.

IRE1 Modulates Expression of the Genes Involved in ER Proteostasis and Classic Oncogenes in Luminal Breast Cancer Cells
(A) Illustration of the IRE1 functional domains. WT, wild-type; K599A, IRE1 kinase dominant-negative mutant; K907A, IRE1 RNase dominant-negative mutant.
(B) RNA-seq heatmap of differentially expressed genes in SUM52-K599A and SUM52-K907A cells compared with SUM52-GFP control (Ctrl) cells.
(C) qPCR analyses of RAB3B mRNA in the IRE1 dominant-negative mutant (K599A or K907A) or 4μ8C-treated SUM52 cells (n = 3 biological repeats).
(D) Western blot analyses of RAB3B protein levels in the IRE1 dominant-negative mutant or 4μ8C-treated SUM52 cells.
(E) Correlation between expression levels of the IRE1 and RAB3B mRNAs in 1,084 TCGA human breast cancer specimens. Expression of two genes exhibits positive correlation with the Pearson correlation coefficient 0.23 (p < 0.0001).
RAB3B is a member of the large Ras superfamily of small GTPases. Recent evidence indicated that RAB family members play important roles in epithelial neoplasia (Goldenring, 2013). There are more than 60 RAB family members in humans, and most RAB members with elevated expression in cancer are positively associated with cancer progression, especially invasion and metastasis (Goldenring, 2013). We analyzed the genetic alterations and expression of the RAB genes in TCGA breast cancer samples. The RAB3B gene is amplified in only 0.56% of TCGA breast cancer samples. Integrated analysis of copy number and mRNA expression data in TCGA showed that RAB3B expression was not associated with gene CNAs. When we analyzed the correlation between expression levels of IRE1 and RAB3B in TCGA breast cancer specimens, we found that two genes had positive correlation with the Pearson correlation coefficient 0.23 (p < 0.0001) (Figure 2E). We then compared expression levels of the RAB3B gene in different subtypes of breast cancer and found that the expression level of RAB3B mRNA was significantly higher in luminal B and HER2+ subtypes (Figure S5A) (p < 0.0001), which is similar to the expression pattern of the IRE1 gene in the breast cancer subtypes (Figures 1B and S1A). Similarly, increased expression of the RET mRNA was positively correlated with elevated expression of the IRE1 mRNA in luminal B breast cancer cells (Figure S5B). These results suggested that elevation of RAB3B or RET may rely on IRE1, but not on RAB3B or RET gene amplification, in select breast cancer subtypes.
IRE1 modulates expression of a set of tumor suppressor miRNAs in luminal breast cancer cells.
A previous study demonstrated that IRE1 promotes the aggressive phenotype of triple-negative/basal-like breast cancer by activating XBP1, which assembles a transcriptional complex with hypoxia-inducing factor 1α (HIF1α) to promote oncogenesis (Chen et al., 2014). However, this work indicated that IRE1 promotes the aggressive phenotype of triple-negative/basal-like breast cancer, but not in luminal breast cancer, by increasing XBP1's transcriptional activity. Indeed, we found no significant change in the expression of the spliced XBP1 mRNA in luminal breast cancer SUM52 cells when IRE1 RNase was suppressed by the IRE1 dominant-negative approach or by treatment with the IRE1 inhibitor 4μ8C (Figure S6), suggesting that the IRE1-XBP1 pathway in luminal B breast cancer cells may not be active. In addition, we queried the XBP1 chromatin immunoprecipitation sequencing dataset with breast cancers (Chen et al., 2014) and found that XBP1 does not directly bind the RAB3B genomic loci. These analyses suggest that over-expressed IRE1 in select luminal B breast cancer cells unlikely takes XBP1 mRNA as the major target for the IRE1-mediated RNA splicing event and that IRE1 may regulate RAB3B expression through an XBP1-independent pathway.
To explore the regulatory mechanism by which IRE1 regulates oncogenic factors, particularly RAB3B, in luminal breast cancer cells, we blocked IRE1 activity in luminal breast cancer cell lines by using the IRE1 inhibitor 4μ8C or expressing IRE1 dominant-negative for miRNA microarray analysis (miRHuman_20 Version, total >2,500 miRNAs) to determine whether IRE1 regulates the oncogene RAB3B through the miRNA pathway. SUM52 and SUM225 lines with high-level IRE1 expression were treated with 4μ8C for 2 days. The IRE1 kinase dominant-negative mutant K599A or K907A was also used to suppress IRE1 kinase or RNase activity in SUM52 cells. The miRNA microarray analysis revealed a landscape change in miRNA expression profiling in IRE1-inhibited luminal breast cancer cells (Figure 3A). Using a criterion of p < 0.05 in miRNA analysis, we identified 41 miRNAs in both SUM52 and SUM225 cells that were altered after inhibiting IRE1 activity. Among these 41 miRNAs, 25 were upregulated and 16 were downregulated (Figure 3A). Conversely, we exogenously over-expressed wild-type IRE1 in human nontumorigenic mammary epithelial MCF10A cells and then performed miRNA array assays. When we combined miRNA data from both IRE1 inhibition models in breast cancer cells and exogenous over-expression of IRE1 in MCF10A cells (p < 0.05), we identified five miRNAs (3607-3p, 374a-5p, 4764-3p, 516a-3p, and 6073) that were upregulated in IRE1-inhibited breast cancer cells and downregulated in MCF10A-IRE1 cells. The qPCR analysis confirmed our miRNA array findings: the top candidates, miR-3607-3p and miR-374a-5p, were upregulated in SUM52 cells expressing K599A or K907A or treated with 4μ8C (Figures 3B and 3C). Our qPCR assay also identified that miR-96-5p expression was regulated by IRE1 in breast cancer cells (Figure 3D), although our combined miRNA analyses did not single out miR-96-5p, possibly due to the strict cutoff of our array analysis. Recently, we demonstrated that IRE1 processes select miRNAs and promote their degradation in mouse liver or skin endothelium through the RIDD pathway (Wang et al., 2017, 2018). The identification of the miRNAs elevated in IRE1-inhibited breast cancer cells implied that IRE1 might regulate expression of oncogenic factors by repressing miRNAs.
Figure 3.

IRE1 Represses a Set of Tumor Suppressor miRNAs in Luminal Breast Cancer
(A) Heatmaps of the relative abundance of human miRNAs in SUM52 and SUM225 (left lanes), 4μ8C-treated SUM225 or SUM52 cells, as well as IRE1-K599A mutant or IRE1-K907A mutant SUM52 (right lanes). 41 altered miRNAs identified from SUM52-K599A, SUM52-K907A, SUM52-4μ8C, and SUM225-4μ8C cells, compared with GFP, SUM52, and SUM225 controls. Five top upregulated miRNAs (3607-3p, 516a-3p, 6073, 4764-3p, and 374a-5p) are highlighted by red on the right.
(B–D) Expression levels of (B) miR-3607-3p, (C) miR-374-5p, and (D) miR-96-5p in IRE1 dominant-negative mutant (K599A and K907A) and 4μ8C-treated SUM52 cells, determined by qPCR. For each comparison group, the miRNA level of SUM52 control was defined as 1 and was used to calculate the fold changes of miRNA levels for the other groups. Data in all panels are means ± SEM from n = 3 biological replicates. ∗p < 0.05, ∗∗p < 0.01 versus controls by unpaired two-tailed Student's t test.
IRE1 Suppresses Biosynthesis of Select miRNAs by Mediating Degradation of the Pre-miRNAs
It was suggested that IRE1 can process mRNA or pre-miRNAs by recognizing the G/C cleavage site in the RNA stem-loops (Figures 4A and 4B) (Upton et al., 2012; Wang et al., 2017; Yoshida et al., 2001). The RNase activity of IRE1 can cleave selected pre-miRNAs and thus terminate de novo miRNA biosynthesis and maturation, which leads to their degradation, a process that we call “IRE1-dependent pre-miRNA decay” (Upton et al., 2012; Wang et al., 2017, 2018). Bioinformatics analysis indicated that miR-3607 and miR-374, whose levels were upregulated in IRE1-inhibited SUM52 or SUM225 breast cancer cells, possess G/C cleavage sites in the stem-loops or at the joint sites of loops and arms in the secondary structure of the pre-miRNAs (Figure 4C). These pre-miRNAs are potentially recognized and processed by IRE1.
Figure 4.

IRE1 Suppresses Maturation of miR-3607 and miR-374 by Mediating Degradation of the Pre-miRNAs
(A) Illustration of IRE1-mediated miRNA decay by processing pre-miRNA stem-loop structure.
(B) IRE1 cleavage sites within human XBP1 mRNA, pre-miR-374a, and pre-miR-3607.
(C) Predicted mRNA secondary structures for human pre-miR-3607 and pre-miR-374a and their potential IRE1 cleavage sites (G/C sites marked with red arrows).
(D) In vitro cleavage of pre-miR-374 and pre-miR-3607 by recombinant IRE1 protein (1 μg each), incubated at 37°C with ATP (2 mM) or not as controls, and resolved on a 1.2% agarose gel. VH, vehicle buffer containing no IRE1 protein. The image is representative of three experiments.
(E and F) miR-qPCR analyses of the abundance of mature (E) miR-374a and (F) miR-3607-3p in IRE1-KO and WT control MEFs transfected with plasmid vector expressing human pre-miR-374 or pre-miR-3607. Data are means ± SEM (n = 3 biological replicates); ∗p < 0.05.
(G and H) qPCR analyses of expression levels of the mRNAs encoding human primary (pri)-miR-3607 (G) and pri-miR-374 (H) in the IRE1 dominant-negative mutant (K599A or K907A) and control SUM52 cells. The data are means ± SEM (n=3 biological repeats).
To test whether IRE1 can directly process the select miRNAs and lead to their degradation through the RIDD pathway, we performed in vitro cleavage assay by incubating pre-miR-3607 or pre-miR-374 oligo with the purified recombinant human IRE1 protein. In the presence of ATP, IRE1 processed both pre-miR-3607 and pre-miR-374 and led to their degradation (Figure 4D). This result validated the role of IRE1 as an RNase to process pre-miR-3607 and pre-miR-374 and lead to their decay through the RIDD pathway. To further confirm that IRE1 downregulates miR-3607 and miR-374, we over-expressed an expression vector encoding human pre-miR-3607 or pre-miR-374 in wild-type (WT) or IRE1 knockout (KO) mouse embryonic fibroblasts (MEFs) and then measured the levels of mature miR-3607 and miR-374. The levels of miR-3607-3p and miR-374a in IRE1-KO MEFs were considerably higher than those in the WT MEFs (Figures 4E and 4F). In addition, we examined expression levels of the human primary miR-3607 (pri-miR-3607) and primary miR-374 (pri-miR-374) in the IRE1 dominant-negative mutant (K599A or K907A) and control SUM52 cells by qPCR analyses. The results showed that expression levels of the human pri-miR-3607 and pri-miR-374 in SUM52-K599A or SUM52-K907A cells were not significantly changed, compared with that in SUM52 control cells (Figures 4G and 4H), suggesting that elevation of precursor or mature hs-miR-3607 or hs-miR-374 in IRE1-defective SUM52 cells is not due to any change at the level of transcription. Together, these results suggest that IRE1 directly processes pre-miR-3607 and pre-miR-374 and thus suppresses biosynthesis of miR-3607 and miR-374 through the RIDD pathway.
IRE1 Regulates RAB3B Expression via miR-3607 in Luminal Breast Cancer
Next, we tested whether IRE1 regulates expression of the classic oncogene RAB3B in luminal breast cancer by modulating the select miRNAs. Indeed, the 3′ UTR of the human RAB3B gene contains three binding sequences of miR-3607-3p, the IRE1-targeted miRNA (Figure 5A). To determine whether miR-3607-3p can directly repress RAB3B expression, we performed western blot analysis with the IRE1-over-expresssing luminal breast cancer cell line SUM52 transfected with mimics or inhibitors of miR-3607-3p, miR-96-5p, or controls. The expression of RAB3B was suppressed in cells transfected with miR-3607-3p mimics, but not miR-96-5p mimics or miR controls (Figure 5B). Conversely, RAB3B expression was higher in cells transfected with a miR-3607-3p inhibitor, but not miR-96-5p inhibitor or the inhibitor control (Figure 5C), suggesting that IRE1 regulates expression of RAB3B by repressing miR-3607-3p in luminal breast cancer cells. To confirm the regulation of RAB3B expression in luminal breast cancer cells by the IRE1-miR-3607 regulatory axis, we treated SUM52 cells with the specific IRE1 inhibitor 4μ8C, or 4μ8C plus the hs-miR-3607 inhibitor. The results showed that 4μ8C treatment decreased RAB3B expression in SUM52 cells (Figure 5D). However, treatment of the miR-3607 inhibitor increased the levels of RAB3B expression in SUM52 cells when IRE1 was inhibited (Figure 5D), implicating that hs-miR-3607 inhibitor can increase RAB3B expression independent of IRE1. Together, these data suggested that increased IRE1 activity in luminal breast cancer cells leads to increased RAB3B expression by repressing/processing hs-miR-3607.
Figure 5.

IRE1 Regulates RAB3B Expression via miR-3607 in Luminal Breast Cancer
(A) miRNA-binding sequences of miR-3607 in the 3′ UTRs of human RAB3B genes. The analysis was based on the data from microrna.org.
(B and C) Western blot analysis of RAB3B protein levels in SUM52 cells treated with the miR-3607-3p miRNA mimic or inhibitor as indicated. The miR-96-5p miRNA mimic and inhibitor were included as controls.
(D) Western blot analysis of RAB3B levels in SUM52 cells treated with 4μ8C, 4μ8C plus hs-miR-3607 inhibitor, or vehicle (miR-Ctrl). Tubulin levels were detected as the loading control.
(E and F) Relative expression levels of Hs-miR-3607-3p in different groups of TCGA breast cancer samples based on (E) RIDD status and (F) XBP1s status. Based on the “XBP1s” and “RIDD” gene signatures; TCGA breast cancer samples were manually divided into three groups of XBP1s or RIDD activity (high, middle, and low). The levels of miR-3607-3p were negatively correlated (Pearson r = −0.185, p < 0.0001) with “RIDD” activity, but not “XBP1s” activity in breast cancers. RPM: normalized count in reads per million miRNA mapped.
To further verify IRE1-miR-3607-RAB3B regulatory pathway in luminal breast cancer cells, we utilized a bioinformatic approach to validate the correlation between levels of miR-3607-3p and expression of RAB3B in TCGA breast cancer samples. Based on the expression profiles of RAB3B protein in 77 TCGA breast cancer from the Clinical Proteomic Tumor Analysis Consortium portal (Mertins et al., 2016), the levels of miR-3607-3p were negatively associated with RAB3B protein expression in TCGA breast cancer samples (Pearson r = −0.069) (Figure S7A), even though the differences were not statistically significant, possibly due to the small sample size for RAB3B protein data. In addition, IRE1 may regulate RET expression via miR-96-5p in luminal breast cancer cells, as is evidenced by the fact that miR-96-5p was repressed by IRE1 (Figure 3D), that the 3′ UTR of the human RET gene contains two binding sequences of miR-96-5p (Figure S4C), and that the RET mRNA and protein levels were elevated in the IRE1-knockdown breast cancer cells (Figures S4A and S4B). However, whether the IRE1-RIDD-miRNA pathway plays a direct role in promoting RET expression in luminal breast cancer cells remains to be further investigated.
IRE1-RIDD-miRNA Pathway Is Active in Luminal B Breast Cancer Cells
An important question regarding the activity of the IRE1-RIDD pathway versus the IRE1-XBP1 splicing pathway in different breast cancer subtypes remains. To address this question, we utilized gene signature to gauge activation of the IRE1-XBP1 mRNA splicing (XBP1s) and IRE1-RIDD pathways in a number of breast cancer cell lines (Lhomond et al., 2018; Marcotte et al., 2016). Based on the “XBP1s” and “RIDD” signature scores, activities of XBP1 mRNA splicing and RIDD activity status in the breast cancer cell lines are divided into three groups: high, middle, and low. Consistent with previous reports (Chen et al., 2014; Logue et al., 2018), several triple-negative/basal-like breast cancer cell lines, such as SUM149, SUM159, HCC1937, and MDA-MA-468, had higher “XBP1s” activity (Table S1). In contrast, luminal breast cancer cell lines, such as SUM52 and MCF7 with the IRE1 gene gain/amplification, had relatively higher “RIDD” activity (Table S1). Furthermore, we computed the “XBP1s” and “IRE1-RIDD” signatures for more than 1,000 TCGA breast cancer samples. Consistently, triple-negative/basal-like breast cancer samples had relatively higher “XBP1s” activities, compared with the other breast cancer subtypes (Figure S7B). In addition, the levels of miR-3607-3p, the target of IRE1 RNase, were negatively correlated (Pearson r = −0.185, p < 0.0001) with “IRE1-RIDD” activity in 1,062 TCGA breast cancer samples that contain both miRNA and gene signature data (Figure 5E). There was no significant correlation between the levels of miR-3607-3p or miR-374a-5p and “XBP1s” activity score in TCGA breast cancer samples (Figure 5F). Taken together, our data suggest that a set of luminal B breast cancer cells may possess a relatively higher activity of the IRE1-RIDD-miRNA pathway, whereas triple-negative/basal-like breast cancer cells have higher activity of the IRE1-XBP1 splicing pathway, compared with the other breast cancer subtypes.
IRE1 Inhibitor 4μ8C Suppresses Luminal Breast Cancer Cell Growth and Aggressive Phenotypes
We evaluated the functional significance of IRE1 amplification/over-expression in breast cancer growth and aggressive phenotypes. First, we used a small-molecule IRE1 inhibitor, 4μ8C, which attains its selectivity by binding specifically to the K907 residue in the IRE1 RNase catalytic pocket to form a stable imine, to block the IRE1 RNase activity (Cross et al., 2012). We treated two endogenous IRE1-over-expressing breast cancer cell lines, SUM52 and SUM225, and two control lines, SUM159 triple-negative/basal-like breast cancer cells and the non-tumorigenic mammary gland cell line MCF10A, with 4μ8C. Treatment significantly inhibited the growth of IRE1-over-expressing breast cancer cell lines, including SUM52 (luminal B) and SUM225 (luminal/HER2+) cells, in a dose-dependent manner (p < 0.05) (Figure 6A). In contrast, SUM159 and MCF10A cells, which have low expression of IRE1, exhibited significantly higher IC50 (half maximal inhibitory concentration) values for 4μ8C (Figure 6A).
Figure 6.

IRE1 Inhibitor 4μ8C Suppresses Luminal Breast Cancer Cell Growth and Aggressive Phenotypes
(A) Cell survival rates of MCF10A and three breast cancer cells, SUM225, SUM52, and SUM159, after the treatment of 4μ8C at the concentration ranging from 4 to 64 μM for 3 days. The same number of cells was seeded, and the absorbance reading value representing cell viability for each treatment was determined by MTT assay. The relative cell survival rates (percentages) were calculated by comparing the MTT values to those of the cells treated with vehicle (0 concentration). Results are means ± SEM (n = 4 biological samples). The half maximal inhibitory concentration (IC50) values for 4μ8C in four cell lines were calculated.
(B) Representative images of SUM52 cell colonies in soft agar with or without the treatment of 4μ8C (0.25μM) for 3 weeks. The numbers of cell colony in SUM52 cells treated with 4μ8C or vehicle were quantified (n = 3 biological replicates; ∗∗p < 0.01.
(C) Survival rates (MTT assay) of IRE1 knockdown and control SUM225, SUM52, SUM159, and MCF10A cells. The IRE1 gene was knocked down in SUM225, SUM52, SUM159, and MCF10A cells by using pGIPZ lentiviral shRNAs. The cells transduced with non-silencing siRNA were included as the control. Two shRNAs targeting IRE1 were utilized to generate two lines of IRE1 knockdown cells. The relative cell survival rates (percentage) were calculated by comparing the MTT values with those of the knockdown control cells. Results are means ± SEM (n = 4). ∗p < 0.05; ∗∗p < 0.01.
Anchorage-independent growth of cancer cells in vitro has been used to evaluate aggressive tumor phenotypes, particularly with respect to metastatic potential (Mori et al., 2009). We examined whether 4μ8C can suppress anchorage-independent growth of luminal breast cancer cells in soft agar. The luminal B breast cancer cell line SUM52, which can form colonies robustly in soft agar, was treated with 4μ8C. The treatment of 4μ8C significantly inhibited the colony-forming ability of SUM52 in soft agar, and notably, colonies barely formed even with 0.25 μM of 4μ8C (Figure 6B). To confirm the contribution of endogenous IRE1 over-expression on the transformation of human breast cancer, we performed IRE1 knockdown experiments using pGIPZ lentiviral short hairpin RNAs (shRNAs) in four cell lines, SUM225, SUM52, SUM159, and MCF10A. shRNA knockdown of IRE1 significantly inhibited the proliferation of two IRE1-over-expressing breast cancer cell lines, SUM52 and SUM225 (Figure 6C). Conversely, in MCF10A and SUM159 lines that express low levels of IRE1, no significant inhibitory effects of IRE1 knockdown were observed.
Discussion
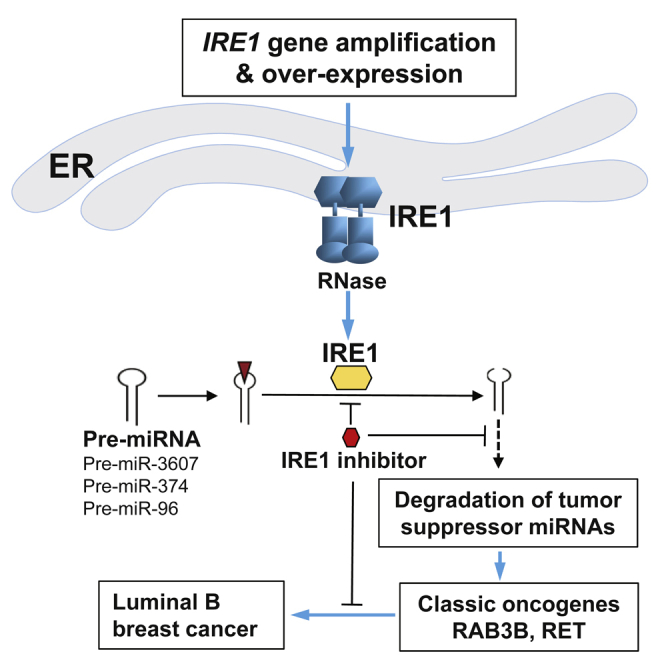
In this work, we demonstrated a causative relationship between the UPR primary transducer IRE1 and luminal breast cancer malignancy through a meta-analysis of cancer gene CNAs, breast cancer subtypes, and clinical outcome. Mechanistically, we revealed that IRE1 processes and mediates degradation of a subset of tumor suppressor miRNAs, including miR-3607-3p and miR-374a-5p, via RIDD mechanism and subsequently promotes expression of the RAS oncogene GTPase, RAB3B, in IRE1-amplified human luminal breast cancer cells (Figure 7). Inhibition of IRE1 with pharmacological and genetic approaches suppresses luminal breast cancer cell proliferation and aggressive cancer phenotypes. In summary, we have uncovered a regulatory mechanism of tumor suppressor miRNAs in human breast cancer, in which the cell stress sensor IRE1 acts as the key driver by mediating unconventional cleavage and degradation of pre-miRNAs for breast tumorigenesis. Identification of the regulatory mechanism for the classic oncogenes RAB3B and the approach to modulate RAB GTPase oncogenic activities by targeting IRE1 has important implications in the treatment of human breast cancers.
Figure 7.

Illustration of the IRE1-RIDD-miRNA Pathway in Luminal Breast Cancer Malignancy
Previous studies on the role of IRE1 in breast cancer were focused exclusively on its activity in processing the XBP1 mRNA, and no work addressing the potential role of IRE1-mediated RIDD pathway in breast cancer malignancy has been reported. Previous works showed that activated XBP1 promotes triple-negative breast cancer progression via HIF1 and MYC oncogenic pathways (Chen et al., 2014; Zhao et al., 2018). Here, our study demonstrated that the RNase activity of IRE1 can cleave selected pre-miRNAs and thus terminate de novo miRNA biosynthesis and maturation, which leads to their degradation, a process we call “IRE1-dependent pre-miRNA decay.” IRE1 induces the degradation of a subset of tumor suppressor miRNAs, particularly miR-3607-3p, which subsequently enhances the expression of the transforming oncogenic factor RAB3B in luminal B breast cancers. miR-3607-3p has been reported to play a tumor suppressive role in lung and prostate cancers (Gao et al., 2018; Saini et al., 2014). RAB proteins are part of the large Ras superfamily of small GTPases. There are more than 60 members in humans, and each of them is specifically localized to a subcellular membrane compartment, where it controls the intracellular traffic (Kelly et al., 2012). Expression levels of most RAB members, particularly RAB2A, RAB3A, RAB3B, RAB3D, RAB5C, RAB22B, RAB25, and RAB27B, are increased in cancer cells and positively associated with cancer progression, invasion, and metastasis (Jacob et al., 2013; Luo et al., 2015; Mitra et al., 2012; Wang et al., 2014). Our data indicate that IRE1 regulates the expression of RAB3B at both mRNA and protein levels by degrading pre-miR-3607 and thus reveal a novel IRE1 regulatory axis in luminal breast cancer cells, IRE1-miR-3607-RAB3B, through which IRE1 promotes breast cancer malignancy.
It has been suggested the RNA sequence and secondary structural features are crucial for IRE1-mediated cleavage of mRNAs or pre-miRNAs (Upton et al., 2012; Wang et al., 2017; Yoshida et al., 2001). The CUGCAG sequence motif, particularly G/C, and the stem-loop structure are critical features of the IRE1-mediated cleavage site in the XBP1 mRNA (Hollien and Weissman, 2006; So et al., 2012; Upton et al., 2012). However, recent studies suggested that the specificity of the IRE1 RNase-targeted sequence motif is less stringent during the RIDD process (Han et al., 2009; Hollien et al., 2009; Hollien and Weissman, 2006). IRE1 can mediate cleavage of RIDD-associated miRNA or mRNA substrates at multiple sites that have loose structural homology to XBP1 stem-loops (Hollien et al., 2009; Oikawa et al., 2010; So et al., 2012). Through the pre-miRNA sequence and secondary structure of the IRE1-downregulated core miRNAs using the miRNA database (http://www.mirbase.org) and Mfold software (Zuker, 2003), we found that 16 of 25 core IRE1-targeted miRNAs, including miR-3607 and miR-374a, contain potential IRE1 G/C cleavage sites at stem-loop structures of their pre-miRNAs. Notably, several pre-miRNAs, including pre-miR-3607, contain a G/C duplex at the joint sites of loops and arms in the secondary structure of the pre-miRNAs (Figure 4), suggesting that alternative IRE1 cleavage modes may exist in select pre-miRNAs. Our in vitro cleavage assays with pre-miR-3607 and pre-miR-374a oligos and purified human IRE1 protein demonstrated that IRE1 indeed processes these pre-miRNAs and leads to their degradation (Figure 4).
Recent studies suggested that IRE1 has a basal RIDD activity without induction of XBP1 mRNA splicing, and this basal RIDD activity is necessary for maintaining ER proteostasis (Hetz et al., 2015; Tam et al., 2014). It is speculated that RIDD is constitutively active and its activity increases progressively with ER stress intensity and/or duration, whereas XBP1 mRNA splicing is activated transiently on ER stress only during the adaptive/pro-survival phase (Maurel et al., 2014). Indeed, we identified 65 genes that were commonly upregulated upon IRE1 inhibition in SUM52 breast cancer cells, and most of the upregulated mRNAs were those encoding enzymes or regulators involved in ER protein folding homeostasis, ER-associated protein degradation, or ER-to-Golgi protein transport (Figures 2 and S2). Notably, among the 37 RIDD mRNA substrates identified in metazoans, Ingenuity Pathway Analysis revealed that 68% are associated with cancer (Maurel et al., 2014). Our work provided important mechanistic basis for the involvement of RIDD pathway in cancer. The IRE1-dependent, RIDD-mediated degradation of mRNAs and miRNAs likely plays a driving role in oncogenesis by maintaining ER proteostasis and boosting the classic oncogenic factors, such as RAB3B and possibly RET (Figures 2 and S4). Our work also demonstrated the therapeutic potential of the small molecule IRE1 inhibitor, 4μ8C, in repressing the aggressiveness of luminal B breast cancers. As the IRE1-RIDD-miRNA regulatory axis simultaneously controls expression of multiple classic oncogenes in breast cancer cells, small molecule inhibition that selectively targets IRE1 RNase activity may represent a highly effective therapeutic approach for a set of breast cancers.
Limitations of the Study
In this study, we employed cellular and biochemical approaches to uncover an IRE1-RIDD-miRNA pathway in luminal B breast cancer cells. However, it should be noted that in vivo studies are needed in the future to address the pathophysiological significance of the newly defined IRE1-RIDD-miRNA pathway in luminal breast cancers. Although we showed that the IRE1-XBP1 pathway is not highly activated to drive oncogenesis in the luminal B subtype of breast cancer, the mechanistic basis governing the differential activation of the IRE1-mediated signaling pathways in different breast cancer subtypes remains unclear. This important question deserves further investigation in the future.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Zeng-Quan Yang (yangz@karmanos.org).
Materials Availability
No new or unique materials were generated by this study.
Data and Code Availability
The accession number for all miRNA array and RNA-seq data reported in this paper are GSE156331 and GSE156332. The datasets and code used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was partially supported by grants from the Department of Defense (DoD) Breast Cancer Program BC095179 and BC161536; DMC Foundation and Molecular Therapeutics Program of Karmanos Cancer Institute to Z.-Q.Y.; and by funding from National Institutes of Health (NIH) grants DK090313 and ES017829, and DoD Breast Cancer Program BC095179P1 to K.Z.. We thank Dr. Stephen P. Ethier for providing the SUM breast cancer cell lines. We thank Lanxin Liu, Wenqi Shan, and Era Cobani for technical contributions.
Author Contributions
K.Z. and Z.-Q.Y. designed and conducted the experiments, analyzed the data, and wrote the manuscript; H.L., Z.S., Y.J., and H.K. performed the experiments and acquired the data; L.S. and H.M.N. provided key reagents or critical comments.
Declaration of Interests
All the authors have no competing interests to declare.
Published: September 25, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101503.
Contributor Information
Kezhong Zhang, Email: kzhang@med.wayne.edu.
Zeng-Quan Yang, Email: yangz@karmanos.org.
Supplemental Information
References
- Ades F., Zardavas D., Bozovic-Spasojevic I., Pugliano L., Fumagalli D., de Azambuja E., Viale G., Sotiriou C., Piccart M. Luminal B breast cancer: molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 2014;32:2794–2803. doi: 10.1200/JCO.2013.54.1870. [DOI] [PubMed] [Google Scholar]
- Albertson D.G. Gene amplification in cancer. Trends Genet. 2006;22:447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Albertson D.G., Collins C., McCormick F., Gray J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehar J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Iliopoulos D., Zhang Q., Tang Q., Greenblatt M.B., Hatziapostolou M., Lim E., Tam W.L., Ni M., Chen Y. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B.C., Bond P.J., Sadowski P.G., Jha B.K., Zak J., Goodman J.M., Silverman R.H., Neubert T.A., Baxendale I.R., Ron D. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. U S A. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A., Griffith O.L., Heiser L.M., Wang N.J., Enache O.M., Sanborn Z., Pepin F., Durinck S., Korkola J.E., Griffith M. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Dejeans N., Manie S., Hetz C., Bard F., Hupp T., Agostinis P., Samali A., Chevet E. Addicted to secrete - novel concepts and targets in cancer therapy. Trends Mol. Med. 2014;20:242–250. doi: 10.1016/j.molmed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Dufey E., Bravo-San Pedro J.M., Eggers C., Gonzalez-Quiroz M., Urra H., Sagredo A.I., Sepulveda D., Pihan P., Carreras-Sureda A., Hazari Y. Genotoxic stress triggers the activation of IRE1alpha-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020;11:2401. doi: 10.1038/s41467-020-15694-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi T.A., Horlings H.M., Ten Hoeve J.J., Mihailovic A., Halfwerk H., Morozov P., Brown M., Hafner M., Reyal F., van Kouwenhove M. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011;71:4443–4453. doi: 10.1158/0008-5472.CAN-11-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P., Wang H., Yu J., Zhang J., Yang Z., Liu M., Niu Y., Wei X., Wang W., Li H. miR-3607-3p suppresses non-small cell lung cancer (NSCLC) by targeting TGFBR1 and CCNE2. PLoS Genet. 2018;14:e1007790. doi: 10.1371/journal.pgen.1007790. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Goldenring J.R. A central role for vesicle trafficking in epithelial neoplasia: intracellular highways to carcinogenesis. Nat. Rev. Cancer. 2013;13:813–820. doi: 10.1038/nrc3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M., Kim V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- Han D., Lerner A.G., Vande Walle L., Upton J.P., Xu W., Hagen A., Backes B.J., Oakes S.A., Papa F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C., Chevet E., Oakes S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J., Lin J.H., Li H., Stevens N., Walter P., Weissman J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Jacob A., Jing J., Lee J., Schedin P., Gilbert S.M., Peden A.A., Junutula J.R., Prekeris R. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J. Cell Sci. 2013;126:4647–4658. doi: 10.1242/jcs.126573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen A., Silber J., Harinath G., Huse J.T., Schultz N., Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat. Struct. Mol. Biol. 2013;20:1325–1332. doi: 10.1038/nsmb.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E.E., Horgan C.P., Goud B., McCaffrey M.W. The Rab family of proteins: 25 years on. Biochem. Soc. Trans. 2012;40:1337–1347. doi: 10.1042/BST20120203. [DOI] [PubMed] [Google Scholar]
- Lhomond S., Avril T., Dejeans N., Voutetakis K., Doultsinos D., McMahon M., Pineau R., Obacz J., Papadodima O., Jouan F. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018;10:e7929. doi: 10.15252/emmm.201707929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Korennykh A.V., Behrman S.L., Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U S A. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.H., Li H., Yasumura D., Cohen H.R., Zhang C., Panning B., Shokat K.M., Lavail M.M., Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Bollig-Fischer A., Kreike B., van de Vijver M.J., Abrams J., Ethier S.P., Yang Z.Q. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491–4500. doi: 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. MicroRNAs in breast cancer initiation and progression. Cell Mol. Life Sci. 2012;69:3587–3599. doi: 10.1007/s00018-012-1128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Liu L., Holowatyj A., Jiang Y., Yang Z.Q. Integrated genomic and functional analyses of histone demethylases identify oncogenic KDM2A isoform in breast cancer. Mol. Carcinog. 2016;55:977–990. doi: 10.1002/mc.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S.E., McGrath E.P., Cleary P., Greene S., Mnich K., Almanza A., Chevet E., Dwyer R.M., Oommen A., Legembre P. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018;9:3267. doi: 10.1038/s41467-018-05763-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.L., Gong C., Chen C.H., Hu H., Huang P., Zheng M., Yao Y., Wei S., Wulf G., Lieberman J. The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via Erk signaling activation. Cell Rep. 2015;11:111–124. doi: 10.1016/j.celrep.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R., Sayad A., Brown K.R., Sanchez-Garcia F., Reimand J., Haider M., Virtanen C., Bradner J.E., Bader G.D., Mills G.B. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel M., Chevet E., Tavernier J., Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014;39:245–254. doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- McGrath E.P., Logue S.E., Mnich K., Deegan S., Jager R., Gorman A.M., Samali A. The unfolded protein response in breast cancer. Cancers (Basel) 2018;10:344. doi: 10.3390/cancers10100344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S., Cheng K.W., Mills G.B. Rab25 in cancer: a brief update. Biochem. Soc. Trans. 2012;40:1404–1408. doi: 10.1042/BST20120249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S., Chang J.T., Andrechek E.R., Matsumura N., Baba T., Yao G., Kim J.W., Gatza M., Murphy S., Nevins J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene. 2009;28:2796–2805. doi: 10.1038/onc.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve R.M., Chin K., Fridlyand J., Yeh J., Baehner F.L., Fevr T., Clark L., Bayani N., Coppe J.P., Tong F. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D., Tokuda M., Hosoda A., Iwawaki T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 2010;38:6265–6273. doi: 10.1093/nar/gkq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Qiu Q., Zheng Z., Chang L., Zhao Y.S., Tan C., Dandekar A., Zhang Z., Lin Z., Gui M., Li X. Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013;32:2477–2490. doi: 10.1038/emboj.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray M.E., Yang Z.Q., Albertson D., Kleer C.G., Washburn J.G., Macoska J.A., Ethier S.P. Genomic and expression analysis of the 8p11-12 amplicon in human breast cancer cell lines. Cancer Res. 2004;64:40–47. doi: 10.1158/0008-5472.can-03-1022. [DOI] [PubMed] [Google Scholar]
- Riaz M., van Jaarsveld M.T., Hollestelle A., Prager-van der Smissen W.J., Heine A.A., Boersma A.W., Liu J., Helmijr J., Ozturk B., Smid M. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013;15:R33. doi: 10.1186/bcr3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini S., Majid S., Shahryari V., Tabatabai Z.L., Arora S., Yamamura S., Tanaka Y., Dahiya R., Deng G. Regulation of SRC kinases by microRNA-3607 located in a frequently deleted locus in prostate cancer. Mol. Cancer Ther. 2014;13:1952–1963. doi: 10.1158/1535-7163.MCT-14-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So J.S., Hur K.Y., Tarrio M., Ruda V., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Lichtman A.H., Iwawaki T., Glimcher L.H. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012;16:487–499. doi: 10.1016/j.cmet.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecklein S.R., Jensen R.A., Pal A. Genetic and epigenetic signatures of breast cancer subtypes. Front Biosci. (Elite Ed.) 2012;4:934–949. doi: 10.2741/E431. [DOI] [PubMed] [Google Scholar]
- Tam A.B., Koong A.C., Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014;9:850–858. doi: 10.1016/j.celrep.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S.F., Alarcon C., Oskarsson T., Padua D., Wang Q., Bos P.D., Gerald W.L., Massague J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W., Welihinda A.A., Kaufman R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran B., Bedard P.L. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011;13:221. doi: 10.1186/bcr2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton J.P., Wang L., Han D., Wang E.S., Huskey N.E., Lim L., Truitt M., McManus M.T., Ruggero D., Goga A. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.M., Qiu Y., Yang Z., Kim H., Qian Q., Sun Q., Zhang C., Yin L., Fang D., Back S.H. IRE1alpha prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018;11:eaao4617. doi: 10.1126/scisignal.aao4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.M., Qiu Y., Yang Z.Q., Li L., Zhang K. Inositol-requiring enzyme 1 facilitates diabetic wound healing through modulating MicroRNAs. Diabetes. 2017;66:177–192. doi: 10.2337/db16-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang J. MicroRNA-mediated breast cancer metastasis: from primary site to distant organs. Oncogene. 2012;31:2499–2511. doi: 10.1038/onc.2011.444. [DOI] [PubMed] [Google Scholar]
- Wang T., Gilkes D.M., Takano N., Xiang L., Luo W., Bishop C.J., Chaturvedi P., Green J.J., Semenza G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U S A. 2014;111:E3234–E3242. doi: 10.1073/pnas.1410041111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- You J.S., Jones P.A. Cancer genetics and epigenetics: two sides of the same coin? Cancer cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Wang S., Malhotra J., Hassler J.R., Back S.H., Wang G., Chang L., Xu W., Miao H., Leonardi R. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011;30:1357–1375. doi: 10.1038/emboj.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Wang X., Huo Q., Sun M., Cai C., Liu Z., Hu G., Yang Q. MicroRNA-30a suppresses breast tumor growth and metastasis by targeting metadherin. Oncogene. 2013;33:3119–3128. doi: 10.1038/onc.2013.286. [DOI] [PubMed] [Google Scholar]
- Zhao N., Cao J., Xu L., Tang Q., Dobrolecki L.E., Lv X., Talukdar M., Lu Y., Wang X., Hu D.Z. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Invest. 2018;128:1283–1299. doi: 10.1172/JCI95873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for all miRNA array and RNA-seq data reported in this paper are GSE156331 and GSE156332. The datasets and code used and/or analyzed during the current study are available from the corresponding authors on reasonable request.