

Abstract
Abstract
Based on the fact that a search for influenza antivirals among nucleoside analogues has drawn very little attention of chemists, the present study reports the synthesis of a series of 1,2,3-triazolyl nucleoside analogues in which a pyrimidine fragment is attached to the ribofuranosyl-1,2,3-triazol-4-yl moiety by a polymethylene linker of variable length. Target compounds were prepared by the Cu alkyne-azide cycloaddition (CuAAC) reaction. Derivatives of uracil, 6-methyluracil, 3,6-dimethyluracil, thymine and quinazolin-2,4-dione with ω-alkyne substituent at the N1 (or N5) atom and azido 2,3,5-tri-O-acetyl-D-β-ribofuranoside were used as components of the CuAAC reaction. All compounds synthesized were evaluated for antiviral activity against influenza virus A/PR/8/34/(H1N1) and coxsackievirus B3. The best values of IC50 (inhibiting concentration) and SI (selectivity index) were demonstrated by the lead compound 4i in which the 1,2,3-triazolylribofuranosyl fragment is attached to the N1 atom of the quinazoline-2,4-dione moiety via a butylene linker (IC50 = 30 μM, SI = 24) and compound 8n in which the 1,2,3-triazolylribofuranosyl fragment is attached directly to the N5 atom of the 6-methyluracil moiety (IC50 = 15 μM, SI = 5). According to theoretical calculations, the antiviral activity of the 1,2,3-triazolyl nucleoside analogues 4i and 8n against H1N1 (A/PR/8/34) influenza virus can be explained by their influence on the functioning of the polymerase acidic protein (PA) of RNA-dependent RNA polymerase (RdRP).
Graphic abstract

Electronic supplementary material
The online version of this article (10.1007/s11030-020-10141-y) contains supplementary material, which is available to authorized users.
Keywords: Nucleoside analogues; 1,2,3-Triazole; Influenza virus; Coxsackievirus; Click chemistry
Introduction
Naturally occurring nucleosides represent a unique scaffold for drug design due to their involvement in numerous biological processes as well as the fact that they serve as essential building blocks for DNA and RNA synthesis [1]. Nucleosides play important roles in the replication and transcription of genetic information and, as such, have been utilized for decades for antibacterial or antiviral therapeutics [2, 3]. In the early 1960s, the idea appeared that various nucleoside derivatives (or analogues) can also affect the biochemical processes in the cells of bacteria and viruses. Over the next 50 years, a diverse series of numerous nucleoside analogues have been synthesized and evaluated for their biological activities. Numerous modifications to the nucleosides included alterations to the sugar, nucleobase and glycosidic bond [2–5].
Overwhelming majority of synthesized nucleoside analogues displayed anticancer or antiviral activity. For example, Gemcitabine was approved for the treatment of breast cancer, ovarian cancer, non-small cell lung cancer, pancreatic cancer and bladder cancer, and Floxuridine is an oncology drug to treat colorectal cancer [2–5]. Zalcitabine, Didanosine, Zidovudine, Stavudine, Carbovir and Entecavir were approved for the treatment of HIV/AIDS [2–6]. Trifluridine and Idoxuridine are used to treat herpes simplex virus [2, 4]. Entecavir was approved drug for treating hepatitis B virus (HBV) [2, 4]. Brivudine is an antiviral drug used in the treatment of herpes zoster (VZV) [2, 4, 7]. As to antiviral activity, the literature has provided just a few examples of nucleoside analogues which are influenza A virus inhibitors [8–13]. Among approved influenza drugs (Amantadine, Rimantadine, Zanamivir, Oseltamivir, Laninamivir octanoate, Peramivir, Favipiravir and Ribavirin [4]), there is only one nucleoside analogue, namely Ribavirin (1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide) [4, 7, 14–17]. Ribavirin possesses non-interferon-inducing, broad-spectrum antiviral properties against a wide range of RNA viruses [7, 14–17]. These facts have drawn much attention of chemists and pharmacologists who began to synthesize Ribavirin derivatives for developing novel antiviral agents to treat various viral diseases including influenza [18–23]. Note that Ribavirin possesses the 1,2,4-triazole moiety. Triazole heterocyclic compounds are paid special attention due to their potential applications as medicinal agents since 1,2,4- and 1,2,3-triazole units which are not present in natural products are remarkably stable to metabolic transformations that allow to utilize them in various drugs [24, 25]. The introduction of “click chemistry” reactions using Cu alkyne-azide cycloaddition (CuAAC) in medicinal chemistry has provided a great number of 1,2,3-triazolyl nucleoside analogues demonstrating various bioactivities. Nucleoside analogues containing substituted 1,2,3-triazole moieties at the C5′ position of the ribofuranose residue demonstrated significant anticancer activity against cancer cell lines A549, HT-29, MCF-7, A-375 and/or antibacterial and antifungal activities [26, 27]. Pyrimidine nucleoside analogues in which a 1,2,3-triazole ring was attached either directly to the C5 position of 2′-deoxyuridine or via a methylene unit were synthesized and exhibited both antiviral activity against herpes simplex viruses, varicella-zoster virus, human cytomegalovirus, vaccinia virus [28–31] and significant anticancer effects against cancer cell lines PC-3, MDA-MB-231, ACHN [28]. Recently, a series of nucleoside analogues in which the pyrimidine fragment was attached to the ribose moiety at the C1′ carbon via a 1,2,3-triazolyl bridge has been synthesized [32–35]. No inhibitory activity against HCV virus was observed with any of these compounds, but several C5-substituted 1-β-d-ribofuranosyl-1,2,3-triazolidomethyluracils showed potent inhibitory activity against RNase A [34] and completely inhibited the angiogenic activity of hAng in vivo [35].
The above brief review of the literature reveals that all natural nucleosides and their analogues, including 1,2,3-triazole ones, evaluated primarily for their anticancer and antiviral activities. As a result, many nucleoside analogues were approved as drugs for the treatment of HIV/AIDS, HBV, HCV, HSV [2–7]. Unfortunately, the search for influenza drugs among nucleoside analogues has drawn very little attention of chemists and pharmacologists. This is despite the fact that influenza virus infection constitutes a significant health problem in need of more effective therapies [8]. The worldwide spread of drug-resistant influenza strains poses an urgent need for novel antiviral drugs, particularly with a different mechanism of action [10]. In this regard, 1,2,3-triazole nucleoside analogues are a promising scaffold for creating new anti-influenza drugs. Herein, we report the synthesis of a series of 1,2,3-triazolyl nucleoside analogues using the Cu alkyne-azide cycloaddition (CuAAC) reaction and evaluation of their ability to inhibit the in vitro growth of influenza virus A/PR/8/34 (H1N1) and coxsackievirus B3. In these compounds the pyrimidine fragment is attached to the ribofuranosyl-1,2,3-triazol-4-yl moiety via polymethylene linker of variable length.
Results and discussion
Synthetic chemistry
At first, we focused on the synthesis of alkyne components of the CuAAC reaction, namely pyrimidine derivatives 1a–c, 2a–c, 3a–c, 4a–c, 6a–c with ω-alkyne substituents at the N1 atom. They were prepared according to the method reported previously [36]. Starting pyrimidines uracil (1), 6-methyluracil (2), thymine (3) and condensed uracil derivative, namely quinazoline-2,4-dione (4), were reacted with an excess of hexamethyldisilazane (HMDS) in toluene in the presence of H2SO4 at reflux for 8 h to afford bissilylated derivatives 5 which were then engaged in the alkylation with ω-iodo-alk-α-ynes without purification (Scheme 1). Alkyne derivatives of uracil (1b,c; 2b,c), thymine (3b,c) and quinazolin-2,4-dione (4a–c) were obtained in 33–64% yield. As we have reported [36], in the case of 6-methyluracil 2 the reaction of its bissilylated derivative 5 with a propargyl bromide unexpectedly led to the mixture of N1- and N3-substituted products. Therefore, N1-propargyl derivatives of uracil 1, 6-methyluracil 2, thymine 3, as well as N1-ω-alkynyl derivatives of 3,6-dimethyluracil 6 (compounds 1a, 2a, 3a, 6a–c, respectively) were synthesized by the alkylation of monosodium salts 7 [36] (Scheme 1).
Scheme 1.

Synthesis of pyrimidine derivatives containing an ω-alkyne substituent at the N1 or the C5 position of the pyrimidine ring
6-Methyluracil derivatives 8g,h,i with a ω-alkyne substituent at the C5 atom were prepared in three steps by analogy with procedures earlier described [37, 38]. Initially, ethylacetoacetate was alkylated in a solution of sodium in ethanol by treatment with (a) propargyl bromide to prepare ethyl 2-acetyl-4-pentynoate 8a; (b) 5-iodo-pent-1-yne to prepare ethyl 2-acetyl-6-heptynoate 8b; (c) 6-iodo-hex-1-yne to prepare ethyl 2-acetyl-7-octynyloate 8c (Scheme 1). Then, β-keto esters 8a, 8b, 8c were condensed with thiourea to give 6-methyl-5-(ω-alkyne)-2-thio-(1H,3H)pyrimidine-4-ones. These thiopyrimidines were next hydrolyzed with chloroacetic acid in EtOH/H2O to afford target 6-methyl-5-(ω-alkyne)pyrimidine-2,4(1H,3H)-diones in good yields (85%, 65%, 80%, respectively).
The azide component of the CuAAC reaction, namely azido ribofuranose 9c, was prepared based on the published procedure [36] (Scheme 2). D-ribose was converted into methyl b-ribofuranoside 9a by reacting with methanol and subsequent acetylation. The methoxyl group of the obtained monosaccharide 9a was replaced by an acetoxy group which then was converted to an azido one by the reaction of 9b with trimethylsilyl azide (TMSN3). The target compound 9c was obtained in 95% yield.
Scheme 2.

Synthesis of 2,3,5-tri-O-acetyl-β-d-ribofuranosyl azide 9c
Coupling the alkyne components 1a–c, 2a–c, 3a–c, 4a–c, 6a–c, 8 with the azido component 9c was accomplished by a CuAAC reaction in according with a procedure already described [36] (Scheme 3). The 1,2,3-triazolyl analogues 1–4, 6, 8 with acetyl protection were obtained in good yields (60–96%). The 1H NMR spectra of these compounds showed a singlet within the range 7.50–7.97 ppm corresponding to the triazolyl proton C5-H [29–32, 34, 36]. The signals within the range 142.9–147.8 ppm and 120.1–123.5 ppm in the 13C NMR spectra were certainly assigned to the triazolyl carbons C4 and C5, respectively [29–32, 34, 36]. The anomeric protons of 1,2,3-triazolyl analogues 1–4, 6, 8 resonated in the 1H NMR spectra as doublets within the range 5.98–6.20 ppm with vicinal coupling constants of 3.5–4.2 Hz that confirmed β-orientation of the glycoside bonds in a full agreement with the literature [39]. Removal of the acetyl protection of 1–4, 6, 8 provided the target 1,2,3-triazolyl nucleoside analogues 1–4, 6, 8 with free hydroxy groups in good yields (76–98%) (Scheme 3).
Scheme 3.

Synthesis of 1,2,3-triazolyl nucleoside analogues with uracil, 6-methyluracil, 3,6-dimethyluracil, thymine and quinazoline-2,4-dione moieties
Biological evaluation. Antiviral activity
The in vitro antiviral activity of the compounds synthesized was evaluated regarding A/Puerto Rico/8/34 (H1N1) strain of influenza virus and coxsackievirus B3. The resulting data expressed as virus-inhibiting activity (IC50), cytotoxicity (CC50) as well as selectivity index (SI), which is the ratio CC50/IC50, are presented in Tables 1 and 2. The analysis of Table 1 leads to the following four important conclusions. First of all, 1,2,3-triazolyl derivatives of natural nucleosides uridine (1) and thymidine (3) lack antiviral activity against influenza virus A/PR/8/34/(H1N1). On the contrary, the antiviral activity was demonstrated by 1,2,3-triazolyl nucleoside analogues in which nucleic base has been replaced with 6-methyluracil (compounds 2h, 2i, 8j, 8k, 8m, 8n) or quinazoline-2,4-dione (compounds 4g, 4i). The high antiviral activity (IC50 = 30 µM), low cytotoxicity (CC50 > 719 µM) and the high selectivity index (SI) value of 24 were shown by 1,2,3-triazole nucleoside analogue 4i possessing the quinazoline-2,4-dione moiety instead of a nucleic base. Secondly, the antiviral activity depends both on the nature of the pyrimidine moiety and the length of the linker coupling it with the 1,2,3-triazolylribofuranosyl fragment. Thus, in a series of the derivatives of 6-methyluracil 2g, 2h, 2i and quinazoline-2,4-dione 4g, 4h, 4i the best antiviral activity was exhibited by compounds 2h, 2i and 4i having propylene and butylene linker, respectively. Among the derivatives of 6-methyluracil 2g, 2h, 2i, compound 2g with methylene linker appeared to be inactive (no virus inhibition was achieved even at highest concentrations used), while among the derivatives of quinazoline-2,4-dione 4g, 4h, 4i, compound 4g with the same methylene linker showed a good activity (IC50 = 42 µM). Thirdly, the antiviral activity depends on the position of attachment of the 1,2,3-triazolylribofuranosyl fragment to the pyrimidine moiety. Thus, if compound 2i in which the 1,2,3-triazolylribofuranosyl fragment was attached to the 6-methyluracil moiety at the N1 atom via butylene linker showed a good activity (CC50 = 311 µM, CC50 = 48 µM, SI = 6), then compound 8o in which the same fragment was attached to the 6-methyluracil moiety at the C5 atom was completely inactive (IC50 > 787 µM). Fourthly, 1,2,3-triazolyl nucleoside analogues based on 3,6-dimethyluracil in which both nitrogen atoms of the pyrimidine fragment are alkylated (compounds 6g, 6h, 6i) completely devoid of antiviral activity against influenza virus A H1N1. Summarizing the data presented in Table 1, one can see that the 1,2,3-triazole nucleoside analogue 4i seems to be the most promising one among others combining high antiviral potency (IC50 = 30 µM) and very low cytotoxicity (CC50 > 719 µM) with the high selectivity index (SI) of 24. Nucleoside 8n, although possessing more higher cytotoxicity (CC50 = 79 µM), can also be considered a lead compound because it had the highest antiviral activity among the studied ones (IC50 = 15 µM).
Table 1.
Antiviral activity against H1N1 A/Puerto Rico/8/34 influenza virus and cytotoxicity of the synthesized 1,2,3-triazolyl nucleoside analogues
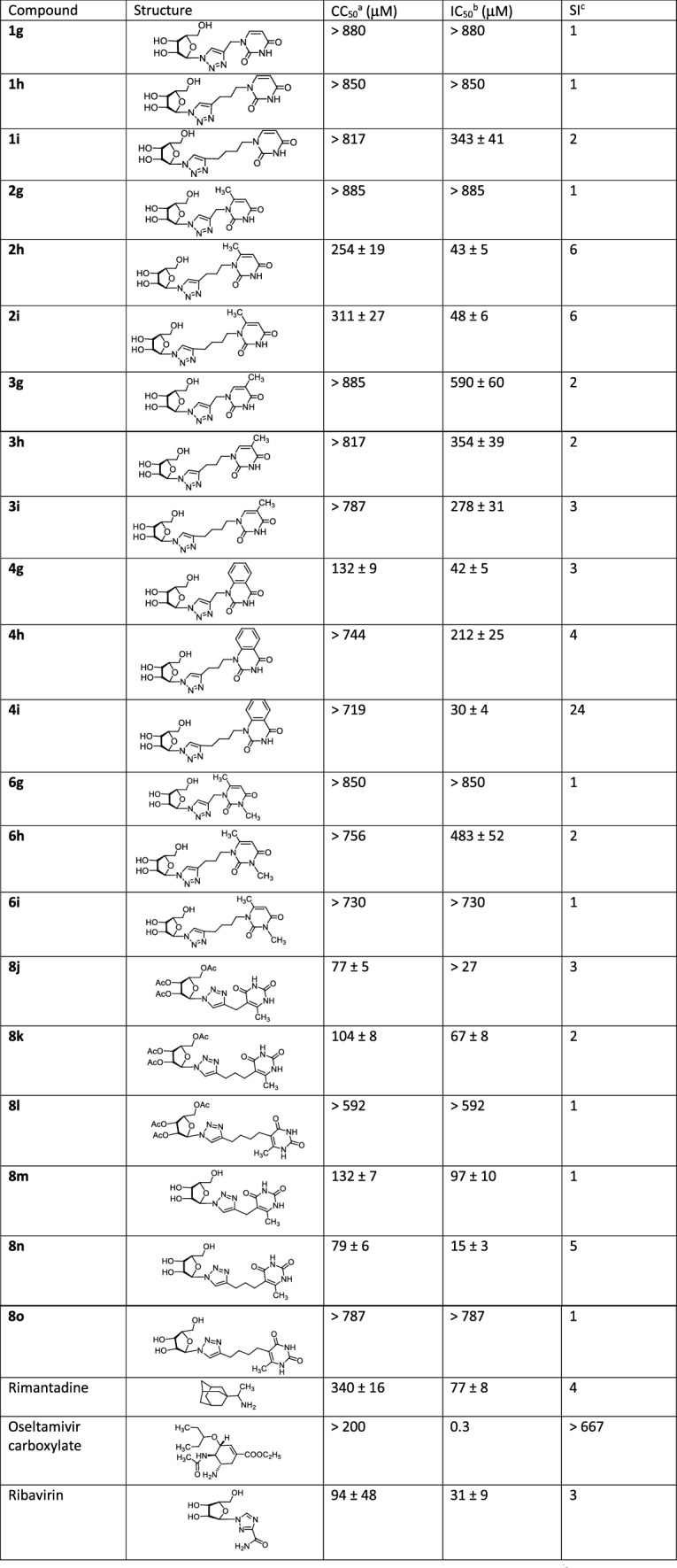
aCC50 is the median cytotoxic concentration, i.e., the concentration causing 50% cell death
bIC50 is the concentration causing 50% inhibition of virus replication
cSI is the selectivity index, which is the CC50/IC50 ratio
Table 2.
Antiviral activity against Coxsackie B3 virus and cytotoxicity of synthesized 1,2,3-triazolyl nucleoside analogues

aCC50 is the median cytotoxic concentration, i.e., the concentration causing 50% cell death
bIC50 is the concentration causing 50% inhibition of virus replication
cSI is the selectivity index, which is the CC50/IC50 ratio
It is interesting to compare antiviral activity of the lead compound 4i and antiviral activity of the reference compounds which differ according to both their structure and viral targets. Rimantadine blocks the transport of H+ ions through the M2 protein channels, Oseltamivir is a highly selective inhibitor of influenza A and B virus neuraminidases and Ribavirin in the triphosphate form efficiently inhibits the RNA polymerase of those viruses [4]. And as for the structure, Ribavirin is the only drug approved for the treatment of influenza viruses which is a nucleoside analogue and its structure is the closest to the structure of the compounds synthesized in this study. Table 1 demonstrates that the IC50 value of the lead compound 4i is very close to the IC50 value of Ribavirin that can be an indirect indication that 4i affects the same viral target as Ribavirin does.
The results of an in vitro study of cytotoxic and antiviral properties of several synthesized compounds against Coxsackie B3 virus are presented in Table 2. It can be seen that almost all the compounds studied were inactive against the Coxsackie B3 virus. The only exception is the 1,2,3-triazolyl pyrimidine analogue 8k in which the 1,2,3-triazolylribofuranosyl residue is attached via propylene linker to the C5 atom of the 6-methyluracil moiety. This lead compound showed both high antiviral activity (IC50 = 9 μM) and good selectivity index (SI = 16).
The analysis of Table 2 enables to note the following two interesting features. Firstly, it was namely compound 8k with protected hydroxy groups of sugar residue showed maximum antiviral activity against coxsackievirus B3. In our opinion, this is the first case when antiviral activity was exhibited by a nucleoside analogue with protected hydroxy groups of the sugar residue. Secondly, Ribavirin having the structure similar to that of compound 8k, in contrast to 8k, appeared to be inactive against this enterovirus. At the same time, compound 8k showed antiviral activity against coxsackie B3 virus which was three times higher than that of Pleconaril having the structure significantly different from that of 8k. As to Pleconaril, it prevents the interaction of Coxsackievirus genome with surface glycoproteins ICAM-1 of cells of the human immune system thus inhibiting the viral replication [40]. Does this mean that compound 8k affect the same target of Coxsakie B3 virus? The question is certainly interesting and requires a special research.
Molecular docking
Current researches on developing new antiviral drugs focus on a limited number of potential targets [41]. Most approved antivirals target surface proteins of influenza A virus [41] such as the M2 ion channel, neuraminidase and hemagglutinin which allow the virus to interact with the cell surface. However, the future belongs to antivirals inhibiting proteins which drive the virus replication cycle [42] such as viral polymerase, non-structural proteins and nucleoproteins. Great hopes are pinned on new inhibitors of RNA-dependent RNA polymerase (RdRp) [43] because of its critical role in virus replication and high degree of sequence conservation in influenza A and B viruses, particularly in the active sites for RNA binding, cleavage or elongation [44]. The RdRp consists of three separate polypeptides called polymerase basic 2 (PB2), polymerase basic 1 (PB1) and polymerase acidic (PA) [45]. If these proteins will be inhibited or their mutual interaction will be disrupted, the RNA-dependent RNA polymerase will not be able to function and virus replication will be stopped [43, 45]. It has been shown that the antiviral activity of nucleoside and nucleobase analogues can be due to interactions with the fragment of PA containing the N-terminal endonuclease domain (PA-Nter) of RdRp [43, 46]. This is also true for compounds having similar structural motif with the lead compound 4i, e.g., BMS-183355, BMS-183021 [47] and Ribavirin (RBV) [4, 7, 14–17]. Therefore, we decided to test the ability of the lead compounds 4i, 8n as well as RBV which was chosen as one of the reference compounds to bind to RNA-dependent RNA polymerase (RdRp) in molecular docking simulations. As a potential target we chose the ligand-binding domain (LBD) of acidic polymerase (PA) of RNA-dependent RNA polymerase. The PA-Nter domain has a cation-dependent endonuclease active site core, the catalytic residues His41, Glu80, Asp108 and Glu119 being conserved among all influenza A subtypes and strains [42]. In addition to synthesized compounds 4i, 8n and reference compound RBV, the ability of their 5′-triphosphate (TP) derivatives 4i-TP, 8n-TP and RBV-TP to bind to the LBD has been also evaluated, because nucleoside analogues undergo an intracellular phosphorylation and inhibit RdRp in a 5′-triphosphate form [48–50].
We have evaluated the binding of 4i, 8n and Ribavirin (RBV) as well as their 5′-triphosphate derivatives 4i-TP, 8n-TP and RBV-TP to the PA-Nter endonuclease domain (PDB code 4AWK). The positions of the optimized docking models of compounds 4i, 8n, 4i-PPP, 8n-PPP demonstrating the best binding energy in the PA-Nter active site are shown in Fig. 1. According to the docking simulations, all ligands are located deep in the cavity of the active site of PA-Nter, being in the same amino acid environment. Compounds 4i, 8n and RBV do not show a similar binding motif in LBD PA-Nter. The lead compound 4i is retained in the PA-Nter cavity by hydrogen bonding of the C5′-OH group with the amino acid residues Arg82, Glu23 and by the π–π interaction between the quinazoline-2,4-dione moiety and Tyr24. The electrostatic interaction of the amino acid residues Ala20, Ile38 and His41 with the polar part of the quinazoline-2,4-dione moiety also contributes to the binding. However, the lead compound 8n binds to the protein cavity mainly due to nonspecific interactions, and the only electrostatic interaction is observed with the Glu23. RBV binding occurs due to the hydrogen bond of the C5′-OH group with Tyr130 and the π–π interaction of the 1,2,4-triazole ring of RBV with the imidazole moiety of His41, as well as hydrophobic interactions of RBV with Ala20, Ile120 and Val122. At the same time, 4i-TP and 8n-TP bind to the PA-Nter cavity in a similar manner due to the hydrogen bonds of their heterocyclic moieties with the Glu80` residue and the triphosphate anion moiety with the Lys134 residue. Besides, 8n-TP forms additional hydrogen bonds with the Glu23 and Leu106 residues of the protein molecule and 4i-TP binds by electrostatic interactions with amino acid residues Glu23, Tyr24 and Lys137. The binding pattern of RBV-TP and triphosphates 4i-TP and 8n-TP in the PA-Nter cavity differs significantly. The RBV-TP molecule interacts with the protein through the hydrogen bonds of its heterocyclic moiety with Pro107 and the sugar residue with Glu119.
Fig. 1.


Molecular docking simulations and two-dimensional interaction map of the optimized docking model of compounds 4i (a), 4i-TP (b), 8n (c), 8n-TP (d) RBV (e), RBV-TP (f) in the PA-Nter (PDB code 4AWK) active site obtained in the lowest-energy conformations
The lead compounds 4i and 8n were found to bind equally well to the PA-Nter active site with a high binding energy (− 8.1 and − 7.9 kcal/mol, respectively), while RBV binding is worser (− 6.1 kcal/mol) (Table 3). At the same time, their 5′-triphosphate derivatives 4i-TP, 8n-TP and RBV-TP bind to the LBD PA-Nter better than their precursors demonstrating binding energies − 8.3, − 8.5 and − 8.0 kcal/mol, respectively. It should be noted that if there is no a correlation between the antiviral activity and the binding energy of the ligand–protein complex for the lead compounds 4i, 8n and reference compound RBV, then for their 5′-triphosphate derivatives 4i-TP, 8n-TP and RBV-TP such a correlation is clearly visible in Table 3. The IC50 values decrease in the series of 5′-triphosphates RBV-TP > 4i-TP > 8n-TP from 32 to 15 μM, that is, the antiviral activity increases in this row, and in the same direction the binding energy increases from 8.0 to 8.5 kkal/mol, that is, the binding of these 5′-triphosphates with the active site of the protein becomes more stronger when going from RBV-TP to 8n-TP. Since Ribavirin inhibits RNA-dependent RNA polymerase (RdRp) in the 5′-triphosphate form [4, 48, 49], the observed correlation testifies that the cause of the antiviral activity of the lead compounds 4i and 8n against influenza virus A/PR/8/34 can also be associated with the inhibition of RdRp by their 5′-triphosphate derivatives.
Table 3.
Inhibitory activity and binding energies of compounds 4i, 8n, RBV and their hypothetical 5′-triphosphate derivatives 4i-TP, 8n-TP, RBV-TP obtained by molecular docking simulations

Conclusion
In summary, a series of 1,2,3-triazolyl nucleoside analogues have been synthesized using “click” chemistry methodology. It has been observed that 1,2,3-triazolyl derivatives of natural nucleosides uridine (1g, 1h, 1i) and thymidine (3g, 3h, 3i) lack antiviral activity against influenza virus A/PR/8/34/(H1N1). On the contrary, the antiviral activity was demonstrated by 1,2,3-triazolyl nucleoside analogues in which the nucleic bases were replaced by 6-methyluracil (compounds 2h, 2i, 8j, 8k, 8m, 8n) and quinazoline-2,4-dione (compounds 4g, 4h, 4i) moieties. The best values of IC50 and SI were demonstrated by the lead compound 4i in which the 1,2,3-triazolylribofuranosyl fragment is attached to the N1 atom of the quinazoline-2,4-dione moiety via butylene linker (IC50 = 30 μM, CC50 > 719 μM, SI = 24). Compound 8n in which the 1,2,3-triazolylribofuranosyl fragment is attached to the N5 atom of the uracil moiety via propylene linker showed the highest antiviral activity among the studied compounds (IC50 = 15 µM) but more higher cytotoxicity (CC50 = 79 µM) and very low selectivity index (SI = 5). It should be noted that alkylation of the N3 atom of the 6-methyluracil moiety in the 1,2,3-triazole nucleoside analogues, that is, going from compounds 2g, 2h, 2i to compounds 6g, 6h, 6i leads to the complete loss of antiviral activity against influenza virus A H1N1. Almost all the compounds studied appeared to be inactive against the coxsackievirus B3. The only exception is the 1,2,3-triazolyl pyrimidine analogue 8k in which the 1,2,3-triazolylribofuranosyl residue is attached via propylene linker to the C5 atom of the 6-methyluracil moiety. This lead compound showed both high antiviral activity (IC50 = 9 μM) and a good selectivity index (SI = 16). According to theoretical calculations, the antiviral activity of the 1,2,3-triazolyl nucleoside analogues 4i, 8n against H1N1 (A/PR/8/34) influenza virus can be explained by their influence on the functioning of the polymerase acidic protein (PA) of RNA-dependent RNA polymerase (RdRp).
Experimental
Chemicals and instrumentations
The 1H NMR spectra were recorded on 400 MHz and 600 MHz Brucker Advance. 13C NMR spectra were obtained in the above instruments operating at 100.6 MHz. Melting points were obtained on an Electrothermal IA 9000 instrument (Electrothermal, Great Britain). Mass spectra (MALDI) were recorded in a positive ion mode on a Bruker Ultraflex III TOF/TOF mass spectrometer for 10−3 mg/ml solutions. The ESI MS measurements were performed using an AmazonX ion trap mass spectrometer (Bruker Daltonic GmbH, Germany) in positive mode in the mass range of 70–3000. The capillary voltage was 3500 V, nitrogen drying gas 10 L min−1, desolvation temperature 250 °C. A methanol/water solution (70:30) was used as a mobile phase at a flow rate of 0.2 mL/min by binary pump (Agilent 1260 chromatograph, USA). The sample was dissolved in methanol to a concentration of 10−6 g L−1. Flash chromatography was performed on silica gel 60 (40–63 μm, Buchi, Sepacore). Thin-layer chromatography was carried out on plates with silica gel (Sorbfil, Russia). Spots of compounds were visualized by using ultraviolent fluorescence under a short wavelength (254 nm) followed by heating the plates (at ca. 150 °C) after immersion in a solution of 5% H2SO4 and 95% H2O. All reactions sensitive to air and/or moisture were carried out under argon atmosphere with anhydrous solvents. Anhydrous solvents were purified and dried (where appropriate) according to standard procedures.
Starting ω-alkyne substituted pyrimidines
Pyrimidine derivatives 1a–c; 2a–c; 3a–c; 4a,c; 6a–c with ω-alkyne substituents at the N1 atom were prepared as described earlier [36]. Spectral data of 1b,c, 2a–c, 3b,c, 4a,c, 6a–c were in keeping with published ones [36]. Spectral data of 1a, 3a agreed with the literature [51]. Pyrimidine derivatives 8d, 8e, 8g were synthesized according to the known protocols [37, 38]. Spectral parameters of 8e and 8g agreed with those presented in Refs. [37, 38], respectively.
N1-(Pent-4′-yn-1′-yl)quinazoline-2,4-dione (4b)
Concentrated sulfuric acid (0.15 mL) was added under stirring at room temperature to a suspension of quinazoline-2,4(1H,3H)-dione (3 g, 19.0 mol) and hexamethyldisilazane (HMDS) (6.86 g, 47.0 mmol) in toluene (100 mL). The mixture was refluxed for 4 h, the solvent and excessive HMDS were distilled off, and a solution of 5-iodo-1-pentyne (5.53 g, 28.5 mol) in DMF (3 mL) were added in the residue. The mixture was stirred at 110–120 °C for 12 h. The solvent was distilled off, chloroform (150 mL) was added to the residue, and the mixture was filtered. The filtrate was concentrated to 15–20 mL, and the residue was subjected to column chromatography on silica gel using first petroleum ether, second petroleum ether-ethyl acetate (1.5:1) and then ethyl acetate as eluent. Compound 4b was isolated from the fraction of ethyl acetate as a cream-colored powder. Yield: 3.54 g (82%); mp: 140 °C. 1H NMR (400 MHz, CDCl3): δ 8.42 (s, 1H, NH), 8.23 (dd, 1H, J = 1.8, 7.9 Hz, ArH.), 7.74–7.68 (m, 1H, ArH), 7.36–7.27 (m, 2H, ArH.), 4.24 (t, 2H, J = 7.7 Hz, CH2-11), 2.41–2.33 (m, 2H, CH2-13), 2.07 (t, 1H, J = 2.8 Hz, CH-15), 2.02–1.93 (m, 2H, CH2-12). 13C NMR (100 MHz, CDCl3): δ 161.89 (C-4), 150.33 (C-2), 140.94 (C-9), 135.55 (C-10), 128.99, 123.08, 116.15, 113.88 (C-5, C-6, C-7, C-8), 82.85 (C-14), 69.50 (C-15), 41.95 (C-11), 25.72 (C-13), 16.03 (C-12). MALDI-MS calcd. for C13H12N2O2: [M+H]+ 229.1, [M+Na]+ 251.1, [M+K]+ 267.1; found: [M+H]+ 229.2, [M+Na]+ 251.2, [M+K]+ 267.2. Anal. calcd. for C13H12N2O2, %: C, 68.41; H, 5.30; N, 12.27; found, %: C, 68.31; H, 5.39, N, 12.34. M 228.25.
5-(Pent-4′-yn-1′-yl)-6-methyluracil (8h)
The mixture of 6-methyl-5-(pent-4′-yn-1′-yl)-2-thio-4-oxo(1H,3H)-pyrimidine 8e (4.36 g, 21.0 mol) and chloroacetic acid (3.97 g, 40.5 mol) in 200 ml of water was refluxed for 20 h. The reaction mixture was cooled, and the precipitate was filtered off and recrystallized from water. After drying compound 8h was obtained as a white powder. Yield: 2.62 g (65%); mp: 225 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.85 (s, 1H, NH), 10.57 (s, 1H, NH), 2.74 (t, 1H, J = 2.6 Hz, CH-11), 2.28 (t, 2H, J = 7.7 Hz, CH2-9), 2.15–2.11 (m, 2H, CH2-7), 2.05 (s, 3H, CH3), 1.54–1.48 (m, 2H, CH2-8). 13C NMR (100 MHz, DMSO-d6): δ 164.20 (C-4), 150.63 (C-2), 147.92 (C-6), 107.97 (C-5), 84.30 (C-10), 71.05 (C-11), 27.28 (C-7), 23.16 (C-9), 17.34 (C-8), 15.73 (CH3). MALDI-MS calcd. for C10H12N2O2: [M+H]+ 193.1, [M+Na]+ 215.1, [M+K]+ 231.1; found: [M+H]+ 193.2, [M+Na]+ 215.2, [M+K]+ 231.2. Anal. calcd. for C10H12N2O2, %: C 62.49; H 6.29; N 14.57; found, %: C 62.59; H 6.20, N 14.47. M 192.22.
Ethyl 2-acetyl-7-octynoate (8c)
Acetoacetic ester (20 g, 153.8 mol) was added dropwise with stirring to a solution of sodium (3.54 g, 153.9 mol) in 200 ml of ethanol at 0–5 °C, the mixture then was treated with 6-iodo-hex-1-yne (13.12 g, 160.0 mol) at temperature not above than 10 °C. The reaction mixture was stirred for 2.5 h at room temperature and then refluxed for 10 h.The solvent was removed, and the residue was treated with chloroform and filtered. Distillation of the dried (MgSO4) chloroform solution gave the product 8c as a colorless oil. Yield: 21.00 g (65%); bp: 90–95 °C/1.00 mm. 1H NMR (400 MHz, CDCl3): δ 4.11 (q, 2H, J = 7.2 Hz, CH2-9), 3.32 (t, 1H, J = 7.6 Hz, CH-2), 2.13 (s, 3H, CH3-12), 2.12–2.07 (m, 2H, CH2-3), 1.86 (t, 1H, J = 5.2 Hz, CH-8), 1.80–1.72 (m, 2H, CH2-6), 1.50–1.41 (m, 2H, CH2-5), 1.36–1.28 (m, 2H, CH2-4), 1.19 (t, 3H, J = 7.2 Hz, CH3-10). 13C NMR (100 MHz, CDCl3): δ 202.64 (C-1), 169.50 (C-11), 83.76 (C-7), 68.40 (C-8), 61.08 (C-9), 59.47 (C-2), 28.54 (C-3), 27.89 (C-6), 27.35, 26.19 (C-5, C-4), 17.90 (C-12), 13.88 (C-10). Anal. calcd. for C12H18O3, %: C 68.54; H 8.63; found, %: C 68.59; H 8.70. M 210.27.
6-Methyl-5-(Hex-5-yn-1-yl)-2-thio-4-oxo(1H,3H)-pyrimidine (8f)
The mixture of 8c (7.27 g, 34.6 mol), thiourea (2.63 g, 34.6 mol) and K2CO3 (4.77 g, 34.6 mol) in 150 ml of methanol was refluxed for 15 h. The solvent was distilled off, the residue was soluted in water, and the solution was acidified to approximately pH 2–3. The precipitate was recrystallized from methanol to afford compound 8f. Yield: 4.60 g (60%); mp: 216 °C. 1H NMR (600 MHz, DMSO-d6): δ 12.22 (s, 1H, NH), 12.03 (s, 1H, NH), 2.71–2.62 (m, 1H, CH-12), 2.27–2.21 (m, 2H, CH2-10), 2.20–2.13 (m, 2H, CH2-7), 2.11 (s, 3H, CH3-6), 1.47–1.37 (m, 4H, CH2-8, CH2-9). 13C NMR (100 MHz, DMSO-d6): δ 173.76 (C-4), 161.18 (C-2), 148.15 (C-6), 114.42 (C-5), 84.25 (C-11), 70.92 (C-12), 27.50 (C-7), 26.98 (C-10), 23.25 (C-9), 17.40 (C-8), 15.53 (CH3). MALDI-MS calcd. for C11H14N2OS: [M+H]+ 223.1, [M+Na]+ 245.1, [M+K]+ 261.1; found: [M+H]+ 223.1, [M+Na]+, 245.1, [M+K]+ 261.1. Anal. calcd. for C11H14N2OS, %: C 59.43; H 6.35; N 12.60; S 14.42; found, %: C 59.55; H 6.27, N 12.50; S 14.50. M 222.31.
5-(Hex-5′-yn-1′-yl)-6-methyluracil (8i)
The mixture of compound 8f (2.52 g, 11.3 mol) and chloroacetic acid (2.10 g, 22.2 mol) in 100 ml of water was refluxed for 20 h. The reaction mixture was cooled, and the precipitate was filtered off and recrystallized from water to afford the target compound 8i as a white powder. Yield: 1.88 g (80%); mp: 203 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.83 (s, 1H, NH), 10.55 (s, 1H, NH), 2.67 (m, 1H, CH-12), 2.23–2.13 (m, 4H, CH2-7, CH2-10), 2.04 (s, 3H, CH3), 1.45–1.37 (m, 4H, CH2-8, CH2-9). 13C NMR (100 MHz, DMSO-d6): δ 164.24 (C-4), 150.64 (C-2), 147.57 (C-6), 108.60 (C-5), 84.36 (C-11), 70.96 (C-12), 27.54 (C-7), 23.21 (C-10), 17.43 (C-9, C-8), 15.74 (CH3). MALDI-MS calcd. for C11H14N2O2: [M+H]+ 207.1, [M+Na]+ 229.1, [M+K]+ 245.1; found: [M+H]+ 207.1, [M+Na]+, 229.1, [M+K]+ 245.1. Anal. calcd. for C11H14N2O2, %: C 64.06; H 6.84; N 13.58; found, %: C 64.15; H 6.87, N 13.50. M 206.25.
Target 1,2,3-triazolyl nucleosides analogues
1,2,3-Triazolyl nucleoside analogues 1g, h, i; 2g, h, i; 3g, h, i; 4g, h, i; 6g, h, i; 8m, n, o were synthesized according to the protocol described in Ref. [36]. Spectral data of 1h, i; 2g, h, i; 3h, i; 4g, i; 6g, h, i were in keeping with published ones [36]. The spectroscopic data of 1g, 3g were found to be in agreement with those previously reported [32].
N1-{[1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]propyl}-2,4(1H,3H)-quinazolinedione (4e)
A white foam, yield: 0.38 g (83%). 1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H, NH), 8.20 (dd, 1H, J = 7.8, 1.5 Hz, ArH.), 7.73–7.67 (m, 1H, ArH), 7.67 (s, 1H, CH-5″), 7.31–7.23 (m, 2H, ArH), 6.16 (d, 1H, J = 3.6 Hz, CH-1′), 5.82 (dd, 1H, J = 5.2, 3.9 Hz, CH-3′), 5.62 (t, 1H, J = 5.2 Hz, CH-2′), 4.49–4.44 (m, 1H, CH-4′), 4.41 (dd, 1H, J = 12.1, 3.2 Hz, CH-5′a), 4.27–4.18 (m, 3H, CH-5′b, CH2-11), 2.88 (t, 2H, J = 7.1 Hz, CH2-13), 2.23–2.15 (m, 2H, CH2-12), 2.12 (s, 3H, OAc), 2.11 (s, 3H, OAc), 2.07 (s, 3H, OAc). 13C NMR (100 MHz, CDCl3): 170.35, 169.40, 169.23 (COCH3), 161.88 (C-4), 150.60 (C-2), 146.91 (C-4″), 140.80 (C-9), 135.61, 128.76, 123.05, 116.09, 114.20 (C-5, C-6, C-7, C-8, C-10,), 120.72 (C-5″), 89.85 (C-1′), 80.71 (C-4′), 74.24 (C-3′), 70.79 (C-2′), 63.01 (C-5′), 42.11 (C-11), 26.31, 22.59 (C-12, C-13), 20.64, 20.41, 20.34 (COCH3). MALDI-MS calcd. for C24H27N5O9: [M+H]+ 530.1, [M+Na]+ 552.1, [M+K]+ 568.1; found: [M+H]+ 530.1, [M+Na]+ 552.1, [M+K]+ 568.0. Anal. calcd. for C24H27N5O9, %: C 54.44; H 5.14; N 13.23; found, %: C 54.41; H 5.19, N 13.26. M 529.50.
N1-{[1-(β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]propyl}-2,4(1H,3H)-quinazolinedione (4h)
A white foam, yield: 0.14 g (92%). 1H NMR (400 MHz, DMSO-d6): δ 11.49 (s, 1H, NH), 8.04 (s, 1H, CH-5″), 7.98 (dd, 1H, J = 7.9, 1.5 Hz, ArH.), 7.75–7.70 (m, 1H, ArH), 7.39 (d, 1H, J = 8.4 Hz, ArH), 7.26 (t, 1H, J = 7.5 Hz, ArH), 5.86 (d, 1H, J = 4.8 Hz, CH-1′), 4.32 (t, 1H, J = 4.8 Hz, CH-3′), 4.12–4.05 (m, 3H, CH-2′, CH2-11), 3.96–3.93 (m, 1H, CH-4′), 3.58 (dd, 1H, J = 12.1, 3.9 Hz, CH-5′a), 3.48 (dd, 1H, J = 12.1, 4.4 Hz, CH-5′b), 2.73 (t, 2H, J = 7.5 Hz, CH2-13), 1.98–1.89 (m, 2H, CH2-12). 13C NMR (100 MHz, DMSO-d6): 162.43 (C-4), 150.63 (C-2), 146.99 (C-4″), 141.10 (C-9), 136.00, 128.09, 123.13, 115.98, 114.99 (C-5, C-6, C-7, C-8, C-10,), 120.96 (C-5″), 92.30 (C-1′), 85.99 (C-4′), 75.36 (C-3′), 70.73 (C-2′), 61.77 (C-5′), 42.01 (C-11), 26.79, 22.56 (C-12, C-13). MALDI-MS calcd. for C18H21N5O6: [M+H]+ 404.1, [M+Na]+ 426.1, [M+K]+ 442.1; found: [M+H]+ 404.1, [M+Na]+ 426.1, [M+K]+ 442.1. Anal. calcd. for C18H21N5O6, %: C 53.59; H 5.25; N 17.36; found, %: C 53.56; H 5.29, N 17.38. M 403.39.
6-Methyl-5-{[1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]methyl}-2,4(1H,3H)-pyrimidinedione (8j)
A white foam, yield: 0.41 g (75%). 1H NMR (600 MHz, CD3OD): δ 7.88 (s, 1H, CH-5″), 6.21 (d, 1H, J = 3.5 Hz, CH-1′), 5.85–5.83 (m, 1H, CH-3′), 5.62 (t, 1H, J = 5.5 Hz, CH-2′), 4.48–4.45 (m, 1H, CH-4′), 4.35 (dd, 1H, J = 12.3, 3.5 Hz, CH-5′a), 4.18 (dd, 1H, J = 12.5, 4.3 Hz, CH-5′b), 3.77 (s, 2H, CH2-7), 2.22 (s, 3H, CH3), 2.09 (s, 3H, OAc), 2.08 (s, 3H, OAc), 1.99 (s, 3H, OAc). 13C NMR (100 MHz, CD3OD): δ 172.19, 171.38, 171.08 (COCH3), 166.54 (C-4), 152.91 (C-2), 151.74 (C-6), 147.87 (C-4″), 123.36 (C-5″), 108.68 (C-5), 91.35 (C-1′), 82.22 (C-4′), 75.49 (C-3′), 72.25 (C-2′), 63.94 (C-5′), 21.71 (C-7), 20.64, 20.42, 20.32 (COCH3), 16.79 (CH3). MALDI-MS calcd. for C19H23N5O9: [M+Na]+ 488.1, [M+K]+ 504.1; found: [M+Na]+ 488.1, [M+K]+ 504.1. Anal. calcd. for C19H23N5O9, %: C 49.03; H 4.98; N 15.05; found, %: C 48.99; H 4.99, N 15.09. M 465.41.
6-Methyl-5-{[1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]propyl}-2,4(1H,3H)-pyrimidinedione (8k)
A white foam, yield: 0.56 g (68%). 1H NMR (400 MHz, CDCl3): δ 9.66 (s, 1H, NH), 8.96 (s, 1H, NH), 7.61 (s, 1H, CH-5″), 6.13 (d, 1H, J = 4.0 Hz, CH-1′), 5.83–5.79 (m, 1H, CH-3′), 5.61 (t, 1H, J = 5.1 Hz, CH-2′), 4.48–4.39 (m, 2H, CH-4′, CH-5′a), 4.22 (dd, 1H, J = 11.9, 4.0 Hz, CH-5′b), 2.77 (t, 2H, J = 7.3 Hz, CH2-9), 2.44 (t, 2H, J = 8.2 Hz, CH2-7), 2.15 (s, 3H, CH3), 2.12 (s, 3H, OAc), 2.11 (s, 3H, OAc), 2.08 (s, 3H, OAc), 1.91–1.82 (m, 2H, CH2-8). 13C NMR (100 MHz, CDCl3): δ 170.48, 169.46, 169.30 (COCH3), 164.44 (C-4), 151.71 (C-2), 148.21 (C-6), 147.66 (C-4″), 120.30 (C-5″), 110.74 (C-5), 89.85 (C-1′), 80.74 (C-4′), 74.25 (C-3′), 70.85 (C-2′), 63.03 (C-5′), 27.92 (C-9), 25.09 (C-7), 24.06 (C-8), 20.72, 20.46, 20.40 (COCH3), 16.79 (CH3). MALDI-MS calcd. for C21H27N5O9: [M+H]+ 494.5, [M+Na]+ 516.5; found: [M+H]+ 494.5, [M+Na]+ 516.5. Anal. calcd. for C21H27N5O9, %: C 51.11; H 5.51; N 14.19; found, %: C 51.03; H 5.55, N 14.21. M 493.47.
6-Methyl-5-{[1-(2′,3′,5′-tri-O-acetyl-β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]butyl}-2,4(1H,3H)-pyrimidinedione (8l)
A white foam, yield: 0.55 g (65%). 1H NMR (400 MHz, CDCl3): δ 9.93 (s, 1H, NH), 9.17 (s, 1H, NH), 7.58 (s, 1H, CH-5″), 6.13 (d, 1H, J = 3.8 Hz, CH-1′), 5.81–5.77 (m, 1H, CH-3′), 5.63 (t, 1H, J = 5.4 Hz, CH-2′), 4.48–4.39 (m, 2H, CH-4′, CH-5′a), 4.23 (dd, 1H, J = 11.6, 4.1 Hz, CH-5′b), 2.77 (t, 2H, J = 7.5 Hz, CH2-10), 2.38 (t, 2H, J = 7.7 Hz, CH2-7), 2.16 (s, 3H, CH3), 2.12 (s, 3H, OAc), 2.11 (s, 3H, OAc), 2.08 (s, 3H, OAc), 1.76–1.67 (m, 2H, CH2-9), 1.54–1.45 (m, 2H, CH2-8). 13C NMR (100 MHz, CDCl3): δ 170.52, 169.48, 169.30 (COCH3), 164.25 (C-4), 151.66 (C-2), 148.29 (C-6), 147.11 (C-4″), 120.16 (C-5″), 111.22 (C-5), 89.94 (C-1′), 80.65 (C-4′), 74.35 (C-3′), 70.81 (C-2′), 63.07 (C-5′), 28.72 (C-10), 27.91 (C-7), 24.99, 24.10 (C-9, C-8), 20.71, 20.48, 20.41 (COCH3), 16.80 (CH3). MALDI-MS calcd. for C22H29N5O9: [M+H]+ 508.4, [M+Na]+ 530.4; found: [M+H]+ 508.4, [M+Na]+ 530.4. Anal. calcd. for C22H29N5O9, %: C 52.07; H 5.76; N 13.80; found, %: C 52.01; H 5.79, N 13.85. M 507.4.
6-Methyl-5-{[1-(β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]methyl}-2,4(1H,3H)-pyrimidinedione (8m)
A white powder, yield: 0.1 g (83%); mp: 259–260 °C. 1H NMR (600 MHz, DMSO-d6): δ 7.94 (s, 1H, CH-5″), 5.83 (d, 1H, J = 4.7 Hz, CH-1′), 5.57 (br s, 1H, OH), 5.28 (br s, 1H, OH), 5.05 (br s, 1H, OH), 4.35–4.29 (m, 1H, CH-3′), 4.11–4.04 (m, 1H, CH-2′), 3.96–3.91 (m, 1H, CH-4′), 3.6 (s, 2H, CH2-7), 3.58–3.43 (m, 2H, CH2-5′), 2.11 22 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δ 164.45 (C-4), 151.09 (C-2), 149.86 (C-6), 146.01 (C-4″), 120.95 (C-5″), 106.88 (C-5), 92.07 (C-1′), 85.89 (C-4′), 75.09 (C-3′), 70.57 (C-2′), 61.60 (C-5′), 20.61 (C-7), 16.51 (CH3). MALDI-MS calcd. for C13H17N5O6: [M+Na]+ 362.1; found: [M+Na]+ 362.2. Anal. calcd. for C13H17N5O6, %: C 46.02; H 5.05; N 20.64; found, %: C 45.99; H 5.09, N 20.62. M 339.30.
6-Methyl-5-{[1-(β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]propyl}-2,4(1H,3H)-pyrimidine-dione (8n)
A white powder, yield: 0.36 g (78%); mp: 191–193 °C. 1H NMR (600 MHz, CD3OD): δ 8.08 (s, 1H, CH-5″), 6.00 (d, 1H, J = 3.8 Hz, CH-1′), 4.47 (t, 1H, J = 4.2 Hz, CH-3′), 4.29 (t, 1H, J = 4.7 Hz, CH-2′), 4.13–4.10 (m, 1H, CH-4′), 3.81 (dd, 1H, J = 12.4, 3.1 Hz, CH-5′a), 3.68 (dd, 1H, J = 12.3, 4.1 Hz, CH-5′b), 2.74 (t, 2H, J = 7.3 Hz, CH2-9), 2.40 (t, 2H, J = 7.6 Hz, CH2-7), 2.11 (s, 3H, CH3), 1.84–1.78 (m, 2H, CH2-8). 13C NMR (100 MHz, CD3OD): δ 167.01 (C-4), 152.94 (C-2), 150.37 (C-6), 148.92 (C-4″), 122.24 (C-5″), 110.97 (C-5), 94.58 (C-1′), 87.25 (C-4′), 77.12 (C-3′), 71.96 (C-2′), 62.91 (C-5′), 29.35 (C-9), 25.86 (C-7), 24.86 (C-8), 16.39 (CH3). ESI-MS calcd. for C15H21N5O6: [M+H]+ 368.2, [M+Na]+ 390.2; found: [M+H]+ 368.2, [M+Na]+ 390.2. Anal. calcd. for C15H21N5O6, %: C 49.04; H 5.76; N 19.06; found, %: C 49.00; H 5.79, N 19.09. M 367.36.
6-Methyl-5-{[1-(β-d-ribofuranosyl)-1H-1,2,3-triazol-4-yl]butyl}-2,4(1H,3H)-pyrimidinedione (8o)
A white powder, yield: 0.11 g (76%); mp: 179–181 °C. 1H NMR (400 MHz, CD3OD): 8.00 (s, 1H, CH-5″), 5.98 (d, 1H, J = 4.0 Hz, CH-1′), 4.46 (t, 1H, J = 4.6 Hz, CH-3′), 4.30 (t, 1H, J = 5.1 Hz, CH-2′), 4.13–4.08 (m, 1H, CH-4′), 3.80 (dd, 1H, J = 12.1, 3.3 Hz, CH-5′a), 3.68 (dd, 1H, J = 12.1, 4.4 Hz, CH-5′b), 2.73 (t, 2H, J = 7.5 Hz, CH2-10), 2.36 (t, 2H, J = 7.7 Hz, CH2-7), 2.12 (s, 3H, CH3), 1.74–1.65 (m, 2H, CH2-9), 1.51-1.42 (m, 2H, CH2-8). 13C NMR (100 MHz, CD3OD): δ 167.02 (C-4), 152.96 (C-2), 150.10 (C-6), 149.22 (C-4″), 122.01 (C-5″), 111.42 (C-5), 94.35 (C-1′), 87.14 (C-4′), 77.06 (C-3′), 71.98 (C-2′), 62.97 (C-5′), 30.09 (C-10), 29.29 (C-7), 26.03, 25.02 (C-9, C-8), 16.41 (CH3). MALDI-MS calcd. for C16H23N5O6: [M+H]+ 382.2; found: [M+H]+ 382.2. Anal. calcd. for C16H23N5O6, %: C 50.39; H 6.08; N 18.36; found, %: C 50.36; H 6.11, N 18.34. M 381.4.
Docking study
Molecular docking was carried out using the AutoDock 4.2 Vina software and AutoDock Tools (ADT 1.5.6) [52]. The three-dimensional (3D) crystal structure of N-terminal endonuclease domain of polymerase acidic protein (PA) of RNA-dependent RNA polymerase (PDB code 4AWK) [53] was obtained from the RCSB Protein Data Bank [54]. The standard 3D structures of 4i, 8n, RBV, 4i-TP, 8n-TP and RBV-TP were constructed using the HyperChem 8.0 [55] and converted into an pdb file by Open Babel [56], and a cubic grid box of 16 × 16 × 18 Å (x, y, z) with a spacing of 1.000 Å and grid maps were generated. The docking parameters were used as the default settings. The enzyme–ligand interactions were detected using ADT 1.5.6 and have been presented as 2D diagrams.
Biology
Cytotoxicity assay
MDCK (ATCC CCL-34) and Vero (ATCC CCL-81) cells initially obtained from American Type Culture Collection (Rockville, MD, USA) were seeded into 96-well plates and incubated for 24 h at 36 °C at 5% CO2 until confluent monolayer is formed. Threefold dilutions (400–4 μg/mL) were prepared on Eagle’s minimal essential medium (MEM) from the compounds under investigation, added to the cells and incubated for 24 h at 36 °C at 5% CO2. The cell monolayer was washed twice with saline (0.9% NaCl), and 100 μL of MTT solution [3-(4,5-dimethylthiazole-2)-2,5-diphenyltetrazolium bromide], 0.5 μg/mL in MEM, was added into each well. The plates were incubated for 1 h at 36 °C, then the medium was removed and formazan pellets were dissolved in dimethyl sulfoxide (0.1 mL per well). The optical density in the wells was measured on a spectrophotometer Thermo Multiskan FC at the wavelength of 540 nm. The results obtained were used for calculating the concentration of the compound resulting in death of 50% cells in the culture (CC50) using GraphPad Prism software employing the four-parameter logistic curve model. The values of CC50 were then converted from μg/mL to μM.
Cell protection assay
The compounds in appropriate concentrations were added to cells (0.1 mL per well). Cells were further infected with either A/Puerto Rico/8/34 (H1N1) influenza virus (for MDCK cells) or Coxsackie B3 virus (for Vero cells) (m.o.i 0.01 in both cases) and incubated for 48 h at 36 °C at 5% CO2. After that, cell viability was assessed by MTT test (see above). The cytoprotective activity of compounds was considered as their ability to increase the values of OD comparing to control wells (with virus only, no drugs). Based on the results obtained, the values of IC50, i.e., concentration of compounds that result in 50% cells protection, were calculated using GraphPad Prism software.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the Assigned Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for technical assistance in research.
Abbreviations
- PA
Polymerase acidic protein
- RdRP
RNA-dependent RNA polymerase
- PA-Nter
N-terminal endonuclease domain
- LBD
Ligand-binding domain
Author contributors
VEK and VES performed design of the compounds and analyses of chemical and biological experiments; LFS and MMS contributed to syntheses of ω-alkyne-substituted pyrimidines; OVA and MGB were involved in syntheses of 1,2,3-triazolyl nucleosides analogues; BFG helped in molecular modeling; VVZ, AVS and ILY conducted antiviral activity studies.
Funding
This work was financially supported by the Russian Science Foundation (Grant No. 19-13-00003). In part of synthesis initial compounds, Marina M. Shulaeva is grateful to the Ministry of Education and Science of the Russian Federation (theme no AAAA-A18-118040390114-8) for financial support.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lieberman MA, Ricer R. Biochemistry, molecular biology, and genetics. Philadelphia: Wolters Kluwer Health; 2020. [Google Scholar]
- 2.Seley-Radtke KL, Yates MK (2018) The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir Res 154:66–86 (and references herein cited). 10.1016/j.antiviral.2018.04.004 [DOI] [PMC free article] [PubMed]
- 3.Yates MK, Seley-Radtke KL (2019) The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: complex modifications to the nucleoside scaffold Antivir Res 162:5–21 (and references herein cited). 10.1016/j.antiviral.2018.11.016 [DOI] [PMC free article] [PubMed]
- 4.Clercq E, Li G (2016) Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29:695–747 (and references herein cited). 10.1128/CMR.00102-15 [DOI] [PMC free article] [PubMed]
- 5.Clercq E (2009) The discovery of antiviral agents: ten different compounds, ten different stories. Med Res Rev 28:929–953 (and references herein cited). 10.1002/med.20128 [DOI] [PubMed]
- 6.Matyugina ES, Khandazhinskaya AP, Kochetkov SN (2012) Carbocyclic nucleoside analogues: classification, target enzymes, mechanisms of action and synthesis. Russ Chem Rev 81:729–746 (and references herein cited). 10.1070/RC2012v081n08ABEH004314 [DOI]
- 7.Clercq E (2009) Another ten stories in antiviral drug Discovery (Part C): ‘‘Old’’ and “new” antivirals, strategies, and perspectives Med Res Rev 29:611–645 (and references herein cited). 10.1002/med.20153 [DOI] [PubMed]
- 8.Vedula MS, Jennepalli S, Aryasomayajula R, Rondla SR, Musku MR, Kura RR, Bandi PR. Novel nucleosides as potent influenza viral inhibitors. Bioorg Med Chem. 2010;18:6329–6339. doi: 10.1016/j.bmc.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Saladino R, Neri V, Checconi P, Celestino I, Nencioni L, Palamara AT, Crucianelli M. Synthesis of 2′-deoxy-1′-homo-N-nucleosides with anti-influenza activity by catalytic methyltrioxorhenium (MTO)/H2O2 oxyfunctionalization. Chem Eur J. 2013;19:2392–2404. doi: 10.1002/chem.201201285. [DOI] [PubMed] [Google Scholar]
- 10.Wang G, Wan J, Hu Y, Wu X, Prhavc M, Dyatkina N, Rajwanshi VK, Smith DB, Jekle A, Kinkade A, Symons JA, Jin Z, Deval J, Zhang Q, Tam Y, Chanda S, Blatt L, Beigelman L. Synthesis and anti-influenza activity of pyridine, pyridazine, and pyrimidine C-nucleosides as Favipiravir (T-705) analogues. J Med Chem. 2016;59:4611–4624. doi: 10.1021/acs.jmedchem.5b01933. [DOI] [PubMed] [Google Scholar]
- 11.Lin C, Sun C, Liu X, Zhou Y, Hussain M, Wan J, Li M, Li X, Jin R, Tu Z, Zhang J. Design, synthesis, and in vitro biological evaluation of novel 6-methyl-7-substituted-7-deaza purine nucleoside analogs as anti-influenza A agents. Antivir Res. 2016;129:13–20. doi: 10.1016/j.antiviral.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Yoon J-J, Toots M, Lee S, Lee M-E, Ludeke B, Luczo JM, Ganti K, Cox RM, Sticher ZM, Edpuganti V, Mitchell DG, Lockwood MA, Kolykhalov AA, Greninger AL, Moore ML, Painter GR, Lowen AC, Tompkins SM, Fearns R, Natchus MG, Plempera RK. Orally efficacious broad-spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses antimicrob. Antimicrob Agents Chemother. 2018;62:e00766-18. doi: 10.1128/AAC.00766-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeuchi T, Sriwilaijaroen N, Sakuraba A, Hayashi E, Kamisuki S, Suzuki Y, Ohrui H, Sugawara F. Design, synthesis, and biological evaluation of EDAP, a 4′-ethynyl-2′-deoxyadenosine 5′-monophosphate analog, as a potent influenza A inhibitor. Molecules. 2019;24:2603. doi: 10.3390/molecules24142603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnussen CR, Douglas RG, Jr, Betts RF, Roth FK, Meagher MP. Double-blind evaluation of oral Ribavirin (Virazole) in experimental influenza a virus infection in volunteers. Antimicrob Agents Chemother. 1977;12:498–502. doi: 10.1128/AAC.12.4.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simons C, Wu Q, Htar TT. Recent advances in antiviral nucleoside and nucleotide therapeutics. Curr Top Med Chem. 2005;5:1191–1203. doi: 10.2174/156802605774463051. [DOI] [PubMed] [Google Scholar]
- 16.Crotty S, Cameron C, Andino R. Ribavirin’s antiviral mechanism of action: lethal mutagenesis? J Mol Med. 2002;80:86–95. doi: 10.1007/s00109-001-0308-0. [DOI] [PubMed] [Google Scholar]
- 17.Pauly MD, Lauring AS. Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J Virol. 2015;89:3584–3597. doi: 10.1128/JVI.03483-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clercq E, Cools M, Balzarini J, Snoeck R, Andrei G, Hosoya M, Shigeta S, Ueda T, Minakawa N, Matsuda A. Antiviral activities of 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboxamide and related compoundst. Antimicrob Agents Chemother. 1991;35:679–684. doi: 10.1128/AAC.35.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung D-H, Kumarapperuma SC, Sun Y, Li Q, Chu Y-K, Arterburn JB, Parker WB, Smith J, Spik K, Ramanathan HN, Schmaljohn CS, Jonsson CB. Synthesis of 1-β-d-ribofuranosyl-3-ethynyl-[1,2,4]triazole and its in vitro and in vivo efficacy against Hantavirus. Antivir Res. 2008;79:19–27. doi: 10.1016/j.antiviral.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krajczyk A, Kulinska K, Kulinski T, Hurst BL, Day CW, Smee DF, Ostrowski T, Januszczyk P, Zeidler J. Antivirally active ribavirin analogues—4,5-disubstituted 1,2,3-triazole nucleosides: biological evaluation against certain respiratory viruses and computational modelling. Antivir Chem Chemother. 2014;23:161–171. doi: 10.3851/IMP2564. [DOI] [PubMed] [Google Scholar]
- 21.Ostrowski T, Zeidler J. Synthesis of 5-ethynyl-1-β-d-robofuranosyl-1H-[1,2,3]triazole-4-carboxylic acid amide (isosteric to EICAR) and its derivatives. Nucleic Acids Symp Ser. 2008;52:585–586. doi: 10.1093/nass/nrn296. [DOI] [PubMed] [Google Scholar]
- 22.McDowell M, Gonzales SR, Kumarapperuma SC, Jeselnik M, Arterburn JB, Hanleya KA. A novel nucleoside analog, 1-β-d-ribofuranosyl-3-ethynyl-[1,2,4]triazole (ETAR), exhibits efficacy against a broad range of flaviviruses in vitro. Antivir Res. 2010;87:78–80. doi: 10.1016/j.antiviral.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhurilo NI, Chudinov MV, Matveev AV, Smirnova OS, Konstantinova ID, Miroshnikov AI, Prutkov AN, Grebenkina LE, Pulkova NV, Shvets VI. Isosteric ribavirin analogues: synthesis and antiviral activities. Bioorg Med Chem Lett. 2018;28:11–14. doi: 10.1016/j.bmcl.2017.11.029. [DOI] [PubMed] [Google Scholar]
- 24.Petrova KT, Potewar TM, Correia-da-Silva P, Barros MT, Calhelha RC, Ciric A, Sokovic M, Ferreira ICFR. Antimicrobial and cytotoxic activities of 1,2,3-triazole-sucrose derivatives. Carbohydr Res. 2015;417:66–71. doi: 10.1016/j.carres.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Zhou C-H, Wang Y. Recent researches in triazole compounds as medicinal drugs. Curr Med Chem. 2012;19:239–280. doi: 10.2174/092986712803414213. [DOI] [PubMed] [Google Scholar]
- 26.Ruddarraju RR, Murugulla AC, Kotla R, Tirumalasetty MCB, Wudayagiri R, Donthabakthuni S, Maroju R, Baburao K, Parasa LS. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur J Med Chem. 2016;123:379–396. doi: 10.1016/j.ejmech.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 27.Chaudhary PM, Chavan SR, Shirazi F, Razdan M, Nimkar P, Maybhate SP, Likhite AP, Gonnade R, Hazara BG, Deshpande MV, Deshpande SR. Exploration of click reaction for the synthesis of modified nucleosides as chitin synthase inhibitors. Bioorg Med Chem. 2009;17:2433–2440. doi: 10.1016/j.bmc.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 28.Park SM, Yang H, Park S-K, Kim HM, Kim BH. Design, synthesis, and anticancer activities of novel perfluoroalkyltriazole-appended 2′-deoxyuridines. Bioorg Med Chem Lett. 2010;20:5831–5834. doi: 10.1016/j.bmcl.2010.07.126. [DOI] [PubMed] [Google Scholar]
- 29.Montagu A, Roy V, Balzarini J, Snoeck R, Andrei G, Agrofoglio LA. Synthesis of new C5-(1-substituted-1,2,3-triazol-4 or 5-yl)-2′-deoxyuridines and their antiviral evaluation. Eur J Med Chem. 2011;46:778–786. doi: 10.1016/j.ejmech.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Shmalenyuk ER, Chernousova LN, Karpenko IL, Kochetkov SN, Smirnova TG, Andreevskaya SN, Chizhov AO, Efremenkova OV, Alexandrova LA. Inhibition of Mycobacterium tuberculosis strains H37Rv and MDR MS-115 by a new set of C5 modified pyrimidine nucleosides. Bioorg Med Chem. 2013;21:4874–4884. doi: 10.1016/j.bmc.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Alexandrova LA, Efremenkova OV, Andronova VL, Galegov GA, Solyev PN, Karpenko IL, Kochetkov SN. 5-(4-Alkyl-1,2,3-triazol-1-yl)methyl derivatives of 2′-deoxyuridine as inhibitors of viral and bacterial growth. Russ J Bioorg Chem. 2016;42:677–684. doi: 10.1134/S1068162016050022. [DOI] [Google Scholar]
- 32.Elayadi H, Mesnaoui M, Korba BE, Smietana M, Vasseur JJ, Secrist JA, Lazrek HB. Preparation of 1,4-disubstituted-1,2,3-triazolo ribonucleosides by Na2CuP2O7 catalyzed azide-alkyne 1,3-dipolar cycloaddition. ARKIVOC. 2012;viii:76–89. doi: 10.3998/ark.5550190.0013.807. [DOI] [Google Scholar]
- 33.St. Amant AH, Bean LA, Guthrie JP, Hudson RHE. Click fleximers: a modular approach to purine base-expanded ribonucleoside analogues. Org Bioorg Chem. 2012;10:6521–6525. doi: 10.1039/c2ob25678a. [DOI] [PubMed] [Google Scholar]
- 34.Parmenopoulou V, Chatzileontiadou DSM, Manta S, Bougiatioti S, Maragozidis P, Gkaragkouni D-N, Kaffesaki E, Kantsadi AL, Skamnaki VT, Zographos SE, Zounpoulakis P, Balatsos NAA, Komiotis D, Leonidas DD. Triazole pyrimidine nucleosides as inhibitors of Ribonuclease A. Synthesis, biochemical, and structural evaluation. Bioorg Med Chem. 2012;20:7184–7193. doi: 10.1016/j.bmc.2012.09.067. [DOI] [PubMed] [Google Scholar]
- 35.Chatzileontiadou DSM, Tsika AC, Diamantopoulou Z, Delbe J, Badet J, Courty J, Skamnaki VT, Parmenopoulou V, Komiotis D, Hayes JM, Spyroulias GA, Leonidas DD. Evidence for novel action at the cell-binding site of human angiogenin revealed by heteronuclear NMR spectroscopy, in silico and in vivo studies. ChemMedChem. 2018;13:259–269. doi: 10.1002/cmdc.201700688. [DOI] [PubMed] [Google Scholar]
- 36.Andreeva OV, Belenok MG, Saifina LF, Shulaeva MM, Dobrynin AB, Sharipova RR, Voloshina AD, Saifina AF, Gubaidullin AT, Khairutdinov BI, Zuev YF, Semenov VE, Kataev VE. Synthesis of novel 1,2,3-triazolyl nucleoside analogues bearing uracil, 6-methyluracil, 3,6-dimethyluracil, thymine, and quinazoline-2,4-dione moieties. Tetrahedron Lett. 2019;60:151276. doi: 10.1016/j.tetlet.2019.151276. [DOI] [Google Scholar]
- 37.Wilson JG. Synthetic approaches to a carboranyl thiouracil. Pigment Cell Res. 1989;2:297–303. doi: 10.1111/j.1600-0749.1989.tb00208.x. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds RC, Trask TW, Sedwick WD. 2,4-Dichloro-5-(l-O-carboranylmethyl)-6-methylpyrimidine: a potential synthon for 5-(1-O-carboranylmethyl)pyrimidines. J Org Chem. 1991;56:2391–2395. doi: 10.1021/jo00007a026. [DOI] [Google Scholar]
- 39.Nisic F, Speciale G, Bernardi A. Stereoselective synthesis of α- and β-glycofuranosyl amides by traceless ligation of glycofuranosyl azides. Chem Eur J. 2012;18:6895–6906. doi: 10.1002/chem.201200309. [DOI] [PubMed] [Google Scholar]
- 40.Thibaut HJ, De Palma AM, Neyts J. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem Pharm. 2012;83:185–192. doi: 10.1016/j.bcp.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 41.Krug RM, Aramini JM. Emerging antiviral targets for influenza A virus. Trends Pharm. 2009;30:269–277. doi: 10.1016/j.tips.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das K, Aramini JM, Ma L-C, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat SMB. 2010;17:530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stevaert A, Naesens L. The influenza virus polymerase complex: an update on its structure, functions, and significance for antiviral drug design. Med Res Rev. 2016;36:1127–1173. doi: 10.1002/med.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogolino D, Carcelli M, Sechi M, Neamati N. Viral enzymes containing magnesium: metal binding as a successful strategy in drug design. Coord Chem Rev. 2012;256:3063–3086. doi: 10.1016/j.ccr.2012.07.006. [DOI] [Google Scholar]
- 45.Stubbs TM, Velthuis AJW. The RNA-dependent RNA polymerase of the influenza A virus. Future Virol. 2014;9:863–876. doi: 10.2217/fvl.14.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pala N, Stevaert A, Dallocchio R, Dessì A, Rogolino D, Carcelli M, Sanna V, Sechi M, Naesens L. Virtual screening and biological validation of novel influenza virus PA endonuclease inhibitors. ACS Med Chem Lett. 2015;6:866–871. doi: 10.1021/acsmedchemlett.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clanci C, Chung TDY, Meanwell N, Putz H, Hagen M, Oclonno RJ, Krystal M. Identification of N-hydroxamic acid and N-hydroxyimide compounds that inhibit the influenza virus polymerase. Antivir Chem Chemother. 1996;7:353–360. doi: 10.1177/095632029600700609. [DOI] [Google Scholar]
- 48.Wray SK, Gilbert BE, Knight V. Effect of ribavirin triphosphate on primer generation and elongation during influenza virus transcription in vitro. Antivir Res. 1985;5:39–48. doi: 10.1016/0166-3542(85)90013-0. [DOI] [PubMed] [Google Scholar]
- 49.Wu JZ, Larson G, Walker H, Shim JH, Hong Z. Phosphorylation of Ribavirin and Viramidine by adenosine kinase and cytosolic 5′-nucleotidase II: implications for ribavirin metabolism in erythrocytes. Antimicrob Agents Chemother. 2005;49:2164–2171. doi: 10.1128/AAC.49.6.2164-2171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furuta Y, Komeno T, Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B. 2017;93:449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazrek HB, Taourirte M, Oulih T, Barascut JL, Imbach JL, Pannecouque C, Witrouw M, Clercq E. Synthesis and anti-HIV activity of new modified 1,2,3-triazole acyclonucleosides. Nucleosides Nucleotides Nucleic Acids. 2001;20:1949–1960. doi: 10.1081/NCN-100108325. [DOI] [PubMed] [Google Scholar]
- 52.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OHJ, Ruigrok RWH, Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS. 2012;8:e1002831. doi: 10.1371/journal.ppat.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bermsn HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.HyperChem Professional 8.0 (2007). Hypercube, Inc. http://www.hyper.com/?tabid=360. Accessed 14 Sept 2020
- 56.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.