

Abstract
Migalastat (1-deoxygalactonojirimycin) is approved for the treatment of Fabry disease (FD) in patients with an amenable mutation. Currently, there are at least 367 amenable and 711 non-amenable mutations known, based on an in vitro good laboratory practice (GLP) assay. Recent studies demonstrated that in vitro amenability of mutations did not necessarily correspond to in vivo amenability of migalastat-treated patients. This discrepancy might be due to (methodological) limitations of the current GLP-HEK assay. Currently, there are several published comparable cell-based amenability assays, with partially different outcomes for the same tested mutation, leading to concerns in FD-treating physicians. The aim of this review is to elucidate the idea of amenability assays from their beginning, starting with patient-specific primary cells to high-throughput assays based on overexpression. Consequently, we compare methods of current assays, highlighting their similarities, as well as their pros and cons. Finally, we provide a literature-based list of α-galactosidase A mutations, tested by different assays to provide a comprehensive overview of amenable mutations as a good basis for the decision-making by treating physicians. Since in vitro amenability does not always correspond with in vivo amenability, the treating clinician has the responsibility to monitor clinical and laboratory features to verify clinical response.
Graphical Abstract
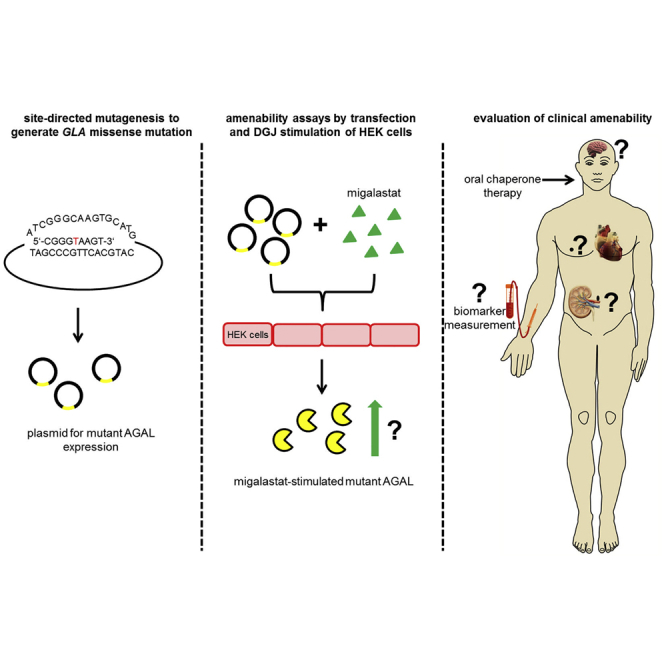
Fabry disease patients with amenable mutations can be treated with migalastat. Lenders and colleagues compare methods of current amenability assays, highlighting their similarities, as well as their pros and cons. They provide a literature-based list of α-galactosidase A mutations, tested by different assays to provide a comprehensive overview of amenable mutations as a good basis for the decision-making by treating physicians.
Main Text
Fabry disease (FD; Online Mendelian Inheritance in Man [OMIM] #301500) is an X chromosome-linked inborn error due to various mutations within the α-galactosidase A (GLA/AGAL) gene, resulting in deficient enzymatic AGAL activity. Fabry-specific manifestations are a consequence of systemic accumulation of glycolipids (mainly globotriaosylceramide [Gb3]) in various tissues and cell types.1 The progressive Gb3 accumulation is accompanied by a high risk of early onset of stroke, life-threatening arrhythmia, myocardial infarction, or cardiac and renal failure. Since 2001, enzyme replacement therapy (ERT) with agalsidase-alfa (Replagal, Shire/Takeda) and agalsidase-beta (Fabrazyme, Sanofi Genzyme) is available for treatment, resulting in intracellular Gb3 reduction, leading to a clinical stabilization or at least a slowed disease progression in males and females.2, 3, 4, 5, 6, 7, 8, 9 A second treatment option based on a pharmaceutical chaperone (migalastat, 1-deoxygalactonojirimycin [DGJ]; Galafold, Amicus Therapeutics) has been approved in Europe since May 2016, in Canada since September 2017, in Japan since March 2018, and in the United States since August 2018 for long-term treatment of FD in adults (≥18 years of age in United States and Canada, ≥16 years in other countries) for patients with an amenable mutation and an estimated glomerular filtration rate (eGFR) ≥30 mL/min per 1.73 m2. Amenability, which means the response in terms of increasing enzymatic activities of an AGAL mutation to migalastat, is currently being tested in a cell culture-based good laboratory practice (GLP) assay.10 Several amenability assays have been published during the recent years, partially with different outcomes for the same mutation. This review aims to provide background information, including the history of migalastat, and focus on the current referred amenability assays to explain their function, similarities, and differences and their pros and cons, which might explain observed inter-assay results.
Identification of Migalastat as a Pharmaceutical Chaperon to Treat Patients with FD
Most missense GLA mutations result in an unstable and misfolded protein, leading to reduced AGAL activities within affected cells. Misfolded proteins will not pass the protein quality control mechanism within the endoplasmic reticulum (ER), resulting in a premature degradation before reaching the lysosomes.11,12 To restore folding and stability of the protein, pharmacological chaperones can be used, which bind reversibly to the active center of the protein.13 The first in vitro studies on FD were performed by adding galactose as a chaperone to fibroblasts carrying the mutation p.Q279E, resulting in an increase of AGAL activity by 15%.14 Infusion of galactose (1 g/kg body weight) in a patient with p.G328R and a cardiac phenotype was effective but not practically implementable due to the high frequency and durations of infusions (every other day, 4 h).15 The small molecule DGJ is an iminosugar, which was initially identified as a competitive inhibitor of the AGAL enzyme. However, at sub-inhibitory concentrations (extracellular, 20–100 μM), a binding to the enzyme’s catalytic center (wild-type and amenable mutations) can facilitate proper protein folding in the ER and an accelerated maturation and trafficking to the lysosome.16,17 This resulted in an increase of enzymatic AGAL activity in healthy control cells and, importantly, in Fabry patient-derived lymphoblasts carrying the first described responsive mutations p.R301Q and p.Q279E.16,17 Daily oral intake resulted in increased AGAL activities in the heart, kidneys, liver, and spleen in heterozygous as well as homozygous transgenic mice with a mutant p.R301Q background.18,19 Stimulation of these transgenic mice with daily orally-applied DGJ during 2 weeks finally led to a decreased Gb3 storage in kidneys.19 Prolonged treatment durations and dose optimization resulted in a Gb3 clearance of additional FD-relevant tissues such as the brain and the heart, especially in aged transgenic mice, indicating a superior effect of every other day treatment over a daily intake.20 In contrast to ERT, increased AGAL activities and decreased Gb3 concentrations within the brain further demonstrated the ability of DGJ to cross the blood-brain barrier.20 Of note, the co-administration of ERT and migalastat resulted in an increased AGAL tissue uptake and improved Gb3 reduction in Fabry mice21 as well as higher systemic exposures and tissue levels of AGAL in Fabry patients.22 Unfortunately, this potentially very effective combination strategy was not followed up.
Impact of Migalastat on Clinical Outcomes
The safety and efficacy of oral migalastat at 123 mg every other day in patients with genetically confirmed FD were assessed in two randomized, multi-center, placebo-controlled (FACETS)23 or active comparator-controlled (ATTRACT)24 phase III trials. Migalastat was efficacious in ERT-naive and ERT-experienced patients with amenable AGAL mutations, resulting in reduced cardiac mass, stable renal function, and increased and sustained endogenous AGAL activity levels, with reductions in Gb3 accumulation in renal tissue. Plasma globotriaosylsphingosine (lyso-Gb3) levels in previously untreated patients decreased23 or remained constant in ERT-pretreated patients24 under migalastat. Real-world data from a single center as well as a multi-center study confirmed the safety, a reduction of cardiac mass, and biochemical outcomes after 12 months of migalastat treatment.25,26 In contrast, a stabilization of renal function was not observed in either of the two studies. However, due to the currently limited number and duration of studies,27 short-term observations and heterogeneous and small patient cohorts might have influenced partially different outcomes, and results should be interpreted carefully. Future studies including well-characterized patient cohorts (separated by sex, phenotype, and other factors) and longer observational periods are now warranted to further clarify the impact of migalastat in patients with amenable mutations.
First Amenability Assays
A response (currently termed amenability) of AGAL mutations to sub-inhibitory concentrations of DGJ had (and still has) to be checked individually for every (missense) mutation. First characterizations were performed using patient-specific cell lines identifying p.R301Q, p.Q279E,16 as well as p.T194I and p.V390fsX812 as amenable. Later on, Benjamin et al.28 additionally analyzed the amenability of 75 different AGAL mutations in patient-derived lymphoblasts. However, the large variety of individual mutations required a more feasible method with a less time- and money-consuming approach to measure the amenability of AGAL variations to DGJ. In this respect, Shimotori et al.29 used COS7 cells, individually transfected with AGAL mutants identified in their patient cohort and incubated with 10 μM DGJ during 3 days, to identify 11 missense mutations as amenable. Since a response of overexpressed AGAL mutants in cell culture to DGJ did not necessarily reflect a comparable situation in FD-specific cells, another cell-based assay in human embryonic kidney (HEK)293 cells was designed, and outcomes were compared to appropriate male Fabry patient-derived lymphoblasts also incubated with DGJ.30 Overexpression and subsequent incubation of 81 mutants with different DGJ concentrations for 4–5 days identified 49 mutations with a significant response.30 The combination of HEK293 cells and overexpression constructs is the basis for the currently used amenability assays. Two open-label uncontrolled phase II studies of 12 and 24 weeks in nine males with FD documented that a treatment with 150 mg of migalastat every other day led to increased AGAL activities and decreased Gb3 concentrations in six of nine patients.31 As inclusion criteria, a missense GLA mutation, residual AGAL activity of at least 3% of normal, and an increase in AGAL activity by at least 20% in the presence of 20 μM migalastat in patients’ cultured lymphocytes were required.31 Of note, the three patients who did not show a consistent response in vivo after migalastat intake had AGAL mutations that did not respond to migalastat in transfected HEK293T cells.31 However, the positive biochemical outcomes were subsequently confirmed in females32 and further supported by reduced plasma lyso-Gb3 levels in patients with amenable mutations from a phase II study.33
Current Amenability Assays
Evolving numbers of newly identified GLA mutations over time required high-throughput (HTP) assays, which are able to measure a response of an AGAL mutation without the need of patient-specific material. In addition, inconsistencies between in vitro and in vivo amenability required further improved assays. In general, all currently used assays are based on HEK293 cells, which are transfected with individual GLA mutations and subsequently incubated with DGJ to measure an increase of enzymatic activity (Figure 1). In the following discussion, the most recent HTP assays are presented and discussed concerning their pros and cons (Table 1).
Figure 1.

Schematic Overview of the Basic Principles of HEK293 Amenability Assays
HEK assays can be separated into five steps. Step 1: cloning of GLA mutations using site-directed mutagenesis. Step 2: transfection of seeded HEK293 cells with a mock control (negative control; necessary for AGAL background substraction), wild-type AGAL (positive control; reference activity is set to 100%), and the GLA mutation of interest. Step 3: incubation of cells with or without DGJ. Step 4: cell lysis to release overexpressed AGAL. Step 5: AGAL activities are measured and compared to each other. Depending on the assay protocols, various sub-steps can be included in between, such as wash-outs, determination of transfection efficiencies, among others. AGAL/GLA, α-galactosidase A; DGJ, 1-deoxygalactonojirimycin; WT, wild-type.
Table 1.
Comparison of the Currently Used Cited In-House and Good Laboratory Practice Amenability Assays
| Assay | Lukas et al.34,35 | Benjamin et al.10(GLP-HEK Assay) | Oommen et al.36 | Lenders et al.37(HEK293T GLA Knockout Assay) | Lenders et al.37(Patient-Derived Urinary Cells) |
|---|---|---|---|---|---|
| Cells | HEK293H | HEK293 | HEK293H (GripTite 293 MSR) | HEK293T | fibroblast-like primary immortalized cell line |
| Duration (days) | 2.5 | 5 | 5 | 2 | 2 |
| DGJ (μM) | 20 | 10 | 10 | 10 (20) | 10 + 20 |
| Expression plasmid | pcDNA3.1/V5-His6 | pcDNA6 | pcDNA6/V5-His | pcDNA3.1 | not required |
| Transfection efficiency | quantitative western blots | qPCR | commercial SEAP | commercial luciferase | not required |
| Amenability criteria | 1.5-fold over baseline or >5% compared to untreated value | ≥1.2-fold over baseline + absolute increase of ≥3% wild-type activity | |||
| Pros | high-throughput screening | no overexpression model | |||
| no patient samples required | patient-specific mutation in appropriate genetic background | ||||
| identification of amenability in mutations with high residual activity | identification of amenability in mutations with high and low residual activity | ||||
| DGJ wash-out (2 h) | DGJ wash-out | ||||
| no endogenous AGAL activity | assessment of potential Gb3 depletion | ||||
| no heterodimerization between wild-type and mutant AGAL | |||||
| Cons | overexpression model | requires patients’ urine | |||
| high background due to endogenous AGAL activity | immortalization process | ||||
| heterodimerization between wild-type and mutant AGAL | time consuming | ||||
| no NAGA inhibition mentioned | overexpression of mutant AGAL might affect SEAP trafficking and secretion | overexpression of mutant AGAL might affect luciferase trafficking and secretion | expensive | ||
| no DGJ wash-out mentioned | no DGJ wash-out mentioned | ||||
AGAL, α-galactosidase A; DGJ, 1-deoxygalactonojirimycin; Gb3, globotriaosylceramide; HEK, human embryonic kidney; NAGA, α-galactosidase B; SEAP, secreted embryonic alkaline phosphatase.
One of the first assays was established by Lukas et al.34 using HEK293H cells transfected with GLA mutations, which were subsequently incubated with 20 μM DGJ for 60 h (Table 1). The amount of 20 μM chaperone did not reflect the average maximum concentration in the plasma following a single dose (150 mg) of migalastat, which has been calculated as 10 μM.38 Although not directly stated, the authors used 20 μM DGJ probably according to the original studies by Fan et al.16 Amenability criteria were defined as a 1.5-fold increase of AGAL activity over baseline or >5% activity compared to the untreated control.34 In their recent meta study, the authors adopted the current GLP-HEK assay criteria (absolute increase by ≥3% of wild-type AGAL activity and a ≥1.2-fold increase of baseline enzymatic activity) and identified 89 out of 178 mutations as amenable.34,35,39 Although used as a basis for future assays, the setup has some limitations that need to be considered. Since migalastat is an inhibitor, a wash-out step after incubation is very important.12,16 Wash-out to remove traces of DGJ was not reported,34 nor was a specific inhibition of α-galactosidase B (NAGA). NAGA was demonstrated to degrade the artificial substrate 4-methylumbelliferyl-α-d-galactopyranoside,40 which was used in the fluorimetric-based enzyme activity assay. An insufficient competitive inhibition of NAGA may lead to slightly increased AGAL activities,41 increasing the risk for false-positive results, especially for AGAL mutations with low residual activities. A further critical issue is the lack of proper transfection efficacy controls. Only a semiquantitative western blot to detect protein level changes was applied,34 which is not a proper tool to assess transfection efficiencies in general.
Due to the lack of comprehensive transfection controls in their initial work, a GLP-HEK assay was established by Benjamin et al.10 based on the same assay but featuring some improvements (Table 1). This GLP-HEK assay is the basis for the accreditation, and only patients with amenable mutations according to this assay are approved to be treated with migalastat. Most importantly, the transfection efficacy for every transfection was now generally controlled by qPCR, and cells were stimulated with 10 μM chaperone. However, the stimulation duration was now prolonged to 5 days combined with a wash-out of 2 h. Of note, considering the approved medication (every other day) for migalastat, the prolonged stimulation (5 days) seems to be a long period and does not reflect the in vivo situation correctly. However, due to the small-scale setup in 96-well plates, these changes were probably required to detect sufficient activity changes. In contrast to amenability criteria by Lukas et al.,34,35 mutations were, and still are, defined as amenable when AGAL activity is ≥1.2-fold over baseline with an additional absolute increase of ≥3% wild-type AGAL activity in the presence of 10 μM migalastat.10 Based on this assay and definition, 1,384 amenable and 754 non-amenable theoretical mutations have been identified so far, for which migalastat treatment is approved (https://www.galafoldamenabilitytable.com/; last updated in May 2020).
Recently, a comparable amenability assay was implemented by Oommen et al.36 to detect potential inter-assay variability among AGAL mutations (Table 1). In accordance with the GLP-HEK assay, the authors used the same experimental setup, including stimulation with 10 μM DGJ for 5 days.36 Using the same amenability criteria, the authors identified 6 out of 59 tested mutations that did not match the classification of amenability reported using the GLP-HEK assay.10 In contrast to the GLP-HEK assay, four mutations were identified as not amenable and two mutations were identified as amenable.36 A comparison to the GLP-HEK assay-derived data revealed poor assay reproducibility. In contrast to qPCR, secreted embryonic alkaline phosphatase (SEAP) was used to measure and correct for transfection efficacy.36 However, a possible impact of the overexpressed mutant AGAL on trafficking and secretion of SEAP and vice versa was not completely excluded until now and is discussed as a limitation of the assay.42 In addition, a wash-out of chaperone was not explicitly mentioned by the authors.
Although all three assays share the advantage of an HTP tool, without the need of patient-specific material, they share a major disadvantage. They are based on an artificial overexpression model, not reflecting the in vivo situation in a patient or of patient-derived cells. Furthermore, the assays are performed in wild-type HEK cells with endogenous AGAL activity, requiring the subtraction of background activity from transfected cells. In addition, the presence of a wild-type AGAL might also lead to a heterodimerization of the wild-type and the mutated AGAL, as observed for other proteins previously.43 This might decrease assay sensitivity and lead to false-positive results, especially for mutations with very low AGAL activities, as recently demonstrated for some mutations.26,37 The latter is addressed by a recent assay based on CRISPR-Cas9-mediated GLA-knockout HEK293T cells (Table 1).37 Although this assay also depends on overexpression, an effect of endogenous AGAL can be excluded, increasing the sensitivity especially for mutations with low AGAL activities. Furthermore, cells were stimulated for 2 days, making this setup more comparable to the in vivo situation.23,24 A luciferase assay system to examine transfection efficacy was used and, as criticized for the SEAP, it is not clear whether overexpressed mutant AGAL might impair the reporter protein and vice versa. However, based on this next-generation assay, mutations of patients who did not biochemically respond to migalastat were identified as not amenable.26,37 To further substantiate these data, a more specific assay based on patient-derived urinary cells37 was designed (Table 1). Comparable to the assays of the pre-HTP area, this assay uses patient-derived (urinary fibroblast-like cells) incubated either with 10 or 20 μM chaperone for 2 days. Using the same amenability criteria as for the GLP-HEK assay, it could be demonstrated that AGAL activity of an amenable mutation increased and intracellular Gb3 inclusions decreased after DGJ stimulation, whereas a mutation previously termed amenable did not respond to DGJ, and appropriate cells accumulated Gb3.37 Due to the time-consuming extraction and cultivation of primary cells and subsequent immortalization, this assay does not provide the advantages of HTP assays. Similar to the pre-HTP assays, this assay can be used to investigate the real AGAL activity of a specific mutation and provides an impression on the potential biochemical outcomes in terms of intracellular Gb3 inclusions as well. Hence, this assay seems to be a patient- and mutation-specific tool for mutations with low AGAL activities.
Comparisons between the amenability assays in wild-type HEK cells demonstrated significant inter-assay discrepancies. Table 2 provides a comprehensive overview of amenable AGAL mutations (according to the GLP-HEK assay) in comparison to the other three in-house assays. Interestingly, when comparing outcomes for amenable mutations (according to the GLP-HEK assay) with data from the in-house assays by Lukas et al.39 and Oommen et al.,36 the in-house assay by Lukas et al. fits better to the GLP-HEK assay (Figure 2). Amenability data by Lenders et al.26 correlated best with the GLP-HEK assay, but results should be carefully interpreted due to the limited number of tested mutations. In conclusion, all observed discrepancies between the assays are probably due to several reasons, including different plasmids used for overexpression, distinct concentrations of DGJ, differences in incubation times, missing wash-outs and absent or different transfection controls, and the potential heterodimerization of endogenous wild-type AGAL (functional) and overexpressed mutant AGAL, which might result in slightly higher measured AGAL activities. This is especially important for AGAL mutations with very low enzymatic activities, in which cases the mutant activity will be measured incorrectly high.
Table 2.
Comparison of Amenability Outcomes Between a GLP-HEK Assay and In-House Assays
| Assay | Benjamin et al.10 | Lukas et al.39 | Oommen et al.36 | Lenders et al.26 | Benjamin et al.10 | Lukas et al.39 | Oommen et al.36 | Lenders et al.26 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acid | w/o DGJ | Absolute Increase %WT, (n-Fold) | w/o DGJ | Absolute Increase %WT, (n-Fold) | w/o DGJ | Absolute Increase %WT, (n-Fold) | w/o DGJ | Absolute Increase %WT, (n-Fold) | Amenable (Yes/No) | |||
| p.L3P | 71.9 | 20.3 (1.3) | 117.7 | 11.3 (1.1) | 69.4 | 37.1 (1.5) | yes | yes | yes | |||
| p.A13T | 51.7 | 10.4 (1.2) | 61.9 | 19.6 (1.3) | yes | yes | ||||||
| p.A15G | 19.0 | 9.0 (1.5) | 15.5 | 15.5 (2.0) | yes | yes | ||||||
| p.A15T | 39.1 | 18.4 (1.5) | 12.4 | 85.2 (7.8) | yes | yes | ||||||
| p.A20D | 4.3 | 5.7 (2.4) | 5.8 | 10.6 (2.8) | yes | yes | ||||||
| p.A20P | 11.5 | 4.4 (1.4) | 13.6 | 22.6 (2.7) | yes | yes | ||||||
| p.W24R | 52.6 | 10.9 (1.2) | 50.5 | 20.6 81.4) | yes | yes | ||||||
| p.D33G | 29.3 | 41.3 (2.4) | 37.4 | 24.6 (1.7) | yes | yes | ||||||
| p.N34S | 0.6 | 16.1 (29.2) | 2.0 | 14.1 (8.2) | 0.9 | 1.5 (2.7) | yes | yes | no | |||
| p.L36W | 0.7 | 15.9 (23.7) | 2.3 | 20 (9.7) | yes | yes | ||||||
| p.A37T | 48.9 | 47.5 (2.0) | 69.6 | 63.3 (1.9) | yes | yes | ||||||
| p.M42L | 38.8 | 21.3 (1.5) | 16.5 | 26.5 (2.6) | yes | yes | ||||||
| p.M42V | 0.5 | 3.8 (8.6) | 0 | 7.2 (n/c) | yes | yes | ||||||
| p.M42T | 2.5 | 17.8 (8.1) | 2.9 | 18.5 (7.4) | yes | yes | ||||||
| p.H46P | 31 | 75.9 (3.4) | 40.1 | 58.7 (2.5) | yes | yes | ||||||
| p.M51K | 6.3 | 15.8 (3.5) | 0 | 8.7 (n/c) | yes | yes | ||||||
| p.M51I | 22.3 | 24.8 82.1) | 37.4 | 24.6 (1.7) | yes | yes | ||||||
| p.C56Y | N/A | 7.3 (n/c) | 0.0 | 6.6 (n/c) | yes | yes | ||||||
| p.Q57L | 71.6 | 21.7 (1.3) | 52.9 | 28.4 (1.5) | yes | yes | ||||||
| p.E59K | 8.6 | 8.9 (2.0) | 2.2 | 16.3 (8.4) | yes | yes | ||||||
| p.P60L | 21.7 | 39.3 (2.8) | 15.6 | 17.5 (2.1) | yes | yes | ||||||
| p.E66G | 34.2 | 11.9 (1.4) | 28.9 | 37.0 (2.3) | yes | yes | ||||||
| p.E66K | 4.8 | 8.1 (2.7) | 6.8 | 11.5 (2.7) | 6.9 | 8.8 (2.3) | yes | yes | yes | |||
| p.M72I | 54.7 | 22.8 (1.4) | 106.5 | 101.9 (2.0) | yes | yes | ||||||
| p.A73V | 53.6 | 33.3 (1.6) | 44 | 20.7 (1.5) | yes | yes | ||||||
| p.D83N | 69.2 | 23.8 (1.3) | 62.9 | 8.7 (1.1) | 187.5 | 54.3 (1.3) | yes | yes | yes | |||
| p.G85M | 7.9 | 4.0 (1.5) | 6.5 | 15.1 (3.3) | yes | yes | ||||||
| p.G85S | 12.4 | 6.7 (1.5) | 7.6 | 12.2 (2.6) | yes | yes | ||||||
| p.I91T | 0.9 | 11.7 (14.0) | 0.7 | 6.3 (10.0) | yes | yes | ||||||
| p.S102L | 19.9 | 42.9 (3.2) | 71.6 | 7.3 (1.1) | yes | yes | ||||||
| p.A108T | 57.1 | 23.7 (1.4) | 73.3 | 11.2 (1.15) | yes | no | ||||||
| p.R112H | 2.6 | 14.8 (6.7) | 1.6 | 17.8 (12.1) | yes | yes | ||||||
| p.R118C | 24.0 | 5.5 (1.3) | 5.3 | 5.8 (2.1) | 45.4 | 20.1 (1.4) | yes | yes | yes | |||
| p.A121T | 18.9 | 49.0 (3.6) | 50 | 5.5 (1.1) | yes | yes | ||||||
| p.S126G | 83.7 | 30.2 (1.4) | 51.3 | 16.1 (1.3) | 143.7 | 26.9 (1.19) | yes | yes | no | |||
| p.G128E | 45.2 | 13.4 (1.3) | 26.4 | 17.4 (1.7) | yes | yes | ||||||
| p.A135V | 0 | 3.7 (n/c) | 0 | 6.9 (n/c) | 0.1 | 0.1 (2.0) | yes | yes | no | |||
| p.D136E | 1.4 | 11.5 (9.2) | 0 | 31.3 (n/c) | yes | yes | ||||||
| p.N139S | 65.5 | 13.6 (1.2) | 64.6 | 9.7 (1.2) | 70.1 | 47.6 (1.7) | 103.4 | 24.0 (1.2) | yes | yes | yes | yes |
| p.A143T | 21.4 | 22.4 (2.0) | 31.3 | 18.1 (1.6) | yes | yes | ||||||
| p.G144D | 50.2 | 26.3 (1.5) | 28.8 | 16.1 (1.6) | yes | yes | ||||||
| p.P146S | 41.9 | 22.2 (1.5) | 17.9 | 22.7 (2.3) | yes | yes | ||||||
| p.A156V | 1.2 | 11.6 (10.7) | 4.3 | 12.5 (3.9) | yes | yes | ||||||
| p.W162G | 0.8 | 5.1 (7.4) | 0 | 5.2 (n/c) | 1.4 | 1.5 (2.1) | yes | yes | no | |||
| p.D165H | 1.3 | 7.0 (6.4) | 3.4 | 8.5 (3.5) | yes | yes | ||||||
| p.D175E | 44.3 | 9.1 (1.2) | 57.9 | 9.6 (1.17) | yes | no | ||||||
| p.G183A | 22.4 | 34.0 (2.5) | 10 | 36.6 (4.6) | yes | yes | ||||||
| p.M187V | 1.3 | 13.6 (11.5) | 22.8 | 44.2 (2.9) | 12.2 | 26.5 (3.2) | yes | yes | yes | |||
| p.M187I | 5.1 | 25.6 (6.0) | 3.1 | 28.1 (10.1) | yes | yes | ||||||
| p.I198T | 64.7 | 30.8 (1.5) | 38.7 | 11.7 (1.3) | 58.3 | 58.5 (2.0) | yes | yes | yes | |||
| p.E203V | 43.0 | 21.5 (1.5) | 144.6 | 72.1 (1.5) | yes | yes | ||||||
| p.P205T | 14.4 | 34.4 (3.4) | 10.2 | 60.2 (6.9) | yes | yes | ||||||
| p.P210S | 75.2 | 38.2 (1.5) | 41.9 | 49.5 (2.2) | yes | yes | ||||||
| p.K213M | 43.2 | 12.7 (1.3) | 31.5 | 12.9 (1.4) | 61.6 | 21.6 (1.4) | yes | yes | yes | |||
| p.P214S | 22.4 | 60.1 (3.7) | 18.1 | 43.5 (3.4) | yes | yes | ||||||
| p.P214L | 33 | 58.6 (2.8) | 19.4 | 44.7 (3.3) | yes | yes | ||||||
| p.N215D | 43.8 | 14.5 (1.3) | 41.1 | 22.4 (1.5) | yes | yes | ||||||
| p.N215S | 15.6 | 20.0 (2.3) | 36.7 | 24.9 (1.7) | 27.1 | 28.1 (2.0) | 26.8 | 57.5 (3.1) | yes | yes | yes | yes |
| p.Y216C | 2 | 18.7 (10.4) | 2.3 | 23.6 (11.2) | yes | yes | ||||||
| p.I219T | 55.8 | 37.8 (1.7) | 53.3 | 32 (1.6) | yes | yes | ||||||
| p.R220Q | 45.2 | 16.5 (1.4) | 115.5 | 38.7 (1.3) | yes | yes | ||||||
| p.N224S | 10.3 | 19.4 (2.9) | 31.1 | 51.1 (2.6) | yes | yes | ||||||
| p.H225D | 43.8 | 66.8 (2.5) | 32.2 | 28.3 (1.9) | yes | yes | ||||||
| p.N228S | 124.5 | 44.7 (1.4) | 59.5 | 11.1 (1.2) | 89.6 | 54.9 (1.6) | yes | yes | yes | |||
| p.I232T | 15 | 70.0 (5.7) | 11.5 | 50.1 (5.4) | yes | yes | ||||||
| p.S238N | 37.1 | 59.3 (2.6) | 36 | 58.3 (2.6) | yes | yes | ||||||
| p.I239T | 37.7 | 55.1 (2.5) | 26.6 | 58.3 (3.2) | yes | yes | ||||||
| p.K240N | N/A | > 3% (≥1.2) | 33.4 | 24.4 (1.7) | yes | yes | ||||||
| p.I242N | 7.6 | 59.8 (59.8) | 3.1 | 46.7 (16.1) | yes | yes | ||||||
| p.L243F | 7.9 | 34.4 (5.4) | 11.4 | 59.4 (6.2) | yes | yes | ||||||
| p.W245G | 44.1 | 18.9 (1.4) | 51.3 | 229.9 (5.5) | yes | yes | ||||||
| p.N249K | 17.9 | 17.3 (2.0) | 23.7 | 30.9 (2.3) | yes | yes | ||||||
| p.Q250P | 24.8 | 33.9 (2.4) | 18.5 | 42.5 (3.3) | yes | yes | ||||||
| p.I253T | 38.9 | 41.3 (2.1) | 73 | 42.8 (1.6) | yes | yes | ||||||
| p.I253S | 3.3 | 27.9 (9.5) | 4.4 | 49 (12.1) | yes | yes | ||||||
| p.P259R | 23.3 | 37 (2.6) | 20.5 | 19.5 (2.0) | yes | yes | ||||||
| p.N263S | 15.8 | 64.7 (5.1) | 6.7 | 57.8 (9.7) | yes | yes | ||||||
| p.D264Y | 0.5 | 5.7 (12.9) | 0 | 5.4 (n/c) | 2.8 | 8.6 (4.0) | yes | yes | yes | |||
| p.V269M | 4.4 | 21.5 (5.9) | 0 | 17.3 (n/c) | yes | yes | ||||||
| p.V269A | 0 | 7.8 (n/c) | 9 | 36 (5.0) | yes | yes | ||||||
| p.G271D | 1.5 | 30.7 (21.5) | 3.1 | 34.8 (12.3) | yes | yes | ||||||
| p.S276N | 2.3 | 7.0 (3.9) | 4.6 | 40.4 (9.9) | yes | yes | ||||||
| p.T282I | 5.2 | 18.5 (4.6) | 5 | 42.7 (9.5) | 12.6 | 9.2 (1.7) | yes | yes | yes | |||
| p.M284V | 25.2 | 37.9 (2.5) | 16.2 | 27.2 (2.7) | yes | yes | ||||||
| p.M290L | 58.6 | 52.8 (1.9) | 69.4 | 53.1 (1.8) | yes | yes | ||||||
| p.A291T | 16.5 | 24.0 (2.5) | 13.2 | 42.5 (4.2) | yes | yes | ||||||
| p.L294S | 0 | 4.9 (n/c) | 0 | 12.4 (n/c) | 0.2 | 0.3 (2.5) | yes | yes | no | |||
| p.R301G | 19.1 | 45.6 (3.4) | 19.3 | 37.2 (2.9) | yes | yes | ||||||
| p.R301Q | 5.5 | 39.0 (8.1) | 8.5 | 39.5 (5.6) | yes | yes | ||||||
| p.R301P | 0 | 4.2 (n/c) | 0 | 5 (n/c) | yes | yes | ||||||
| p.S304N | 94.1 | 27.7 (1.3) | 77.8 | 12.5 (1.16) | yes | no | ||||||
| p.S304T | 76.4 | 40.5 (1.5) | 56.7 | 23.4 (1.4) | yes | yes | ||||||
| p.L310F | 0.8 | 10.8 (14.5) | 1.6 | 26.1 (17.3) | 1.8 | 24.5 (14.6) | yes | yes | yes | |||
| p.L311V | 2 | 16.0 (9.0) | 1.9 | 38.2 (21.1) | yes | yes | ||||||
| p.D313G | 25.5 | 14.9 (1.6) | 25.1 | 25.4 (2.0) | yes | yes | ||||||
| p.D313Y | 59 | 21.9 (1.4) | 83.9 | 16.4 (1.2) | 54.2 | 28.9 (1.5) | yes | yes | yes | |||
| p.V316I | 92.1 | 34.0 (1.4) | 99.8 | 33.4 (1.3) | yes | yes | ||||||
| p.I317T | 6.5 | 17.0 (3.4) | 2.7 | 15.2 (6.7) | yes | yes | ||||||
| p.I319T | 10.3 | 17.7 (2.7) | 20.2 | 38.1 (2.9) | yes | yes | ||||||
| p.N320I | 1.4 | 15.7 (12.2) | 2 | 29.8 (15.9) | 6.0 | 14.3 (3.4) | yes | yes | yes | |||
| p.Q321H | 1.9 | 17.9 (10.4) | 3.3 | 22.0 (7.7) | yes | yes | ||||||
| p.D322N | 36.1 | 10.3 (1.3) | 17.8 | 13.3 (1.8) | yes | yes | ||||||
| p.G325S | 24.7 | 37.8 (2.5) | 25.6 | 29.8 (2.2) | yes | yes | ||||||
| p.Q327E | 22.9 | 27.5 (2.2) | 21.5 | 59.4 (3.8) | yes | yes | ||||||
| p.G328A | 6.9 | 21.8 (4.2) | 6.2 | 23.8 (4.8) | yes | yes | ||||||
| p.G334E | 86.7 | 18.8 81.2) | 57.5 | 23.4 (1.4) | yes | yes | ||||||
| p.P343L | 36.6 | 13.0 (1.4) | 39.6 | 22.6 (1.6) | yes | yes | ||||||
| p.S345P | 0 | 3.7 (n/c) | 0 | 9.7 (n/c) | yes | yes | ||||||
| p.R356W | 11 | 38.1 (4.5) | 16.9 | 45.8 (3.7) | yes | yes | ||||||
| p.R356Q | 36.1 | 39.0 (2.1) | 33.3 | 33.0 (2.0) | yes | yes | ||||||
| p.G360C | 8.7 | 8.1 (1.9) | 16.2 | 16.7 (2.0) | 5.3 | 9.9 (2.9) | yes | yes | yes | |||
| p.R363H | 20 | 30.5 (2.5) | 31.9 | 26.0 (1.8) | yes | yes | ||||||
| p.A368T | 54.6 | 18.0 (1.3) | 79.6 | 21.3 (1.3) | yes | yes | ||||||
| p.T385A | 57.3 | 16.1 (1.3) | 28.6 | 17.3 (1.6) | 68.1 | 9.6 (1.1) | yes | yes | no | |||
| p.G395A | 24.4 | 6.3 (1.3) | 13.4 | 5.2 (1.4) | yes | yes | ||||||
| p.L403S | 14.0 | 4.9 (1.4) | 13.2 | 13.0 (2.0) | yes | yes | ||||||
| p.T410I | 0.4 | 11.8 (30.5) | 2.3 | 13.8 (7.0) | yes | yes | ||||||
| p.E418G | 67.5 | 21.9 (1.3) | 74.6 | 14.5 (1.2) | 140.7 | 51.8 (1.4) | yes | yes | yes | |||
| p.M421V | 83.0 | 25.6 81.3) | 58.3 | 26.0 (1.5) | yes | yes | ||||||
Only amenable mutations according to the GLP-HEK assay were included. According to the GLP-HEK assay, an amenable mutation responds with an absolute increase by ≥3% of wild-type AGAL activity and a ≥1.2-fold increase of baseline enzymatic activity in the presence of 10 μM migalastat. w/o, without; WT, wild-type; NA, not available; n/c, not calculated.
Figure 2.

Comparison of the GLP-HEK Assay with In-House Assays
(A–C) Pearson correlation of α-galactosidase A activities presented from (A) baseline activities without DGJ, (B) total activities after incubation with DGJ, and (C) total activity increase over baseline in % of wild-type (WT) activity after incubation with DGJ. (D) Bland-Altman analysis of the in-house assays compared to the GLP-HEK assay for total activity increase over baseline. The dotted lines represent the 95% confidence intervals. Only amenable mutations according to the GLP-HEK assay were included and compared to data available from in-house assays (Lukas et al.,39 n = 77 mutations; Oommen et al.,36 n = 54 mutations; Lenders et al.,26 n = 12 mutations). Data were extracted from Benjamin et al.,10 Lukas et al.,39 Oommen et al.,36 and Lenders et al.26 GraphPad Prism v8.0 software (GraphPad, La Jolla, CA, USA) was used for statistical analysis and visualization.
In Vivo Amenability
Despite their advantages and disadvantages, current amenability assays can be used to identify potentially amenable mutations. However, they do not represent in vivo conditions in patients, since measurement of AGAL activity is based on an artificial substrate, and stimulation is performed in a cell culture background. The GLA-knockout HEK293T assay seems to be a more patient-specific approach, especially for mutations with very low enzymatic activities, although no direct conclusion can be drawn about the extent of Gb3 depletion in individual patients. Furthermore, future studies are warranted to generate data for additional mutations. Unfortunately, the cutoff value of required intracellular and lysosomal AGAL activities after migalastat stimulation to achieve a beneficial therapeutic response in patients remains elusive. Currently, low baseline activities, absent or low biochemical responsiveness, and inter-assay discrepancy are alarm signals for the potential misclassification of a genetic variant/mutation.39 Therefore, amenability assays can only give an indication of a potential treatability of the patient with a possible therapy efficiency.
According to current knowledge, “amenable” mutations can be divided into four groups as follows: (1) non-pathogenic amenable genetic variants where patients should not be treated, (2) good responding mutations (e.g., p.N215S) where patients benefit very well from migalastat therapy, (3) mutations (e.g., p.N34S, p.W162G, p.L294S) that do not respond or respond poorly, and (4) theoretically a group of mutations is conceivable, in which migalastat acts as an inhibitor with a very strong affinity to the active center, leading to a potential increase of (lyso-)Gb3, with the clinical consequence of therapy discontinuation. As a consequence, we propose the following workup for the treatment of patients with migalastat (Figure 3).
Figure 3.

Work-Up to Assess Clinical Amenability in Patients Treated with Migalastat
Biochemical response after migalastat initiation should already be measurable after 3 months of treatment by increasing AGAL activities and decreasing plasma lyso-Gb3 levels. Regular follow-up examinations should include echocardiography of the heart to assess left ventricular hypertrophy (LVH) and left ventricular masses (LVMs), determination of eGFR and proteinuria/albuminuria measures for kidney function, brain MRI to assess cerebrovascular events (CVEs) and white matter lesions (WMLs), a pain questionnaire for FD-specific pain and gastrointestinal symptoms, and α-galactosidase A (AGAL) activity and globotriaosylsphingosine (lyso-Gb3) measurements in blood.
After diagnostic confirmation of a patient in need of treatment44,45 with a FD-causing mutation, the physician should check the amenability of the GLA mutation via the appropriate website (https://www.galafoldamenabilitytable.com/hcp; last accessed on June 10, 2020). If amenable, a decision needs to be made either for ERT or migalastat treatment. If migalastat is chosen, at least after 6 and 12 months a follow-up examination should be performed to control whether current therapeutic goals46 of a Fabry-specific therapy are fulfilled, which, in the context of multimodal care, should include (1) prevention/delay of the progression of organ manifestations (especially in the kidney, heart, and central nervous system), (2) reduction of complaints (especially neuropathic pain reduction), and (3) improvement of quality of life.45,46
Furthermore, the biochemical response to migalastat should be accessed by measuring AGAL activity and plasma lyso-Gb3 in the blood, since levels already change after 3 months.25,37 Increasing AGAL activities are associated with a decrease of myocardial mass25 so that AGAL activity (and lyso-Gb3 values) might serve as potential markers for therapy efficiency. If most of these aims are achieved, the patient is clinically amenable (in vivo amenability) and treatment should be continued. If a patient is clinically not amenable, the patient’s adherence must be checked. In case of nonadherence, the physician should encourage the patient to take the oral chaperone consistently. If adherence is ensured, a physician (Fabry expert) of an interdisciplinary Fabry center should verify the in vitro amenability of the GLA mutation using secondary literature and comparing in vitro data (if possible). If the in vitro amenability is ambiguous, the physician should consider switching from migalastat to ERT. In addition, control for comorbidities and/or adjustment for concomitant medications such as renin-angiotensin-aldosterone system (RAAS) blockers and others are warranted.
In conclusion, migalastat seems to be an attractive alternative to ERT for many FD patients with amenable mutations.23, 24, 25, 26 Amenability assays can only give an indication of a potential treatability of the patient with possible therapy efficiency. The physician has the responsibility to verify the clinical responsiveness of the patient through regular clinical follow-ups in an interdisciplinary Fabry center by an examination of the three main organ systems (kidney, heart, and central/peripheral nervous system) as well as AGAL activity and (lyso-)Gb3 levels.
Conflicts of Interest
M.L. has received speaker honoraria, travel funding, and research grants from Amicus Therapeutics, Sanofi Genzyme, and Shire/Takeda. E.B. has received research grants and speaker honoraria from Sanofi Genzyme, Shire/Takeda, and Amicus Therapeutics. F.S. has received speaker honoraria from Sanofi Genzyme.
References
- 1.Zarate Y.A., Hopkin R.J. Fabry’s disease. Lancet. 2008;372:1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]
- 2.Eng C.M., Guffon N., Wilcox W.R., Germain D.P., Lee P., Waldek S., Caplan L., Linthorst G.E., Desnick R.J., International Collaborative Fabry Disease Study Group Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry’s disease. N. Engl. J. Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 3.Schiffmann R., Kopp J.B., Austin H.A., 3rd, Sabnis S., Moore D.F., Weibel T., Balow J.E., Brady R.O. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 4.Banikazemi M., Bultas J., Waldek S., Wilcox W.R., Whitley C.B., McDonald M., Finkel R., Packman S., Bichet D.G., Warnock D.G., Desnick R.J., Fabry Disease Clinical Trial Study Group Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann. Intern. Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 5.Hughes D.A., Elliott P.M., Shah J., Zuckerman J., Coghlan G., Brookes J., Mehta A.B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart. 2008;94:153–158. doi: 10.1136/hrt.2006.104026. [DOI] [PubMed] [Google Scholar]
- 6.Mehta A., Beck M., Elliott P., Giugliani R., Linhart A., Sunder-Plassmann G., Schiffmann R., Barbey F., Ries M., Clarke J.T., Fabry Outcome Survey investigators Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986–1996. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- 7.Lenders M., Karabul N., Duning T., Schmitz B., Schelleckes M., Mesters R., Hense H.W., Beck M., Brand S.M., Brand E. Thromboembolic events in Fabry disease and the impact of factor V Leiden. Neurology. 2015;84:1009–1016. doi: 10.1212/WNL.0000000000001333. [DOI] [PubMed] [Google Scholar]
- 8.Lenders M., Canaan-Kühl S., Krämer J., Duning T., Reiermann S., Sommer C., Stypmann J., Blaschke D., Üçeyler N., Hense H.W. Patients with Fabry disease after enzyme replacement therapy dose reduction and switch—2-year follow-up. J. Am. Soc. Nephrol. 2016;27:952–962. doi: 10.1681/ASN.2015030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenders M., Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. J. Am. Soc. Nephrol. 2018;29:2265–2278. doi: 10.1681/ASN.2018030329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benjamin E.R., Della Valle M.C., Wu X., Katz E., Pruthi F., Bond S., Bronfin B., Williams H., Yu J., Bichet D.G. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017;19:430–438. doi: 10.1038/gim.2016.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romeo G., D’Urso M., Pisacane A., Blum E., De Falco A., Ruffilli A. Residual activity of alpha-galactosidase A in Fabry’s disease. Biochem. Genet. 1975;13:615–628. doi: 10.1007/BF00484919. [DOI] [PubMed] [Google Scholar]
- 12.Yam G.H., Zuber C., Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005;19:12–18. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- 13.Boyd R.E., Lee G., Rybczynski P., Benjamin E.R., Khanna R., Wustman B.A., Valenzano K.J. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013;56:2705–2725. doi: 10.1021/jm301557k. [DOI] [PubMed] [Google Scholar]
- 14.Ishii S., Kase R., Sakuraba H., Suzuki Y. Characterization of a mutant α-galactosidase gene product for the late-onset cardiac form of Fabry disease. Biochem. Biophys. Res. Commun. 1993;197:1585–1589. doi: 10.1006/bbrc.1993.2659. [DOI] [PubMed] [Google Scholar]
- 15.Frustaci A., Chimenti C., Ricci R., Natale L., Russo M.A., Pieroni M., Eng C.M., Desnick R.J. Improvement in cardiac function in the cardiac variant of Fabry’s disease with galactose-infusion therapy. N. Engl. J. Med. 2001;345:25–32. doi: 10.1056/NEJM200107053450104. [DOI] [PubMed] [Google Scholar]
- 16.Fan J.-Q., Ishii S., Asano N., Suzuki Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999;5:112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- 17.Asano N., Ishii S., Kizu H., Ikeda K., Yasuda K., Kato A., Martin O.R., Fan J.Q. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 2000;267:4179–4186. doi: 10.1046/j.1432-1327.2000.01457.x. [DOI] [PubMed] [Google Scholar]
- 18.Ishii S., Yoshioka H., Mannen K., Kulkarni A.B., Fan J.-Q. Transgenic mouse expressing human mutant α-galactosidase A in an endogenous enzyme deficient background: a biochemical animal model for studying active-site specific chaperone therapy for Fabry disease. Biochim. Biophys. Acta. 2004;1690:250–257. doi: 10.1016/j.bbadis.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Ishii S., Chang H.-H., Yoshioka H., Shimada T., Mannen K., Higuchi Y., Taguchi A., Fan J.Q. Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. J. Pharmacol. Exp. Ther. 2009;328:723–731. doi: 10.1124/jpet.108.149054. [DOI] [PubMed] [Google Scholar]
- 20.Khanna R., Soska R., Lun Y., Feng J., Frascella M., Young B., Brignol N., Pellegrino L., Sitaraman S.A., Desnick R.J. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. 2010;18:23–33. doi: 10.1038/mt.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamin E.R., Khanna R., Schilling A., Flanagan J.J., Pellegrino L.J., Brignol N., Lun Y., Guillen D., Ranes B.E., Frascella M. Co-administration with the pharmacological chaperone AT1001 increases recombinant human α-galactosidase A tissue uptake and improves substrate reduction in Fabry mice. Mol. Ther. 2012;20:717–726. doi: 10.1038/mt.2011.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warnock D.G., Bichet D.G., Holida M., Goker-Alpan O., Nicholls K., Thomas M., Eyskens F., Shankar S., Adera M., Sitaraman S. Oral migalastat HCl leads to greater systemic exposure and tissue levels of active α-galactosidase A in Fabry patients when co-administered with infused agalsidase. PLoS ONE. 2015;10:e0134341. doi: 10.1371/journal.pone.0134341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Germain D.P., Hughes D.A., Nicholls K., Bichet D.G., Giugliani R., Wilcox W.R., Feliciani C., Shankar S.P., Ezgu F., Amartino H. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016;375:545–555. doi: 10.1056/NEJMoa1510198. [DOI] [PubMed] [Google Scholar]
- 24.Hughes D.A., Nicholls K., Shankar S.P., Sunder-Plassmann G., Koeller D., Nedd K., Vockley G., Hamazaki T., Lachmann R., Ohashi T. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017;54:288–296. doi: 10.1136/jmedgenet-2016-104178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müntze J., Gensler D., Maniuc O., Liu D., Cairns T., Oder D., Hu K., Lorenz K., Frantz S., Wanner C., Nordbeck P. Oral chaperone therapy migalastat for treating Fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin. Pharmacol. Ther. 2019;105:1224–1233. doi: 10.1002/cpt.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenders M., Nordbeck P., Kurschat C., Karabul N., Kaufeld J., Hennermann J.B., Patten M., Cybulla M., Müntze J., Üçeyler N. Treatment of Fabry’s disease with migalastat: outcome from a prospective observational multicenter study (FAMOUS) Clin. Pharmacol. Ther. 2020;108:326–337. doi: 10.1002/cpt.1832. [DOI] [PubMed] [Google Scholar]
- 27.Feriozzi S., Hughes D.A. New drugs for the treatment of Anderson-Fabry disease. J. Nephrol. 2020 doi: 10.1007/s40620-020-00721-4. Published online March 20, 2020. [DOI] [PubMed] [Google Scholar]
- 28.Benjamin E.R., Flanagan J.J., Schilling A., Chang H.H., Agarwal L., Katz E., Wu X., Pine C., Wustman B., Desnick R.J. The pharmacological chaperone 1-deoxygalactonojirimycin increases α-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009;32:424–440. doi: 10.1007/s10545-009-1077-0. [DOI] [PubMed] [Google Scholar]
- 29.Shimotori M., Maruyama H., Nakamura G., Suyama T., Sakamoto F., Itoh M., Miyabayashi S., Ohnishi T., Sakai N., Wataya-Kaneda M. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 2008;29:331. doi: 10.1002/humu.9520. [DOI] [PubMed] [Google Scholar]
- 30.Wu X., Katz E., Della Valle M.C., Mascioli K., Flanagan J.J., Castelli J.P., Schiffmann R., Boudes P., Lockhart D.J., Valenzano K.J., Benjamin E.R. A pharmacogenetic approach to identify mutant forms of α-galactosidase A that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011;32:965–977. doi: 10.1002/humu.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Germain D.P., Giugliani R., Hughes D.A., Mehta A., Nicholls K., Barisoni L., Jennette C.J., Bragat A., Castelli J., Sitaraman S. Safety and pharmacodynamic effects of a pharmacological chaperone on α-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: report from two phase 2 clinical studies. Orphanet J. Rare Dis. 2012;7:91. doi: 10.1186/1750-1172-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giugliani R., Waldek S., Germain D.P., Nicholls K., Bichet D.G., Simosky J.K., Bragat A.C., Castelli J.P., Benjamin E.R., Boudes P.F. A phase 2 study of migalastat hydrochloride in females with Fabry disease: selection of population, safety and pharmacodynamic effects. Mol. Genet. Metab. 2013;109:86–92. doi: 10.1016/j.ymgme.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 33.Young-Gqamana B., Brignol N., Chang H.H., Khanna R., Soska R., Fuller M., Sitaraman S.A., Germain D.P., Giugliani R., Hughes D.A. Migalastat HCl reduces globotriaosylsphingosine (lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients. PLoS ONE. 2013;8:e57631. doi: 10.1371/journal.pone.0057631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukas J., Giese A.K., Markoff A., Grittner U., Kolodny E., Mascher H., Lackner K.J., Meyer W., Wree P., Saviouk V., Rolfs A. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in Fabry disease. PLoS Genet. 2013;9:e1003632. doi: 10.1371/journal.pgen.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukas J., Scalia S., Eichler S., Pockrandt A.M., Dehn N., Cozma C., Giese A.K., Rolfs A. Functional and clinical consequences of novel α-galactosidase A mutations in Fabry disease. Hum. Mutat. 2016;37:43–51. doi: 10.1002/humu.22910. [DOI] [PubMed] [Google Scholar]
- 36.Oommen S., Zhou Y., Meiyappan M., Gurevich A., Qiu Y. Inter-assay variability influences migalastat amenability assessments among Fabry disease variants. Mol. Genet. Metab. 2019;127:74–85. doi: 10.1016/j.ymgme.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Lenders M., Stappers F., Niemietz C., Schmitz B., Boutin M., Ballmaier P.J., Zibert A., Schmidt H., Brand S.M., Auray-Blais C., Brand E. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019;56:548–556. doi: 10.1136/jmedgenet-2019-106005. [DOI] [PubMed] [Google Scholar]
- 38.Johnson F.K., Mudd P.N., Jr., Bragat A., Adera M., Boudes P. Pharmacokinetics and safety of migalastat HCl and effects on agalsidase activity in healthy volunteers. Clin. Pharmacol. Drug Dev. 2013;2:120–132. doi: 10.1002/cpdd.1. [DOI] [PubMed] [Google Scholar]
- 39.Lukas J., Cimmaruta C., Liguori L., Pantoom S., Iwanov K., Petters J., Hund C., Bunschkowski M., Hermann A., Cubellis M.V., Rolfs A. Assessment of gene variant amenability for pharmacological chaperone therapy with 1-deoxygalactonojirimycin in Fabry disease. Int. J. Mol. Sci. 2020;21:956. doi: 10.3390/ijms21030956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kusiak J.W., Quirk J.M., Brady R.O. Purification and properties of the two major isozymes of alpha-galactosidase from human placenta. J. Biol. Chem. 1978;253:184–190. [PubMed] [Google Scholar]
- 41.Mayes J.S., Scheerer J.B., Sifers R.N., Donaldson M.L. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin. Chim. Acta. 1981;112:247–251. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 42.Schiffmann R., Bichet D.G., Benjamin E., Wu X., Giugliani R. The migalastat GLP-HEK assay is the gold standard for determining amenability in patients with Fabry disease. Mol. Genet. Metab. Rep. 2019;20:100494. doi: 10.1016/j.ymgmr.2019.100494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Förg T., Hafner M., Lux A. Investigation of endoglin wild-type and missense mutant protein heterodimerisation using fluorescence microscopy based IF, BiFC and FRET analyses. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0102998. e102998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biegstraaten M., Arngrímsson R., Barbey F., Boks L., Cecchi F., Deegan P.B., Feldt-Rasmussen U., Geberhiwot T., Germain D.P., Hendriksz C. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J. Rare Dis. 2015;10:36. doi: 10.1186/s13023-015-0253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ortiz A., Germain D.P., Desnick R.J., Politei J., Mauer M., Burlina A., Eng C., Hopkin R.J., Laney D., Linhart A. Fabry disease revisited: management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018;123:416–427. doi: 10.1016/j.ymgme.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 46.Wanner C., Arad M., Baron R., Burlina A., Elliott P.M., Feldt-Rasmussen U., Fomin V.V., Germain D.P., Hughes D.A., Jovanovic A. European expert consensus statement on therapeutic goals in Fabry disease. Mol. Genet. Metab. 2018;124:189–203. doi: 10.1016/j.ymgme.2018.06.004. [DOI] [PubMed] [Google Scholar]