

Abstract
Nonhematopoietic stromal cells in lymph nodes such as fibroblastic reticular cells (FRCs) can support the survival of plasmablasts and plasma cells (together, antibody-forming cells (AFCs)). However, a regulatory function for the stromal compartment in AFC accumulation has not been appreciated. Here, we show that CCL2-expressing stromal cells limit AFC survival. FRCs express high levels of CCL2 in vessel-rich areas of the T cell zone and the medulla, where AFCs are located. FRC CCL2 is upregulated during AFC accumulation, and we use lymph node transplantation to show that CCL2 deficiency in BP3+ FRCs and lymphatic endothelial cells increases AFC survival without affecting B or germinal center cell numbers. Monocytes are key expressers of the CCL2 receptor, CCR2, as monocyte depletion and transfer late in AFC responses increases and decreases AFC accumulation, respectively. Monocytes express reactive oxygen species (ROS) in an NADPH oxidase 2 (NOX2)-dependent manner, and NOX2-deficient monocytes fail to reduce AFC numbers. Stromal CCL2 modulates both monocyte accumulation and ROS production, and is regulated in part by manipulations that modulate vascular permeability. Together, our results reveal that the lymph node stromal compartment, by influencing monocyte accumulation and functional phenotype, has a regulatory role in AFC survival. Our results further suggest a role for inflammation-induced vascular activity in tuning the lymph node microenvironment. The understanding of stromal-mediated AFC regulation in vessel-rich environments could potentially be harnessed to control antibody-mediated autoimmunity.
One Sentence Summary:
Lymph node stromal CCL2 limits plasma cell survival via monocyte accumulation and reactive oxygen species and is tuned by vascular permeability.
Introduction
Lymphocytes in lymph nodes are supported by a non-hematopoietic stromal compartment comprised of mesenchymal cells, blood vessels, and lymphatic sinuses. The mesenchymal cells, comprised mainly of fibroblastic reticular cells (FRCs) that are marked by the expression of podoplanin (PDPN), ensheathe and produce the matrix components that make up a reticular network of collagen-rich fibrils (1–3). FRCs have additional functions in regulating immune cell positioning and lymphocyte survival and activity, and they interact closely with the blood vessels and lymphatic sinuses that transport oxygen, micronutrients, cells, and antigens to and from lymph nodes. During immune responses, the stromal compartment undergoes proliferative expansion and phenotypic alterations as lymph nodes grow (4, 5). Fully understanding this dynamic compartment and how it shapes immune responses could aid in the development of stromal-focused approaches to modulate immunity in disease.
Plasmablasts and plasma cells (collectively referred to as antibody-forming cells (AFCs)) in secondary lymphoid organs are thought to contribute to autoantibody titers in diseases such as lupus (6–8). During T cell-dependent B cell responses, an initial burst of short-lived plasmablasts is followed by the accumulation of long-lived plasma cells (9, 10). Plasmablasts in spleen are considered extrafollicular in origin, but in lymph nodes, they may also derive in part from germinal center responses. Both short- and long-lived cells are thought to migrate through the T cell zone (T zone) to accumulate in the medulla where most die and some, especially during secondary responses, will egress and home to the bone marrow to further mature and contribute to a long-lived pool (9–12).
Relatively little is known about the contributions of the lymph node microenvironment to regulating AFCs. We have shown that depletion of ZBTB46+ dendritic cells (DCs) at day 8 after immunization with OVA-Alum leads to a 75% loss of AFCs at day 9 and that this was at least partly attributable to the loss of FRCs (13). The AFC loss was rescued by BAFF supplementation, suggesting that FRCs support AFCs by ligating BAFF-binding receptors on AFCs (13). Recently, T zone stromal cells bordering follicles were shown to express APRIL and BAFF that can promote AFC survival upon AFC exit from the germinal center (14). In addition, medullary FRCs support medullary cord AFCs via IL6 production (15). Myeloid cells colocalize with AFCs as AFCs traverse the T zone to the medulla, and these myeloid cells express APRIL and IL-6 that could support AFCs (12). However, there is also evidence that at least some myeloid cells play regulatory roles. Depletion of LysM-Cre+ or CCR2+ cells at the initiation of, or early after immunization and deletion of Myd88 or FcεR1γ in presumably myeloid cells increased AFC numbers (16–18). Similarly, CCR2 deficiency or monocyte depletion upon viral infection increased AFC numbers, and iNOS expressed by monocytes or monocyte-derived cells has been identified as one mediator (17, 19). Together, studies suggest that FRCs promote AFC development and survival while myeloid cells such as monocytes may play a regulatory role. Whether there is an FRC-AFC regulatory axis is unknown.
In this study, we show that the stromal compartment, and especially FRCs, in AFC-rich areas in the T zone and medulla express high levels of CCL2 and limit AFC survival. Monocytes are key CCL2-responsive cells that regulate AFCs in a manner dependent on NOX2, which is needed for reactive oxygen species (ROS) generation. We show that stromal CCL2 modulates both monocyte accumulation and ROS production and is regulated by manipulations that modulate vascular permeability. These results suggest a model whereby the lymph node stromal compartment, in addition to supporting AFCs, also functions to limit AFC responses and is in part regulated by the vasculature.
Results
CCL2 is highly expressed by lymph node FRCs in the T cell zone and medulla
In examining for CCL2 expression, we analyzed CCL2 reporter mice produced by BAC-mediated transgenesis that express CCL2 linked to GFP. The GFP is clipped off in the cytosol and remains there to mark CCL2-producing cells (“M1R” mice from (20)). In homeostatic lymph nodes, GFP was expressed in the T cell zone and medulla and excluded from B cell follicles (Fig. 1A). Within the T cell zone, vascular-rich regions under the follicles known as the cortical ridge (21) and vascular cords running toward the medulla (22) are recognizable by the high density of ER-TR7+ vessels, and GFP was most brightly expressed in these areas (Fig. 1A). Bone marrow chimeras repopulating CCL2-GFP hosts with WT bone marrow (WT→CCL2-GFP chimeras) showed a similar pattern of GFP expression (Fig. 1B), suggesting that CCL2hi-expressing cells in the T zone and medulla could be stromal in origin. Consistent with this idea, GFP was mostly expressed in a reticular pattern (Fig. 1C), although round, likely hematopoietic, GFP-expressing cells were also seen (Fig. 1C, arrowheads). Flow cytometric analysis confirmed that both CD45+ hematopoietic and CD45- non-hematopoietic cells expressed GFP (Fig. 1D). CD45+ GFP+ cells were mostly CD11b+ myeloid cells and could be divided into Ly6C+ presumed monocytes and Ly6C- cells (Fig. 1D). The majority of CD45- GFP+ cells were CD31-PDPN+ FRCs and under 20% were CD31+PDPN+ lymphatic endothelial cells (LECs) (Fig. 1D). FRCs expressed the highest level of GFP when compared to LECs and CD11b+ cells (Fig. 1E). Together, these results suggested that FRCs are major CCL2 expressers in homeostatic lymph nodes, with LECs and myeloid cells expressing lower levels of CCL2.
Fig. 1. Lymph node stromal cells in the T cell zone and medulla express CCL2.
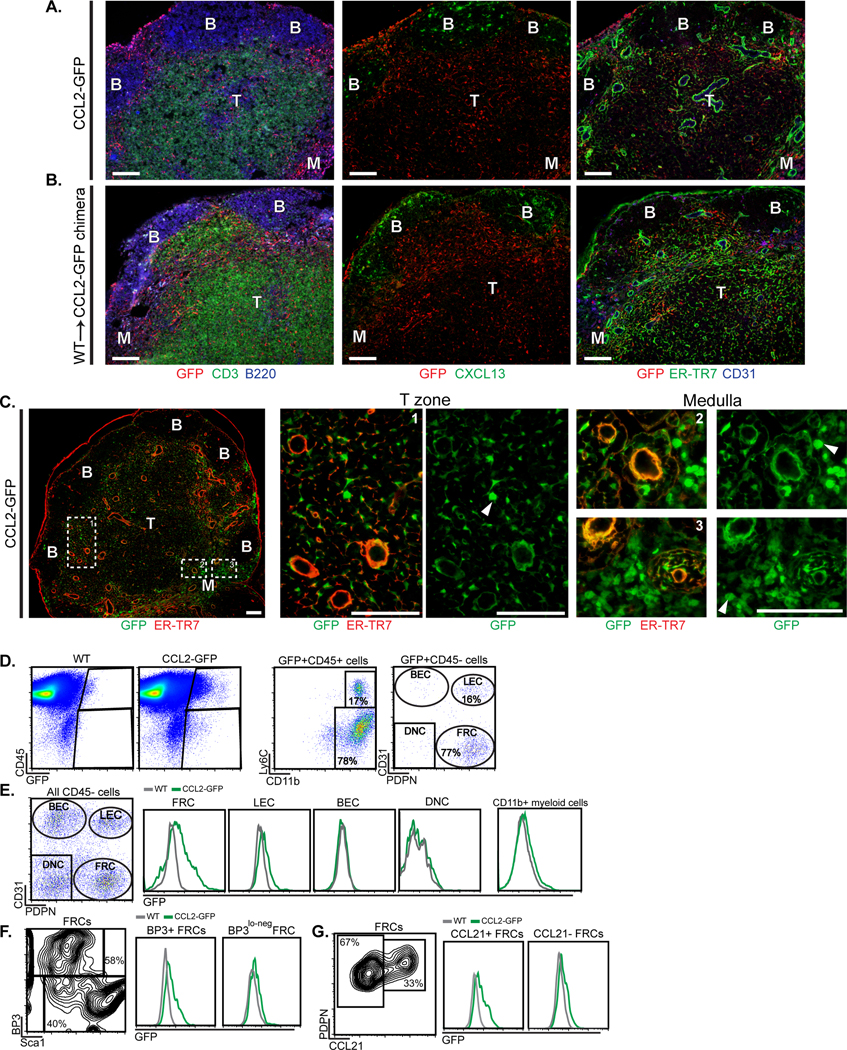
(A-G) Homeostatic brachial lymph nodes from indicated mice were examined. (A-B) Sections from (A) CCL2-GFP mice and (B) WT→CCL2-GFP chimeras were stained for GFP and indicated markers. (B) B cell follicles, (T) T cell zone, (M) medulla. (C) Magnified views of GFP-expressing cells. Arrowheads point to round cells. (D-G) Flow cytometric characterization of cells from CCL2-GFP mice. Fluorescence scale is log10. (D) Characterization of GFP+ cells. (BEC) blood endothelial cells, (LEC) lymphatic endothelial cells, (FRC) fibroblastic reticular cells, (DNC) double negative cells. (E) Histograms depicting GFP levels in indicated cell populations. (F, G) Density plots and histograms showing GFP expression in indicated FRC subsets. (A-C) Scale bars=100 μm. (A-G) Results are representative of n≥3 mice/condition.
We further examined the characteristics of the GFP-expressing FRCs. BP3/CD157/BST-1 marks well-differentiated CCL21-expressing T zone FRCs (fig. S1A) as well as CXCL13-expressing marginal reticular cells (MRCs) and follicular dendritic cells (FDCs) (21, 23, 24). Consistent with recent findings (15), the medulla is generally dimmer for BP3, although stromal BP3 staining is detectable within medullary cords in both homeostatic and immunized lymph nodes (fig. S1B). (PDPN+) BP3lo-neg cells are comprised mainly of CD34+ reticular cells that are also Sca1+ (fig. S1A), and have been shown to be perivascular and have progenitor potential (25, 26). GFP was expressed at higher levels by BP3+ cells than by BP3lo-neg cells in CCL2-GFP mice (Fig. 1F), as was intracellular CCL2 protein (fig. S1C). The BP3+ CCL2hi expressers were also found in the CCL21+ population (Fig. 1G), supporting the idea that the high CCL2 expression in the cortical ridge and paracortical vascular cords was by T zone FRCs. Together, these results indicated that CCL2 is expressed most highly by BP3+ FRCs, some of which are T zone FRCs.
Stromal CCL2 is upregulated with immunization and co-localizes with AFCs
The regions of high stromal CCL2 are also areas of AFC accumulation (12, 14), leading us to ask whether stromal CCL2 regulated AFCs. A kinetic analysis of B cell responses in popliteal lymph nodes after OVA-Alum immunization showed that germinal center B cells and IgG+ AFCs were detectable in large numbers by day 9 (fig. S2, A and B). The AFCs showed a high proliferative rate at day 9, suggesting that many were plasmablasts (fig. S2C). By day 12, AFC numbers had dropped (fig. S2 A and B), consistent with the apoptosis of plasmablasts seen in spleen (10, 27) and the drop seen in lymph nodes (11, 12), and remained at day 12 levels at least through day 15 (fig. S2, A and B). At days 12–15, the AFC proliferation rate was lower than at day 9 but still at about 12% (fig. S2C), suggesting the steady AFC numbers between days 12 and 15 reflected continuous cell turnover, with a balance mainly between proliferation and apoptosis. Because this day 12–15 window allowed for investigation of AFC proliferation and survival, we focused our efforts on studying this time period.
To assess the role of stromal CCL2 in regulating AFCs, we examined for immunization-induced alterations in CCL2 expression in reporter mice and co-localization of AFCs and CCL2 in WT→CCL2-GFP chimeras. BP3+ FRCs upregulated GFP expression by day 9 after immunization (Fig. 2A), as did CCL21+ FRCs (Fig. 2, A and B), suggesting that T zone FRCs were among the cells that upregulated CCL2. Interestingly, CCL21- FRCs, some of which are medullary and/or inter-follicular cells (26, 28, 29), showed an early upregulation of CCL2 at day 2 which decreased by day 15 (Fig. 2B). LECs but not myeloid cells also showed CCL2 upregulation after immunization, although LEC CCL2 expression remained quite low compared to that of FRCs and had returned to nearly homeostatic levels by day 15 (Fig. 2A and B). At both days 10 and 15 after OVA-Alum immunization, CCL2 expression was highest in regions of AFC localization (Fig. 2C and fig. S2D). The co-localization of CCL2-expressing FRCs with AFCs suggested a potential functional interaction between the two.
Fig. 2. Stromal CCL2 is upregulated upon immunization and co-localizes with AFCs.

(A-C) CCL2-GFP mice or WT→CCL2-GFP chimeras were immunized in footpads with OVA-Alum on day 0 (D0), and popliteal nodes harvested on indicated days. (A) Contour plots and histograms show GFP levels in the indicated cells. Fluorescence scale is log10. (B) Percentage of FRCs and LECs that are GFP+ over time. Each symbol represents one mouse; n=3–8/condition over 5 experiments. *P < 0.05, **P < 0.01 using 2-tailed unpaired Student’s t test. Error bars represent SD. (C) GFP and AFC localization. Sections from day 15 WT→CCL2-GFP chimeras were stained for GFP, mouse IgG, and CD31. (B) B cell follicles, (T) T cell zone, (M) medulla. Representative of n≥3 mice. Scale bars=100 μm.
Lymph node stromal CCL2 regulates AFC numbers and survival
We examined the effect of CCL2 deficiency on AFC responses. While B cell numbers were similar in homeostatic WT and Ccl2−/− mice (fig. S3A), Ccl2−/− mice at day 15 after OVA-Alum showed increased numbers of total B cells, germinal center B cells, and AFCs with no change in T cell numbers (Fig. 3A). The increased AFCs in CCL2-deficient mice was accompanied by increased anti-OVA-secreting cells and anti-OVA serum IgG (Fig. 3, B and C). Since the increase in AFCs in Ccl2−/− mice could be a consequence of increased germinal center B cell numbers, we further characterized the AFCs. AFCs in Ccl2−/− mice showed no change in ki67 expression but had decreased activated caspase-3 levels (Fig. 3, D and E), suggesting that they were proliferating at similar rates but undergoing less apoptosis than WT AFCs. Anti-OVA-secreting cell numbers in bone marrow were similar (Fig. 3F), suggesting that the increased lymph node AFC accumulation was not due to reduced emigration from lymph node to bone marrow. AFCs localized to the T cell zone and medulla in both WT and Ccl2−/− lymph nodes (Fig. 3G). These data suggested that CCL2 limits B cell responses and AFC survival.
Fig. 3. Ccl2 −/− mice show increased AFC accumulation and survival.

(A-G) WT and Ccl2−/− mice were immunized on day 0 and examined on day 15. (A) Numbers of indicated cell type/lymph node by flow cytometric analysis. (B) Anti-OVA IgG spots/lymph node using ELISPOT. (C) Anti-OVA IgG serum titers. (D-E) Percentages of lymph node AFCs positive for (D) ki67 and (E) activated caspase-3. (F) Anti-OVA IgG spots in bone marrow using ELISPOT. (G) Representative lymph node sections stained for mouse IgG and ER-TR7. Scale bars=100 μm. (H-I) WT and Ccl2−/− mice were injected with LPS in footpads on day 0 and examined on day 8. (A-F, H-I) Each symbol represents one mouse; n=3–12/condition, data are from 5–6 (A-C,E) and 2 (D,F,H,I) experiments. **P<0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation.
We further assessed the role of CCL2 on a germinal center-independent lymph node AFC response and on splenic responses. At 8 days after footpad LPS immunization, Ccl2−/− lymph nodes showed unchanged T and B cells, an almost two-fold increase in AFC numbers (Fig. 3H), and reduced AFC activated caspase-3 ( Fig. 3I). However, splenic responses to OVA-Alum and NP-Ficoll were similar in WT and Ccl2−/− mice (fig. S3, B and C). These results suggested that CCL2 can regulate lymph node AFC survival independent of an effect on germinal centers and that CCL2 does not play the same role in splenic responses in our models.
We asked about the role of stromal-derived CCL2. We considered crossing the Ccl19-Cre driver (30) with Ccl2f/f mice (20) to delete FRC CCL2. However, Ccl19-Cre; YFPf/STOP/f mice showed that only 52% of BP3+ FRCs (+/−12%; n=3 mice) were YFP+ and BP3lo-neg FRCs expressed very little YFP at day 15 after immunization (fig. S4), suggesting that FRC CCL2 would not be fully deleted in our model. We thus used a lymph node transplant model (31) where we transplanted (CD45.2) WT and Ccl2−/− popliteal lymph nodes into CD45.1 mice (Fig. 4 A and B). In similar systems, transplanted lymph node tissue is repopulated by recipient hematopoietic cells while the stromal compartment remains donor-derived (32, 33). While we initially performed bilateral transplantations (Fig. 4A), recovery rate of transplanted lymph nodes was only 32% (Fig. 4C), leading us to perform unilateral transplantations (Fig. 4B). Unilateral transplantations improved lymph node recovery to 93% (Fig. 4C), and the results of unilateral and bilateral transplantations were pooled as indicated.
Fig. 4. Lymph node stromal CCL2 limits AFC survival.

(A-F) WT CD45.1+ hosts received either bilateral (A) or unilateral (B) lymph node transplants, as indicated, prior to immunization with OVA/Alum and examination 15 days later. (C) Recovery rate of bilateral or unilateral transplanted lymph nodes. (D) Percentages of B, T, and CD11b+ cells that were host (CD45.1+) and donor (CD45.2+)-derived. Data are from bilateral transplants. (E) Normalized numbers of indicated cells/lymph node. (F) Normalized percentage of AFCs that are activated caspase-3+ or ki67+. (G-I) WT nodes were transplanted into left side of CCL2-GFP mice, as depicted in (G). (H) Histograms showing GFP expression in indicated cells from WT donor or CCL2-GFP host lymph nodes. “B6” node is from an untransplanted WT mouse. Fluorescence scale is log10. (I) Percentages of indicated cells that are GFP+. (D,E,F,I) Each symbol represents one lymph node; n=3–17 mice/condition from 2 (D), 11 (3 bilateral, 8 unilateral transplants)(E,F), and 1(I) independent experiments. *P < 0.05, **P < 0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation.
As early as 4 weeks after transplantation, recovered homeostatic lymph nodes showed normal organization, with robust B cell follicles, FDCs, and reticular pattern of ER-TR7 staining in the T zone (fig. S5A). After immunization, germinal centers and AFCs, when they were seen in sections, appeared normal in location (fig. S5B). The T, B, and myeloid cells in the transplanted lymph nodes were almost entirely CD45.1+ (i.e. recipient-derived) (Fig. 4D), as expected.
Of the recovered lymph nodes, we further examined for optimal and suboptimal transplants. Lymph node B cell numbers increase disproportionately relative to T cell numbers upon immunization (34, 35)(fig. S6A), but we found that some transplanted immunized lymph nodes had an abnormally low B:T cell ratio of less than 1 (fig. S6A). This phenotype suggested that the signals from the immunized footpad did not reach the transplanted lymph node and likely reflected incomplete reconstitution of the vascular connections, and we termed these lymph nodes as “suboptimal transplants.” The low B:T cell ratio occurred in both unilateral or bilateral transplants (fig. S6B) and in both WT and Ccl2−/− genotypes, consistent with the idea that this phenotype reflected poor transplant quality(fig. S6C). We excluded these suboptimal transplants from further analysis.
Relative to the WT controls, immunized transplanted Ccl2−/− lymph nodes showed no difference in the numbers of total, T, B, or germinal center B cells. AFCs, however, showed increased numbers, decreased activated caspase-3 expression, and no change in proliferation (Fig. 4E and F, fig. S6D–G). These AFC-specific effects pointed to a key role for the lymph node stromal compartment and its expression of CCL2 in limiting AFC survival.
To assess the degree to which different FRC subpopulations in our system were donor-derived, we transplanted WT popliteal lymph nodes into one side of CCL2-GFP reporter mice (Fig. 4G) and assessed for recipient GFP+ FRCs in the (GFP-) donor lymph nodes. In transplanted WT lymph nodes, BP3+ FRCs showed very low levels of GFP while BP3lo-neg FRCs were comparable in GFP expression to native (CCL2-GFP) lymph node BP3lo-neg FRCs (Fig. 4H and I). These results suggested that, in transplanted nodes, the CCL2 hi-expressing BP3+ FRCs remain largely donor-derived while the CCL2lo-expressing BP3lo-neg FRCs are replaced by host cells. Additionally, transplanted lymph node LECs did not show GFP expression, suggesting that they remain entirely donor-derived (Fig. 4H and I). In summary, the transplanted lymph nodes retain BP3+ FRCs and LECs but not BP3lo-neg FRCs. Our results together supported a role for CCL2 expressed by lymph node BP3hi FRCs and/or LECs in regulating AFC accumulation and survival.
Monocytes are key CCR2+ cells that regulate AFC survival late in immune responses
CCL2 interacts with CCR2 (36), and we sought to identify CCR2+ cells that regulated AFC survival in our system. We did not observe CCR2 expression by AFCs using either Ccr2-GFP mice (37) or CCR2 antibody staining (fig. S7A), suggesting that lymph node stromal CCL2 regulated AFCs indirectly. GFP was expressed mostly by CD11b+ myeloid cells, the majority of which consisted of Ly6Chi presumed monocytes (38, 39) and Ly6Clo cells (Fig. 5A). The Ly6Clo cells were comprised of 1) MHCIIhiEpCAM-CD103- cells (Fig. 5A) that were CCR7+ (fig. S7B), consistent with their identity as dermal or monocyte-derived DCs that migrated from skin (39), and 2) CD11chi CD8- cells that could be resident DCs or monocyte-derived cells (40, 41). Ly6Chi cells uniformly expressed GFP (Fig. 5B) and at higher levels than other GFP+ populations (fig. S7C), consistent with their identity as Ly6Chi monocytes (38, 39, 42). These results suggested that key CCR2+ cells could be myeloid cells.
Fig. 5. Monocytes late in immune responses are key CCR2+ cells that limit AFCs.

(A-C) CCR2-expressing cells in day 12 immunized lymph nodes were characterized using Ccr2-GFP reporter mice. (A) Representative flow cytometry plots show GFP+ subsets. (B) Percentage of CCR2+ cells in indicated myeloid populations. (C) Frozen section stained for indicated markers. Arrowhead points to round CCR2hi cell; arrow points to dendritic cell-shaped CCR2med cell. Scale bar=100 μm. (D-E) Numbers of indicated myeloid populations at day 15 after immunization in (D) WT and Ccl2−/− popliteal nodes and (E) WT and Ccl2−/− popliteal nodes transplanted into WT recipients. (F-H) CCR2-DTR-CFP mice were immunized with OVA-Alum on day 0, treated with DT on days 12 and 14, and examined on day 15. (F) Flow cytometry plots showing CCR2+ cell depletion. (G) Numbers of indicated cells. (H) Percentages of AFCs that are activated caspase-3+ and ki67+. (I-L) WT mice were injected with anti-Gr1 or control IgG on days 12,13, and 14 after OVA-Alum and examined on day 15. (I) Flow cytometry plots showing CCR2+ cell depletion. (J) Numbers of indicated myeloid populations. (K) Numbers of indicated cells. (L) Percentage of AFCs that are activated caspase-3+. (A,F,I) Fluorescence scale is log10. (B,D,E,G,H,J-L) Each symbol represents one mouse; n=3–17/condition; data are from 6–11 (D,E) and 2 (B,G,H,J-L) experiments. *P < 0.05, **P < 0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation.
Ly6Chi monocyte accumulation paralleled the two waves of FRC CCL2 upregulation seen after immunization. Monocyte numbers first increased at day 2 when CCL21- FRCs upregulated CCL2 and further increased at day 9 when CCL21+ FRCs upregulated CCL2 (Fig. 2B and fig. S7D). GFP+ cells in Ccr2-GFP mice were mainly in the T cell zone and medulla and co-localized with AFCs at all time points examined (fig. S7E–F). The GFP+ cells in these regions were comprised of both round GFPhi cells likely to be monocytes and elongated GFPmed cells presumed to be DCs (Fig. 5C). These results are consistent with a role for stromal CCL2 in positioning CCR2+ myeloid cells to promote interactions with AFCs.
We asked the extent to which stromal CCL2 promoted lymph node accumulation of monocytes and other CCR2+ cells. At day 15 after immunization, Ccl2−/− mice had reduced lymph node Ly6Chi and Ly6Cmed monocytes without an effect in other myeloid populations (Fig. 5D). Homeostatic popliteal and brachial Ccl2−/− lymph nodes showed fewer Ly6Chi monocytes (fig. S8A). While these results could reflect the critical role of bone marrow stromal CCL2 in mobilizing monocytes from bone marrow into circulation (20, 42, 43), transplanted Ccl2−/− lymph nodes also showed a specific reduction in Ly6Chi and Ly6Cmed monocytes (Fig. 5E). These results suggested a distinct role for lymph node stromal CCL2 in mediating lymph node monocyte accumulation, either by entry or retention, and supported the possibility that CCR2+ monocytes limit AFC survival.
We confirmed that CCR2+ cells regulated AFCs during days 12–15 by treating Ccr2-DTR mice (44) with diphtheria toxin (DT) during this window (fig. S8B). DT depleted 90% of CCR2+ cells (Fig. 5F), and, consistent with the work of others (16) (17), led to increased AFC numbers (Fig. 5G). Total B and germinal center B cell numbers were not affected (Fig. 5G), and the AFC increase was associated with decreased apoptosis and unchanged proliferation (Fig. 5G and H). Our results together suggested that stromal CCL2 limits AFC survival late during immune responses at least in part by mediating lymph node accumulation of CCR2+ cells.
To better understand the importance of monocytes as key CCR2+ cells, we depleted monocytes with anti-Gr1, which recognizes Ly6C and Ly6G (45, 46), between days 12 and 15 (fig. S8C). Ly6Chi monocytes and Ly6CmedLy6G+ neutrophils were well-depleted while Ly6Cmed Ly6G- monocytes were partially depleted (Fig. 5I and J). This led to increased AFC numbers and decreased AFC apoptosis without affecting the numbers of B and germinal center B cells (Fig. 5K and L). These results were not attributable to neutrophil depletion, as their depletion with anti-Ly6G had no effect on AFCs (fig. S8D–G). These results point to monocytes as key CCR2+ cells that limit lymph node AFC survival during the later stages of antibody responses.
We asked whether monocytes are sufficient to limit AFC numbers in our model. Ccr2−/− mice showed greatly reduced lymph node monocyte numbers (Fig. 6A), increased B cell, germinal center B cell, and AFC numbers, and increased AFC survival (Fig. 6B and C), which further supported a role for monocytes in regulating AFCs. The effects on B cell responses were greater than in Ccl2−/− mice, potentially reflecting additive roles of CCL2 with other CCR2 ligands such as CCL7 (43, 47). We transferred CD45.1 Ly6Chi monocytes on day 11 after OVA-Alum and examined the Ccr2−/− recipients at day 15 (Fig. 6D–F). Transferred cells that were recovered from the immunized lymph nodes (Fig. 6G) expressed medium to low levels of Ly6C and were CD11cmed-hi and MHCIImed (Fig. 6H), suggesting some degree of differentiation. Monocyte transfer reduced AFCs without affecting germinal center B or T cell numbers (Fig. 6I). These results complement previous findings showing that monocyte transfer at the time of viral infection could reduce AFC responses (19). Together, our results suggested that CCR2+ monocytes limit AFC accumulation in the later stages of antibody responses and are key mediators of the stromal CCL2 effect on AFCs.
Fig. 6. Monocytes limit AFCs in a NOX2-dependent manner.

(A-C) WT and Ccr2−/− mice were immunized on day 0 and draining popliteal nodes were examined on day 15. Numbers of (A) Ly6Chi monocytes and (B) indicated cells. (C) Percentage of AFCs that are ki67+ and activated caspase-3+. (D-I) CD45.1+ Ly6Chi monocytes were transferred into Ccr2−/− recipients at day 11 after OVA-Alum, as in (D). (E) Gating strategy for monocyte sorting. (F) sorted monocyte characterization. (G-H) Recovered donor cell numbers (G) and characterization (H). (I) Numbers of indicated cells. (J-L) Cells from day 15 nodes were loaded with the fluorescent ROS indicator CM-H2DCFDA. (J) Fluorescence in indicated cells. (K-L) Relative ROS expression. MFI in each population was normalized to the MFI of (K) Ly6Chi monocytes or (L) each WT population. Dashed line represents the fluorescence of WT B cells relative to that of WT Ly6Chi monocytes. (M-O) WT or NOX2-deficient monocytes were transferred into Ccr2−/− recipients on day 11 post immunization and analyzed on day 15. Absolute numbers and numbers normalized to those of Ccr2−/− mice that received no cells are both shown. Lines connecting the symbols denote the matched mice from a given experiment. (M) B cells, (N) germinal center B cells, and (O) AFCs. (A-C,G,I,K-O) Each symbol represents one mouse; n=3–7/condition. Data are from 1 (A-C) experiment, representative of 4 similar experiments (see (I)). Data are from 2 (K), 3 (L), 4 (M-O), 5 (G), and 7(I) experiments. *P < 0.05, **P < 0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation.
Monocyte NOX2 contributes to ROS production and AFC regulation
Myeloid cells can limit T cell responses via ROS (48), and monocytes showed high levels of ROS when compared to lymphocytes (Fig. 6J and K). NADPH oxidase is a major contributor to the generation of myeloid cell ROS that is released extracellularly (49) and, interestingly, global deficiency of the Nox2 gene that encodes the NOX2/gp91phox subunit of NADPH oxidase in a lupus model increased plasmablast numbers (50). Given that monocytes from Nox2−/− mice (51) showed reduced ROS levels at day 15 after immunization (Fig. 6L), we examined their ability to limit AFC responses. Nox2−/− monocytes were less able than WT monocytes to limit AFCs when transferred into Ccr2−/− mice at day 11 (Fig.6M–O). Our results suggested that monocytes limit AFC accumulation via NOX2-dependent ROS.
Stromal CCL2 modulates monocyte ROS production
In addition to regulating monocyte accumulation and positioning in lymph nodes, CCL2 could potentially modulate monocyte ROS expression. Indeed, monocytes and potential monocyte-derived cells that can express CCR2 such (CD11b+) Ly6Cmed or Ly6Clo cells showed decreased ROS levels in Ccl2−/− mice (Fig. 7A and B), leading us to ask whether lymph node stromal CCL2 can directly regulate monocyte ROS production. Cultured FRCs expressed CCL2 (Fig. 7C), and we added supernatant from WT or Ccl2−/− FRC cultures to sorted monocytes and observed that monocytes had lower intracellular ROS when exposed to Ccl2−/− FRC supernatant (Fig. 7D). In addition, WT FRC supernatant increased extracellular ROS levels when added to monocytes (Fig. 7E) while Ccl2−/− FRC supernatant was less able to do so (Fig. 7E), consistent with the idea that FRC-derived CCL2 can modulate monocyte ROS generation. These in vitro results suggested that, in addition to monocyte accumulation and positioning, stromal CCL2 can directly modulate monocyte function, inducing ROS generation and release that can limit AFC survival.
Fig. 7. FRC CCL2 regulates monocyte ROS production.

(A,B) Day 15 OVA-Alum immunized WT or Ccl2−/− lymph node cells were loaded with CM-H2DCFDA. (A) Fluorescence in indicated cells. (B) Relative ROS levels as indicated by MFI of each Ccl2−/− population normalized to MFI in WT cells. Data are from 2 experiments. Dashed line represents fluorescence of WT B cells relative to that of WT Ly6Chi monocytes. (C) GFP expression by FRCs cultured from WT and CCL2-GFP mice. (D-F) Monocytes were incubated with WT or Ccl2−/− FRC supernatants and then cells and supernatants were assayed as indicated. (D) Relative intracellular ROS in monocytes, as indicated by MFI after CM-H2DCFDA-loading. (E) Extracellular ROS in supernatants, as indicated by fluorescence of the superoxide indicator dihydroethidium (DHE) in each supernatant normalized to the value of the WT supernatant without monocytes. (F) Normalized MFI of anti-NOX2 staining in monocytes. Dashed line represents the level of NOX2 antibody staining in Nox2−/− sorted monocytes which was used as a negative control and to which the other MFIs were normalized to. (A,C) Fluorescence scale is log10. (D-F) Data are from 5 (D) and 3 (E,F) independent experiments. (D,F) each symbol is an individual well with different symbols denoting independent experiments; (E) each symbol represents the average of 2–3 wells from an experiment. *P < 0.05, **P < 0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation. .
We next tried to understand how CCL2 modulated monocyte ROS levels and how ROS might regulate AFCs. Ccl2−/− FRC supernatant induced a slight reduction in monocyte NOX2 levels (Fig. 7F), suggesting that CCL2 may potentially regulate ROS monocyte production at least in part by modulating NOX2 expression. We also found that FRCs sorted from immunized WT and Ccl2−/− lymph nodes expressed BAFF at similar levels (fig. S8H), suggesting that the larger B cell response with CCL2 deficiency is not due to FRC BAFF overexpression.
Stromal CCL2 expression is regulated by altering vascular permeability
We examined for factors that upregulated stromal CCL2 in stimulated lymph nodes. Type I interferon limits B cell responses early during viral infections (52, 53), but IFNαR antibody blockade between days 5 to 9 or days 11 to 15 after immunization did not alter FRC CCL2 expression (fig. S9A–D).
Because stromal CCL2 was high in vascular-rich areas, we asked whether inflammation-associated vascular permeability increases that occur in lymph nodes (54) can modulate stromal CCL2, potentially by increasing exposure to intravascular contents. VE-cadherin mediates endothelial barrier integrity, and anti-VE-cadherin (55) injected into hind footpads could induce local permeability changes, increasing interstitial accumulation of systemically-injected Evans blue dye in popliteal but not brachial nodes (Fig. 8A). Anti-VE-cadherin increased CCL2 expression in FRCs and LECs but not in monocytes or CD11b+Ly6Clo cells (Fig. 8B–C), and this was associated with increased monocytes, without affecting the numbers of total lymph node cells, other myeloid cells, or lymphocytes (Fig. 8D, fig. S9E and F). Interestingly, FRCs had an altered phenotype upon anti-VE-cadherin treatment, with modestly increased PDPN and a larger increase in Sca-1 expression (fig. S9G and H). FRC numbers were unchanged (fig. S9I). The Sca-1 upregulation was mainly in the BP3+ population (fig. S9J), and other markers such as CD34, SMA and CCL21 did not change (fig. S9K and L). These results suggested that increasing vascular permeability upregulated stromal CCL2, which increased monocyte accumulation.
Fig. 8. Vascular permeability regulates stromal CCL2 expression.

(A-D) Homeostatic WT (A) or CCL2-GFP (B-D) mice were injected with anti-VE-cadherin or control IgG in footpads, and draining popliteal (A-D) and non-draining brachial (A) lymph nodes analyzed 24 hours later. (A) Vascular permeability measurement. (B) GFP in FRCs and LECs of CCL2-GFP mice. (C) Normalized GFP MFI in indicated populations. (D) Numbers of indicated cells. (E-I) CCL2-GFP (F) or WT (G-I) mice received Angiopietin-1 (Ang1) in footpads at days 13 and 14 after OVA-Alum and popliteal nodes were harvested on day 15, as in (E). (F) Normalized GFP MFI in indicated populations. (G,H) Numbers of indicated cells. (I) Percentage of AFCs that are activated caspase-3+. (A,C,D,F-I) Each symbol represents one mouse; n=3–6/condition. Data are from 2 (A,C,D,F) and 3 (G-I) independent experiments. *P < 0.05, **P < 0.01 by 2-tailed unpaired Student’s t test. Error bars represent standard deviation..
Angiopoietin1 (Ang1) reduces vascular permeability by acting on endothelial cell junctions (56, 57), and Ang1 in immunized mice reduced CCL2 expression by FRCs but not by LECs, monocytes, or CD11b+Ly6Clo cells (Fig. 8E and F). Ang1 also increased B cell and AFC numbers and AFC survival (Fig. 8G–I). These results are consistent with the idea that reducing vascular permeability reduced stromal CCL2 expression and consequently increased AFC survival.
While anti-VE-cadherin and Ang1 can modulate the permeability of both blood and lymphatic vessels, blood serum caused upregulation of both FRC PDPN and CCL2 (fig. S9M), similar to the effects of anti-VE-cadherin in vivo (Fig. 8B–C). These results are consistent with the idea that increased blood vessel permeability and consequent exposure to serum may contribute to upregulating FRC CCL2 in the vascular-rich areas of lymph nodes.
Discussion
Here we showed that the lymph node stromal compartment can function to limit AFC survival. High stromal CCL2 expression co-localized with AFCs and CCR2+ cells in the T zone and medulla, and lymph node transplantation experiments indicated the importance of stromal CCL2. FRCs express higher levels of CCL2 than LECs and upregulated CCL2 during the later stages of the antibody response. Taken together with previous findings that FRCs have AFC-supportive functions (13–15), our current results suggest that the stromal compartment plays dual roles in AFC regulation.
Our finding that FRCs express high levels of CCL2 is in agreement with recent analyses of FRC gene expression patterns (58)(Immgen.org)(5). Furthermore, Cyster and colleagues’ recent single cell RNA sequencing analysis of lymph node stromal cells at day 0 and day 15 after LCMV-Armstrong infection (26) showed that CCL2 is one of the differentially expressed genes that mark the CXCL9+ subset. CXCL9-expressing FRCs were suggested to represent an activated FRC population and CXCL9 is found in the interfollicular regions, the T zone, and the medulla (26, 59). That CCL2 is upregulated upon lymph node activation and high expression is localized to some of the same areas as CXCL9 suggests that at least some CCL2-expressing FRCs are in the CXCL9-expressing population (26). We speculate that differential cytokine expression by different FRC subsets likely contributes to the dual nature of FRCs in both supporting and limiting AFCs.
In addition to a role in modulating AFC survival, there are likely other roles for stromal CCL2 in lymph nodes. For example, CCL2 expressed at lower levels by FRCs at homeostasis may contribute to the CCR2-dependent accumulation of monocyte-derived macrophages in the T cell zone (60). Interestingly, these macrophages expanded by proliferation rather than recruitment during immune responses, further supporting the idea that the upregulated FRC CCL2 during immune responses has a distinct function in part by recruiting bloodborne Ly6Chi monocytes to limit AFC accumulation. We also observed that CCL2 is rapidly upregulated at day 2 in CCL21- FRCs presumed to be part of interfollicular or medullary compartments. This early stromal CCL2 upregulation coincides with an early wave of Ly6C+ monocyte infiltration which is involved in stimulating the initial lymph node stromal proliferation (41) while the delayed CCL2 upregulation in CCL21+ FRCs is temporally associated with the AFC response. This suggests that there may be distinct roles for CCL2 expressed by different subsets of FRCs over the course of immune responses.
Our work also complements and extends recent data showing roles for myeloid cells in regulating AFC responses. Giordano et al (17) and Sammichelli et al (19) showed that monocyte manipulation at the initiation of immune responses increased antibody responses, suggesting that monocytes could act on the nascent B cell response and/or subsequent AFCs. Our experiments showed that CCR2+ cell depletion, anti-Gr1 treatment, and monocyte transfer at day 11 or 12 after OVA-Alum immunization affected AFC but not total B cell and germinal center cell numbers, suggesting that monocytes may regulate AFCs specifically at this later stage. Interestingly, these recent studies also implicated monocyte-derived iNOS in regulating AFCs, and our finding that monocyte NOX2 is important may reflect the multiple mechanisms by which monocytes can regulate AFC responses. Fooksman et al (16) also observed that CCR2+ cell depletion at 4 days into a secondary response led to greater AFC numbers by day 7, the time of peak AFC accumulation in their system, although they did not detect a role for apoptosis (by annexin V staining) or monocytes (by anti-Gr1 treatment). We speculate that our results differ because of differences in the time window being examined. Collectively, our study in conjunction with these previous studies suggest that different CCR2+ cells may play distinct roles in modulating AFCs at different time points in antibody responses.
Our results suggest that vessel-rich areas of lymph nodes such as the cortical ridge, the T cell zone vascular cords, and the medulla can be specialized microenvironments in part due to the dynamic activity of the vasculature. The lymph node vasculature can regulate immunity by controlling cellular trafficking (61) and by direct effects on lymphocytes (62). Our results suggest that vascular functions such as altered permeability also offer opportunities to affect immune responses, in part by modulating stromal function.
Our results have implications for better treating autoimmune and inflammatory diseases. Although CCL2 and CCR2-expressing cells have been implicated in tissue damage in conditions such lupus and inflammatory arthritis (63–68), targeting a CCL2-CCR2 axis globally has not been a successful strategy (69). Disrupting the regulatory role of lymph node stromal CCL2 could have been a contributing factor, and bone marrow mesenchymal stromal cell CCL2 can limit bone marrow plasma cell antibody production in a direct manner (70, 71). Additionally, swift production of CCL2 by FRCs in omental fat-associated lymphoid clusters is crucial for the induction of peritoneal immunity (72), and disrupting CCL2 globally may be detrimental to protective immunity. Interestingly, bone marrow mesenchymal stromal cells from lupus patients expressed lower levels of CCL2 (71), suggesting the possibility that lymph node FRC CCL2 expression may also be reduced, contributing to autoimmunity. Better understanding how the source and context of CCL2 production determines its function and how factors such as vascular permeability shape the stromal microenvironment will better inform potential targeting of CCL2 and other CCR2 ligands in disease. Consistent with this idea, our results point to a need to consider potential dual roles of stromal elements when considering how to design stromal-targeting strategies.
Materials and Methods
Study Design
The purpose of this study was to understand expression, function, and regulation of lymph node stromal CCL2. The subjects were laboratory mice. We used immunofluorescence microscopy to visualize cell localization and flow cytometry to quantify cell numbers. For in vivo experiments, sample size of n=3–17 animals per condition evaluated in 1 to 11 independent experiments was found to be optimal for statistical analysis. For in vitro experiments, sample size of n=2–3 wells per condition per experiment in 3–5 independent experiments was used.
Mice
Mice between 6–12 weeks were used unless otherwise specified. We used C57Bl/6, CCL2−/− (73), and Nox2−/− (51) mice mice from Jackson Laboratory (JAX)(Bar Harbor, Maine) and CD45.1+ (B6.SJL-PtprcaPepcb/BoyCrl) mice from Charles River (Wilmington, MA) or our own breeding colony. CCL2-GFP (20) and CCR2-DTR (44) mice were bred at our facility. Ccl19-Cre mice (30) were crossed at our facility with ROSA26-YFPf/STOP/f mice (74)(JAX). All animal procedures were performed in accordance with the regulations of the Institutional Animal Care and Use Committee at Weill Cornell Medicine (New York, NY).
Mouse immunization and treatments
Mice were immunized in the hind footpads with 30 μg OVA adsorbed to 30ul of Alum. DT (Enzo Life Sciences, Farmingdale, NY)(250 ng DT/dose) was injected IP. Anti-Gr1 (RB6–8C5),anti-Ly6G (1A8), and isotype controls (LTF-2, 2A3)(all BioXCell, West Lebanon NH)( 250 μg/dose) were injected IP. Anti-VE-Cadherin (BV13) or rat IgG (both Thermo Fisher Scientific, Waltham, MA)(25 μg) and Ang1 (Peprotech, Rocky Hill, NJ)(5 μg) were injected in footpad.
Lymph node transplantation
For lymph node transplantations (31),donor popliteal nodes were harvested after euthanasia, prior to transplantation. Recipient mice were anesthetized, injected with 1% Evans blue in dorsal footpads to localize popliteal lymph nodes, which were removed through incisions in the popliteal fossa and with minimal disruption to the surrounding fat pad and blood vessels. Donor lymph nodes were placed into the fossa and skin was closed using 3–0 non-absorbable sutures
Vascular permeability assay
Mice were injected retro-orbitally with 2% Evans blue, euthanized after 90 minutes, and perfused with 30 ml of PBS before lymph node harvest. Evans blue was extracted in 200μl formamide at 60°C overnight and quantified by spectrophotometry (680nm fluorescence emission intensity, 620nm excitation) with titration curve.
Flow cytometry staining and quantification
Lymph nodes were harvested, minced, and digested with type II collagenase (Worthington, Lakewood NJ) as described (75). The following antibodies were used: anti- CD45, CD31 (both BD Biosciences, San Jose, CA), PDPN,, BP3, Sca1, CD11b, CD11c, I-Ab, Ly6C, Ly6G, CD3, B220 (all BioLegend, San Diego, CA), CD34 and GFP (both Thermo Fisher Scientific), CCL21, CCL2, CCR2, and activated caspase-3 (all R&D Systems, Minneapolis, MN), rabbit and goat IgG (Jackson Immunoresearch, West Grove, PA). PNA was from Vector Laboratories, Burlingame, CA. BD Cytofix/Cytoperm kit was used for intracellular staining. The Foxp3 buffer set (Thermo Fisher Scientific) was used for ki67 staining after AFC staining.
Cells per lymph node was calculated by multiplying the % of total of a gated population to lymph node cell count. For normalized experiments where there was more than one control sample, the control values were averaged and the individual control and experimental samples were normalized to this average value.
Reactive oxygen species assay
Intracellular and extracellular ROS were measured using CM-H2DCFDA and dihydroethidium (DHE), respectively (both Thermo Fisher Scientific) according to manufacturer specifications.
Tissue section staining and microscopy
CCL2-GFP tissues were fixed in 4% paraformaldehyde (1 hour on ice), cryoprotected in 30% sucrose, and frozen in optimal cutting temperature (OCT) embedding medium (Tissue-Tek, Torrance, CA). Other tissues were fresh-frozen in OCT. Antibodies are as used for FACS except anti-CXCL13 (R&D Systems), ERTR7 (Santa Cruz Biotechnologies, Santa Cruz, CA), GFP-Alexa488 and FITC-Alexa488 (both Thermo Fisher Scientific), and goat-Alexa488, rat-rhodamine, armenian hamster-AMCA, rabbit-rhodamine, mouse IgG-biotin, and streptavidin-rhodamine/AMCA (all Jackson Immunoresearch).
ELISPOT Assay
ELISPOT detection of anti-OVA-secreting cells was performed as described (13). Wells were coated with 0.1% ovalbumin (Sigma Aldrich, St. Louis, MO),cells were incubated at 37°C for 4 hours, and secreted anti-OVA was detected using anti-mouse IgG-biotin (Jackson Immunoresearch), streptavidin-alkaline phosphatase (Jackson Immunoresearch), and 5-bromo-4chloro-3-indolyl-phosphate (Sigma-Aldrich).
Cell sorting
For monocyte isolation, cells from long bones and spleen were pooled, depleted with anti-CD3/B220/ Ly6G via magnetic selection (Miltenyi-Biotec, Bergisch Gladbach, Germany), and Ly6Chi CD11b hi cells were sorted using a BD Influx (BD Biosciences). For FRC sorting, cells from draining lymph nodes 15 days after immunization were pooled, depleted with anti-CD45/CD31, and CD45- CD31- PDPN+ cells were sorted into RLT buffer (Qiagen, Venlo, Netherlands) for RNA extraction.
Real time PCR
RNA extracted (RNeasy Minikit, Qiagen), cDNA was synthesized (iScript kit, Bio-Rad, Hercules, CA) and real-time PCR (iQ SYBR-Green Supermix kit, Bio-Rad)) was performed using primers for BAFF and GAPDH (as in (13)).
In vitro experiments
Peripheral lymph node FRCs from homeostatic mice were cultured as described (13). Collagenase-digested lymph node cells were plated in RPMI/10% fetal calf serum, washed of non-adherent cells at day 5, harvested at day 7, and depleted with anti-CD45/CD31, resulting in FRC purity over 97%. FRCs were cultured in 96-well plates at 7500 cells/well/100μl. Supernatants were collected at 2 days and stored at −20°C until use. For experiments, supernatant was added to monocytes for 24 hours prior to harvest.
Statistics
Statistical significance was determined using 2-tailed unpaired Student’s t test; p<0.05 was considered significant. Error bars represent standard deviation.
Supplementary Material
Fig. S1. Characterization of BP3+ and BP3lo-neg FRCs.
Fig. S2. Characterization of lymph node AFC responses and localization after OVA-Alum.
Fig. S3. Characterization of immune cell populations in homeostatic and immunized Ccl2−/− lymph nodes and spleens.
Fig. S4. YFP expression in Ccl19-Cre;YFPf/STOP/f FRCs at day 15 after OVA-Alum.
Fig. S5. Transplanted lymph nodes show normal organization
Fig. S6. Analysis of optimal transplants and immune responses.
Fig. S7. Further characterization of CCR2 expression and CCR2+ cell identity, numbers, and localization after immunization
Fig. S8. Monocytes numbers are lower in homeostatic Ccl2−/− lymph nodes, and neutrophils do not play a role in limiting antibody responses.
Fig. S9. Effects of interferon receptor blockade on stromal CCL2, anti-VE-cadherin on cell populations and FRC phenotype, and serum on FRC CCL2
Acknowledgements:
We thank Selina Chen-Kiang and Carl Blobel for insightful discussions, Jackie Finik for statistical consultation, Eric Pamer for CCL2-GFP, CCR2-GFP and CCR2-DTR mice, Clifford Lowell for Ccl19-Cre breeders, and Jason Cyster and the Lu lab for critical reading of manuscript. Ccl19-Cre (Tg(Ccl19-cre)489Biat) mice are available from Burkhard Ludewig under a material transfer agreement with the Kantonsspital St.Gallen, Institute of Immunobiology. Babak Mehrara has grant funding from Atyr Corp and Puretech Corp and serves as an advisor for Puretech.
Funding:
Supported by MSTP T32GM007739 to the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program (WDS), T32AR071302 to the Hospital for Special Surgery Research Institute Rheumatology Training Program (WDS), National Health and Medical Research Council (Australia) Fellowship 1060675 (DT), NIH/NCI Cancer Center Support Grant P30 CA008748 (BJM), NIH R01AI079178 (TTL), Alliance for Lupus Research (TTL), St. Giles Foundation (TTL), Scleroderma Foundation (TTL), O’Neill Foundation from Barbara Volcker Center for Women and Rheumatic Diseases (TTL); NIH Office of the Director grant S10OD019986 to Hospital for Special Surgery.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and notes
- 1.Chang JE, Turley SJ, Stromal infrastructure of the lymph node and coordination of immunity. Trends Immunol 36, 30–39 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Alexandre YO, Mueller SN, Stromal cell networks coordinate immune response generation and maintenance. Immunological reviews 283, 77–85 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Fletcher AL, Acton SE, Knoblich K, Lymph node fibroblastic reticular cells in health and disease. Nat Rev Immunol 15, 350–361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dasoveanu DC, Shipman WD, Chia JJ, Chyou S, Lu TT, Regulation of Lymph Node Vascular-Stromal Compartment by Dendritic Cells. Trends Immunol, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregory JL et al. , Infection Programs Sustained Lymphoid Stromal Cell Responses and Shapes Lymph Node Remodeling upon Secondary Challenge. Cell Reports 18, 406–418 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Hoyer BF et al. , Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 199, 1577–1584 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.William J, Euler C, Christensen S, Shlomchik MJ, Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Chyou S, Tian S, Ekland EH, Lu TT, Normalization of the lymph node T cell stromal microenvironment in lpr/lpr Mice is associated with SU5416-induced reduction in autoantibodies. PLoS ONE 7, e32828 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM, The generation of antibody-secreting plasma cells. Nat Rev Immunol 15, 160–171 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Smith KG, Hewitson TD, Nossal GJ, Tarlinton DM, The phenotype and fate of the antibody-forming cells of the splenic foci. Eur J Immunol 26, 444–448 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Fooksman DR et al. , Development and migration of plasma cells in the mouse lymph node. Immunity 33, 118–127 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohr E et al. , Dendritic cells and monocyte/macrophages that create the IL-6/APRIL-rich lymph node microenvironments where plasmablasts mature. J Immunol 182, 2113–2123 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Kumar V et al. , A Dendritic-Cell-Stromal Axis Maintains Immune Responses in Lymph Nodes. Immunity 42, 719–730 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y et al. , Plasma cell output from germinal centers is regulated by signals from Tfh and stromal cells. The Journal of Experimental Medicine, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang HY et al. , Identification of a new subset of lymph node stromal cells involved in regulating plasma cell homeostasis. Proc Natl Acad Sci U S A 115, E6826-E6835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fooksman DR, Nussenzweig MC, Dustin ML, Myeloid Cells Limit Production of Antibody-Secreting Cells after Immunization in the Lymph Node. The Journal of Immunology 192, 1004–1012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giordano D, Draves KE, Li C, Hohl TM, Clark EA, Nitric Oxide Regulates BAFF Expression and T Cell–Independent Antibody Responses. The Journal of Immunology 193, 1110–1120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweet RA, Nickerson KM, Cullen JL, Wang Y, Shlomchik MJ, B Cell-Extrinsic Myd88 and Fcer1g Negatively Regulate Autoreactive and Normal B Cell Immune Responses. J Immunol 199, 885–893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sammicheli S et al. , Inflammatory monocytes hinder antiviral B cell responses. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi C et al. , Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 34, 590–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katakai T et al. , A novel reticular stromal structure in lymph node cortex: an immuno-platform for interactions among dendritic cells, T cells and B cells. Int Immunol 16, 1133–1142 (2004). [DOI] [PubMed] [Google Scholar]
- 22.von Andrian UH, Mempel TR, Homing and cellular traffic in lymph nodes. Nat Rev Immunol 3, 867–878 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Gunn MD et al. , A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 391, 799–803 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Link A et al. , Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol 8, 1255–1265 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Sitnik KM et al. , Context-Dependent Development of Lymphoid Stroma from Adult CD34(+) Adventitial Progenitors. Cell Rep 14, 2375–2388 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Rodda LB et al. , Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity 48, 1014–1028.e1016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kallies A et al. , Plasma Cell Ontogeny Defined by Quantitative Changes in Blimp-1 Expression. The Journal of Experimental Medicine 200, 967–977 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schumann K et al. , Immobilized Chemokine Fields and Soluble Chemokine Gradients Cooperatively Shape Migration Patterns of Dendritic Cells. Immunity 32, 703–713 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Braun A et al. , Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol 12, 879–887 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Chai Q et al. , Maturation of lymph node fibroblastic reticular cells from myofibroblastic precursors is critical for antiviral immunity. Immunity 38, 1013–1024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang JJ et al. , Lymph Node Transplantation Decreases Swelling and Restores Immune Responses in a Transgenic Model of Lymphedema. PLoS ONE 11, e0168259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahrendt M, Hammerschmidt SI, Pabst O, Pabst R, Bode U, Stromal cells confer lymph node-specific properties by shaping a unique microenvironment influencing local immune responses. J Immunol 181, 1898–1907 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Mebius RE, Breve J, Kraal G, Streeter PR, Developmental regulation of vascular addressin expression: a possible role for site-associated environments. Int Immunol 5, 443–449 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Angeli V et al. , B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 24, 203–215 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Soderberg KA et al. , Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc Natl Acad Sci U S A 102, 16315–16320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charo IF et al. , Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A 91, 2752–2756 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serbina NV, Hohl TM, Cherny M, Pamer EG, Selective Expansion of the Monocytic Lineage Directed by Bacterial Infection. The Journal of Immunology 183, 1900–1910 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geissmann F, Jung S, Littman DR, Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Tamoutounour S et al. , Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39, 925–938 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Merad M, Sathe P, Helft J, Miller J, Mortha A, The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annual Review of Immunology 31, 563–604 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benahmed F et al. , Multiple CD11c+ cells collaboratively express IL-1beta to modulate stromal vascular endothelial growth factor and lymph node vascular-stromal growth. J Immunol 192, 4153–4163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serbina NV, Pamer EG, Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Tsou CL et al. , Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117, 902–909 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohl TM et al. , Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6, 470–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleming TJ, Fleming ML, Malek TR, Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6–8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. The Journal of Immunology 151 2399–2408 (1993. ). [PubMed] [Google Scholar]
- 46.Dunay IR, Fuchs A, Sibley LD, Inflammatory Monocytes but Not Neutrophils Are Necessary To Control Infection with Toxoplasma gondii in Mice. Infection and Immunity 78, 1564–1570 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia T et al. , Additive Roles for MCP-1 and MCP-3 in CCR2-Mediated Recruitment of Inflammatory Monocytes during Listeria monocytogenes Infection. The Journal of Immunology 180, 6846–6853 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI, Antigen-Specific Inhibition of CD8+ T Cell Response by Immature Myeloid Cells in Cancer Is Mediated by Reactive Oxygen Species. The Journal of Immunology 172, 989–999 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Bedard K, Krause K-H, The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiological Reviews 87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Campbell AM, Kashgarian M, Shlomchik MJ, NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 4, 157ra141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollock JD et al. , Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nature genetics 9, 202–209 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Sammicheli S et al. , Inflammatory monocytes hinder antiviral B cell responses. Science Immunology 1, eaah6789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moseman EA, Wu T, de la Torre JC, Schwartzberg PL, McGavern DB, Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson ND, Anderson AO, Wyllie RG, Microvascular changes in lymph nodes draining skin allografts. Am J Pathol 81, 131–160 (1975). [PMC free article] [PubMed] [Google Scholar]
- 55.Corada M et al. , Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A 96, 9815–9820 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thurston G et al. , Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 6, 460–463 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Gavard J, Patel V, Gutkind JS, Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 14, 25–36 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Malhotra D et al. , Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol 13, 499–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Groom JR et al. , CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baratin M et al. , T Cell Zone Resident Macrophages Silently Dispose of Apoptotic Cells in the Lymph Node. Immunity, (2017). [DOI] [PubMed] [Google Scholar]
- 61.Girard JP, Moussion C, Forster R, HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol 12, 762–773 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Mendoza A et al. , Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature 546, 158–161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raghu H et al. , CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Annals of the rheumatic diseases 76, 914–922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kulkarni O et al. , Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. Journal of the American Society of Nephrology : JASN 18, 2350–2358 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Mizoguchi F et al. , Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nature Communications 9, 789 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rao DA et al. , Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR, Monocyte Chemoattractant Protein 1–Dependent Leukocytic Infiltrates Are Responsible for Autoimmune Disease in Mrl-Faslpr Mice. The Journal of Experimental Medicine 190, 1813–1824 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stephenson W et al. , Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nature Communications 9, 791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Proudfoot AE, Is CCR2 the right chemokine receptor to target in rheumatoid arthritis? Arthritis and rheumatism 58, 1889–1891 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Rafei M et al. , Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood 112, 4991–4998 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Che N et al. , Impaired B Cell Inhibition by Lupus Bone Marrow Mesenchymal Stem Cells Is Caused by Reduced CCL2 Expression. The Journal of Immunology, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez-Shibayama C et al. , Fibroblastic reticular cells initiate immune responses 2 in visceral adipose tissues and secure peritoneal immunity. Sci Immunol, (In press). [DOI] [PubMed] [Google Scholar]
- 73.Lu B et al. , Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 187, 601–608 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srinivas S et al. , Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC developmental biology 1, 4 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chyou S et al. , Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells. J Immunol 187, 5558–5567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of BP3+ and BP3lo-neg FRCs.
Fig. S2. Characterization of lymph node AFC responses and localization after OVA-Alum.
Fig. S3. Characterization of immune cell populations in homeostatic and immunized Ccl2−/− lymph nodes and spleens.
Fig. S4. YFP expression in Ccl19-Cre;YFPf/STOP/f FRCs at day 15 after OVA-Alum.
Fig. S5. Transplanted lymph nodes show normal organization
Fig. S6. Analysis of optimal transplants and immune responses.
Fig. S7. Further characterization of CCR2 expression and CCR2+ cell identity, numbers, and localization after immunization
Fig. S8. Monocytes numbers are lower in homeostatic Ccl2−/− lymph nodes, and neutrophils do not play a role in limiting antibody responses.
Fig. S9. Effects of interferon receptor blockade on stromal CCL2, anti-VE-cadherin on cell populations and FRC phenotype, and serum on FRC CCL2