

Abstract
Environmental pollution of heavy metals is increasingly becoming a problem and has become of great concern due to the adverse effects it is causing around the world. These inorganic pollutants are being discarded in our waters, soils and into the atmosphere due to the rapidly growing agriculture and metal industries, improper waste disposal, fertilizers and pesticides. This review shows how pollutants enter the environment together with their fate. Some metals affect biological functions and growth, while other metals accumulate in one or more different organs causing many serious diseases such as cancer. The pharmacokinetics and toxicological processes in humans for each metal is described. In summary, the review shows the physiological and biochemical effects of each heavy metal bioaccumulation in humans and the level of gravity and disquieting factor of the disease.
Keywords: Heavy metals, Soil, Air, Water, Agricultural science, Earth sciences, Environmental science, Food science, Toxicology
Heavy metals; soil; Air; Water; Agricultural Science; Earth Sciences; Environmental Science; Food Science, Toxicology
1. Introduction
The environment is the surroundings where humans, plants, animals and micro-organisms live or work. It is composed of the land, the Earth's atmosphere and the water. The Earth's system is defined by the four spheres: the biosphere (living things), the atmosphere (air), the lithosphere (land) and the hydrosphere (water) which all work in harmony together as shown in Figure 1. Environmental contaminants, as well as pollutants, are chemicals that are present at higher levels than in any section of the environment [1, 2, 3].
Figure 1.

Relationship of all the spheres.
During the last hundred years, industrialisation has grown at a fast rate. It has thus increased the demand for exploitation of the Earth's natural resources at a careless rate, which has exacerbated the world's problem of environmental pollution [4]. The environment has been seriously polluted by several pollutants such as inorganic ions, organic pollutants, organometallic compounds, radioactive isotopes, gaseous pollutants and nanoparticles [2]. Heavy metal pollution will be discussed further in this article.
There has been an ongoing discussion with regards to the definition of the term ‘heavy metals’.
They are defined as heavy metals either due to their high atomic weight or because of their high density. Nowadays, the word ‘heavy metal’ has been used to describe metallic chemical elements and metalloids which are toxic to the environment and humans. Some metalloids and also lighter metals such as selenium, arsenic and aluminium are toxic. They have been termed heavy metals while some heavy metals are typically not toxic such as the element gold [5, 6, 7, 8].
A list of heavy metals according to their density of being greater than 5 g/cm3 and which are more common in our everyday life are:
-
•
Titanium
-
•
Vanadium
-
•
Chromium
-
•
Manganese
-
•
Iron
-
•
Cobalt
-
•
Nickel
-
•
Copper
-
•
Zinc
-
•
Arsenic
-
•
Molybdenum
-
•
Silver
-
•
Cadmium
-
•
Tin
-
•
Platinum
-
•
Gold
-
•
Mercury
-
•
Lead
The objective of this review is to highlight the pollution of heavy metals and how they are brought about in our environment, their pharmacokinetic mechanisms, together with their toxicological effects on humans.
2. Sources of heavy metal pollution
These heavy metals are found naturally on the Earth's crust since the Earth's formation. Due to the astounding increase of the use of heavy metals, it has resulted in an imminent surge of metallic substances in both the terrestrial environment and the aquatic environment [4]. Heavy metal pollution has emerged due to anthropogenic activity which is the prime cause of pollution, primarily due to mining the metal, smelting, foundries, and other industries that are metal-based, leaching of metals from different sources such as landfills, waste dumps, excretion, livestock and chicken manure, runoffs, automobiles and roadworks. Heavy metal use in the agricultural field has been the secondary source of heavy metal pollution, such as the use of pesticides, insecticides, fertilisers, and more. Natural causes can also increase heavy metal pollution such as volcanic activity, metal corrosion, metal evaporation from soil and water and sediment re-suspension, soil erosion, geological weathering [1, 2, 4, 5, 9, 10, 11].
3. Properties of heavy metals
Metalloids tend to form covalent bonds, making them show toxicological properties. The two most important consequences of this property are that they can bind covalently with organic groups. Hence they form lipophilic ions and compounds, and they can generate toxic effects when they bind to nonmetallic elements of cellular macromolecules. Due to becoming lipophilic, the metalloids distribution within the biosphere and their toxic response vary from the action of simple ionic forms of the same element. Examples of lyophilic compounds are tributyltin oxide and methylated forms of arsenic which are highly toxic. Examples of binding to nonmetallic elements are the binding of lead and mercury to sulphydryl groups of the protein. Heavy metals may enter a human in four ways from; ingestion of contaminated food; inhalation from the atmosphere, drinking contaminated water; and due to skin contact from agriculture, pharmaceutical, manufacturing, residential and industrial areas [1, 2].
Metals cannot be broken down and are nonbiodegradable. Organisms may detoxify metal ions by hiding the active element within a protein or depositing them in intracellular granules in an insoluble form to be excreted in the organism's faeces or for long-term storage. When the heavy metals are swallowed or inhaled into our bodies, they bioaccumulate in our system. Thus they are classified as dangerous. This bioaccumulation causes biological and physiological complications. Some heavy metals are necessary for life and are called essential elements which are required for a variety of biochemical and physiological functions. However, they can be toxic when present in large amounts as seen in Figure 2 [4, 5, 6, 7, 8]. They have been widely used in agriculture, industry, medicine and other sectors, to the effect that they have been dispersed into the environment including our atmosphere, waters and soils [5, 6, 7, 8].
Figure 2.

The relationship between the person's performances concerning the concentration of the essential element in the diet.
Essential elements are grouped into three groups; the major elements needed for the body, macrominerals and trace elements. Four important major elements are needed for the building blocks of most living matter. These are hydrogen, carbon, nitrogen and oxygen according to their atomic number. There are seven other major elements called the macrominerals, which are integral elements that maintain the ionic balance of structural compounds, amino acids and nucleic acids. These include sodium, magnesium, phosphorous, sulfur, chlorine, potassium and calcium according to their atomic number. The last group are the trace elements which is made up of thirteen elements; silicon, vanadium, chromium, manganese, iron, cobalt, nickel, copper, zinc, arsenic, selenium, molybdenum and iodine according to their atomic number. The essential elements are important for the maintenance of skeletal structure formation, acid-base equilibrium regulation, colloidal system maintenance. They are also important as constituents of key enzymes, structural proteins and hormones such as zinc being a constituent for many enzymes, iron is important for haemoglobin, selenium being essential for the glutathione peroxidase enzyme [2, 5, 12]. Nonessential metals do not have any key role in the body, but they may also cause toxicity as they can affect the level of an essential element in the body [2].
Cellular organelles and components of the cell have been reported to be affected by heavy metals, such as the mitochondria, nuclei, lysosomes, cell membrane and enzymes. It has emerged that metal ions interact with DNA and nuclear proteins, thus causing DNA damage, consequently leading to cell cycle modulation, apoptosis or carcinogenesis [5]. A pathway showing the consequences of heavy metal pollution can be seen in Figure 3 (see Figures 4, 5, 6, 7).
Figure 3.

Heavy metal source pathway and human exposure [13].
Figure 4.

Metal-induced oxidative stress pathways [14].
Figure 5.

Chromium formation of free radicals [14].
Figure 6.

Cobalt reactions [14].
Figure 7.
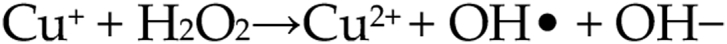
Copper Fenton reaction [14].
Heavy metals have been noted to interact with nuclear proteins together with DNA which cause site-specific damage. Two types of damages may be caused, “direct” and “indirect” damage. In the “direct” damage, conformational changes occur to the biomolecules, due to the metal. On the other hand the heavy metal causes “indirect” damage, which is a result of the production of reactive oxygen and nitrogen species which comprise of the hydroxyl and superoxide radicals, hydrogen peroxide, nitric oxide and other endogenous oxidants. Heavy metals have been noted to activate signaling pathways [14].
Metal toxicity causes the formation of free radicals which causes DNA damage, alteration of sulphydryl homeostasis, and lipid peroxidation. Alterations have also been noted in metal-mediated calcium homeostasis due to membrane damage which causes a variety of calcium dependent systems to be activated including endonucleases. Free radical formation has mostly been investigated for iron, copper, nickel, chromium and cadmium. The last three metals are recognised for the carcinogenic properties [14].
Iron, copper, vanadium, chromium and cobalt follow the Fenton reaction of the superoxide and the hydroxyl radical (Eqs. (1) and (2)). Fenton reactions are primarily linked to mitochondria, microsomes and peroxisomes [14].
| Me(n+1)+ + O2•–→ Men+ + O2 |
| 2O2•– + 2H+→ H2O2 + O2 |
| (Fenton reaction) Men+ + H2O2→ Me(n+1)+ + •OH + OH- [14] |
Metal-mediated free radicals cause the mutagenicity of DNA base alterations revealing the link between carcinogenesis and oxidative damage. The free radicals formed, cause a variety of DNA base modifications where most of them are pro-mutagenic, thus showing the vital link between the oxidative damage caused by the metals together with their carcinogenicity. The metals cadmium, nickel and arsenic, are known to inhibit the DNA repair mechanisms. Oxidative effects in DNA include (i) base modification which is seen by chromium and nickel; (ii) crosslinking which is seen by nickel, copper and oxidant, iron and oxidant; (iii) strand scission which is seen by nickel, cadmium, chromium and oxidant; and (iv) depurination which is seen by copper, chromium and nickel (Equation 2) [14].
Protection is provided against free radical attacks mediated by the metals, by the variety of antioxidants which can be enzymatic and non-enzymatic. Iron toxicity is generally protected by antioxidants through (i) the prevention of molecular oxygen and/or peroxides reactions, and the chelation of ferrous ion; (ii) the chelation of iron and the redox state being kept, making the iron incapable of reducing molecular oxygen; and (iii) the trapping of radicals formed. Thiol compounds are one of the classes classified as being most effective, including glutathione which traps radicals, maintains the cell's redox state and reduce peroxide, thus protecting the cell. Vitamin E is a non-enzymatic antioxidant which can prevent damage caused by the metals in vitro systems and animals loaded with iron, copper and cadmium, as long as the daily dose does not exceed 400IU which might cause death. Metal-induced toxicity, together with carcinogenicity, can be determined by a common factor by the enriched formation of free radicals and other reactive species [14].
4. Entry, effects and transport of pollutants into the ecosystem
Pollutants may enter the ecosystem in various ways and will enter into the hydrosphere, lithosphere and atmosphere. Apart from also entering through natural ways as previously said, through volcanic activity and weathering of rocks, anthropogenic activity is a big cause of pollutants entering the ecosystem. They can be an unintended release such as in shipwrecks, oil spills, mining and fires; in the intended application of biocides such as vector controls; and waste disposals such as industrial effluents and sewage disposal. Movement of heavy metals or any other pollutants depends on temperature, movements and direction of surface waters, circulation of air masses and the speed of the wind. Apart from these, there are other factors which influence the distribution and movement of the pollutants, such as partition coefficient, polarity, vapour pressure and molecular stability [2].
4.1. Soil pollution
Soil pollution can be both deliberate or not. Deliberate pollution includes wastewater irrigation, pesticides, animal manures, fertilisers, leaded paint, mine ore waste (mine tailing), sewage sludge, spillage of petroleum distillates, coal combustion residues, waste dumpings. Using sewage and wastewater that are not treated have caused a lot of heavy metals in our agricultural lands and thus have been absorbed by the crops that tend to be eaten by humans themselves. Non-deliberate pollution may be brought about through flooding of seas and rivers which brings sewage and contaminated water to the land and accidents involving vehicles transporting toxic chemicals. Since heavy metals are non-degradable, since they cannot undergo and microbial or chemical degradation, they stay in the soil for a very long time.
The ecosystem is being ruined to the fact that the heavy metals are entering the food chain. Heavy metals also affect the biodegradability of organic pollutants, making them less degradable and thus causing double the effect of polluting the environment. These metals present in the soil cause risks to all the biosphere and are taken up through direct ingestion, absorbed by plants which can be hazardous both to the plant and also to the food chain that eats the plant, altering the properties of the soil such as the pH, colour, porosity and natural chemistry thus impacting the quality of the soil, and also contaminating the water [1, 2, 15, 16, 17, 18].
4.2. Water pollution
Two major origins are the culprits of water contamination: urbanisation and industrialisation. The metals are transported by the runoffs from villages, towns, cities and industries which accumulate in the sediments of water bodies. Even if traces are transported to water bodies, they might still be very toxic to human beings and other ecosystems. Toxicity of heavy metals depends on a lot of factors such as which metal is present, the nature of the metal, the biological role of the metal, the organism exposed and the period of the organisms life when it is exposed. If one organism is affected, this will affect all the food chain. Since humans are usually the last of the food chain, this will affect us more as we would have accumulated more heavy metal as the concentration increases along the food chain. Both industrial and domestic wastes usually are expelled into the sewage system.
Heavy metals are found in high concentrations in raw sewage, and these are not degraded in the sewage treatment. They are removed either in the final effluent or else in the sludge produced. The properties and contaminants of the sewage that enters the water depend on the treatment of the sewage. Several controls have been set up due to the problems caused by sewage elimination into the rivers and seas without being treated. Stringent regulations have been placed, and better technology have been developed to decrease the amounts of pollutants that are thrown in the waters.
Sewage treatment is divided into three stages, primary, secondary and tertiary. Primary stage involves the sedimentation of the solid waste found in the wastewater which occurs after filtering the larger contaminants in the wastewater. The water is directed through various tanks and filters which will separate the contaminants from the water, which will then form a sludge which is fed into a digester which is further processed. The sludge at this stage contains approximately half the suspended solids present in water. Secondary treatment involves the use of oxidation which helps to purify the wastewaters and can be done in three ways, biofiltration, aeration or oxidation ponds. The tertiary treatment is the last step and consists of the removal of phosphates and nitrates from the water supply. This process usually uses activated carbon and sand to help in removing the contaminants. These are the basic steps used in sewage treatments depending on what the sewage contains and where it is being processed. Many controls have been set up due to the problems caused by sewage elimination into the rivers and seas without being treated. Stringent regulations have been placed, and better technology have been developed to decrease the amounts of pollutants that are thrown in the waters [1, 2, 19, 20, 21].
Pollutants can exist in diverse states: in surface waters; in solution or suspension form. They can be transported over an extensive distance by water, where particulate materials can descend to the bottom. Liquid droplets can either descend to the sediment or rise to the surface. Distance travelled in rivers depends on the currants, stability and physical state of the pollutant. When transported into the sea and oceans, wind and currents transport the pollutant further. The difference in density of the seawater plays another factor in transportation to a higher concentration of salt or due to a fall in temperature. The persistent pollutants such as heavy metals can then enter the food chain through marine life such as fish which can then affect predators such as bigger fish, birds and mammals, including humans, which migrate and transport the pollutant to different ecosystems [2].
4.3. Air pollution
Like water contamination, air pollution has been caused due to urbanisation and industrialisation. Pollutants enter the atmosphere in different forms. They can enter as particles, droplets, or in the gaseous form, or association with particles or droplets. Particles and droplets do not travel long distances and usually fall on the ground after a short distance, though if small in size can travel a longer distance. Particles in the gaseous state can be transported over long distances due to air masses.
Natural and anthropogenic activity has caused the release of particulate matters (PMs), especially fine particles, and dust. Particulate matters that are present through natural activity are released through sand storms, volcanic activity, soil erosion and the weathering of rocks. While particulate matters that are present to human activity are released through industrial activity, burning of fossil fuels, vehicle exhaust, smelting and more. The particulate matters can precipitate severe health problems and also cause infrastructure deterioration, the formation of acid rain, corrosion, eutrophication due to particulate matters falling in the water when it rains, and it can cause haze.
Chimneys are one of the main sources of atmospheric pollution where a number of gases are released. The height of the chimney and the weather, make a difference of how far the pollutant travels. The higher the chimney, the further the pollutant travels. The warmer the climate is and the windier it is, the farther away the pollutants travel since convection currents occur and the side currents help it move further away. In cold and foggy weather the pollutants travel very short distances.
Other sources of atmospheric pollution are internal combustion and jet engines. Catalytic converters and unleaded petrol have helped to reduced pollution from vehicles, apart from improvement of the engines. Though diesel engines, old cars and too many cars still cause a problem. Pesticides application is another source of pollutants together with refrigerators, aerosols and radioactive pollution [1, 2].
The atmosphere is divided into five main layers, though the troposphere and stratosphere are essential for pollutant transportation. The troposphere is the first layer closest to the Earth and stratosphere is above it where at the top the ozone layer lies. In the troposphere, vigorous vertical mixing occurs, with a consistent air pattern of circulation, and pollutants can be transported in a small amount of time. There is little vertical mixing in the stratosphere. Pollutants released close to the Earth tend not to travel far due to turbulence and confined airflow. Though pollutants eliminated at a higher distance can travel further due to the circulating air. Air pollutants can thus travel far when entering air circulation and cause global issues. Soluble particles can then react with the rain and fall into waters and onto land [2].
5. The fate of heavy metals in the ecosystem
Increase in toxicity of heavy metal is caused by localisation of a high amount of the metal. In some areas, chimneys have been built higher to dilute the emissions of the metal as they are dispersed more and thus not fall in a localised spot. Although sometimes this still has other effects, as being emitted higher make it more prone to acid rain. Even though the Earth is seen as one compartment, it can be subdivided into many other compartments such as an organism or individual cells. Potential toxins on organisms may be compartmentalised into insoluble deposits; thus, this prevents any interactions with essential biochemical reactions that occur in the cytoplasm.
Since metals are non-biodegradable and hence remain persistent in the environment for a very long time, they cannot be broken down. Heavy metals present in soils and sediments remain present for an extended period until they are eluted to other compartments. They can also react with other elements in the soil or sediment and form or degrade to become more toxic. An example of this is the formation of poisonous methyl mercury from the inorganic mercury and activity of bacteria found in water, sediment and soil [2].
Anthropogenic activity has left a very high concentration of metals in contaminated sites such as disused mining sites, or previously used metal-containing pesticides. In these areas, vegetation is sparse, and only metal-tolerant strains grow in the area. In these zones, sometimes capping is introduced, meaning that an impermeable layer is placed on top of the contaminated site, and new soil is put on top of it. Capping will help the vegetation not absorb any metals and also help the water going down not to take heavy metals into the groundwater. Metal containing pesticide contained arsenic, copper, lead and chromium and these may still be found in some areas where it had been used [2, 22].
Farmers sometimes use sewage sludge and mix it in the soil, though this may contain heavy metals, primarily if the sludge has been produced by industries. Heavy metals, such as copper, zinc, lead, cadmium and chromium, have been found in the soil of these agricultural lands at a high concentration [15, 16, 17, 18, 19]. Smelting causes localised pollution through atmospheric pollution, which then deposits on the soil. Some areas were smelting occurs shows dead vegetation and absence of life such as earthworms and woodlice, which help in vegetation to be decomposed. Lead-contaminated gasoline was used with a high concentration of lead, lead shotgun pellets, and lead fishing weights all contributed to lead being found in our environment. Some have been banned in certain parts of the world. Shotgun pellets have been taken up by birds, and this then moves through the food chain, the weights have caused lead to be found in wetlands also. Metals are bound more to the soil if the clay content, organic matter, and the pH, are higher. The more acidic the soil, the less elemental elements have been found as these become more soluble and leach lower in the ground where the roots do not reach causing nutrient deficiency to the plants [2, 23].
In the water sector, most rivers are polluted especially those that pass from near industries and mining areas. These then flow down to the sea where they mostly descend to the bottom and since the current slows down. The solubility of the metals depends mostly on the pH of the water. As soon as the streams containing heavy metals flow into the sea, the acid rises, and the solubility of the metals decrease and thus precipitate downwards towards the bed [2, 24, 25].
6. The heavy metals
The heavy metals which are described in further detail include aluminium, vanadium chromium, manganese, cobalt, nickel, copper, zinc, arsenic, selenium, molybdenum, silver, cadmium, mercury and lead which are placed in ascending order according to their atomic number. Substances may be classified in different groups according to whether they are classified as carcinogenic or not. The International Agency for Research on Cancer (IARC) has classified them into four groups [26] (see Table 1).
Table 1.
| Group | Carcinogenicity level in humans | Evidence | Heavy metal classification |
|---|---|---|---|
| Group 1 | Carcinogenic | Sufficient evidence in humans |
|
| Group 2A | Probably carcinogenic | Limited evidence in humans, enough evidence in animals |
|
| Group 2B | Possibly carcinogenic | Limited evidence in humans, not enough evidence in animals |
|
| Group 3 | Carcinogenicity not classifiable | Insufficient evidence in humans, insufficient enough evidence in animals |
|
| Group 4 | Probably not carcinogenic | Evidence suggests no carcinogenic properties in humans or animals |
|
Excess of all metals can be the source of disease. Essential metals are essential to the body, and both excess and deficiency can affect the human body. Deficiencies are caused usually through malabsorption can be caused by a diarrhoeal state. On the contrary excess of the metal and its toxicity effects are dependent on certain factors such as the ingestion or inhalation of the metal, entry rate of the metal, tissue distribution and the concentration achieved, and lastly the excretion rate of the metal. Toxicity mechanisms comprise of the inhibition of enzyme activity, protein synthesis, variations in nucleic acid function, and the changes in the permeability of the cell membrane [27].
Metals having toxic and carcinogenic properties can interact with nuclear proteins and DNA, which causes biological macromolecules to deteriorate oxidatively. Evidence is seen to back this up by the wide range of nucleobase products which are typical of DNA subjected to oxygen attack, in animals and cultured cells which are to the carcinogenic metals. Some metals can produce reactive radicals which go on to result in DNA damage, lipid peroxidation and depletion of protein sulphhydryls, together with other effects. These reactions can be seen by copper, iron, cadmium, chromium, nickel, mercury, and vanadium. Reactive radical species involves a variety of radicals (oxygen-, sulphur-, carbon-) which are derived from the superoxide radical, lipid peroxides, and hydrogen peroxide, together with chelates of amino acids, proteins and peptides which are complexed with the toxic metals. Metal toxic adverse effects include neurotoxicity, hepatoxicity, and nephrotoxicity.
6.1. Aluminium (Al)
Table 2.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 3.
Aluminium thresholds [29].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | No data |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of Al in air | 15 mg/m3 total dust 5 mg/m3 respirable fraction |
| Drinking water | WHO | Drinking-Water Quality Guidelines in large water treatment facilities | ≤0.1 mg/L |
| WHO | Drinking-Water Quality Guidelines in small water treatment facilities | ≤0.2 mg/L | |
| EPA | SMCL for AL, based on taste, odour and colour, and no adverse effects | 0.05–0.2 mg/L | |
| Bottled Water | FDA | Bottled water limit | 0.2 mg/L |
6.1.1. Pharmacokinetic processes
Aluminium mechanism of action is not known, though it is noted that aluminium competes with cations in biological systems, especially magnesium. It competes even though there is a difference in oxidation state, where it binds with citrate and transferrin in the blood. Second messenger systems may also be affected by aluminium, together with calcium availability. Aluminium binds irreversibly to the cell nucleus components. Neuronal microtubule formation was observed to be inhibited due to the metal. Absorption through the gastrointestinal tract is usually low. Absorption is regulated according to aluminium's ionic availability in the gut's aqueous conditions, including the pH, chemical form of the metal, and the existence if complexing ligands in the gut where the metal would be able to form an absorbable aluminium species. Aluminium usually is found in the stomach acidic conditions as Al3+ (monomolecular species). Soluble Al3+ can then re-complex with the original aluminium compound anion ingested, or it can form another complex with dietary ligands. Complexation processes occur due to dietary constituents with mono-, di-, and tri-, carboxylic acids, though most solubilized aluminium is not usually complexed. Gut bioavailability is increased by citrate as it enhances the permeability of the paracellular channels, probably due to a disruption in calcium homeostasis. The citrate aids in aluminium absorption by maintaining the aluminium in a form which is incorporated readily into one or more absorption mechanisms. Aluminium hydroxyl complexes form in the duodenum since pH rises, and consecutive deprotonation occurs, forming aluminium hydroxide as the predominant form. The aluminium hydroxide is swiftly precipitated in the close to neutral pH, as insoluble, which is then excreted in the faeces. Studies suggest that absorption of aluminium is mostly by passive diffusion as neutral complexes, through the intercellular tight junction pathways. Though it might also be absorbed through transcellular transport, where both transcellular and paracellular pathways might be involved, transcellular transport might probably include a passive process by cell-mediated endocytosis, cation specific channel via facilitated diffusion, and simple diffusion or lipophilic and neutral aluminium complexes. Another pathway might be active transport via the iron absorption pathways. Apart from citrate, studies show that parathyroid hormone and vitamin D might also enhance aluminium absorption [29].
Inhalation mechanisms of the metal are not characterized well though it is thought that large particles are removed through the mucociliary cells and deposited into the gastrointestinal tract. In contrast, small particles are taken up from alveoli to the bloodstream by dissolution, or through macrophage phagocytosis [29].
6.1.2. Toxicology processes
Organs that seem to be the target of aluminium poisoning are the lungs, central nervous system and bone. Aluminium exposure has been noted to cause two types of osteomalacia. The first type of osteomalacia is seen in healthy individuals who use antacids. The aluminium present in the antacids binds to dietary phosphorus which then impairs the absorption of phosphorus in the gastrointestinal tract. Since phosphorus decreases, osteomalacia and rickets are seen. Dialyzed uremic patients have shown to show osteomalacia since they are exposed to aluminium present in the dialysis fluid. Aluminium in bones is increased in these patients and aluminium is found in between the junction of non-calcified and calcified bones. Patients suffering from hyperphosphatemia may also show osteomalacia since aluminium is administered orally to control it. There will be an increased mineralization lag time, osteoid area, osteoid surface, slightly elevated calcium levels in the serum, and low parathyroid hormone levels due to osteomalacia [29].
Effects of neurotoxicity due to aluminium have been prompted in animals. More than one mechanism probably exists for aluminium neurotoxicity which causes neurodevelopmental and neurobehavioural alterations, including neurodegenerative brain pathological changes. Aluminium in certain species and exposure situations has produced changes to the cytoskeletal proteins. These changes manifest themselves as hyperphosphorylated neurofilamentous aggregates inside the brain neurones. Many neurodegenerative disorders are associated with comparable neurofibrillary pathological changes, thus showing that abnormal neuronal function caused by aluminium might involve cytoskeleton protein function changes in the affected cells. The aggregates result from altered phosphorylation, due to posttranslational alterations in protein synthesis. However, it might also comprise of proteolysis, transport and synthesis, where the interactions cause the generation of the phosphorylated neurofilaments. Each process can be affected by kinases, which are initiated by second messenger systems. Aluminium influences calcium homeostasis together with calcium-dependent processes found in the brain through the damage of the phosphoinositide second messenger-producing system. It also seems to influence calcium-activated proteinases causing the alteration of the concentration and the distribution of cytoskeletal proteins. Neurodegeneration is also caused by apoptosis. Studies in rats have shown that aluminium affects blood-brain barrier permeability, signal transduction pathways, cholinergic activity, lipid peroxidation, impairment of the neuronal glutamate nitric oxide-cyclic GMP pathway, and the interference of essential trace elements metabolism [29].
6.2. Vanadium (V)
Table 4.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 5.
Vanadium thresholds [32].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines Time-weighted average over 24 h, excluding cancer and smell/irritation |
1 μg/m3 |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of vanadium pentoxide respirable dust | 0.5 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of vanadium pentoxide fumes | 0.1 mg/m3 | |
| Drinking-Water | WHO | Drinking-Water Quality Guidelines | No data |
| Bottled Water | FDA | Bottled water limit | No data |
6.2.1. Pharmacokinetic processes
Two oxidation states of vanadium are found in the body, which are vanadyl (V4+) and vanadate (V5+). Vanadium can bind reversibly to the protein transferrin present in the blood, which is then taken up by erythrocytes [32]. Vanadium enters the cells through a two-phase mechanism. It involves the fast equilibration of vanadate by transport through the anion channels. It then involves a slower phase which includes the reduction of vanadate to vanadyl [14]. Vanadate is a potent inhibitor of the plasma membrane Na+K+ ATPase and is reactive with a variety of enzymes, making it more toxic than vanadyl. Vanadate is also taken up at a higher rate by erythrocytes than vanadyl since vanadyl has to be oxidised to vanadate [32]. Vanabins (vanadium-binding proteins) which are present in the cytoplasm can assist the transport and accumulation of the metal in the vacuoles [14].
6.2.2. Toxicology processes
In vivo studies in mice, showed a decrease in NADPH, NADH and glutathione-SH after an hour of being injected with sodium vanadate. Vanadium was seen to act as a phosphate analogue during in vitro studies. It interfered with a variety of ATPases, phosphate-transfer enzymes and phosphatases. The metal was noted to inhibit:
-
•
Na+K+ ATPase
-
•
Ca2+ ATPase
-
•
Ca+Mg+ ATPase
-
•
H+K+ ATPase
-
•
K+ ATPase
-
•
Actomyosin ATPase
-
•
Dynein ATPase
-
•
Glucose-6-phosphatase
-
•
Acid and alkaline phosphatase
-
•
Ribonuclease
-
•
Phosphodiesterase
-
•
Phosphotryosyl-phosphatase
On the other hand, it was noted to stimulate:
-
•
Tyrosine kinase phosphorylase
-
•
Adenylate cyclase
-
•
NADPH oxidase [32].
The metal has also been noted to mimic insulin properties, it can stimulate the uptake of glucose an oxidation, together with glycogen synthesis and inducement of cell proliferation. Adverse effects seen in vanadium toxicity are due to the effects the metal has on several enzymes. Mechanism of toxicity in vivo has not been researched well. It was shown that vanadium pentoxide in the respiratory tract, induced production of mucin in the epithelial cells of mice. Though the production of mucin was induced by the independent pathways of EGFR- and MAPK-, and the dependent pathways of RAF1-1KK-NF-кB. An increase in collagen and/or fibroblasts were seen around the airways due to airway fibrosis caused by the vanadium pentoxide. mRNA levels are increased, with the presence of vanadium, which encodes:
-
•
pro-fibrogenic growth factors such as PDCF-C, CTGF and TGF-β1
-
•
chemokines such as CXCL9, CXCL10, IFN-α, and IFN-β [32].
Mice exposed to vanadium had a higher amount of collagen mRNA levels. Inflammation and an increase in apoptosis were seen when sodium metavanadate was inhaled, where a small amount of lung cell necrosis occurred. These occurrences are probably due to the generations of reactive oxygen species, predominantly hydrogen peroxide [32].
Vanadate is reduced to vanadyl in the plasma by both enzymatic and non-enzymatic antioxidants, which are then transported and bound to the plasma proteins, where peroxovanadyl is formed. Vanadyl hydroperoxide is converted by a dismutation reaction with dismutase (SOD) into hydrogen peroxide [14] as seen in the following reactions.
| V5+ + NADPH -> V4+ + NADP+ + H+ |
| V4+ + O2 -> V5+ + O2•- |
| V5+ + O2•- -> [V4+-OO•] [14] |
| V4+ + H2O2 -> V5+ +OH− + •OH (Fenton reaction) [14]. |
Cell injury due to the presence of vanadium causes the Fenton-like reaction. The free radicals produced in the reaction will further produce the radical superoxide, which will then be reduced by the dismutase, to oxygen and hydrogen peroxide [14].
6.3. Chromium (Cr)
Table 6.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 7.
Chromium thresholds [36].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines Chromium (VI) |
1 μg/m3 for lifetime risk of 4 × 10−2 |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of Cr (VI) | 0.005 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of Cr (III) | 0.5 mg/m3 | |
| OSHA | Legal limit over an 8-hour workday of Cr (0) | 1 mg/m3 | |
| NIOSH | Legal limit over a workday of 8 h for Cr (0), Cr (II) and Cr (III) | 0.5mg/m3 | |
| NIOSH | Exposure limit over a workday of 10 h for Cr (VI) | 0.001 mg/m3 | |
| Drinking water | WHO | Drinking-Water Quality Guidelines for total chromium | 0.05 mg/L |
| EPA | Maximum contaminant level for total chromium | 0.1 mg/L | |
| Bottled Water | FDA | Not to exceed the total chromium concentration | 0.1 mg/L |
6.3.1. Pharmacokinetic processes
Chromium particle properties affect the absorption of inhaled chromium. Apart from the physical and chemical properties of chromium, the activity of macrophages in the alveoli also affect the absorption of the metal. Cr6+ is absorbed at a faster rate than Cr3+ in the bloodstream. Chromium which is not absorbed through the lungs, may then enter the gastrointestinal tract since it is cleared by the mucociliary clearance. It is primarily absorbed in the jejunum. Oxidation state and formulation of the metal are factors that affect the extent of absorption [36].
Inorganic Cr3+ is not absorbed well by the gastrointestinal tract. Cr6+ is absorbed to a higher degree than Cr3+ after oral exposure, though it is seen to be reduced to Cr3+ by gastric juices in the gastrointestinal tract. Cr6+ can be reduced to Cr3+by the liver, blood, lung, erythrocytes, epithelial fluid, peripheral parenchyma cells and alveolar macrophages [36]. Once it enters into the bloodstream, Cr3+ binds to transferrin, and Cr6+ is taken up by erythrocytes after it has been absorbed and is then reduced to the trivalent form. It is taken up mostly by the spleen, bone marrow, lungs, lymph nodes, liver and kidney. The lungs are the organs that take up the most chromium. The metal is excreted mostly through urine but is also eliminated by bile excretion, and smaller quantities in nails, hair, milk and sweat [14].
6.3.2. Toxicology processes
Toxicity mechanism and carcinogenicity of chromium is a complex process. Mechanisms are mediated by the intermediates during the intracellular reduction of Cr6+ and Cr3+ together with oxidative reaction. It is then partly mediated by the trivalent form, which is formed by the reduction of the hexavalent form and forms toxic complexes with vital target macromolecules [36]. DNA lesions such as DNA-protein crosslinks, Cr-DNA adducts, DNA-DNA crosslinks, changes in cellular signalling pathways, and DNA strand breaks are caused by complexes made up of Cr3+ and peptides, DNA and proteins inside the cell, which all may be a factor to toxicity and carcinogenicity by the chromium compounds [14, 36]. Cr6+ is more toxic than the trivalent oxidative state due to (i) having a higher redox potential; and (ii) being able to enter the cells more. Cr6+ at physiological pH is found in a tetrahedral chromate anion which has a similar structure to other natural anions such as phosphate and sulphate, which are permeable through the nonselective membrane channels [14].
On the other hand, Cr3+ has an octahedral complex structure which does not cross the channels easily. The difference in structures shows why the trivalent form of chromium is less toxic than the hexavalent form. Once the hexavalent form of chromium enters the cell, through redox reactions, Cr6+ is reduced to Cr3+ together with the intermediates Cr4+ and Cr5+. The reaction contributes to Cr4+ which has a higher toxic potency than Cr3+. Ascorbate, amino acids and glutathione can be involved in the reactions [36] (Equation 4).
In vitro studies have shown that chromium, especially Cr4+, produces free radicals from H2O2. In vivo studies have shown that free radicles are detected in animals due to chromium present in the liver and blood. It was observed that one-electron reduction was generated from Cr5+ intermediates.
Chromate compounds were noted to induce DNA damage in a variety of ways leading to the production of DNA adducts, changes in replication sister chromatid exchanges, chromosomal aberrations, and transcription of DNA as seen in, in vivo and in vitro studies [13].
In vitro studies have shown that some compounds were shown to reduce Cr6+. Among others the most common are:
-
⁃
GSH
-
⁃
Cysteine
-
⁃
Ascorbate
-
⁃
Lipoic acid
-
⁃
NAD(P)H
-
⁃
Ribose
-
⁃
Fructose [14].
Formation of the oxygen radical species leads to an oxidative stress which may be the cause for the variety if deleterious effects by the chromium on the cells. This also includes effects on the lipid peroxidation, signalling pathways, modifications in cellular communications and cellular cytoskeleton. Inhibition and induction, the result of the generation of ROS which produces Cr6+-induced oxidative stress, occurs of the transcription factors, AP-1 and NF-кB, activation of hypoxia-inducible factor 1 (HIF-1), activation of p53, p53-dependent apoptosis, and cell-cycle arrest. Radical scavengers have been noted to block cellular damage by chromium, showing that oxygen radicals are an important key role in the toxicity of chromium [36].
Functional damage due to chromium includes RNA polymerase arrest, DNA polymerase arrest, altered gene expression, and mutagenesis. Cr-DNA ternary adducts causes repair errors together with collapsed replication forks which causes DNA double strand breaks. Cellular communication alteration, outcomes on signaling pathways and cytoskeleton, can be caused by double strand breaks. Chromium-induced chromosome instability, which leads to carcinogenicity, has been seen to mediate through spindle and centrosome assembly checkpoint bypass. Another factor is location of the lung particle deposition and the dissolution of extracellular Cr6+ [36].
6.4. Manganese
Table 8.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 9.
Manganese thresholds [46].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | 0.15 μg/m3 |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of manganese in air | 5 mg/m3 |
| Drinking-Water | WHO | Drinking-Water Quality Guidelines | 0.4 mg/L |
| EPA | 1–10 day exposure of Mn at this concentration does not expect to produce any adverse effects in a youngster | 1 mg/L | |
| EPA | Lifetime exposure of Mn at this concentration is not expected to produce any adverse effects in humans | 0.3 mg/L | |
| Bottled Water | FDA | Legal limit not to be exceeded | 5 mg/m3 |
6.4.1. Pharmacokinetic processes
Manganese is absorbed by the gut by non-saturable simple diffusion via the mucosal layer of the gut, or through the high affinity, rapidly storable, low capacity active transport mechanism. Particles of the metal that are too large to diffuse through the alveoli are coughed up through mucociliary action and then swallowed. Diets that have a high concentration of iron tend to suppress the absorption of manganese while low concentration of iron tends to increase uptake of manganese. Phosphorous and calcium are other elements that tend to suppress manganese uptake if present. Manganese (II) is noted to enter the gastrointestinal tract and bind to albumin or α2-macroglobulin in the plasma. Most manganese is secreted in bile through the liver, though some Mn2+ will be oxidised to Mn3+ by ceruloplasmin [46].
6.4.2. Toxicology processes
Mn3+ conjugates with transferrin in plasma and enter neurones where it dissociates and is transported to axon terminals. Other studies have noted that Mn2+ enters the brain through saturable mechanism when entering the choroid plexus and cerebral cortex while using the non-saturable mechanism to other brain regions and the cerebrospinal fluid due to passive diffusion. Recent studies have shown that the metal enters the brain through three pathways, from the:
-
•
Nasal mucosa, through the olfactory neural connection and then the brain olfactory bulb;
-
•
The blood which then leads to the capillary endothelial cells which lead to the blood-brain barrier cells;
-
•
The blood which then leads to the choroid plexus and thus to the cerebral spinal fluid [46].
Manganese was noted to accumulate the mitochondria present in neurons, oligodendrocytes and astrocytes cells. It was seen to disrupt ATP synthesis through the inhibition of the F1/F0 ATP synthase or the complex 1 (NADH dehydrogenase) in the mitochondrial respiration chain. Manganese was also seen to inhibit ATP synthesis in the brain mitochondria at the glutamate/aspartate exchanger site or the complex II (the succinate dehydrogenase) site, subject to the mitochondrial energy source. Oxidative stress is thus increased due to the disruption of ATP synthesis caused by manganese which causes a decrease in the intracellular ATP levels together with the generation of free radicles. The increase of oxidative stress caused may play a part in the cellular manganese toxicity. Animal studies have shown that the dopaminergic system can be disrupted due to the dopamine being oxidised by manganese and reacting to quinone species. The dopamine transporter (DAT1) takes up the reactive dopamine species causing dopaminergic neurotoxicity [13].
6.5. Cobalt
Table 10.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 11.
Cobalt thresholds [49].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Workplace Air | OSHA | Legal limit over an 8-hour workday of Co | 0.1 mg/m3 |
| Drinking water | WHO | Drinking-Water Quality Guidelines | No Data |
6.5.1. Pharmacokinetic processes
Biological solubility plays a vital role in cobalt inhalation absorption. Phagocytosis and mucociliary transport clear particles of physiologically insoluble cobalt, thus having a low systemic macrophage. Cobalt particles, to some degree, maybe dissolved within the alveolar macrophages. Soluble forms of the metal end up in the bloodstream through bronchial and alveolar walls. Cobalt oral absorption can depend on other factors such as iron deficiency and fasting, which increases the absorption of cobalt. Iron and cobalt in the intestine and compete for the absorptive pathway, though ferritin is not present for cobalt absorption. Oral absorption can also be affected by the solubility of the metal. The more the metal is soluble, the more it is absorbed. Dermal absorption depends if the skin is intact or not. If the skin is intact, absorption is less, while if the skin is broken, the absorption of cobalt is higher [49]. Cobalt is absorbed mostly by the liver and secondly by the kidney, and in the lungs when inhaled. Cobalt is excreted mostly through urine and to a lower extent in the faeces and bile [51].
6.5.2. Toxicology processes
Cobalt has been noted to have a high affinity to the sulphydryl group, thus causing inhibition of crucial enzymes. This may cause mitochondrial respiration; transposition of the divalent cations in the metal-activated enzyme ion centre; effects on the Ca2+ channel antagonist; inhibition of the calcium to enter, calcium signalling, and competition for the calcium ion for the intracellular binding sites; formation of ROS in cells by Fenton reaction thus causing oxidative stress which will lead to oxidative damage to the proteins, DNA and lipids [51].
Cobalt has been seen to impede with the process of DNA repair and can cause direct induction of DNA-protein crosslinking DNA damage, and sister-chromatid exchange. Carcinogenic effects have been seen due to Cr2+ in animal studies. Formation of the cobalt-mediated free radical has contributed to toxicity and carcinogenicity caused by the cobalt [14].
The peptides in the presence of Co2+ may function as pro-oxidants was noted to cause cellular damage. The Co2+-mediated Fenton reaction was noted to induce DNA cleavage at all the bases. During this reaction, experiments showed the presence of singlet oxygen in the presence of chelators. ROS generation in hepatocytes showed that the sites of cytotoxic ROS generation due to Co2+ are the lysosomes [14].
Metal-induced activation by cobalt causes hypoxia-inducible factor (HIF) in nearly all the cells, with the transcription of a variety of hypoxia-responsive HIF-target genes which probably promotes tumour development and also growth [51]. During studies, mDNA have been seen to be damaged and cell morphology altered due to the presence of cobalt (II) chloride. Levels of protein were changed, which is indicative of cellular response due to DNA damage and hypoxia; HIF-1 alpha, p21, p53, and PCNA were modulated [14].
The metal and the ions induce apoptosis and are thus classified as cytotoxic. At higher concentrations, the metal causes necrosis, including inflammatory response. In vitro tests have shown that cobalt metals and its salts are genotoxic in mammalians, mainly due to the oxidative DNA damage caused by the reactive oxygen species, and inhibition repair. Although it is considered as carcinogenic in animals, according to the IARC, it has not been considered carcinogenic in humans. Cobalt is seen to cause cardiomyopathy and stimulate production erythropoietin. Hypoxia is the physiological stimulus for the production of erythropoietin, and reduced delivery of oxygen to the kidney. Studies have shown that cobalt mimics hypoxia through a direct action in the kidney and liver, and on the erythropoietin producing cells, hypothetically by the metal-induced activation of the transcriptional activator HIF. Hyperoxia suppresses the erythropoietin effect of cobalt while hypoxia enhances the effect [51].
6.6. Nickel
Table 12.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 13.
Nickel thresholds [52].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | 3.8 × 10−4 (μg/m3)−1 |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of Ni in air | 1 mg/m3 |
| Drinking water | WHO | Drinking-Water Quality Guidelines | 0.02 mg/L |
| EPA | Drinking water threshold | 0.1 mg/L |
6.6.1. Pharmacokinetic processes
Nickel is absorbed as a lipophilic compound with a low molecular weight through the gastrointestinal tract. Ions and various ligands present in the gut will affect the absorption of nickel in the gut. Studies in animals have shown that nickel present in low amounts will be absorbed by the active transport together with facilitated diffusion. In contrast, if nickel is present in high amounts, carriers become saturated, and thus nickel is absorbed through passive diffusion. In vitro studies show similar results were nickel was absorbed by the jejunum, and passively diffused through the ileum. Nickel binds to albumin together with ultra-filterable ligands, including amino acids and small polypeptides, and is transported in the blood. Nickel competes with copper at the albumin site. Nickel is taken up in the liver through calcium channels found in hepatocytes, as seen during in vitro studies in rats. Excretion of nickel usually takes place through the urine. The metal is also eliminated from the hair and skin, sweat and milk [52].
Nickel carbonyl, soluble nickel, is fat-soluble and can thus cross the cell membrane. Usually, it crosses through calcium channels and diffusion. Free calcium is released from the intracellular store due to nickel blocking the calcium channel and thus less calcium entering the intracellular space. The lack of calcium entering the intracellular space has seen to cause cell growth and differentiation together with apoptosis due to signal gene expression changes. It is transported in the blood mostly by the protein albumin and is noted to be present at high concentrations in the brain, lungs, kidneys, adrenals and liver. It is excreted through the urine and faeces and has also been noted in sweat and saliva [14].
6.6.2. Toxicology processes
During the inflammatory process of nickel together with the apoptosis process, the transcription factor NF- κB was noted to be activated by nickel. Another transcription factor that is activated by nickel is ATF-1 (belonging to the ATF/CREB family), which due to an increase in calcium causes a protein kinase cascade activation which in turn mediated the ATF/CREB phosphorylation. It has been seen that activation of ATF-1 by nickel causes a diminution in the regulator of TSP I, which goes on to enhance the angiogenesis, which then stimulates tumour growth [14].
The metal nickel has a wide extent of carcinogenic mechanisms which comprise of transcription factors, production of free radicals and controlled expression of particular genes. Nickel was noted to be involved in regulating the expression of certain long non-coding RNAs, particular mRNAs and microRNAs. The heavy metal can stimulate methylation of the promoter and prompt the downregulation of maternally expressed gene 3 known as MEG3. The down-regulation causes an upregulation in the two proteins, hypoxia-inducible factor-1α, which has been seen to be implicated in carcinogenesis. Since nickel has been seen to generate free radicals, it is also implicated in the process of carcinogenesis [13, 14].
6.7. Copper
Table 14.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 15.
Copper thresholds [55].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Workplace Air | OSHA | Legal limit over an 8-hour workday of Cu fumes in the air | 0.1 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of Cu dust | 1 mg/m3 | |
| Drinking-Water | EPA | Drinking water threshold | 1.3 mg/L |
6.8. Pharmacokinetic processes
Copper is needed by many enzymes to function normally and is thus classified as an essential element. It can change states from Cu2+to Cu+ by cuproenzymes which are involved in redox reactions. This change of state can also make it toxic as superoxide, and hydroxyl radicals can be formed. A variety of homeostatic mechanisms in normal circumstances keeps a physiologically essential amount of copper. Homeostasis of copper involves the control of absorption, intracellular transport, cellular uptake and efflux, sequestration/storage, and excretion of copper from the body. Copper absorption by the gastrointestinal tract was seen to be inversely proportionate to the dietary intake of the metal. Studies have shown that uptake is saturable, and uptake or efflux are swayed by intracellular amounts of copper [55].
A possible mechanism of absorption is through the copper transporters which regulate the uptake, hCtr1 and hCtr2. The MNK protein may also be involved to transport copper through the basolateral membrane found in the intestinal wall. MNK is a copper translocating P-type ATPase. MNK delivers copper to the enzymes that are dependent on the metal and the efflux the metal out of the cell. Efflux of copper is regulated by the concentration of copper in the cell. The MNK protein has a conformational change when exposed to copper which results in a copper cluster, thus allowing access to the phosphorylation site. Once phosphorylation occurs, translocation of copper occurs. Once copper reaches the bloodstream from the gastrointestinal tract, it binds to albumin and histidine where it is transported to the liver through the portal circulation. Copper then complexes with copper chaperones which are small cytoplasmic proteins in the hepatic cells. The copper chaperone is included in the distribution of intracellular copper ions. WND protein which is also a P-type ATPase found in the liver, deliver copper to the ceruloplasmin which goes on to be released into the blood where it is dispersed to other tissues and organs. WND releases copper when there is an elevated amount of copper, at the canalicular membrane with the resulting biliary excretion of the metal [55]. Copper is eliminated mostly through the bile, but it can also be excreted in small amounts through the faeces, sweat and urine [14].
6.8.1. Toxicology processes
Reactive oxygen species (ROS) are formed by the participation of copper ions. The ROS which can contribute to the redox reactions includes Cu2+ (cupric) and Cu+ (cuprous). The cupric ROS can be reduced to the cuprous ROS if biological reductants like ascorbic acid and glutathione are present. Cuprous ROS can catalyse the hydrogen peroxide decomposition, through the Fenton reaction, which forms OH•, which can react with several biomolecules [13].
Copper was confirmed through studies to being able to induce DNA strands to break and cause bases to oxidise by oxygen free radicals and hydroxyl radical. Cupric and cuprous states of copper enhance DNA breakage though the genotoxic benzene metabolite (1,2,4-benzenetriol), more than iron [14]. In vitro studies, unlike in vivo studies, have shown that copper induces low-density lipoprotein (LDL) oxidation [13]. It was recently noted that the upper limit of copper “free” pools is much less than a single atom for each cell, signifying that substantial overloading for copper chelation in the cell [14].
6.9. Zinc
Table 16.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 17.
Zinc thresholds [58].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Workplace Air | OSHA | Legal limit over an 8-hour workday of zinc chloride fumes in the air | 1 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of zinc oxide and fumes dust | 5 mg/m3 | |
| NIOSH | Legal limit over a 10-hour workday of zinc chloride fumes in the air | 1 mg/m3 | |
| Drinking-Water | WHO | Drinking water threshold | No data |
6.9.1. Pharmacokinetic processes
Zinc is classified as an essential element where it is needed by over 300 enzymes. Some of the enzymes are:
-
•
alcohol dehydrogenase,
-
•
alkaline phosphatase,
-
•
Cu, Zn-superoxide dismutase
-
•
Carbonic anhydrase,
-
•
DNA polymerases
-
•
RNA transcriptase
-
•
Carboxypeptidase
Zinc function in these metalloenzymes is to participate in catalytic functions, regulatory functions, and maintenance of the stability of the structure. Zinc is implicated in DNA and RNA synthesis, together with cell proliferation [59].
Zinc is bound to the intestinal mucosa through CRIP, which is a diffusible intracellular zinc carrier, through a saturable process. Metallothionein influences the transport of zinc through the intestinal lumen where zinc absorption can be inhibited by competing with CRIP. CRIP is noted to have a limited binding capacity for the metal zinc and is saturated when high concentrations of zinc are present in the intestine. Zinc in the lumen can cause damage to the brush border membrane making absorption of the metal higher as it binds to non-specifically to cell protein and to other ligands. Intestinal uptake of zinc can be influenced by a number of factors such as methionine, cysteine, histidine, citrate, reduced glutathione, and prostaglandin E2. Other factors inhibit the absorption of zinc such as cadmium, calcium, copper and ferrous iron. Phytate and some dietary fibre components decrease zinc availability by complexing with the metal itself. Albumin is the primary carrier in the plasma for zinc. Other carriers are amino acids and α2-macroglobulin. After ingestions, zinc is concentrated mostly at the liver and then distributed throughout the whole body. Major storage sites for zinc are the liver, bone, pancreas, kidney and the muscles [58].
Zinc–Iron related transporter Proteins (ZIP) are transmembrane proteins that transport Zn2+ in eukaryotes. These transmembrane proteins are found in the plasma membrane and are noted to shape an eight transmembrane domain channel which is involved in the regulation of zinc concentrations in the cell. Zinc has been implicated in second messenger metabolism, extracellular signal recognition, dephosphorylation, protein phosphorylation, and in transcription factors activity. Zinc interacts with second messenger metabolism such as the interference of the metal with calcium regulation and with cyclic nucleotide metabolism. Zn2+ is influxed into the cell when the heart cells are electrically stimulated, through the voltage-dependent Ca2+ channels [14].
Interaction of zinc with cyclic nucleotide metabolism has also been seen. The metal was noted to mediate second messenger cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP), occurring by activities of cyclic nucleotide phosphodiesterase (PDE). Protein kinase C (PKC) is also regulated by the metal zinc, which causes translocation of the PKC to the cytoskeleton. High zinc concentrations outside the cell cause a stimulation of the activity of protein tyrosine phosphorylation and the mitogen-activated protein kinases (MAPK). Zinc is noted to stimulate mitogenic signalling since zinc and calcium have a synergistic effect on the MAPK, mitogenic signalling and DNA synthesis were detected in NH3T3 cells. The metal response element-binding transcription factor-1 (MTF-1) is a zinc-activated transcription factor. MTF-1 induces zinc transporter ZnT-1 and the metallothionein genes in reaction to cellular zinc [14].
Apart from zinc being involved in cell proliferation as a structural element of the protein, it also takes part in growth regulation. If zinc is deprived than cells die, where apoptosis in a variety of cell types will occur. If zinc is found in high concentrations extracellularly and exceeds the ability of zinc homeostasis, apoptosis occurs as enhanced intracellular zinc levels are triggered due to cytotoxicity. Necrosis of cells occurs at very high zinc concentrations [14].
Oxidative stress can be reduced due to the properties of zinc being and antioxidant. This may be due to the (i) oxidation being prevented by the protection of sulfhydryl group of enzymes and proteins; or (ii) the prevention of the formation of free radicals, which causes the reduction of the formation of the hydroxyl radical from the hydrogen peroxide molecule. Interaction of zinc with mechanisms of the immune system also occurs. Zinc and amyloid-β peptide (Aβ) uncontrolled accumulation have been proposed to lead to Aβ-mediated and zinc-induced oxidative stress together with cytotoxicity [14].
6.10. Molybdenum
Table 18.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 19.
Molybdenum thresholds [61].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | No data |
| NIOSH | Dangerous limits for insoluble molybdenum which causes death or delayed adverse effects | 5,000 mg/m3 | |
| NISOH | Dangerous limits for soluble molybdenum which causes death or delayed adverse effects | 1,000 mg/m3 | |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of soluble molybdenum dust in the air | 5 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of insoluble molybdenum dust in the air | 15 mg/m3 | |
| NIOSH | Legal limit over a 10-hour workday of molybdenum dust in the air | No data | |
| Drinking-Water | WHO | Drinking water threshold | No data |
| EPA | Drinking water threshold | 0.08 mg/L |
6.10.1. Pharmacokinetic processes
Studies in rats have shown that molybdate and sulphate show mutual competitive inhibition in the intestine for absorptive transport, which may be the Na+/SO42- symporter, known as NaS1 or SLC13A1. Since NaS1 is found in the kidneys instead of the intestine in humans, a different absorption transport occurs. Molybdenum transporters ate expressed in bacteria and eukaryotes membrane. MoT2 is a molybdenum transporter which is also found in humans. Molybdate is transported into cells by MoT2 in eukaryotes, for the incorporation of the element into molybdopterin-cofactor (Moco) which is the biologically prosthetic active group found in the molybdenum-dependent enzymes. The transport if inhibited by sulphate, therefore, showing that there is a common carrier for both the molybdate and the sulphate. Molybdate is taken up by red blood cells which involve the involvement of the anion exchanger Cl−/HCO3-. Molybdate is inserted into the Moco through a displacement of copper from the binding site of molybdate. This displacement can provide a mechanism for the interaction of Cu–Mo in the regulation of the Moco synthesis together with the copper-induced deficiency in the molybdenum-dependent enzymes. Excretion mechanism of molybdenum has not been characterised [61].
6.10.2. Toxicology processes
Molybdenum toxicity mechanisms have not been established properly. Data of molybdenum toxicity between animals and humans is limited. Rabbits were seen to be more sensitive than rats, though studies cannot be compared since there are differences in copper content together with other dietary constituents. Since there is no data to the contrary, molybdenum toxicity is assumed to be similar in all species, excluding ruminants [61].
Mode of action though may involve an alteration of copper utilization, though other mechanisms are probably involved such as direct molybdenum alterations. In copper-deficient diets, molybdenum induced copper level alterations in the liver, kidneys, and plasms. Reversal of the adverse effects occurred when high copper doses were administered. High doses of molybdenum caused anaemia and decrease in body weight in animal studies which symptoms are similar to copper-deficient animals. High copper doses reversed the effect. In rats with a regular diet and exposed to high amounts of molybdenum showed high copper plasma levels which are “tightly bound form” as a majority, and are not associated with ceruloplasmin since no increase in this copper-carrying protein is seen. Copper concentrations were seen to increase in the liver and kidney on high molybdenum exposure [61].
Ruminants tend to be more susceptible to molybdenum toxicity, and the metal is seen to react with sulphate, which is generated in the rumen, which forms thiomolybdates. Copper then can bind to the thiomolybdates formed, which impairs copper absorption. Functional copper deficiency can be seen in cupric thiomolybdates which are unavailable to ceruloplasmin [61].
Sodium molybdate exposed to monogastric animals has shown that when sulphate is administered, molybdenum toxicity is decreased. However, when rats were fed molybdate and sulphide in their diets, an increase in the molybdenum and copper plasma level and molybdenum levels in the liver was noted, while a decrease in the ceruloplasmin activity was noted. A change in the fraction of copper in the plasma was associated with ceruloplasmin and albumin, which were seen in rats administered with tetrathiomolybdates but not in rats administered with molybdates without the sulphide. Tetrathiomolybdate was seen to form a tripartite complex in the gastrointestinal tract, with copper and protein, which can stop the absorption of copper [61].
6.11. Arsenic
Table 20.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 21.
Arsenic thresholds [62].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | 1.5 × 10−3 μg/m3 |
| WHO | Air Quality Guidelines for estimation of cancer risk for a lifetime exposure | 1 μg/m3 | |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of airborne arsenic in places that use inorganic arsenic | 10 μg/m3 |
| Drinking-Water | WHO | Drinking water threshold | 0.01 mg/L |
| EPA | Drinking water threshold | 0.05 mg/L |
6.11.1. Pharmacokinetic processes
Absorption of the metal arsenic is subjected to its chemical form, wherein humans, arsenic is orally absorbed by more than 75% in the forms As3+, As5+, methylarsinic acid (MMA) and dimethylarsinic acid (DMA). Arsenic can be orally taken, inhaled and dermally absorbed, though, dermal absorption is much less than the inhalation and oral route. Humans and mice have been noted to absorb arsenic through passive diffusion. However, it has been seen to be absorbed in rats through a process by the saturable carrier-mediated cellular transport mechanism. Arsenic in soil has a lower bioavailability through the oral route than the arsenic sodium salts. Water-insoluble compounds of arsenic are formed in the soil which are absorbed poorly, such as sulphides. Distribution of arsenic, together with its metabolites occurs to all the bodies’ organs, and no preferential distribution, has been observed in both human tissue during autopsies or animal species. Inorganic arsenic methylation occurs mostly at the liver, thought the “first pass” which is possible after absorption from the gastrointestinal tract has not been investigated. The metal and its metabolites are excreted mostly through the urine and are also excreted through faeces and minimally through the nails and hair [62].
The main metabolic pathway is the methylation of inorganic arsenic. Metabolite proportion found in the urine is approximately consistent no matter the scenario of exposure in humans. Duration of exposure to arsenic and the dose of the metal are the main factors in arsenic toxicity. Doses of 2 mg/kg of arsenic and higher have caused fatalities in humans. Doses taken orally of 0.001 mg/kg of arsenic taken over long term daily has been seen to cause skin diseases and cancer of the skin, bladder, liver and kidney. Lung cancer is seen when arsenic is inhaled at low doses of 0.05–0.07 mg/m3. Arsenic compounds were seen to induce metallothionein in mice studies. Arsenic compounds differ in their potency to induce the metallothionein protein; As3+ > As5+ > MMA > DMA. Unlike cadmium, arsenic binds to the metallothionein protein with a lesser affinity, and thus only a small percentage of the metal is bound. Due to the reduction of affinity, it is thought that arsenic toxicity is decreased because of the metallothionein, which acts as an antioxidant protecting against oxidative arsenic injury [62].
6.11.2. Toxicology processes
Free radicals are generated by arsenic, though the generation mechanism for these reactive species is still not clear. The free radicals generated include the superoxide O2-•, nitric oxide NO•, singlet oxygen 1O2, hydrogen peroxide H2O2, peroxyl radical ROO•, dimethylarsinic radical (CH3)2As•, and dimethylarsinic peroxyl radical (CH3)2AsOO• [13].
Toxicity and carcinogenicity caused by arsenic have been noted to be associated with metabolic processes. Pentavalent arsenic (As5+) in the blood is reduced or partially reduced, to trivalent arsenic (As3+) when absorbed. As3+ is distributed to tissues and is absorbed mostly by the hepatocytes. As3+, as arsenite, is absorbed more than As5+, as arsenate, by the cells and is considered more toxic than arsenate. Since arsenite is more toxic than arsenate, the reduction step can be observed to be a bio-activation instead of a detoxification reaction. Glutathione is noted to participate in the reduction process before methylation. MMA and DMA formed through the methylation of arsenic are less toxic compared to the inorganic arsenic, and both methylated arsenic compounds are excreted through urine. Methylation process occurs through the reduction of As5+ and a methyl group addition from adenosylmethionine. The methylation reaction produces intermediates (MMA3+ and DMA3+) which some of them have been seen to be more toxic than the inorganic arsenic. MMA3+ has shown that it is toxic to hepatocytes, bronchial epithelial cells, and epidermal keratinocytes in humans, more than both arsenate and arsenite. Both intermediates have proved to be genotoxic and damage the DNA. Arsenate has proved to:
-
•
during in vitro studies, phosphate is replaced in glucose-6-phosphate and in 6-phosphogluconate;
-
•
substitute phosphate in the anion exchange transport system together with the sodium pump found in the human red blood cells;
-
•
replace the phosphate in the enzymatic reaction which diminishes the ATP formation in vitro;
-
•
diminish the ATP found in some cellular systems, though not in the human erythrocytes.
It has become more evident that the main source of carcinogenicity and toxicity of arsenic is due to the reduction to arsenite. Arsenite was noted to react with thiol-containing molecules in vitro, such as cysteine and glutathione. MMA3+ are potent inhibitors of glutathione and thioredoxin reductases. This thiol binding with arsenite and the methylated intermediates could lead to the inhibition of crucial biochemical reactions, modification of cellular redox status, and this may lead to cytotoxicity. In vitro studies have shown the methylated intermediates to bind to proteins [62].
Pyruvate dehydrogenase (PDH) is a complex which oxidizes pyruvate into acetyl-CoA. This oxidation reaction is inhibited by arsenite. Acetyl-CoA is a precursor to intermediates in the citric acid cycle which is vital to reduce counterparts to the electron transport system in the production of ATP. In vitro studies of arsenite in rats, this has shown the depletion of carbohydrates due to this inhibition. Arsenic was seen to induce alterations in the metabolism of nitric oxide and endothelial function in people who ingest high doses of arsenic in drinking water, where concentrations in serum and urine had a decreased amount of nitric oxide metabolites, which was reversed when drinking water had lower amounts of arsenic present [62].
Recent studies in animal models showed carcinogenic activity to be noted in the skin, liver, urinary bladder, and lung, which is similar to arsenic-induced cancer seen in humans. Thus common mechanisms of action might be involved [62]. Cancer through arsenic poisoning is also seen in the prostate and Kupffer cells. Carcinogenic mechanisms caused by arsenic include epigenetic alterations, generation of ROS and damage to the maintenance system of DNA [13].
Epigenetic changes include alterations to histones, micro RNA (miRNA) and DNA methylations. These alterations have shown that they can cause malignant growth. P53 expression has been seen to be altered in, in vitro studies, which have been seen to cause a decreased expression in the protein p21. Genotoxicity in leukocytes has been noted to be caused in humans and mice by arsenic compounds. DNA repair processes were seen to be inhibited by methylated arsenic, together with a generation of ROS in the spleen and liver as metabolic products. Carcinogenesis risk is increased due to arsenic binding to DNA-binding proteins together with the disruption of the DNA repair processes [13].
6.12. Silver
Table 22.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 23.
Silver thresholds [63].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Workplace Air | OSHA | Legal limit over an 8-hour workday of silver in the air | 0.01 mg/m3 |
| NIOSH | Legal limit over a 10-hour workday of silver in the air | 0.01 mg/m3 | |
| ACGIH | Legal limit over an 8-hour workday of silver metal in the air | 0.1 mg/m3 | |
| ACGIH | Legal limit over an 8-hour workday of soluble silver compounds in workplace | 0.01 mg/m3 | |
| Drinking-Water | EPA | Drinking water threshold | 0.05 mg/L |
6.12.1. Pharmacokinetic processes
Absorption of silver through ingestion is established according to the ionization of the silver source, and the availability of the silver ion, which can interact with the protein receptors located on the cell membranes. Silver ion is highly reactive and not taken up passively. It also has a high binding capacity to sulphydryl, hydroxyl, carboxyl, and protein ligands found on the mucosal surfaces and in the cell debris. Absorption of silver ions is reduced since it binds readily and precipitates with organic food constituents, together with inorganic cations, including phosphate and chloride. Inhalation of silver has caused argyria and argyrosis tough the uptake if the metal through the respiratory membranes is still unclear. Dermal absorption is considered low due to the skin barrier [64].
Ag+ binds to albumins found in the serum and to macroglobulins where they are metabolized and distributed to the soft tissues and the bone. When silver is used in wounds, it tends to induce and bind to the cysteine-rich proteins found in the wound margin, such as metallothionein I and II. These metallothioneins serve as cytoprotectants where they bind the toxic metal ion and thus reduce the risk of cytoplasmic damage. Most silver is excreted through the biliary route, followed by the urinary route [64].
6.12.2. Toxicology processes
Silver toxicity manifests itself when the kidney and the liver cannot eliminate the silver anymore as they exceed the elimination capacity. Argyria is seen by silver toxicity which is characterized by the accumulation of silver sulphide and silver selenide inert precipitates in the connective tissue around the glands and vascular tissue found in the papillary layer of the dermis, though not the epidermis. Argyria is not associated with cellular damage and is not life-threatening [64].
Argyrosis is also not associated with cellular damage. It is a silvery pigmentation of the conjunctiva and cornea after exposure to silver and its salts. Silver particles present in the deep corneal stroma were bound to lysosomes in the connective tissue, or were free in the intercellular spaces and linked with tissue debris [64].
Silver was noted not to be absorbed by the brain or by the central nervous system or the peripheral nervous system. It was noted not to pass the blood-brain barrier or the blood-CSF barrier [64].
Nanoparticles can lead to the inhibition of the proliferation of cells as they can act together with membrane proteins, causing the activation of signalling pathways. Diffusion and endocytosis are other methods where nanosilver particles enter the cell, causing mitochondria dysfunction, generation of ROS which leads to the damage of proteins and nucleic acids found in the cell, and cell proliferation inhibition. If ROS generation exceeds the aptitude of the cellular antioxidant defence system, oxidative stress then occurs. Oxidative damage has been noted to be caused by the reduction of glutathione and the depletion of protein-bound sulphydryl groups, together with the alterations in a variety of the antioxidant enzyme activity which have been suggestive of lipid peroxidation. Silver has a strong affinity for the element sulphur. Interaction of ionic silver and nanosilver particles with molecules containing sulphur has shown toxicity mechanisms in this interaction. Since mitochondria are affected by silver, and the mitochondrial respiratory chain is disrupted, ROS production is increased, and this interrupts the synthesis of ATP, thus leading to DNA damage. In vitro studies on mammalian cells have shown nanosilver particles to interact with the cell membrane, which may cause an alteration of the permeability of the membrane. The cell membrane is disrupted, and oxidative damage occurs, causing apoptosis. Fibroblast cell membrane damage by the nanosilver particles allows an influx of calcium and indices the calcium overload in the cell, thus causing ROS overproduction and potential variation in the mitochondrial membrane. Some studies have shown that nanosilver has caused lysis of the cell membrane in RT-W1 and red blood cells [65].
6.13. Cadmium
Table 24.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 25.
Cadmium thresholds [66].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | 5 ng/m3 |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of cadmium in air | 5 μg/m3 |
| Drinking-Water | EPA | Drinking water at this concentration for 10 days will not cause adverse effects in youths | 0.04 mg/L |
| EPA | Drinking water at this concentration for a lifetime exposure does not cause adverse effects | 0.005 mg/L | |
| WHO | Drinking water Quality Guidelines | 0.003 mg/L | |
| Bottled Water | FDA | Bottled water threshold | 0.005 mg/L |
6.13.1. Pharmacokinetic processes
Cadmium is absorbed mostly through the inhalation route. There is evidence that intestinal absorption mechanism takes place due to transporter proteins, where a study has shown that the divalent metal transporter I am one of the proteins that take place in cadmium absorption through the gastrointestinal tract. Cadmium absorbed by the lungs depends on the particle size. Large particles tend to be deposited in the nasopharyngeal together with the tracheobronchial airways by impaction, and are mostly removed by the mucociliary processes. Smaller particles though reach the alveoli and are absorbed according to the solubility. The most soluble cadmium salts are chloride, acetate, nitrate and sulphate, while the insoluble salts are the sulphides. Since the lungs are rich in carbon dioxide, a variety of cadmium sulphides can react with the carbon dioxide in the lungs [66].
The liver and the kidneys are the organs with the highest concentration of cadmium present, after absorption. The metal has a high affinity for metallothionein and sulphydryl groups of albumin. Absorption of cadmium by gastrointestinal tract cause the excretion of the metal through the faecal route. Cadmium absorbed through the lungs is excreted in equal amounts by faecal excretion and urinary excretion [66].
6.13.2. Toxicology processes
Cadmium has been associated with the promotion of apoptosis, oxidative stress, methylation of DNA, and DNA damage [13]. Primary targets that cadmium is toxic to are the kidneys, lungs and bone [66]. The metal is also known as a potent carcinogenic which affects the kidney, lung, pancreas, and prostate [14].
Cadmium induces the immediate-early response genes (IEGs), where they influence target gene expression, including genes controlling cell growth and cell division. The IEGs are also involved in cell proliferation and cell differentiation. IEGs overexpression induced by cadmium comprises of mitogenic growth signals which stimulate cell proliferation which can lead to carcinogenesis. Cadmium-induced carcinogenicity is also caused by the induction of the expression of a variety of stress response genes. The response genes can be genes that encode MT synthesis and heat shock proteins (HSPs), genes involved in GSH synthesis and other genes, and genes involved in the response of oxidative stress. Cadmium carcinogenicity has been seen when MT synthesis is not expressed. A variety of transcription factors, and genes regulating translation, are also affected by cadmium [14].
Studies have shown that cadmium causes apoptosis in several organs. Apoptosis induction in testes showed an inverse correlation with p53, thereby showing that cadmium induces tumorigenesis. Studies have shown that 60 % of the cells commit apoptosis, though the remained of the dells were viable and showed to increase the production of MT [14].
Toxicity caused by cadmium is primarily thought to be the deactivation of a vital DNA repair activity. Cadmium was noted to inactivate the mismatch repair (MMR) system when the MMR is disrupted through the mutations in the proteins MutS homolog (MSH), which leads to the considerable increase in the genome instability leading to the increase in the incidence of a variety of human cancer [14].
Cadmium does not generate free radicals itself though it generates free radicals indirectly. These involve the superoxide radical, nitric oxide, ad hydroxyl radical. Studies have shown that the non-radical hydrogen peroxide is also generated, which can also be a source of radicals through Fenton reactions [14].
Cadmium chronic exposure through the oral or inhalation route produces kidney changes such as damage to the proximal tubule cell, proteinuria, aminoaciduria, glycosuria, polyuria, enzymuria, and a decrease in absorption of phosphate, as seen in laboratory animals. Proximal tubule degeneration and atrophy, including interstitial fibrosis in worst-case scenarios, result in clinical symptoms. The lipid composition was shown to be disturbed by cadmium, while lipid peroxidation is enhanced. Antioxidant enzymes, especially the superoxide dismutase and the glutathione peroxidase, are depleted which produces cardiotoxic effects. Metabolism of zinc, copper, iron and selenium are altered by cadmium. These alterations in metabolisms are said to initiate the cadmium-induced toxicity in testes where cadmium interferes with the zinc-protein complexes controlling the DNA transcription and thus leading to apoptosis [66].
Glomerular membrane polyanions were noted to deplete with cadmium, resulting in an increase in the excretion of proteins with a high molecular weight. Tubular damage has also been reported when the charge of the glomerular membrane polyanionic in interfered with by cadmium. Hepatic glycogen stores were reported to be reduced by cadmium which leads to an increase in the levels of glucose levels in the blood. Cadmium toxicity is also seen to cause intralobular fibrosis, focal mononuclear infiltrates, cirrhosis, and an explosion of the smooth endoplasmic reticulum [66].
Decreased bone mineral density is seen due to cadmium toxicity, which increases bone fractures. Osteoblastic activity is seen to be inhibited by the metal, which then results in bone organic matrix synthesis to decrease together with a decrease in mineralization. Osteoblastic activity reduction may cause an effect on the osteoclastic activity leading to an increase in bone resorption [66].
Renal damage caused by cadmium interferes with the 25-hydroxyl-vitamin D hydroxylation process to form 1,25-dihydroxy-vitamin D. When 1,25-dihydroxy-vitamin D is reduced in serum level, apart from the diminished kidney resorptive function, a deficiency in calcium and phosphate results. Deficiency of these elements causes a release in the parathyroid hormone, which enhances bone resorption [66].
6.14. Selenium
Table 26.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 27.
Selenium thresholds [69].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Intake | WHO | Adult daily intake | 0.9 μg/kg body weight |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of selenium in air | 0.2 mg/m3 |
| Drinking-Water | EPA | Drinking-water threshold | 0.05 mg/L |
| WHO | Drinking-water Quality Guidelines | 0.01 mg/L | |
| Bottled Water | FDA | Bottled-water threshold | 0.05 mg/L |
6.14.1. Pharmacokinetic processes
Oral absorption and inhalation of selenium are extensive, though absorption rate depends on the form of the metal. The oral bioavailability of selenium is usually independent of the levels of exposure, though it may increase in selenium-deficient individuals. Absorption through the intestine of selenite and selenomethionine is mainly unchanged. Selenite and selenocysteine are metabolized on absorption. Once selenium is absorbed through the intestinal tract and carried to all the tissues. Once in the blood, selenium becomes protein-bound rapidly. Selenite has been noted in vitro studies to accumulate in the erythrocytes through an active transport mechanism. Selenium is found to be higher in humans in the liver and the kidney. Selenomethionine in humans is not synthesized, though it can be combined into proteins instead of methionine. Through this combination to the protein, selenomethionine is kept within the body for a more extended period when compared to the inorganic forms. Selenate and selenocysteine are not combined with plasma protein, and thus this suggests that these forms of selenium are metabolized through particular selenium metabolic processes [69].
Selenium together with GSH system is vital in the body's antioxidant defence. GSH is implicated in the:
-
•
Direct interception of pro-oxidants
-
•
Other antioxidants being reduced from the oxidized form
-
•
Ancillary functions which can intervene in the defence against anti-oxidants.
GSH redox reactions need the catalysts glutathione disulphide (GSSG) and glutathione peroxidase (GPX). Thioether formation needs the enzymes GSH transferases. Selenoamino acids mediate antioxidant protection, either as selenomethionine or as selenocysteine. GPX-like activity is observed by selenomethionine, and selenocysteine residues are found at the GOX active site. Different types of hydroperoxides are reduced by GPX, where GSH acts as the reducing substrate. The GPX antioxidant action towards hydroperoxides involves an enzymatic catalysis reaction cycle which involves three steps comprising of the enzyme-bound selenocysteine found as the selenol. Steps include the organic hydroperoxide reaction, which yields selenic acid and corresponding alcohol, and the successive reduction by GSH, which leads to selenol and GSSG. Selenium is excreted by the body mainly in the urine and faeces, but also by sweat and expired air. Selenium-deficient people have reduced elimination. Detoxification for selenium occurs through methylation, exhalation of dimethyl selenide, while the significant selenium urinary metabolite is the trimethylselenonium ion. Selenium hepatic toxicity seen in mice studies have been caused by the decrease of liver selenium methylation, and thus resulting in the accumulation of a surplus of selenides [69].
6.14.2. Toxicology processes
Out of all the selenium in the body, the selenocysteine-containing proteins are responsible for the selenium antioxidant properties. GPX, iodothyronine 5′-deiodinases, and thioredoxin reductase are the major selenoproteins, which increase and decrease when the metal is depleted or repleted. Selenium can be combined with methionine as selenomethionine. Water-soluble forms of selenium are more absorbed and are usually more toxic. Selenium is noted to have interactions with nutrients such as vitamin E, a variety of xenobiotics and other toxic metals including cadmium and mercury. When selenium concentration is high, it easily substitutes instead of sulphur in biomolecules and many biochemical reactions, especially when sulphur is low. Selenium toxicity may be contributed to the inactivation of sulphydryl enzymes which are necessary for cellular respiration for oxidative reactions through the effects on mitochondrial and the microsomal electron transport. Hepatic haem metabolism is related to lipid peroxidation or GPX, where selenium plays a role in this. Chronic selenium overexposure may cause hair, skin and nail damage where the mechanism for these is unclear. However, this could be due to high selenium levels in these tissues as a result of the selenium being substituted instead of sulphur in specific amino acids. These also include the disulphide bridges which provide the tertiary structure of the proteins and the function to the proteins. Nail and hair are routes for excess selenium excretion. Oxidative stress is noted to be the critical biochemical lesion for the intoxication of selenium. Reactions between the inorganic selenium forms and the tissue thiols through redox catalysis, result in the formation of the superoxide anion reactive oxygen species. Inhibitory effects are seen on thiol proteins due to selenium through modification of:
-
•
Selenotrisulphide and selenylsulphide bond formation;
-
•
Disulphide bond catalysis, where there is no combination of selenium in the protein;
-
•
Diselenide formation
Proteins containing regulatory cysteine can develop selenium adducts which cause the inactivation of crucial thiol groups resulting in toxicity. Selenium can take part in the redox-regulating GPX activities together with phospholipid hydroperoxides and inflammatory superoxides. Inflammatory hydroperoxides do not occur when endothelial cells have a selenoprotein P-supported plasma GPX which is bound to it. GPX reacts with phospholipid hydroperoxides produces metabolites which can suppress cytokine or activates the growth factor triggered gene. Selenium is modulated by the GPX activity and can inhibit the activation of NF-кB, which is implicated in the regulation of numerous cellular gene expression, especially those intricate in the inflammatory, immune and stress responses. Tumour necrosis factor induces apoptosis but might be inhibited by the overexpression of the cytosolic GPX or by the phospholipid hydroperoxides GPX since the apoptotic signalling cascade can be stimulated by the hydroperoxides. Selenol is shown to generate cellular apoptosis while selenium-methylselenocysteine induces cell death in cancer cells by caspases activation [69].
Selenium has a protective role by inhibiting the carcinogen-induced covalent DNA adduct formation, an impedance of oxidative damage to lipids, proteins and DNA, and alterations of cellular and molecular incidents which are critical in the inhibition of cell growth and the multi-step process of carcinogenesis. Selenium deficiency can affect the intracellular redox function, thus free radicals increases in animals’ deficient of selenium [69].
Free radicals, epoxides and aldehydes are formed in membranes due to polyunsaturated fatty acid peroxidative degradation, causing cytotoxic, genotoxic and hepatotoxic effects. Aldehyde and epoxide haemoglobin adduct levels in animals deficient efficient in selenium were enhanced through the loss of the activity of the selenium-dependent GPX. Selenium can protect against loss during early pregnancy due to the reduced antioxidant protection of the biological membranes and DNA by GPX low concentrations. The loss of activity of the membrane phospholipid hydroperoxides GPX is noted to occur in animals which are selenium deficient and causes degenerative diseases like skeletal in addition to cardiac myopathies. Thyroid hormone function is another factor affected by selenium, by the deiodinase enzymes. The metal is an essential component in iodothyronine 5′-deiodinases where the prohormone thyroxine (T4) is converted to triiodothyronine (T3) which is the active form. GPX is responsible for the protection against oxidative damage in the thyroid cells, where GPX is implicated in the detoxification of hydrogen peroxide produced in the conversion of T4 to T3 in the thyroid [69].
6.15. Mercury
Table 28.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 29.
Mercury thresholds [70].
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Intake | WHO | Permissible weekly intake | 5 μg/kg total 3.3 μg/kg methylmercury |
| Workplace Air | OSHA | Legal limit over an 8-hour workday of organic mercury in air | 0.1 mg/m3 |
| OSHA | Legal limit over an 8-hour workday of metallic mercury vapour in the air | 0.05 mg/m3 | |
| NIOSH | Legal limit over a 10-hour workday of metallic mercury vapour in the air | 0.05 mg/m3 | |
| Drinking-Water | EPA | Drinking water inorganic mercury threshold | 0.002 mg/L |
| WHO | Drinking water Quality Guidelines for health-related organics regarding all mercury forms | 0.001 mg/L | |
| Seafood | FDA | Max legal limit of methylmercury | 1 ppm |
6.15.1. Pharmacokinetic processes
Metallic mercury is absorbed mostly through inhalation, followed by ingestion. Mercury has a great affinity for the sulphydryl group, which bonds to sulphur-containing amino acids present in the body. Metallic mercury passes effortlessly through the blood-brain barrier and placenta by adhering to the red blood cells. Some mercury is taken up by the central nervous system though most of it is oxidized rapidly to mercuric mercury. Metallic mercury is deposited to many tissues in the body including the thyroid, myocardium, breast, muscles, liver, adrenals, kidneys, skin, pancreas, sweat glands, salivary glands, lungs, enterocytes, prostate, testes, and breast milk. The metal also binds with a high affinity onto T cell surfaces and sulphydryl groups which influence the T cell function. Metallic mercury is excreted mostly as mercuric mercury [72].
Calomel (Hg22+) is poorly soluble in water and is poorly absorbed through the intestine, although some amount is oxidized to a more absorbable form. Mercurous mercury is an intermediate form between metallic and mercuric mercury. It does not last long in the body, though some are absorbed as pink disease and acrodynia are associated with calomel [72].
Mercuric mercury (Hg2+) is slightly absorbed by the intestine, though due to its corrosive effect on the gastrointestinal tract, absorbency will increase. Mercuric mercury also bonds to the sulphydryl groups on metallothionein, erythrocytes, or glutathione, or it is suspended in plasma. Mercuric mercury is noted to accumulate in the placenta, amniotic fluid and foetal tissues. However, it does not pass through the blood-brain barrier proficiently, though accumulates in the brain through amino acid transporters such as cysteine. Most of the mercuric mercury is located in the convoluted renal tubule where it is bonded to metallothionein. It is also deposited in the periportal of the liver, and at a lesser degree in the choroidal plexus, epithelial tissues and testes. Excretion of mercuric mercury is mostly done through the urine and faeces. Some of the metal is excreted through sweat, tears, saliva and breast milk [72].
Organic mercury, especially methylmercury, is absorbed with efficiency through inhalation and is also absorbed through ingestion and the skin. Once in the bloodstream adherent of methylmercury to the sulphydryl group occurs, especially to those in cysteine. Distribution occurs through transporters especially, the cysteine transporter where it will possibly adhere to the sulphydryl group on the cysteine. Methylmercury concentrates in the brain, kidneys, liver, foetus, placenta and the foetal brain, bone marrow and peripheral nerves. The methylmercury then undergoes slow demethylation into inorganic mercury. Methylmercury is mostly excreted through the faecal route, followed by breast milk [72].
6.15.2. Toxicology processes
Induction of oxidative stress is usually seen when both forms of mercury are exposed tin animals, as in vivo and in vitro studies have shown. Mercuric ions show a high affinity to the binding sites of thiol, which shows that intracellular thiols are depleted, especially glutathione, causing and indirect or direct cause or predisposition for the proximal tubular cells to have oxidative stress [14]. Intracellular thiol is altered where mercury can promote oxidative stress, mitochondrial dysfunction, lipid peroxidation, and haem metabolism alterations. Hg2+ causes depolarization of the inner membrane of the mitochondria, which causes an increase in H2O2 formation [14,70].
Hg2+ is seen to deplete mitochondrial GSH at low concentration, and in diminished respiratory chain electron transport, it enhances the hydrogen peroxide formation in kidney mitochondria. The increased formation of H2O2 can lead to oxidative tissue damage, which can be seen in mercury-induced nephrotoxicities, such as lipid peroxidation [14]. Alterations caused by mercury in the mitochondrial calcium homeostasis in kidney cells may aggravate Hg2+ oxidative stress. A variety of biochemical changes may occur due to the oxidative damage caused in the kidneys. The changes may include porphyrinuria [70].
Lipid peroxidation and enhanced free radical formation produce epithelial cell damage in the kidneys. Reduction of cellular defence mechanisms is observed against oxidative damage when treated with mercury [70]. Mercury is seen to enhance lipid peroxidation, especially in the kidneys, epididymis, and the testes [14]. Methylmercury chloride targets mitochondria as one of the earliest targets where neurotoxicity occurs. Excess of reactive oxygen species occurs probably at the mitochondrial electron transport chain, which is formed in the brain, causing induction of oxidative stress due to the methylmercury chloride poisoning [14].
Zinc-deficient animals were observed to suffer more from mercy chloride toxicity, showing the benefits of zinc in animals. 2,3-dimercapto-1-propanesulphonate (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) were observed to be effective in reducing mercury concentrations in the kidney. Studies showed that animals who were given mercuric chloride together with vitamin E showed to have lower mercury concentrations in the epididymis and the testes. Thus vitamin E has been seen to provide a protective role in the male reproductive toxicity caused by mercury. Reduction in superoxide dismutase, glutathione peroxidase, catalase and glutathione disulphide reductase, were observed during in vitro studies on rats administered with mercuric chloride in the renal cortex [14].
High levels of inorganic mercury were seen to cause non-lethal effects in kidney cells which are associated with the redistribution of intracellular calcium stores. Cytotoxicity is caused by the escalation of intracellular calcium, which depends on the extracellular calcium presence. Calcium in the cytosol increased primarily from the non-mitochondrial intracellular stores resulting from the endoplasmic reticulum. An intracellular decrease can be seen due to buffering processes. Buffering processes may include an uptake through the microsomal Ca2+, mitochondrial uniporter, or the Mg2+-ATPase [14]. Methylmercury increases free calcium levels in the cytosol due to an influx of calcium from outside the cell, and mobilization of calcium from intracellular stores such as the endoplasmic reticulum. Mercuric chloride increases free calcium levels in the cytosol through an influx of Ca2+ from outside the cell. Inhibition of the production of NO by methylmercury is mediated partly by Ca2+-stimulated by the pathway adenylate cyclase-cAMP-protein kinase A [14].
Taking in mercury through diet has been seen to cause an excess risk of heart complications such as myocardial infarction, and death caused by coronary heart disease or cardiovascular disease. This increase in risk is probably due to the lipid peroxidation caused by the mercury. Although fish is a good source for fish-oil fatty acids to prevent cardiovascular diseases, the content of mercury will diminish the protective effect [14].
6.16. Lead
Table 30.
| Properties |
|
| Uses |
|
| Effects on humans |
|
| Food source |
|
Table 31.
| Compartment | Agency | Description | Limits |
|---|---|---|---|
| Air | WHO | Air Quality Guidelines | No Data |
| Workplace Air | OSHA | Legal limit over an 8-hour workday in general industry for elemental, organic and inorganic lead | 0.5 mg/m3 |
| OSHA | Legal limit over an 8-hour workday in general industry for tetraethyl lead and tetramethyl lead | 0.075 mg/m3 | |
| OSHA | Legal limit over an 8-hour workday in shipyards and construction sites for elemental, organic and inorganic lead | 50 μg/m3 | |
| OSHA | Legal limit over an 8-hour workday in shipyards and construction sites for tetraethyl lead | 0.1 mg/m3 | |
| OSHA | Legal limit over an 8-hour workday in shipyards and construction sites for tetramethyl lead | 0.15 mg/m3 | |
| OSHA | Action level over an 8-hour workday for industry and construction for elemental, organic and inorganic lead | 30 μg/m3 | |
| OSHA | Medical removal protection for general industry - Temporary removal blood lead level | ≥60 μg/m3 | |
| OSHA | Medical removal protection for general industry - Return to work blood lead level | ≤40 μg/m3 | |
| OSHA | Medical removal protection for construction and shipyards - Temporary removal blood lead level | ≥50 μg/m3 | |
| OSHA | Medical removal protection for construction and shipyards - Return to work blood lead level | ≤40 μg/m3 | |
| NIOSH | Recommended exposure limit of lead and its compounds | 0.05 mg/m3 | |
| NIOSH | Recommended exposure limit of tetraethyl lead | 0.075 mg/m3 | |
| NIOSH | Immediately dangerous to life or health concentration of lead and its compounds | 100 mg/m3 | |
| NIOSH | Immediately dangerous to life or health concentration of tetraethyl lead | 40 mg/m3 | |
| Drinking-Water | EPA | Drinking water action level | 0.015 mg/L |
| WHO | Drinking water Quality Guidelines | 0.01 mg/L | |
| Bottled Water | FDA | Legal limit | No data |
6.16.1. Pharmacokinetic processes
Small particles of inorganic lead can be absorbed through the respiratory tract, while larger particles are removed by the mucociliary cells and transported to the oropharynx and then swallowed. Lead taken up by the gastrointestinal tract depends on several factors such as age, nutrition, and diet of the person, including the physiological characteristics of the metal, in the medium ingested. Absorption through the gastrointestinal tract is mostly done in the duodenum through saturable mechanisms. The dermal route is not that efficient when it comes to the absorption of inorganic lead, though absorption increases for hand to mouth behaviour [75].
Lead is distributed throughout the body and is route independent. It is distributed mostly to the bones. Pregnancy, menopause, lactation, and osteoporosis are conditions which can increase bone resorption, thus also increasing the lead in the blood. Lead can be found primarily in the red blood cells. Transferring of lead from mother to foetus and through breastfeeding occurs [75].
Metabolism of the inorganic metal forms complexes with many proteins ligands such as albumen and non-protein ligands such as sulphydryls. ALAD is the principal intracellular ligand present in red blood cells. The metal also complexes with proteins in the cytosol and cell nucleus. When it comes to organic lead, alkyl lead compounds are noted to be actively metabolized through an oxidative dealkylation in the liver, and catalyzed by the cytochrome P-450. People exposed to tetraethyl lead showed that it was excreted through the urinary route, as ethyl lead, diethyl lead, and inorganic lead. Teraalkyl lead exposure in workers showed trialkyl lead metabolites to be present in the kidney, brain and liver. The trialkyl metabolites were noted to be also detected in non-workers [75].
The urine and the faecal route are the primary routes of excretion of inorganic lead. Other routes may include sweat, hair, nails, saliva, seminal fluid and breast milk [75].
6.16.2. Toxicology processes
Chronic exposure to lead contributes to increased blood pressure. Critical hormonal and neuronal systems are affected by lead, which plays a role in the regulation of heart rate, peripheral vascular resistance, and cardiac output. Hypertension caused by lead in rats also accompanies depleted nitric oxide, which is vital for the regulation of blood pressure through the peripheral and central mechanisms. NO is derived from oxidative stress and is associated with the increased activity of ROS and reactivity with NO. Disruption of the NO vasodilatory actions is also caused by lead through the alteration f the cell-signalling mechanisms present in the endothelial cells. Downregulation is caused by lead exposure, of the soluble guanylate cyclase expression. This enzyme produces cyclic GMP, which facilitates vasodilation caused by NO. Hypertension caused by lead is associated with adrenergic system abnormalities, which includes increased activity in the central sympathetic nervous system, decreased density of the vascular β-adrenergic receptors, and elevated plasma norepinephrine. The renin-angiotensin-aldosterone system is activated directly or indirectly, through the sympathetic nervous system stimulation. Plasma renin activity, plasma aldosterone concentrations, and plasma angiotensin-converting-enzyme (ACE) become elevated on chronic exposure to lead. Hypertension is also linked to alterations in kallikrein-kinin system regulation, together with the production of the vasodilator hormones. Vascular smooth muscles experience constrictive effects due to lead, which are caused by the inhibition or the activity of Na–K-ATPase and the elevation of Ca2+ levels in the cells, probably due to the protein kinase C activation [76].
In renal cells, lead binds to several proteins, where some have been associated in lead toxicity. Intra-nuclear inclusion bodies are formed in the proximal renal tubule, which produces lead nephron-toxicity. The nuclear inclusions are made of lead complexed with protein, which is associated with an alteration in lead compartmentalization from the cytosol to the nuclear fraction. Cytosolic proteins such as 2-microglobulin cleavage products can assist as carriers of lead or other intermediary ligands regarding lead uptake into the nucleus. Cytosolic proteins could also be involved in the ligand exchange reactions to other cytosolic binding sites, such as δ-aminolevulinic dehydratase, that binds and which lead inhibits. Metallothionein can also be bound with the lead but does not induce the protein compared to the inducers of zinc and cadmium. Structural abnormalities are found in the mitochondria present in the renal proximal tubule cells, were abnormal respiratory functions are seen, together with a decreased respiratory control ration. At the same time, the pyruvate/malate- or succinate-facilitated respiration. Calcium uptake is inhibited by lead into the isolated renal mitochondria, were lead enters as a substrate for a calcium transporter in the mitochondria. Oxidative metabolism impairment might contribute to cellular degeneration and transport deficits. Oxidative stress also is caused by lead exposure, were secondary responses to lead include the induction of NO synthase, transketolase, and glutathione S-transferase in the kidney. In vitro studies in rats show that lead depresses the glomerular filtration rate together with the renal blood flow. Hypertension might also be caused to the proximal tubule injury and impairment, or glomerulus sclerosis, which affects the release of renin and/or renal insufficiency [76].
Lead has also been observed to affect the haematopoietic system in both animals and humans, where an increase in urinary porphyrins are seen, together with an increase in ALA, FEP, EP, ZPP, coproporphyrin, and anaemia. The haem biosynthesis is altered by lead interfering with three enzyme activities which are ALAS, ferrochelatase, and ALAD. The mitochondrial enzyme ALAS is indirectly stimulated by lead, which goes on to catalyse the reaction of condensation of succinyl-coenzyme A and glycine to form ALA. In heam biosynthesis, ALAS activity is the rate-limiting step, where an increase of activity of ALAS transpires through feedback derepression. The cytosolic enzyme ALAD, which contains zinc, is inhibited through lead, which catalyses the condensation of 2 molecules of ALA which forms porphobilinogen. Inhibition of ALAD is non-competitive, where lead binds to the vicinal sulphydryl in the ALADs active site. ALA is accumulated due to the inhibition of ALAD and due to the feedback derepression of ALAS. The activity of mitochondrial enzyme ferrochelatase containing zinc is decreased by lead in a non-competitive fashion. The decrease in activity catalyzes the introduction of Fe2+ into the protoporphyrin ring, which forms the heam. The binding of lead to the vicinyl sulphydryl group at the active site may inhibit ferrochelatase, which causes an accumulation of the protoporphyrin IX present in the erythrocytes that are circulating, as ZPP. ZPP accumulation is seen only in erythrocytes which are formed in the erythropoietic tissue, in the presence of lead. Haemoglobin concentrations are reduced in the blood, due to the interference with the heam synthesis. A decrease in the production of haemoglobin, together with an increase in the destruction of erythrocytes leads to hypochromic, normocytic anaemia, linked with reticulocytosis [76].
Multiple mechanisms affect the nervous system through the presence of lead, where one mechanism is through the mimicking the calcium action and/or calcium homeostasis disruption. The protein kinase C (PKC) is involved in numerous vital processes for synaptic transmission. These processes are like the synthesis of the neurotransmitters, the conductance of ionic channels, the ligand-receptor interactions, and the dendritic branching. There are 12 isozymes in the PKC family, and each one has different requirements for the enzymatic cofactor, cellular distribution, and tissue expression. One of the forms which are a calcium-dependent form of PKC is the γ-isoform, which is a possible target for lead neurotoxicity. The γ-isoform is neuron-specific and participated in the long-term potentiation, memory processes, and spatial learning. AP-1 transcriptional regulatory complex forms by PKC, and controls the expression of a vast number of target genes through the AP-1 promoter elements. The AP-1 promoter regulated by lead is the gene glial fibrillary acidic protein (GFAP), which is an astrocytic intermediate filament protein and is induced when there is a reactive astrocytic gliosis. Endothelial cells and astrocytes make up the blood-brain barrier. Activation of PKC prematurely by lead was shown to impair the brain microvascular formation and function. At high levels of exposure to lead, gross defects were seen in the blood-brain barrier, which can cause an acute lead encephalopathy, where molecules, ions and water can enter the brain freely. The brain does not have a well-developed lymphatic system and thus clearance of the plasma constituents occur at a slow rate, oedema occurs, and thus the intracranial pressure rises. Since the foetus has an immature blood-brain barrier, there will be a higher risk of lead toxicity. Calmodulin is another enzyme that is altered by lead. Calmodulin is present in eukaryotes and is a major intracellular receptor for the element calcium. Calmodulin undergoes a conformational change due to calcium, which changes the protein into an active form. When lead is present, the enzyme is improperly activated [76].
Lead can substitute the element zinc in zinc-finger proteins and some enzymes. Lead in zinc finger proteins can cause effects on de novo expression of the proteins that are bound, and in genes that are transcriptionally-regulated by a specific protein. Zinc-finger transcriptional regulator Sp1 is effected by lead in its binding to its specific DNA sequence. Irregular expression of the Sp1 target genes occurs like the basic protein and the proteolipid protein. The β-amyloid precursor protein (APP) genes are also regulated by Sp1. Lead was seen to have various effects during the development exposure period where lead being exposed early in life is serious for the production of impaired hippocampal long-term potentiation (LTP). However, lead also affects the LTP after weaning, which affects the synaptic plasticity. LTP magnitude, together with the threshold display a U-shape response when lead concentrations increase. LTP is predominately a glutamatergic phenomenon which can be modulated from the input of extra hippocampal sources which include dopaminergic, noradrenergic, and cholinergic sources. Subsequently, the phenomenon is facilitated mainly by an increased glutamate release; the assumption reached is that glutamate release decreased due to the presence of lead. It is thought that lead present at nanomolar concentrations prevents the maximal activation of PKC, and not due to lead blocking the calcium influx through the voltage-gated calcium channels, into the presynaptic terminal [76].
In patients who have Parkinson's disease, lead is said to impair the regulation of dopamine synthesis together with its release and thus demonstrating a presynaptic site of action. Studies have reported opposing effects of the metal on mesolimbic and nigrostriatal dopamine systems concerning receptor binding, synthesis of dopamine, turnover and uptake. Exposure to lead in rats after weaning has shown a supersensitivity of dopamine receptors D1 and D2. The supersensitivity can be due to a compensatory response to a release of dopamine and/or a decreased synthesis. Perseverative deficits were noted when lesions occurred to the frontal cortex and the nucleus accumbens, signifying that lead affects the mesolimbic system. Studies have shown that lead does not effects learning, but the changes observed in the dopaminergic system due to lead could be the cause of a Fixed-Interval (FI) schedule of reinforcement to have an altered response rate. Due to this alteration in response rate, it was observed to produce a failure in the inhibition of an inappropriate response [76].
Lead has been shown to block an induced release of acetylcholine, and thus cholinergic function is diminished, as demonstrated in the peripheral and central synapses. Lead blocks the calcium entry into the neuromuscular junction terminal, which reduces the release of acetylcholine. However, lead also prevents the repossession of intracellular calcium the organelles; thus, an increase in the spontaneous release of the neurotransmitter takes place. In vitro studies have shown that the nicotinic cholinergic receptors can be blocked by lead. However, this action is unclear if it also occurs in vivo ad if the expression of the nicotinic cholinergic receptors undergoes an altered expression by lead, in a developing brain. A majority of data suggests that the metal does not affect muscarinic receptors directly, except for the visual cortex. In the visual cortex, it is observed that lead affects the inhibition of muscarinic receptors directly from the rods and bipolar present in the retina [76].
7. Discussion
The pollution of heavy metals in our atmosphere, soils and waters are all due to human activity. Each is essential in our daily diet but small amounts. In larger doses, heavy metals symptoms may be reversible but may also be irreversible and life-threatening. Workers that work in industries that release heavy metals should be extra cautious and wear protective gear to reduce the daily intake through inhalation, digestion or contact. Diagnoses should be appropriately made, as many of the symptoms resemble other neurological disorders.
The principal removal of heavy metals should be the prevention of heavy metals entering the human body. Less exhaust and industry pollution are one of the problems which can be targeted. Other ways of heavy metals entering our body are through food. Phytoremediation and intercropping are ways in which heavy metals can be absorbed and removed from the soils, sediments and waters [77]. Hyperaccumulator plants are planted in the soils to remove heavy metals. These types of plants have root systems which have a selective uptake, where the contaminant is translocated, bioaccumulated, and then the plants are entirely degraded to remove the heavy metals [78].
Phytostabilisation and phytoextraction occur for inorganic compounds. Phytoextraction is the processes where the heavy metals are translocated from the roots and into the shoots. Phytostabilisation comprises of only parts of the plant to take part in heavy metal removal. Organic compounds are removed through the processes phytodegradation, rhizofiltration and rhizodegradation. Rhizofiltration includes the adsorption or precipitation of the pollutants which are in solution and surround the root, into the roots, once the land is wet. Rhizodegradation involves microbial activity where the contaminants are degraded in the rhizosphere and improved by the roots [78].
Plant species is a significant factor when it comes to choosing the plant as different species take up different heavy metals. Other factors include the soil's pH, organic matter present, amount of phosphorous in the soil, root zone, the chelating agent added to the soil which influences the bioavailability of the metals such as ethylenediaminetetraacetic acid (EDTA), temperature which influences the vegetative uptake through the root length [78, 79, 80]. Hyperaccumulator plants do not show signs of toxicity when they absorb heavy metals [81].
Pteris vittata L. absorbs arsenic at around 0.7 mg/g in dry weight as the whole plant. Populus nigra absorbs arsenic at a concentration of 0.2 mg/g in dry weight, in the plants' roots. Brassica carinata A. Braun absorbs lead at more than 50 mg/g in dry weigh in the whole plant. Solanum lycopersicum, a tomato plant, absorbs the metal cadmium in the plant's shoots, and lead in the plant's roots. The fruit of this plant absorbs excess cadmium. 10 mg/g of zinc and 0.1 mg/g of cadmium is taken up by the shoots of the plant Thlaspi caerulescens in dry weight [81].
On the other hand, the method of intercropping consists of growing two different species of plants at the same time. Plant biomass is improved, and the accumulation of heavy metals is aided. Phytoremediation is improved if used simultaneously with intercropping, which is more environmentally friendly than using chelators [82,83]. The proper species needs to be selected to be used as an appropriate intercropping plant [84]. Intercropping has been noted to increase diversity and stability to fields and a reduction in fertilizer use. Weeds, disease and insects are reduced due to this method [85]. Three varieties of intercropping exist:
-
•
Mixed intercropping – different crops are harvested at the same time,
-
•
Row intercropping – different crops are cultivated in alternative rows,
-
•
Relay cropping – the second crop is sown when the other species reached the reproductive stage [86].
Maize and chickpea plants are usually intercropped together. Maize absorbs cadmium from the soil with a higher absorption when grown with chickpea [82]. Tomato root absorbs ion, and when intercropped with the Japan clover herb, the absorption of iron increases. Cadmium absorption was also increased in this intercropping [81].
Organic farming can be seen as another method of reducing heavy metals from entering our bodies. No pesticides are used, and water treatment has to be of a purified form with no or very low contaminants. Biodiversity is used instead to remove unwanted insects, together with an increase in wild fauna and flora as a benefit of using this type of method [87].
Heavy metals can be removed from water, where the most important factors for removal are the temperature, pH, ionic strength and the presence of natural organic matter. By products and agricultural waste, such as dairy manure, remaining waster material from rice and peanuts, seem to be the best to remove heavy metals. On the other hand, mineral deposits and natural soil seem to be the least effective in the removal of heavy metals [88].
Once heavy metals enter the body, some foods are known to chelate with them. Cilantro, wild blueberries, garlic, lemon water, chlorella, spirulina, curry, green tea, Atlantic dulse, barley grass, tomatoes, and probiotics are foods or supplements that can aid in the gradual removal of metals from the body [89].
8. Conclusion
The world is being polluted in so many ways with heavy metals, mostly due to human contamination, that it is affecting the health of so many people. All these diagnoses should go on to make us aware of the adverse effects that are being caused by these metals, the symptoms that are seen, and ways to remove some of the contamination we have from all the heavy metals.
Declarations
Author Contribution statement
All authors listed have significantly contributed to the development and the writing of this article.
Funding statement
This work was partially funded by the Endeavour Scholarship Scheme (Malta). Scholarships are part-financed by the European Union – European Social Fund (ESF) – Operational Programme II – Cohesian Policy 2014-20 “Investing in human capital to create more opportunities and promote to well-being of society”.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
References
- 1.Masindi V., Muedi K.L. Heavy Metals. InTech; 2018. Environmental contamination by heavy metals. [Google Scholar]
- 2.Walker C.H., Sibly R.M., Hopkin S.P., D.B.P. Principles of Ecotoxicology; Group, T. And F., Ed.; 4th Edition. CRC Press; 2012. [Google Scholar]
- 3.Martin Y.E., Johnson E.A. Biogeosciences survey: studying interactions of the biosphere with the lithosphere, hydrosphere and atmosphere. Prog. Phys. Geogr. 2012;36:833–852. [Google Scholar]
- 4.Gautam P.K., Gautam R.K., Chattopadhyaya M.C., Banerjee S., Chattopadhyaya M.C., Pandey J.D. Transport, Toxicity And Rem; 2016. Heavy metals in the environment: fate, transport, toxicity and remediation technologies Thermodynamic profiling of pollutants View project Materials for Solid oxide fuel cells View project Heavy Metals in the Environment: Fate. [Google Scholar]
- 5.Tchounwou P.B., Yedjou C.G., Patlolla A.K., Sutton D.J. Heavy metal toxicity and the environment. In EXS. 2012;101:133–164. doi: 10.1007/978-3-7643-8340-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenntech BV Heavy Metals. https://www.lenntech.com/processes/heavy/heavy-metals/heavy-metals.htm Available online:
- 7.Duffus J.H. Heavy metals” a meaningless term? Pure Appl. Chem. 2002;74:793–807. [Google Scholar]
- 8.Wang L.K. CRC Press; 2009. Heavy Metals in the Environment. [Google Scholar]
- 9.Shallari S. Heavy metals in soils and plants of serpentine and industrial sites of Albania. Sci. Total Environ. 1998;209:133–142. [PubMed] [Google Scholar]
- 10.Herawati N., Suzuki S., Hayashi K., Rivai I.F., Koyama H. Cadmium, copper, and zinc levels in rice and soil of Japan, Indonesia, and China by soil type. Bull. Environ. Contam. Toxicol. 2000;64:33–39. doi: 10.1007/s001289910006. [DOI] [PubMed] [Google Scholar]
- 11.He Z.L., Yang X.E., Stoffella P.J. Trace elements in agroecosystems and impacts on the environment. J. Trace Elem. Med. Biol. 2005;19:125–140. doi: 10.1016/j.jtemb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 12.Villanueva R., Bustamante P. Composition in essential and non-essential elements of early stages of cephalopods and dietary effects on the elemental profiles of Octopus vulgaris paralarvae. Aquac. Elsevier. 2006;261:225–240. [Google Scholar]
- 13.Azeh Engwa G., Udoka Ferdinand P., Nweke Nwalo F., Unachukwu N.M. Poisoning in the Modern World - New Tricks for an Old Dog? IntechOpen; 2019. Mechanism and health effects of heavy metal toxicity in humans. [Google Scholar]
- 14.Valko M., Morris H., Cronin M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 15.Aronsson P., Perttu K. Willow vegetation filters for wastewater treatment and soil remediation combined with biomass production. For. Chron. 2001;77:293–299. [Google Scholar]
- 16.Jackson A.P., Alloway B.J. The transfer of cadmium from sewage-sludge amended soils into the edible components of food crops. Water. Air. Soil Pollut. 1991;57–58:873–881. [Google Scholar]
- 17.Muchuweti M., Birkett J.W., Chinyanga E., Zvauya R., Scrimshaw M.D., Lester J.N. Heavy metal content of vegetables irrigated with mixtures of wastewater and sewage sludge in Zimbabwe: implications for human health. Agric. Ecosyst. Environ. 2006;112:41–48. [Google Scholar]
- 18.Gupta N., Khan D.K., Santra S.C. Heavy metal accumulation in vegetables grown in a long-term wastewater-irrigated agricultural land of tropical India. Environ. Monit. Assess. 2012;184:6673–6682. doi: 10.1007/s10661-011-2450-7. [DOI] [PubMed] [Google Scholar]
- 19.Carrondo, M.J.T.; Lester, J.N.; Perry, R.; Stoveland, S. Analysis of Heavy Metals in Sewage Sludge, Sewages And Final Effluent;
- 20.Primary, Secondary, and Tertiary Wastewater Treatment: How Do They Work? - Innovative Solutions for Wastewater Treatment. https://www.organicawater.com/primary-secondary-tertiary-wastewater-treatment-work/ Organica Water Inc. Available online: (accessed on Nov 9, 2019)
- 21.Di Bonito M. Environmental Geochemistry: Site Characterization, Data Analysis and Case Histories. Elsevier Inc.; 2008. Sewage sludge in europe and in the UK. Environmental impact and improved standards for recycling and recovery to land; pp. 251–286. [Google Scholar]
- 22.Lepp N., Dickinson N., Ross S. Fungicide-derived copper in tropical plantation crops. In: Ross S., editor. Toxic Metals in Soil-Plant Systems. Wiley; Chichester: 1994. pp. 367–393. [Google Scholar]
- 23.Chibuike G.U., Obiora S.C. Heavy metal polluted soils: effect on plants and bioremediation methods. Appl. Environ. Soil Sci. 2014;2014 [Google Scholar]
- 24.Grant A., Middleton R. An assessment of metal contamination of sediments in the humber estuary. U.K. Estuar. Coast. Shelf Sci. 1990;31:71–85. [Google Scholar]
- 25.Caeiro S., Costa M.H., Ramos T.B., Fernandes F., Silveira N., Coimbra A., Medeiros G., Painho M. Assessing heavy metal contamination in Sado Estuary sediment: an index analysis approach. Ecol. Indic. 2005;5:151–169. [Google Scholar]
- 26.Brunning, A. A Rough guide to the IARC’s carcinogen classifications | Compound Interest Available online: https://www.compoundchem.com/2015/10/26/carcinogens/(accessed on Jan 19, 2019).
- 27.Bernardo J.F. Aluminum Toxicity. https://emedicine.medscape.com/article/165315-overview#showall Available online: (accessed on Apr 12, 2020)
- 28.WHO Air Quality Guidelines for Europe. second ed. 2000. [Google Scholar]
- 29.ATSDR Toxicological Profile for Aluminium. 2008. [Google Scholar]
- 30.Zrinzo R. University of Malta; 2015. Biochemical and Physiological Effect of Aluminium Bioccumulation. [Google Scholar]
- 31.Shaw C.A., Tomljenovic L. Aluminum in the central nervous system (CNS): toxicity in humans and animals, vaccine adjuvants, and autoimmunity. Immunol. Res. 2013;56:304–316. doi: 10.1007/s12026-013-8403-1. [DOI] [PubMed] [Google Scholar]
- 32.ATSDR Toxicological Profile for Vanadium. 2012. [PubMed] [Google Scholar]
- 33.Pedersen T. Facts about Vanadium. https://www.livescience.com/29155-vanadium.html Available online.
- 34.Browne R.C. Vanadium poisoning from gas turbines. Br. J. Ind. Med. 1955;12:57–59. doi: 10.1136/oem.12.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barnhart J. Occurrences. Uses, and properties of chromium. Regul. Toxicol. Pharmacol. 1997;26:S3–S7. doi: 10.1006/rtph.1997.1132. [DOI] [PubMed] [Google Scholar]
- 36.ATSDR Toxicological Profile for Chromium. 2012. [PubMed] [Google Scholar]
- 37.Martin Sabine. Griswold Wendy Human Health Effects of Heavy Metals. Environ. Sci. Technol. Briefs Citizens. 2009 [Google Scholar]
- 38.Dayan A.D., Paine A.J. Mechanisms of chromium toxicity, carcinogenicity and allergenicity: review of the literature from 1985 to 2000. Hum. Exp. Toxicol. 2001;20:439–451. doi: 10.1191/096032701682693062. [DOI] [PubMed] [Google Scholar]
- 39.Kaye G.W.C., Laby T.H. fifteenth ed. John Wiley and Sons Ltd; United States: 1986. Tables of Physical and Chemical Constants. [Google Scholar]
- 40.Dobson A.W., Erikson K.M., Aschner M. Manganese neurotoxicity. Ann. N. Y. Acad. Sci. 2004;1012:115–128. doi: 10.1196/annals.1306.009. [DOI] [PubMed] [Google Scholar]
- 41.Haynes W.M. In: CRC Handbook of Chemistry and Physics. 92nd ed. Haynes W.M., editor. CRC Press; Florida: 2011. [Google Scholar]
- 42.Marcus J.B. Culinary Nutrition. Elsevier; 2013. Vitamin and mineral basics: the ABCs of healthy foods and beverages, including phytonutrients and functional foods; pp. 279–331. [Google Scholar]
- 43.Iregren A. Psychological test performance in foundry workers exposed to low levels of manganese. Neurotoxicol. Teratol. 1990;12:673–675. doi: 10.1016/0892-0362(90)90085-q. [DOI] [PubMed] [Google Scholar]
- 44.Roels H.A., Ghyselen P., Buchet J.P., Ceulemans E., Lauwerys R.R. Assessment of the permissible exposure level to manganese in workers exposed to manganese dioxide dust. Br. J. Ind. Med. 1992;49:25–34. doi: 10.1136/oem.49.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mergler D., Huel G., Bowler R., Iregren A., Belanger S., Baldwin M., Tardif R., Smargiassi A., Martin L. Nervous system dysfunction among workers with long-term exposure to manganese. Environ. Res. 1994;64:151–180. doi: 10.1006/enrs.1994.1013. [DOI] [PubMed] [Google Scholar]
- 46.ATSDR Toxicological Profile for Manganese. 2012. [PubMed] [Google Scholar]
- 47.Leyssens L., Vinck B., Van Der Straeten C., Wuyts F., Maes L. Cobalt toxicity in humans—a review of the potential sources and systemic health effects. Toxicology. 2017;387:43–56. doi: 10.1016/j.tox.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Lauwerys R., Lison D. Health risks associated with cobalt exposure - an overview. Sci. Total Environ. 1994;150:1–6. doi: 10.1016/0048-9697(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 49.ATSDR Toxicological Profile of Cobalt. 2004. [Google Scholar]
- 50.Cobalt Institute Cobalt in Soils Available online: https://www.cobaltinstitute.org/cobalt-in-soil.html (accessed on Mar 16, 2020).
- 51.Simonsen L.O., Harbak H., Bennekou P. Cobalt metabolism and toxicology-A brief update. Sci. Total Environ. 2012;432:210–215. doi: 10.1016/j.scitotenv.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 52.ATSDR Toxicological Profile for Nickel. 2017. [Google Scholar]
- 53.Denkhaus E., Salnikow K. Nickel essentiality, toxicity, and carcinogenicity. Crit. Rev. Oncol. Hematol. 2002;42:35–56. doi: 10.1016/s1040-8428(01)00214-1. [DOI] [PubMed] [Google Scholar]
- 54.Sharma A.D. Low nickel diet in dermatology. Indian J. Dermatol. 2013;58:240. doi: 10.4103/0019-5154.110846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.ATSDR Toxicological Profile for Copper. 2004. [Google Scholar]
- 56.Tsafrir J. Copper Toxicity: A Common Cause of Psychiatric Symptoms. https://www.psychologytoday.com/intl/blog/holistic-psychiatry/201709/copper-toxicity-common-cause-psychiatric-symptoms Available online:
- 57.Fosmire G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990;51:225–227. doi: 10.1093/ajcn/51.2.225. [DOI] [PubMed] [Google Scholar]
- 58.ATSDR Toxicological Profile for Zinc. 2005. [PubMed] [Google Scholar]
- 59.U.S. EPA . 2005. Toxicological Review of Zinc and Compounds; Washington DC. [Google Scholar]
- 60.Momcilović B. A case report of acute human molybdenum toxicity from a dietary molybdenum supplement–a new member of the "Lucor metallicum" family. Arh. Hig. Rada. Toksikol. 1999;50:289–297. [PubMed] [Google Scholar]
- 61.ATSDR Toxicological Profile for Molybdenum. 2017. [PubMed] [Google Scholar]
- 62.ATSDR Toxicological Profile for Arsenic. 2007. [PubMed] [Google Scholar]
- 63.ATSDR Toxicological Profile for SIlver. 1990. [PubMed] [Google Scholar]
- 64.Lansdown A.B.G. A pharmacological and toxicological profile of silver as an antimicrobial agent in medical devices. Adv. Pharmacol. Sci. 2010;16 doi: 10.1155/2010/910686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McShan D., Ray P.C., Yu H. Molecular toxicity mechansim of nanosilver. J. Food Drug Anal. 2014;22:116–127. doi: 10.1016/j.jfda.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.ATSDR Toxicological Profile for Cadmium; Georgia. 2012. [PubMed] [Google Scholar]
- 67.IARC . IARC; 1993. WHO Beryllium, Cadmium, Mercury, and Exposures in the Glass Manufacturing Industry. [PMC free article] [PubMed] [Google Scholar]
- 68.Yang G.Q., Wang S.Z., Zhou R.H., Sun S.Z. Endemic selenium intoxication of humans in China. Am. J. Clin. Nutr. 1983;37:872–881. doi: 10.1093/ajcn/37.5.872. [DOI] [PubMed] [Google Scholar]
- 69.ATSDR Toxicological Profile for Selenium. 2003. [PubMed] [Google Scholar]
- 70.ATSDR Toxicological Profile for Mercury. 1999. [Google Scholar]
- 71.Li R., Wu H., DIng J., Fu W., Gan L., Li Y. Mercury pollution in vegetables, grains and soils from areas surrounding coal-fired power plants. Sci. Rep. 2017;7:46545. doi: 10.1038/srep46545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bernhoft R. Mercury toxicity and treatment: a review of the literature. J. Environ. Public Health. 2012:1–10. doi: 10.1155/2012/460508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.WHO Lead Poisoning and Health. http://www.who.int/en/news-room/fact-sheets/detail/lead-poisoning-and-health Available online:
- 74.Mayo Clinic Lead Poisoning. https://www.mayoclinic.org/diseases-conditions/lead-poisoning/symptoms-causes/syc-20354717 Available online:
- 75.ATSDR Toxicological Profile for Lead. 2019. [Google Scholar]
- 76.ATSDR Toxicological Profile for Lead. 2007. [Google Scholar]
- 77.Hao H., Zhong R., Miao Y., Wang Y., Liu C. Proceedings of the Proceedings - 2012 International Conference on Biomedical Engineering and Biotechnology, iCBEB 2012; IEEE. 2012. The intercropping influence on heavy metal uptake for hyperaccumulators; pp. 1826–1828. [Google Scholar]
- 78.Tangahu B.V., Sheikh Abdullah S.R., Basri H., Idris M., Anuar N., Mukhlisin M. A review on heavy metals (as, Pb, and Hg) uptake by plants through phytoremediation. Int. J. Chem. Eng. 2011;31 [Google Scholar]
- 79.Chen Y., Li X., Shen Z. Leaching and uptake of heavy metals by ten different species of plants during an EDTA-assisted phytoextraction process. Chemosphere. 2004;57:187–196. doi: 10.1016/j.chemosphere.2004.05.044. [DOI] [PubMed] [Google Scholar]
- 80.Wuana R.A., Okieimen F.E. Heavy metals in contaminates soils: a reveiw of sources, chemistry, risks and best available strategies for remidiation. ISRN Ecol. 2011 [Google Scholar]
- 81.An L., Pan Y., Wang Z., Zhu C. Heavy metal absorption status of five plant species in monoculture and intercropping. Plant Soil. 2011;345:237–245. [Google Scholar]
- 82.Li N.Y., Li Z.A., Zhuang P., Zou B., McBride M. Cadmium uptake from soil by maize with intercrops. Water. Air. Soil Pollut. 2009;199:45–56. [Google Scholar]
- 83.Cui T., Fang L., Wang M., Jiang M., Shen G. Intercropping of gramineous pasture ryegrass (LoliumperenneL.) and leguminous forage alfalfa (medicago sativaL.) increases the resistance of plants to heavy metals. J. Chem. 2018;11 [Google Scholar]
- 84.Wan X., Lei M. Intercropping efficiency of four arsenic hyperaccumulator Pteris vittata populations as intercrops with Morus alba. Environ. Sci. Pollut. Res. 2018;25:12600–12611. doi: 10.1007/s11356-018-1366-y. [DOI] [PubMed] [Google Scholar]
- 85.Lowry C.J., Smith R.G. Non-Chemical Weed Control. Elsevier; 2018. Weed control through crop plant manipulations; pp. 73–96. [Google Scholar]
- 86.Lithourgidis A.S., Dordas C., Damalas C., Vlachostergios D.N. Annual intercrops: an alternative pathway for sustainable agriculture. Aust. J. Crop. Sci. 2011;5:396–410. [Google Scholar]
- 87.FAO Organic Agriculture What Are the Environmental Benefits of Organic Agriculture? http://www.fao.org/organicag/oa-faq/oa-faq6/en/ Available online: (accessed on Mar 20, 2020)
- 88.Joseph L., Jun B.M., Flora J.R.V., Park C.M., Yoon Y. Removal of heavy metals from water sources in the developing world using low-cost materials: a review. Chemosphere. 2019;229:142–159. doi: 10.1016/j.chemosphere.2019.04.198. [DOI] [PubMed] [Google Scholar]
- 89.Anthony K., Sullivan D. Heavy metal detox diet: symptoms and recommended foods. https://www.healthline.com/health/heavy-metal-detox Available online: