

Abstract
The microbiome in the hindgut of wood-feeding termites comprises various species of bacteria, archaea, and protists. This gut community is indispensable for the termite, which thrives solely on recalcitrant and nitrogen-poor wood. However, the difficulty in culturing these microorganisms has hindered our understanding of the function of each species in the gut. Although protists predominate in the termite gut microbiome and play a major role in wood digestion, very few culture-independent studies have explored the contribution of each species to digestion. Here, we report single-cell transcriptomes of four protists species comprising the protist population in worldwide pest Coptotermes formosanus. Comparative transcriptomic analysis revealed that the expression patterns of the genes involved in wood digestion were different among species, reinforcing their division of roles in wood degradation. Transcriptomes, together with enzyme assays, also suggested that one of the protists, Cononympha leidyi, actively degrades chitin and assimilates it into amino acids. We propose that C. leidyi contributes to nitrogen recycling and inhibiting infection from entomopathogenic fungi through chitin degradation. Two of the genes for chitin degradation were further revealed to be acquired via lateral gene transfer (LGT) implying the importance of LGT in the evolution of symbiosis. Our single-cell-based approach successfully characterized the function of each protist in termite hindgut and explained why the gut community includes multiple species.
Subject terms: Community ecology, Symbiosis
Introduction
Wood-feeding termites harbor a complex symbiotic system in their gut, comprising various microorganisms from three domains of life [1]. It has long been recognized that the microbiome in the termite gut is essential to the host thriving only on dead wood, which is recalcitrant for digestion and poor in nitrogen. Their ability of efficient lignocellulolysis makes termites a keystone in the global carbon cycle. However, the detailed function of each microbial species in the termite gut has not yet been elucidated because most of them are very difficult to culture in laboratories. About a decade ago, the sequencing of two bacterial symbiont genomes was achieved by uniting whole genome amplification and next generation sequencing (NGS) techniques [2, 3]. Since then, several symbiotic bacterial genomes in the termites have been analyzed from a small number of, or even single cells [4–6]. These studies have shed light on their contribution to the symbiotic system by different means, such as nitrogen fixation, amino acid/cofactor synthesis, reductive acetogenesis, and partial participation in lignocellulose digestion. In contrast to progress on bacterial sequencing, NGS of individual termite gut protist (unicellular eukaryote) species, which belong to either the phylum Parabasalia or the order Oxymonadida (phylum Preaxostyla), have not been reported—although protists occupy the large volume of the microbiome and actively ingest wood particles [7]. At present, only a handful of protist genes for wood decomposition (e.g., endoglucanase, cellobiohydrolase, and xylanase) and phylogenetic marker (e.g., genes for small subunit of rRNA, α-tubulin, and elongation factor 1α) are identified their organismal origin [8–13] except oxymonad Streblomastix strix of which a draft genome was recently sequenced [14]. Meta-omics analyses in the previous studies revealed the series of genes for wood digestion but the “owner” of them cannot be determined [15–17]. The hypothesis that each protist species has a different role in wood decomposition and that lignocellulose is completely degraded by their collaborative work is attractive for explaining the population complexity in the termite gut. Therefore, data obtained from each species of microbiome are highly desirable, which may give us further clues to infer other roles of protists in the termite gut besides digestion.
Coptotermes formosanus is one of the most hazardous and broadly distributed pest species [18]. Based on morphology, the protists in C. formosanus are classified into three species belonging to phylum Parabasalia [19]: Pseudotrichonympha grassii, Holomastigotoides hartmanni (commonly confused with H. mirabile), and Cononympha leidyi (previously called Spirotrichonympha leidyi [20]). The spatially different distribution of three species in the hindgut enabled to roughly assess the ability of each species for wood digestion [21, 22]. Biochemical experiments and identification of the organismal origins of genes for wood degradation supported the concept of the division of role. For example, P. grassii was found to encode a cellobiohydrolase and decompose high-molecular cellulose in the entrance of hindgut [10, 23] while xylan is mainly degraded by H. hartmannii with its xylanase [12]. However, lignocellulose consists of heterogenous compounds besides cellulose and xylan [24]. Moreover, metatranscriptome studies showed the presence of various genes of glycoside hydrolase families of which organismal origins are remain to be unassigned [15–17].
In this study, we performed single-cell transcriptomes targeting the protists in C. formosanus for further investigation of their functional potentials. Comparative analysis of the transcriptomes showed different expression patterns of genes involved in lignocellulose digestion among the protist species and enabled to overview the whole image of division of roles in wood degradation. We also propose a possible contribution of C. leidyi to nitrogen recycling and/or host defense against fungal infection through chitin degradation.
Materials and methods
Libraries preparation for single-cell transcriptome and sequencing
The termite C. formosanus was collected at Ishigaki Island, Okinawa, Japan. The gut of termite was pulled out with sterilized forceps and suspended in 0.46% NaCl. A protist single cell was manually picked into a drop of 0.46% NaCl and washed by transferring another drop of NaCl three times. A washed cell was then transferred in 0.4 μl of 0.5% NP-40 and submitted to cDNA synthesis and amplification, according to the Quartz-seq protocol [25]. Libraries for the Illumina sequencing platform were prepared from the purified cDNAs using a Nextera XT DNA Library Preparation Kit following the manufacturers’ instructions. Sequencing was preformed using MiSeq with MiSeq Reagent Kit v2. After the organismal origin of each library was re-determined (see below), three representative libraries were selected for each species and deeply sequenced by HiSeq 2500 with HiSeq SBS Kit v4. The raw data of single-cell transcriptomic generated in this study were deposited BioProject accession under PRJDB8546. The decontaminated assemblies and the predicted gene models are available at Dryad Digital Repository (10.5061/dryad.05.qfttf04).
Inspecting the organismal origin of single-cell transcriptome libraries
The organismal origins of libraries were initially assigned to P. grassii, H. hartmanii, and C. leidyi based on cell morphology. Considering that the previous study indicated further protist diversity in C. formosanus [26], the organismal origins of libraries were investigated using the data generated from MiSeq. The generated FASTQ files underwent primer removal and quality trimming by Trimmomatic [27]. The trimmed FASTQ of all libraries were concatenated into one file, irrespective of the putative taxonomic assignment, and assembled by Trinity v2.5.1 [28]. The redundancy of the contigs was reduced by CD-HIT clustering with 95% similarity [29]. The nonredundant contigs were quantified for individual libraries by bowtie2 [30] and RSEM [31] using the script provided with Trinity. The genes for the eukaryotic ribosomal proteins were searched by BLASTX [32]. The organismal origins of libraries were re-identified based on expression value of contigs encoding the eukaryotic marker gene. The sequences used as query in BLAST are summarized in Supplementary Table 1.
Fluorescence in situ hybridization (FISH)
FISH was performed as described previously [33] with slight modifications. The detailed protocol is described in the Supplementary material. Color modification of the obtained images and analysis of cell size were performed using ImageJ software [34].
Bioinfomatic analyses of deeply sequenced transcriptomes
The FASTQ files generated from HiSeq were concatenated by species. Quality trimming and assembly was performed as described above. Gene models of each species were predicted from the resultant assembly using TransDecoder (https://github.com/TransDecoder/TransDecoder/wiki) and annotated by KAAS [35]. The gene models were also submitted to a BLASTP [32] search in the NCBI nonredundant database. The contamination, completeness, and reproducibility of transcriptomes were assessed as described in Supplementary material.
The genes for carbohydrate-active enzymes (CAZYs) were searched by dbCAN2 [36] with manual curation. The genes classified to Glycoside hydrolase family 7 (GH7) were further classified into endo-β-1,4-glucanase or cellobiohydrolase based on sequence alignment [37]. The specificity of the other GH genes was inferred according to the homologs of which substrate were enzymatically identified. The comparison of the highly expressed GHs among the protists was performed as described in Supplementary material.
Enzyme assay
The crude enzymes extracted from the protist pellets from the anterior and posterior parts of the hindgut were subjected to chitinase assays. The detailed procedure is described in Supplementary material.
Identification of the protist origin of the putative N-acetyl glucosamine deacetylase (nodB) gene
Whole-cell in situ hybridization was performed to confirm that nodB gene was encoded by C. leidyi. The procedure of in situ hybridization was performed according to the previous study [38] with oligonucleotide probe (5′-CTGCGTATCCTCACTCTGCGAC-3′) attaching digoxigenin (DIG) to its 5′ end. The specificity of the probe was checked as described in FISH probes (see Supplementary material). The hybridization signal was detected using alkaline phosphatase-conjugated anti-DIG antibodies with colorimetric substrate. We also checked the poly A tail of nodB mRNA by 3′ RACE with oligo (dT) primer to confirm that the gene encoded by eukaryotic organisms.
Phylogenetic analyses
The maximum-likelihood (ML) analyses of chitinase and nodB genes were conducted as described in Supplementary material.
Results
Identification and characterization of new symbiotic protist species in Coptotermes formosanus
In total, 17 libraries of single-cell transcriptomes were prepared from a protist cell in C. formosanus and sequenced by Illumina MiSeq platform. The generated reads were concatenated and assembled together. Based on morphological observation, Koidzumi [19] reported C. formosanus harbors P. grassii, H. hartmanni, and C. leidyi (Fig. 1a–c). However, the later study indicated the existence of an additional Holomastigotoides species [26]. Therefore, we investigated organismal origin of libraries from the expression value distribution of the genes for the eukaryotic ribosomal protein in each library. Coinciding with the previous study, the expression patterns of the ribosomal protein genes classified each library to P. grassii, H. hartmanni, C. leidyi, or the undescribed Holomastigotoides (Supplementary Fig. 1). To characterize the undescribed Holomastigotoides, we performed whole-cell FISH using specific probes for the two Holomastigotoides species and successfully distinguished them (Fig. 1d). Although the morphological characteristics delineating the two Holomastigotoides species could not be found under FISH imaging, they showed different size distributions (Fig. 1e). The cells of H. hartmanni were 34–223 μm in length by 21–64 μm in width, with the respective averages ± SD of 112 ± 41 and 75 ± 26 μm (n = 658 cells), whereas those of the other species were 23–117 μm in length by 24–109 μm in width, with the respective averages ± SD of 60 ± 20 and 61 ± 17 μm (n = 497 cells). Their average cell size was also significantly different (Welch’s t-test, p < 1.22E−115 and < 4.82E−29 for length and width, respectively). Considering differences in SSU rRNA [26] and cell size, we designated the undescribed Holomastigotoides as Holomastigotoides minor sp. nov.
Fig. 1. Symbiotic protists in Coptotermes formosanus.

Bright-field images of (a) Pseudotrichonympha grassii, (b) Holomastigotoides hartmanni, and (c) Cononympha leidyi. d Fluorescence in situ hybridization simultaneously (FISH) staining two distinct Holomastigotoides. The oligonucleotide probes are specific to H. hartmanii (Texas red, shown in cyan) and the undescribed Holomastigotoides (6-carboxyfluorescein, shown in pink), respectively. Yellow and blue arrows indicate the cells of P. grassii and C. leidyi, respectively. e Size distribution of H. hartmanni and the undescribed Holomastigotoides. Cyan and pinks dots represent the cell size of H. hartmanni and the undescribed Holomastigotoides, respectively.
Deep sequencing of single-cell transcriptome and quality assessment
As our single-cell transcriptomic libraries appeared to be successfully constructed, we selected three representative libraries of each protist species for further sequencing by Hiseq 2500. The generated reads from 12 libraries were concatenated by species and assembled (Supplementary Fig. 2a). As indicated by Supplementary Fig. 1, the transcriptomes contain reads from nontargeted protists, particularly among small species. The transcriptomes can also include the reads from bacteria because the termite gut is filled with bacteria and the gut protists also harbor dense colonization of ecto- and/or endosymbiotic bacteria. To assess the contamination, all the reads were concatenated by species and aligned with all the assemblies (Supplementary Fig. 2b). The contigs that aligned with reads from the nontarget species were regarded as contamination. The pair plot of the normalized read counts showed that, although there was a substantial amount of contamination (except for the assembly of P. grassii) most were derived from bacteria and their abundance was low (< 30 counts per million reads, corresponding to ~1.5 in pair plot axes; Fig. 2 and Supplementary Figs. 3–6). The contamination from nontarget protist could also be easily assigned to one species because most of the normalized read counts of the contigs derived from the targeted protists were ten times higher or more than those derived from contamination (Fig. 2 and Supplementary Figs. 3–6). A summary of the pre and postdecontaminated assemblies, is shown in Table 1. We were conscious of the fact that the trimmed assemblies could still include some contamination from bacteria due to the limited information of genome sequences of gut bacteria. Therefore, we considered the possibility of persisting bacterial contamination throughout the downstream analyses.
Fig. 2. Comparison of the abundance of the contaminated contigs.

The x and y axes represent log10 (normalized read counts + 1) of the contig in the respective libraries. Blue and orange dots represent bacterial and parabasalian contigs, respectively. a Pair plot comparison of cross-contaminated contigs assigned to Bacteria or Parabasalia between the libraries targeted Pseudotrichonympha grassii (Pg) and Holomastigotoides hartmanni (Hh). b Pair plot comparison of all the contaminated contigs between Pg and Hh libraries. c Pair plot comparison of the cross-contaminated contigs assigned to Bacteria or Parabasalia between the Cononympha leidyi (Cl) and Hh targeted libraries. d Pair plot comparison of all the contaminated contigs between Cl and Hh libraries. All of the pairwise comparisons between libraries of different species are given in Supplementary Figs. 3–6.
Table 1.
Summary of transcriptomes of the protists in Coptotermes formosanus.
| No. of contig | No. of CDS | ||||
|---|---|---|---|---|---|
| Beforea | Afterb | Beforea | Afterb | Completenessc | |
| Pseudotrichonympha grassii | 41,010 | 39,825 | 27,864 | 27,334 | 79.2% (179) |
| Holomastigotoides hartmanni | 123,108 | 108,262 | 87,978 | 57,788 | 54.4% (123) |
| Holomastigotoides minor | 98,635 | 70,088 | 47,872 | 43,317 | 51.3% (116) |
| Cononympha leidyi | 69,830 | 51,266 | 29,500 | 35,949 | 53.1% (120) |
Each transcriptome was assembled from the paired-end reads generated from three single-cell libraries. The number of contigs and CDSs longer than ≥ 300 bp and CDS ≥ 100 amino acids, respectively are shown.
aThe number of contigs and CDSs in assembly before removal of contamination.
bThe number of contigs and CDSs in assembly after removal of contamination from bacteria and nontarget protists.
cThe ratio of the detected marker gene to those of Trichomonas vaginalis. The numbers of the detected marker genes are shown in parentheses.
Another problem of single-cell transcriptome analysis is that it is not always comparable to the transcriptome using a large amount of starting material with respect to the completeness and reproducibility [39, 40]. To evaluate the completeness of assemblies, we defined 226 genes as the gene set conserved in Parabasalia, based on BUSCO dataset of version 3 [41]. After removal of contamination, 126–198 marker genes were detected in our assemblies, corresponding to 51–79% completeness (Table 1, Supplementary Tables 2 and 3). The completeness of the P. grassii assembly was much higher than those of the others, probably due to its much amount of mRNA within their large cells [40].
The reproducibility among libraries targeting the same species was evaluated by aligning the assembly with the reads that generated themselves (Supplementary Fig. 2c) and calculating trimmed mean of M value (TMM). Figure 3 and Supplementary Figs. 7–10 show that the contigs with low TMM were unstable between replicates, as reported in previous studies [39, 40]. These variants of TMM among replicates are thought to be derived technical or stochastic factors due to the small amount of RNA in the starting material, rather than biological differences. In contrast, the highly expressed contigs showed an obvious correlation in a pairwise comparison, indicating their confident reproducibility. In addition to the reliable reproducibility, the contigs with high TMM were devoid of bacterial contamination.
Fig. 3. Comparison of trimmed mean of M value (TMM) between libraries targeted to the same species.

The x and y axes represent log10 (TMM + 1) in the compared libraries. Blue and orange dots represent bacterial and parabasalian contigs, respectively. a Pair plot comparison of TMM of contigs assigned to Bacteria and Parabasalia between two libraries targeted to Pseudotrichonympha grassii (represented as Pg1 and Pg2). b Pair plot comparison of TMM of all the contigs between Pg1 and Pg2. c Pair plot comparison of TMM of contigs assigned to Bacteria and Parabasalia between two libraries targeted to Cononympha leidyi (Cl1 and Cl2). d Pair plot comparison of TMM of all the contigs between Cl1 and Cl2. All pairwise comparisons among the replicates are given in Supplementary Figs. 7–10.
Taking these facts together, our assemblies, derived from three single cells, captured at least half of the whole transcriptomes and could be used to infer the major functions of each protist as well as the expression abundance. Hereafter, we regarded genes with TMM > 100 in at least two of three replicates as the highly and stably expressed genes and use for the inference of protist functions in the termite gut. Focusing on the highly expressed genes is also helpful to evade amplification bias generated in cDNA synthesis from single cell [42].
Differential expression pattern of genes involving lignocellulose digestion among symbionts in C. formosanus
A series of CAZY were detected from the transcriptome of all species, including those involved in cellulose, hemicellulose, and pectin degradation (Supplementary Table 4). The expression heatmap of the GHs (Fig. 4) indicated that some GHs were highly expressed in multiple species, whereas the expression of other GHs varied among them, prompting us to determine their division of roles. Indeed, some GHs that show species-specific expression have unique substrates. For example, P. grassii and C. leidyi highly express the genes for mannanase (GH26), and/or mannosidase (GH2 and GH92). These GHs degrade glucomannan with endo-β-1,4-glucanase. The extremely high expression of cellobiohydrolase (GH7) in the P. grassii transcriptome suggested that P. grassii actively degrades crystalline cellulose. This is consistent with the previous study which suggested that wood particles that arrive at the hindgut were first attacked by the cellobiohydrolase of P. grassii inhabiting the anterior hindgut [21–23, 43]. α-l-arabinoside residues in arabinoxylan and arabinogalactan are released by enzymes of subfamily 2 of GH43 in H. hartmanni and C. leidyi. Although H. minor and C. leidyi showed active expression of PL1, PL1 alone is not enough to decompose pectin that consists of complex polysaccharides and the role of PL1 is uncertain [44].
Fig. 4. Distribution of glycoside hydrolase (GH) and pectin lyase (PL) families in the transcriptome of the four symbiotic protists in Coptotermus formosanus.
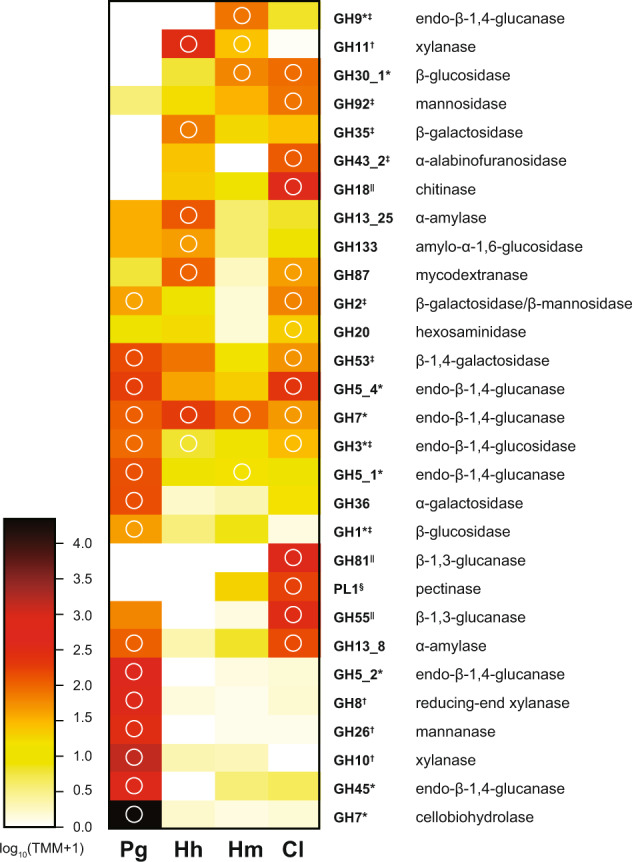
The color intensity of the heatmap shows the expression level of the GH/PL family. The expressions are represented by the average of trimmed mean of M (TMM) of the gene for the GH/PL family in the three cells. GH/PL family was sorted according to the result of clustering. The predicted activities of each GH/PL family are shown. GH/PL families digesting cellulose, hemicellulose, pectin, and fungal cell walls are indicated by an asterisk, dagger, double dagger, and section, respectively. White circles indicate GH/PL families that include the gene(s) of which TMM is larger than 100 at least in 2 of 3 replicates. Pg, Hh, Hm, Cl represents Pseudotrichonympha grassii, Holomastigotoides hartmanni, Holomastigotoides minor, and Cononympha leidyi, respectively.
In contrast, galactosidase, endo-β-1,4-glucanase, and xylanase were encoded by all protists, suggesting that they can all attack the galactose residues in galactoside, as well as the 1,4-β-D-glucosidic linkages in cellulose and xylan. Interestingly, the GH targeting these common substrates and their expression levels, were not always shared among protists. The striking example is xylanase belonging to the GH10 and GH11: P. grassii showed high expression levels of GH10, whereas H. hartmanni and H. minor highly expressed the genes for GH11 xylanase. C. leidyi showed only a low level of expression of GH10 family xylanase and no GH11 gene was detected. The previous study insisted that Holomastigotoides plays a primary role in xylan degradation with GH11 [12] but their evidence did not exclude the existence of another xylan feeder. In fact, our results suggested that P. grassii also highly express the gene for GH10 of which activity to wood xylan were reported [16]. The functional difference between GH10 and GH11 was not evaluated in this study but it can be found in substrate specificity as suggested in elsewhere [45, 46].
Lignin is another main component of wood and it is still under controversy how wood-feeding termites overcome the lignin barrier for cellulose utilization [7]. Lignin degrading enzymes, for example, those belonging to Auxiliary Activity family 3 (AA3), AA4, and AA8, were not detected from any transcriptomes.
Chitin degradation by C. leidyi and the evolutionary origins of chitinase and nodB
The GH expression heatmap indicated that C. leidyi actively expresses chitinase genes (GH18, Fig. 4). Indeed, the GH18 includes the genes with the highest expression of those belonging to GHs in the C. leidyi transcriptome (Supplementary Table 4d). We also assessed the actual enzyme activity using three kinds of chitinase substrate. We collected protist cells separately from the anterior and posterior of the hindgut and successfully prepared fractions that showed different protist composition (Table 2). The posterior fraction showed significantly higher chitinase activity than the anterior, for all assayed chitinase substrates (Table 3). Cells of Holomastigotoides were equally found between the anterior and posterior fraction. On the other hand, the posterior fraction contained more C. leidyi cells than the anterior fraction, whereas the P. grassii cells were reversely distributed. Thus, the higher chitinase activity in the posterior fraction is very likely to be caused by the high density of C. leidyi, consistent with the transcriptome data.
Table 2.
Fractionation of protist density in the anterior and posterior gut content of Coptotermes formosanus.
| Cell density (104 cells/ml) | ||
|---|---|---|
| Species | Anterior | Posterior |
| Pseudotrichonympha grassii | 5.63 | 0.5 |
| Holomastigotoides spp.a | 2.75 | 2.15 |
| Cononympha leidyi | 1.86 | 2.50 |
aAs Holomastigotoides hartmanni and H. minor are difficult to identify, the cells of Holomastigotoides were counted as Holomastigotoides spp.
Table 3.
Chitinase activity of protists collected from the anterior and posterior gut of Coptotermes formosanus.
| Chitinase activity (U/g ± SD, n = 3) | |||
|---|---|---|---|
| Substrate | Anterior | Posterior | p value |
| pNP-(GlcNAc) | 5.16 ± 0.40 | 12.8 ± 0.56 | < 0.006 |
| pNP-(GlcNAc)2 | 0.62 ± 0.01 | 0.88 ± 0.04 | < 0.022 |
| pNP-(GlcNAc)3 | 0.33 ± 0.02 | 0.75 ± 0.06 | < 0.011 |
Every substrate was assayed three times. The activity of posterior fraction was significantly higher than that of the anterior for all the tested substrates (Welch’s t-test). Activity unit was defined as micromoles of the liberated p-nitorophenol per minute per microgram of the extracted protein. Chitinase substrate, 4-nitrophenyl N-acetyl-β-D-glucosamide, 4-nitrophenyl N,N′-diacetyl-β-D-chitobioside and 4-nitrophenyl β-D-N,N′,N″-triacetylchitotriose are shown as pNP-GlcNAc, pNP-(GLcNAc)2, pNP-(GLcNAc)3, respectively. GlcNAc indicates N-acetyl glucosamine.
From the transcriptome of C. leidyi, we further inferred that N-acetyl glucosamine, a degradant of chitin, is converted to ammonium and fructose-6-phosphate, the source of nitrogen compounds and ATP, respectively, by putative NodB, hexokinase (HK), and glucosamine-6-phosphate deaminase (NagB) (Fig. 5a). The four genes involved in these successive reactions were highly expressed among three replicates of C. leidyi transcriptomes. The genes for the chitin degradation pathway were also identified in H. hartmanni and H. minor, but their expression levels were not consistently high in replicates of single-cell transcriptomes. P. grassii also expressed chitinase at a low expression level. Interestingly, BLASTP analyses showed that chitinase genes of Spirotrichonymphea (H. hartmanni, H. minor, and C. leidyi) had affinity to those of fungi, whereas that of P. grassii were similar to those in Trichomonas vaginalis, suggesting vertical inheritance from the common ancestor of Parabasalia. Chitinase genes were also found from some groups of protists [47–50] but their chitinase genes do not show close affinity to the homologs of fungi and those found in our single-cell transcriptomes. We further searched chitinase in the available genome/transcriptomes of the metamonada, which contain Parabasalia and its sister clades [51] but chitinase genes related to fungi, Holomastigotoides, or C. leidyi were not found. To investigate evolutional origin of the chitinase genes in Spirotrichonymphea, we performed a phylogenetic analysis by the ML method. The chitinase genes of C. leidyi formed monophyletic clades, with the sequence reported as the C. formosanus gene with strong statistical value (99% of bootstrap probability, Fig. 5b). The sequence annotated as C. formosanus in this tree was most likely derived from contamination with C. leidyi since it was obtained from the transcriptome using entire termite bodies. The clade of C. leidyi was nested in fungal sequences. Therefore, the result suggests that C. leidyi obtained the chitinase genes from fungi via lateral gene transfer (LGT), although the direct donor lineage could not be determined from the ML tree. In addition, chitinase genes of H. hartmanni and H. minor were included in the fungal clade but located at a separate position from those of C. leidyi, indicating that they independently acquired the chitinase gene by LGT.
Fig. 5. Putative chitin degradation pathway in Cononympha leidyi.

a Predicted pathway of chitin degradation inferred from the transcriptome of C. leidyi. b Maximum-likelihood (ML) tree of chitinase. Only the operational taxonomic units around genes of Coptotermes formosanus symbionts are shown. The sequences of fungi and C. formosanus symbionts are indicated by blue and red fonts, respectively. The nodes supporting 100% bootstrap probability (BP) are indicated by thick branches. The 70–99% BPs are shown on each node. Asterisks indicate the collapsed node includes a few sequences from non-Peizomycotina species. The detailed ML tree is provided in Supplementary Fig. 11. c ML tree of nodB. The sequences from Firmicutes are shown in blue. The collapsed clade comprises miscellaneous organisms and the taxon names cannot be given as in (b). The detailed ML tree is provided in Supplementary Fig. 12. The other details are the same as described in (b). d Identification of the organismal origin of nodB. The antisense probe of nodB mRNA with digoxigenin was hybridized and detected by immunoassay. The signal was exclusively observed in C. leidyi cells. Yellow, light blue, and dark blue triangle indicate the cells of P. grassii, Holomastigotoides, and C. leidyi. Note that it is difficult to classify H. hartmanni and H. minor, but the cell pointed by light blue is probably H. hartmanni, judging from its cell size. Arrows indicate debris. Scale bar: 25 μm.
We also inferred the phylogenetic tree of nodB genes because they were not found in the transcriptome of P. grassii nor the genome and transcriptome of model parabasalids, such as T. vaginalis and Tritrichomonas fetus. In contrast to the eukaryotic origin of the chitinase gene, the ML tree of NodB showed a different perspective. The nodB gene of C. leidyi was grouped with those of H. hartmanni, H. minor, Reticulitermes speratus (termite), and Treponema azotonutricium, with maximum bootstrap support (Fig. 5c). The gene annotated as R. speratus was probably due to contamination of gut symbionts as well as chitinase assigned to C. formosanus. On the other hand, the gene of T. azotonutricium, a bacterium isolated from the gut of the termite Zootermopsis angusticollis [52], is genuinely from the bacterium because it is encoded in the complete genome of T. azotonutricium. In order to confirm that the nodB gene was from C. leidyi and not from contamination of bacteria living in the gut, we conducted in situ hybridization targeting the nodB mRNA. The C. leidyi cells were exclusively stained, confirming that C. leidyi encoded and expressed nodB (Fig. 5d). We also excluded the possibility that bacteria associated with C. leidyi express nodB by checking poly A tail of its mRNA. Considering these facts, we concluded that the common ancestor of Spirotrichonymphea protists in C. formosanus acquired the nodB gene from bacterial neighbor, such as that belonging to Treponema.
Discussion
In this study, we performed single-cell transcriptomes of the gut protists inhabiting in the wood-feeding termite C. formosanus where has been believed to harbor only three protist species for near a century [19]. Despite through morphological observation, the existence of hidden Holomastigotoides species were not suggested until molecular techniques were applied [26]. By using FISH and single-cell transcriptomes, we clearly showed that C. formosanus actually harbors two Holomastigotoides species, which is hardly distinguishable under light microscope except cell size. This finding enforces the importance of evaluating microbial diversity in the termite gut using genetic information even if the community structure looks simple.
Because lignocellulose is a complex compound that comprises cellulose, hemicellulose, pectin, and lignin, the process of wood digestion requires the collaborative action of various enzymes. Several meta-omics studies of wood-feeding termites including C. formosanus detected a number of cellulases, hemicellulases, and pectinases [15–17]. Compared with these meta-omics analyses, our single-cell transcriptomes of the protist species assigned these genes to individual symbionts, resulting the reassignment of GHs that were formerly identified as fungi or bacteria to protists. For example, GH8 and GH26 were identified as bacterial origins in the metatranscriptomic study [16] but they are encoded by P. grassii considering the high and stable expression in the single-cell transcriptomes. As the genes involving in wood degradation can be transferred from bacteria to symbiotic protists in termites [37], similarity-based taxonomic identification of genes found in meta-omics should be interpreted with caution. On the other hands, meta-omics approach using whole gut can circumvent some changes in gene expression caused by single-cell isolation procedure. In this study, the cells of the protists were released from the gut and washed by pipetting before the cDNA synthesis, and the influence of this procedure on gene expression should be evaluated in future.
Our single-cell transcriptomes also showed different expression patterns of GHs among the protists in C. formosanus, giving new insights to understand the division of roles in wood digestion. In the previous studies, Holomastigotoides was regarded as a main wood decomposer because (1) they are equally distributed over the whole hindgut, (2) their cell number increases with host feeding activity, and (3) they ingest wood particles even in the P. grassii-eliminated hindgut [21–23, 43]. The comparative analysis here suggested that, in contrast to P. grassii, Holomastigotoides does not degrade hemicellulose component of which main chain consists of mannan. Therefore, a major role of Holomastigotoides in wood digestion can be derevied from efficient utilization of cellulose and hemicellulose, not accessibility of more various wood components. This indicates that the localization of P. grassii at the entrance of hindgut and utilization of mannose-containing hemicellulose is to avoid an overlap niche with Holomastigotoides. If so, the division of role in C. formosanus has been likely evolved from competition, not collaboration. C. leidyi does not have highly expressed CAZYs digesting crystalline cellulose and main chains of hemicellulose. However, it highly expressed the genes for amorphous cellulose and side chains of hemicellulose. It is not completely matched with the previous assumption that C. leidyi is not involved in the wood digestion and nutritionally dependent on the larger protists [21, 43]. Considering the highly expressed CAZYs in C. leidyi and the fact that tens of C. leidyi cells frequently surround a cell of P. grassii or Holomastigotoides, C. leidyi seems to utilize wood degraded partially by the larger protists. Although a further study is needed to elucidate the degree of C. leidyi’s contribution to wood degradation, it surely participates more or less in wood digestion.
Apart from the genes involved in cellulolysis, it was revealed that C. leidyi actively expresses genes belonging to GH18 (chitinase) and those involved in degradation of chitin. C. leidyi probably converts the chitin degradant to ammonium, then assimilates it into amino acids. Although some GH18 enzymes show lysozyme activity and the enzyme assay we performed here cannot distinguish chitinase and lysozyme activities, we consider that the C. leidyi GH18 works as a chitinase because of its high similarity to chitinases in fungi, of which substrates are characterized. The chitin utilization as a nitrogen source may be essential for C. leidyi to survive in the termite gut, given that dead wood is very poor in nitrogen compounds such as amino acids and that C. leidyi does not possess nitrogen-fixing endosymbiotic bacteria, e.g., Candidatus Azobactroides pseudotrichonymphae in P. grassii [3]. There are two possible sources of chitin in termite guts: (1) shedding skin of termites: it is well observed that the molting skin of termites is eaten by their nestmates; thus, C. leidyi is likely to utilize termite skin as a nitrogen source. Nitrogen compounds in C. leidyi may finally return to the host termites after it is digested, suggesting C. leidyi’s contribution to nitrogen recycling in the symbiotic system. (2) The fungal cell wall: termites are always at the risk of infection from entomopathogenic fungi from their colony environment; however, infected termites are seldomly found in the field. One of the reasons for this is that the fungi attached to the termite cuticle are removed by nestmate grooming and conidial germination of them is inhibited in the gut [53]. Considering this observation, we inferred that C. leidyi degrades the cell wall of the inactivated conidia. Rosengaus et al. [54] also suggested that β-1,3 glucanases derived from protists degrade glucan, another main component of fungal cell wall, and contribute to protection from fungal pathogen. This is consistent with the high expression level of β-1,3 glucanases (GH55 and GH81) in C. leidyi (Fig. 4 and Supplementary Table 4) and thus we propose that C. leidyi plays a role not only in nitrogen recycling but also in host defense. Although the localization of C. leidyi at the posterior hindgut is counterintuitive to this hypothesis, it is still possible that C. leidyi utilized fungal cell wall inactivated by the other symbiont. As a set of genes involved in the chitin degradation pathway are highly expressed only in C. leidyi, nitrogen recycling and/or host defense through chitin degradation is probably a unique function of C. leidyi in the C. formosanus gut.
The phylogenetic analyses clearly indicated that chitinase and NodB encoded in C. leidyi were derived from LGT. In contrast, the genes for NagB and HK, which are responsible for the downstream step of the chitin degradation pathway, are most likely inherited vertically from the common ancestor of Parabasalia. Therefore, laterally transferred genes of separate origins could coordinate the existing system to construct the chitin degradation pathway. Although our transcriptome analyses of H. hartmanni and H. minor did not show high expression levels, they both possess all genes for the chitin degradation pathway, and the phylogenetic analysis indicated that their chitinase and nodB genes were also derived from LGT. If H. hartmanni and H. minor as well as C. leidyi decompose chitin and produce ammonium, the chitin degradation pathway could establish multiple times in the gut of C. formosanus because evolutionary origins of Holomastigotoides chitinase are different from C. leidyi. This assumption may imply the importance of nitrogen recycling and defense against fungi in the termite gut. Finally, as the nodB gene was found in R. speratus where Parabasalia and Oxymonads co-exist, the chitinase degradation pathway can be carried out in R. speratus. It is an interesting question as to which species encodes the genes for chitin degradation, whether their origins are common in C. formosanus and R. speratus symbionts, and to what extent the chitin degradation pathway distributes in the termites, in terms of the evolution of symbiosis in the termite gut microbiome.
In conclusion, our single-cell transcriptomes showed differential expression patterns of GHs among protists in the wood-feeding termite, supporting the concept of their collaborative work in wood digestion. In addition to lignocelluolysis, we speculated that one of the symbionts, C. leidyi, may contribute efficient nitrogen utilization and/or defense against entomopathogenic infection by degrading nestmate skin and fungal cell wall. These insights were achieved by means of single-cell analyses covering all the members of the population, which is in clear contrast to metatranscriptomic approaches that do not determine the exact owners of the genes identified.
Taxonomic summary
Phylum Parabasalia Honigberg 1973; Class Spirotrichonymphea Grassé 1952; Order Spirotrichonymphida Grassé 1952; Family Holomastigotoididae Grassi 1917 emend Čepička et al. 2010; Genus Holomastigotoides Grassi & Foà 1911; Holomasitogotoides minor Nishimura, sp. nov.
Description
Multiflagellate parabasalian. Obligate symbiont of Coptotermes formosanus. Cells 23–117 μm (average 60 μm) in length and 24–109 μm (average 61 μm) in width. Morphologically unidentifiable with H. hartmanii under light microscope but smaller cell size. SSU rRNA gene sequences with 99% identity to JN585011.
Diagnosis
Distinguished from all other Holomastigotoides species by SSU rRNA gene sequence; distinguished from other Holomastigotoides except H. hartmanii by host identity; distinguished from H. hartmanii by its larger cell size (34–223 μm in length by 21–164 μm in width with the respective average 112 and 75 μm).
Locality
Hindgut of Coptotermes formosanus (Isoptera, Rhinotermitidae).
Etymology
The specific epithet minor refers to the smaller cell size compared with the H. hartmanii which lives in the same host.
Holotype
Permanent protargol-stained slide of microscope (TNS-AL-58971), deposited in the herbarium of the National Museum of Nature and Science (TNS), Tokyo.
Supplementary information
Acknowledgements
This work was supported in part by grants from the Japanese Society for Promotion of Science awarded to YN (16H07451 and 18K14783) and MO (17H01447). This work was also supported by RIKEN Competitive Program for Creative Science and Technology (to MO). We thank Prof. Kitade for the preparation of the permanent slide.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-020-0698-z) contains supplementary material, which is available to authorized users.
References
- 1.Ohkuma M. Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends Microbiol. 2008;16:345–52. doi: 10.1016/j.tim.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Hongoh Y, Sharma VK, Prakash T, Noda S, Taylor TD, Kudo T, et al. Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Natl Acad Sci USA. 2008;105:5555–60. doi: 10.1073/pnas.0801389105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hongoh Y, Sharma VK, Prakash T, Noda S, Toh H, Taylor TD, et al. Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science. 2008;322:1108–9. doi: 10.1126/science.1165578. [DOI] [PubMed] [Google Scholar]
- 4.Ohkuma M, Noda S, Hattori S, Iida T, Yuki M, Starns D, et al. Acetogenesis from H2 plus CO2 and nitrogen fixation by an endosymbiotic spirochete of a termite-gut cellulolytic protist. Proc Natl Acad Sci USA. 2015;112:10224–30. doi: 10.1073/pnas.1423979112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuki M, Kuwahara H, Sintani M, Izawa K, Sato T, Starns D, et al. Dominant ectosymbiotic bacteria of cellulolytic protists in the termite gut also have the potential to digest lignocellulose. Environ Microbiol. 2015;17:4942–53. doi: 10.1111/1462-2920.12945. [DOI] [PubMed] [Google Scholar]
- 6.Kuwahara H, Yuki M, Izawa K, Ohkuma M, Hongoh Y. Genome of “Ca. Desulfovibrio trichonymphae”, an H2-oxidizing bacterium in a tripartite symbiotic system within a protist cell in the termite gut. ISME J. 2017;11:766–76.. doi: 10.1038/ismej.2016.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brune A. Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol. 2014;12:168–80. doi: 10.1038/nrmicro3182. [DOI] [PubMed] [Google Scholar]
- 8.Ohkuma M, Iida T, Ohtoko K, Yuzawa H, Noda S, Visogliosi E, et al. Molecular phylogeny of parabasalids inferred from small subunit rRNA sequences, with empasis on Hypermastigea. Mol Phylogenet Evol. 2005;35:646–55. doi: 10.1016/j.ympev.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Noda S, Mantini C, Meloni D, Inoue J, Kitade O, Viscogliosi E, et al. Molecular phylogeny and evolution of Parabasalia with improved taxono sampling and new protein markers of actin and elongation factor-1α. PLoS One. 2012;7:e29938. doi: 10.1371/journal.pone.0029938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe H, Nakashima K, Saito H, Slaytor M. New endo-beta-1,4-glucanases from the parabasalian symbionts, Pseudotrichonympha grassii and Holomastigotoides mirabile of Coptotermes termites. Cell Mol Life Sci. 2002;59:1183–92.. doi: 10.1007/PL00012520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue T, Moriya S, Ohkuma M, Kudo T. Molecular cloning and characterization of a cellulase gene from a symbiotic protist of the lower termite, Coptotermes formosanus. Gene. 2002;11:67–75. doi: 10.1016/j.gene.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 12.Arakawa G, Watanabe H, Yamasaki H, Maekawa H, Tokuda G. Purification and molecular cloning of xylanases from the wood-feeding termites, Coptotermes formosanus Shiraki. Biosci Biotechnol Biochem. 2009;73:710–18. doi: 10.1271/bbb.80788. [DOI] [PubMed] [Google Scholar]
- 13.Duarte S, Nunes L, Borges PAV, Nobre T. A bridge too far? An integrative framewoork linking classical protist taxonomy and metabarcoding in lower termites. Front Microbiol. 2018;9:2620. doi: 10.3389/fmicb.2018.02620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treitli SC, Martin K, Husník F, Keeling PJ, Hampl V. Revealing the metabolic capacity of Streblomastix strix and its bacterial symbionts using single-cell metagenomics. Proc Natl Acad Sci USA. 2019;116:19675–684. doi: 10.1073/pnas.1910793116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tartar A, Wheeler MM, Zhou X, Coy MR, Boucias DG, Scarf ME. Parallel metatransccriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol Biofuels. 2009;2:25. doi: 10.1186/1754-6834-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie L, Zhang L, Zhong Y, Liu N, Long Y, Wang S, et al. Profiling the metatranscriptome of the protistan community in Coptotermes formosanus with emphasis on the lignocellulolytic system. Genomics. 2012;99:246–55. doi: 10.1016/j.ygeno.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Franco Cairo JPL, Carazzolle MF, Leonardo FC, Mofatto LS, Brenelli LB, Gonçalves TA, et al. Expanding the knowledge on lignocellulolytic and redox enzymes of worker and soldier castes from the lower termite Coptotermes gestroi. Front Microbiol. 2016;7:1518. doi: 10.3389/fmicb.2016.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su NY. Overview of the global distribution and control of the Formosan subterranean termites. Sociobiology. 2003;41:7–16. [Google Scholar]
- 19.Koidzumi M. Studies on the intestinal protozoa found found in the termites of Japan. Parasitology. 1921;13:235–309. [Google Scholar]
- 20.Jasso-Selles D, Martini F, Freeman K, Garcia M, Merrell T, Scheffrahn R, et al. The parabasalid symbiont community of Heterotermes aureus: molecular and morphological characterization of four new species and reestablishment of the genus Cononympha. Eur J Protistol. 2017;61:48–63. doi: 10.1016/j.ejop.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Yoshimura T. Distribution of the symbiotic protozoa in the hindgut of Coptotermes formosanus Shiraki (Isoptera; Rhinotermitidae) Jpn J Environ Entomol Zool. 1992;4:115–20. [Google Scholar]
- 22.Yoshimura T, Watanabe T, Tsunoda K, Takahashi M. Distribution of the cellulolytic activities in the lower termite. Coptotermes formosanus Shiraki (Isoptera: Rhinotermitidae) Mater Organismen. 1992;27:273–84. [Google Scholar]
- 23.Yoshimura T, Azuma J, Tsunoda K, Takahashi M. Cellulose metabolism of the symbiotic protozoa in termite, Coptotermes formosanus Shiraki (Isoptera: Rhinotermitidae): I. Effect of degree of polymerization of cellulose. Mokuzai Gakkaishi. 1993;39:221–6. [Google Scholar]
- 24.Terrett OM, Dupree P. Covalent interactions between ligning and hemicelluloses in plant secondary cell walls. Curr Opin Biotechnol. 2019;56:97–104. doi: 10.1016/j.copbio.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 25.Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA-Seq reveals non-genetic gene expression heterogeneity. Genome Biol. 2013;14:R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie L, Liu N, Haung Y, Wang O. Flagellate community structure in Coptotermes formosanus (Isoptera: Rhinotermitidae) and a comparison of three study methods. Acta Entomol Sin. 2011;54:1140–6. [Google Scholar]
- 27.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabherr MG, Haas BJ, Yasssour M, Levin JZ, Thompson DA, Ido A, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Noda S, Ohkuma M, Yamada A, Hongoh Y, Kudo T. Phylogenetic position and in situ identification of ectosymbiotic spirochetes on protists in the termite gut. Appl Environ Microbiol. 2003;69:625–33. doi: 10.1128/AEM.69.1.625-633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Todaka N, Inoue T, Saita K, Ohkuma M, Nalepa CA, Lenz M, et al. Phylogenetic analysis of cellulolytic enzyme genes from representative lingeages of termites and a related cockroach. PLoS One. 2010;5:e8636. doi: 10.1371/journal.pone.0008636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue J, Saita K, Kudo T, Ui S, Ohkuma M. Hydrogen production by termite gut protists: characterization of iron hydrogenases of parabasalian symbionts of the termite Coptotermes formosanus. Eukaryot Cell. 2007;6:1925–32. doi: 10.1128/EC.00251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z, Hu SK, Cambell V, Tatters AO, Heidelberg KB, Caron DA. Single-cell transcriptomics of small microbial eukaryotes: limitations and potential. ISME J. 2017;11:1282–5. doi: 10.1038/ismej.2016.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2017;35:543–8. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiroguchi K, Jia TZ, Peter AS, Xie XS. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc Natl Acad Sci USA. 2012;09:1347–52. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshimura T, Azuma J, Tsunoda K, Takahashi M. Cellulose metabolism of the symbiotic protozoa in termite, Coptotermes formosanus Shiraki (Isoptera: Rhinotermitidae): II. Selective defaunation of protozoa and its effect on cellulose metabolism. Mokuzai Gakkaishi. 1993;39:227–30. [Google Scholar]
- 44.Benoit I, Coutinho PM, Schols HA, Gerlach JP, Herissat B, De Vries RP. Degradation of different pections by fungi: correlations and contrasts between pectinolytic enzyme sets identified in genomes oand growth on pectins of different origin. BMC Genom. 2012;13:321. doi: 10.1186/1471-2164-13-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins T, Gerday C, Feller G. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev. 2005;29:3–23. doi: 10.1016/j.femsre.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Yagi H, Takehara R, Tamaki A, Teramoto K, Tsutsui S, Kaneko S. Functional characterization of the GH10 and GH11 xylanases from Streptomyces olivaceoviridis E-86 provide insights into the advantage of GH11 xylanase in catalyzing biomass degradation. J Appl Glycosci. 2018;66:29–35. doi: 10.5458/jag.jag.JAG-2018_0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joshi MB, Roger ME, Shakarian AM, Yamage M, AI-Harthi SA, Bates PA, et al. Molecular characterization, expression, and in vivo analysis of LmexCht1. J Biol Chem. 2005;280:3847–61. doi: 10.1074/jbc.M412299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gutiérrez Sánchez PA, Alzate JF, Montoya MM. Analysis of carbohydrate metabolisms genes of Spongospora subterranea using 454 pyrosequencing. Rev Fac Nac Agron Medellín. 2013;67:7247–60. [Google Scholar]
- 49.Taira T, Gushiken C, Sugata K, Ohnuma T, Fukamizo T. Unique GH18 chitinase from Euglena gracilis: full-length cDNA cloning and characterization of its catalytic domain. Biosci Biotechnol Biochem. 2018;82:1090–100. doi: 10.1080/09168451.2018.1459463. [DOI] [PubMed] [Google Scholar]
- 50.Cenci U, Sibbald SJ, Curtis BA, Kamikawa R, Eme L, Moog D, et al. Nuclear genome sequence of plastid-lacking cryptomonad Goniomonas avonlea provides insights into the evolution of secondary plastids. BMC Biol. 2018;16:137. doi: 10.1186/s12915-018-0593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leger MM, Kolisko M, Kamikawa R, Stairs CW, Kume K, Čepička I, et al. Organelles that illuminate the origins of Trichomonas hydrogenosomes and Giardia mitosomes. Nat Ecol Evol. 2017;1:0092. doi: 10.1038/s41559-017-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graber JR, Leadbetter JR, Breznak JA. Description of Treponema azotonutricium sp. nov. and Treponema primitia sp. nov., the first spirochetes isolated from termite guts. Appl Environ Microbiol. 2004;70:1315–20. doi: 10.1128/AEM.70.3.1315-1320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanagawa A, Shimizu S. Resistance of the termite, Coptotermes formosanus Shiraki to Metarhizium anisopliae due to grooming. BioControl. 2007;52:75–85. [Google Scholar]
- 54.Rosengaus RB, Schultheis KF, Yalonetskaya A, Bulmer MS, DuComb WS, Benson RW, et al. Symbiont-derived β-1,3-glucanases in a social insect: mutualism beyond nutrition. Front Microbiol. 2014;5:607. doi: 10.3389/fmicb.2014.00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.