

Abstract
Alternative splicing is responsible for much of the transcriptomic and proteomic diversity observed in eukaryotes and involves combinatorial regulation by many cis-acting elements and trans-acting factors. SR and hnRNP splicing regulatory proteins often have opposing effects on splicing efficiency depending on where they bind the pre-mRNA relative to the splice site. Position-dependent splicing repression occurs at spliceosomal E-complex, suggesting that U1 snRNP binds but cannot facilitate higher order spliceosomal assembly. To test the hypothesis that the structure of U1 snRNA changes during activation or repression, we developed a method to structure-probe native U1 snRNP in enriched conformations that mimic activated or repressed spliceosomal E-complexes. While the core of U1 snRNA is highly structured, the 5′ end of U1 snRNA shows different SHAPE reactivities and psoralen crosslinking efficiencies depending on where splicing regulatory elements are located relative to the 5′ splice site. A motif within the 5′ splice site binding region of U1 snRNA is more reactive toward SHAPE electrophiles when repressors are bound, suggesting U1 snRNA is bound, but less base-paired. These observations demonstrate that splicing regulators modulate splice site selection allosterically.
Keywords: allosteric regulation, splice site selection, spliceosomal assembly, splicing regulatory proteins, U1 snRNP
INTRODUCTION
Pre-mRNA splicing is a fundamental RNA processing step that is common to all eukaryotes (Matera and Wang 2014). The combinatorial control that drives alternative pre-mRNA splicing can generate over 200,000 mRNA transcripts from roughly 20,000 human genes and accounts for much of the transcriptomic and proteomic diversity observed in humans and other higher eukaryotes (Hertel 2008; Nilsen and Graveley 2010; Park et al. 2018; Shenasa and Hertel 2019). There are many factors that influence alternative splice site usage which include the strength of splice sites, RNA secondary structure and the presence or absence of splicing regulatory elements (SREs) that recruit trans-acting splicing activators and repressors (Hertel and Maniatis 1998; Buratti and Baralle 2004; Shepard and Hertel 2008; Warf and Berglund 2010; McManus and Graveley 2011; Shepard et al. 2011; Erkelenz et al. 2013; Fu and Ares 2014). All of these features affect which splice sites are selected in any given transcript. There are also examples of each of these factors being the primary drivers of alternative splicing in different cellular and experimental contexts (Shenasa and Hertel 2019).
The spliceosome is a large dynamic macromolecular machine that is composed of small nuclear ribonucleoprotein (snRNP) components, which assemble onto the pre-mRNA in a stepwise fashion, along with other protein factors (Wahl et al. 2009). U1 snRNP recognizes the pre-mRNA through base-pairing interactions between the 5′ end of U1 snRNA and the 9 nt consensus sequence that defines the 5′ splice site. This RNA-RNA base-pairing is the basis for 5′ splice site selection (Wang and Cooper 2007; Wahl et al. 2009; Matera and Wang 2014). The 3′ splice site is initially recognized by the heterodimer U2AF, which binds the polypyrimidine tract and contacts the AG dinucleotide at the 3′ splice site (Wu et al. 1999). Subsequent ATP-dependent rearrangements lead to higher order spliceosomal complex formation, intron excision and exon ligation (Moore et al. 1993; Wahl et al. 2009).
It is known that U1 snRNA participates in splice site pairing and can be the target of splicing regulators. For example, stem–loop IV of U1 snRNA interacts with the U2 snRNP component SF3A1 or the polypyrimidine tract-binding protein to modulate splicing (Sharma et al. 2011, 2014) and mutagenesis of stem–loop IV causes exon skipping in vivo (Sharma et al. 2014). Furthermore, hnRNP L and A1 mediated splicing repression can promote the formation of extended U1 snRNA base-pairing with an exonic pre-mRNA sequence, which is predicted to block U6 snRNA base-pairing (Chiou et al. 2013).
Tissue specific alternative splicing often depends on SREs that recruit differentially expressed splicing regulatory proteins (Zahler et al. 1993; Kamma et al. 1995). These splicing regulators can be divided into two large families that include serine/arginine-rich proteins (SR proteins) and heterogeneous nuclear ribonucleoproteins (hnRNPs). Although SR proteins are best known for splicing activation through binding exonic splicing enhancers (ESEs) and hnRNPs have traditionally been associated with exonic splicing silencers (ESSs), both SR proteins and hnRNPs have been shown to act as activators and repressors in different splicing contexts (Wang and Cooper 2007; Wahl et al. 2009; Matera and Wang 2014). A systematic study showed that SR proteins and hnRNPs display a position-dependent activity at the 5′ splice site (Erkelenz et al. 2013). SR proteins activate splicing when bound upstream of the 5′ splice site, but repress splicing when bound intronically, downstream from the 5′ splice site. An opposite effect was observed for hnRNPs, which repress splicing when bound upstream of the 5′ splice site, but activate splicing when bound intronically, downstream from the 5′ splice site (Fig. 1A). The molecular mechanism for this position-dependent and antagonistic activity by SR proteins and hnRNPs has yet to be elucidated (Erkelenz et al. 2013).
FIGURE 1.

Position-dependent splicing activation and repression. (A) Schematic of the position-dependent activity of SR proteins and hnRNPs. Green arrows indicate splicing activation and red signs indicate splicing repression. (B) Schematic and definition of substrate RNAs used in this study. Exons are depicted as a blue box and introns are depicted as black lines. Binding sites for SRSF7 and TIA-1 are depicted as small white and gray boxes, respectively. For visual aid, green texts indicate substrate RNAs with activators bound, and red text indicates substrate RNAs with repressors bound.
Three related hypotheses can be proposed to explain position-dependent splicing regulation by SREs. It can be postulated that the binding of SR proteins and hnRNPs at activating or repressing positions leads to the recruitment of additional proteins that subsequently cause activation or repression. Alternatively, the interaction of splicing regulators at different positions relative to the 5′ splice site may change the binding affinity of U1 snRNP or U1snRNP-specific proteins with the 5′ splice site of the pre-mRNA. Lastly, it can be hypothesized that splicing regulators change the structure of U1 snRNA in such a way that U1 snRNP binding at the 5′ splice site can occur, but splice site selection or splice site pairing, which is associated with spliceosomal A-complex formation, cannot occur (Lim and Hertel 2004). This last hypothesis is the focus of the present study.
To test whether splicing regulators change the structure of U1 snRNA, we developed a U1 snRNP enrichment and SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) structure-probing approach (Wilkinson et al. 2006; Spitale et al. 2013). The method reliably identifies the binding of the U1 snRNP-specific proteins U1-70K and U1-A. We found the core of U1 snRNA (nucleotides 26–108) to be highly structured across all conditions tested. However, we discovered a motif within the 5′ end of U1 snRNA, referred to as the junction motif, which is predicted to base-pair with the intron–exon junction and is less reactive to SHAPE electrophiles when SRSF7 or TIA-1 bind the pre-mRNA from activating positions. These results suggest that activating splicing regulators promote U1 snRNA base-pairing with the 5′ splice site of the pre-mRNA, a proposal supported by psoralen crosslinking experiments. In contrast, splicing regulators bound at repressive positions prevent base-pairing with the 5′ splice site, thus providing a mechanistic framework for position-dependent splicing activation and repression.
RESULTS
U1 snRNA structure-probing design
We developed an approach to enrich and structure-probe native U1 snRNPs from HeLa cell nuclear extract. Substrate RNAs were derived from pre-mRNAs that were shown to undergo activated or repressed splicing (Erkelenz et al. 2013). Each RNA contained a functional 5′ splice site in addition to verified binding sites for SRSF7 or TIA-1, either upstream or downstream from the 5′ splice site (Table 1; Fig. 1B). The designed substrates were short, excluded a 3′ splice site and a downstream exon to ensure that the 5′ splice site was the only U1 snRNP binding site present and to stall spliceosomal assembly at E-complex without the need to deplete nuclear extract of ATP or use spliceosomal inhibitors (Erkelenz et al. 2013). To confirm that the pull-down approach enriches fully assembled U1 snRNP, western blot analysis was used to verify the presence of U1-specifc proteins U1-70K and U1-C (Fig. 2A). Additionally, enrichment of SRSF7 and TIA-1 binding on substrate RNAs that contain their respective binding sites was also verified (Table 1; Fig. 2B).
TABLE 1.
Substrate RNAs used in pull-down experiments

FIGURE 2.
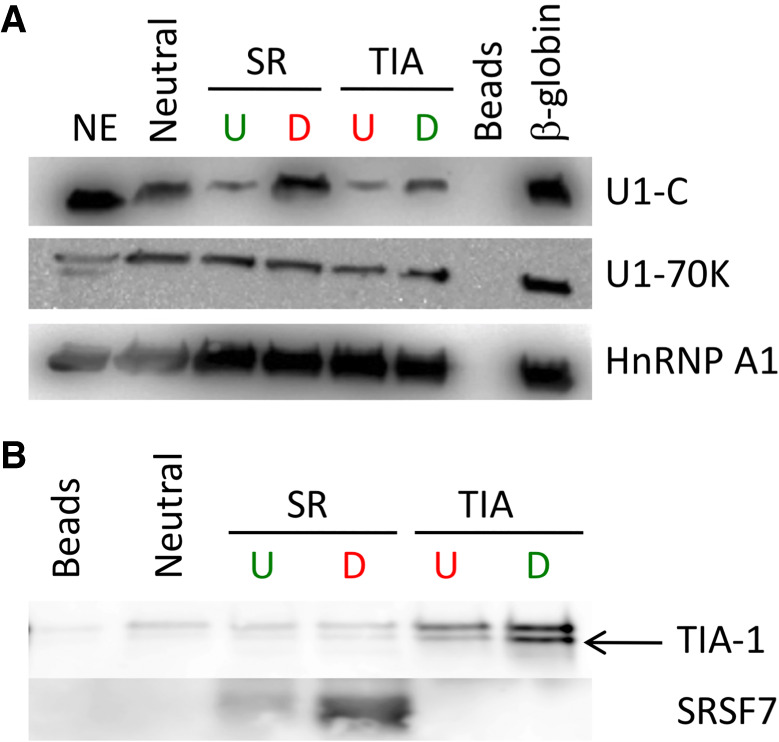
Confirmation of U1 snRNP assembly and splicing regulatory protein enrichment on substrate RNAs. (A) U1 snRNPs bound to substrate RNAs contain both U1-70K and U1-C; hnRNP A1 was used as a loading control. Beads sample contains no RNA and β-globin RNA was used as a positive control. (B) SRSF7 and TIA-1 are enriched on substrate RNAs containing their respective binding sites.
Comparison between in vitro transcribed U1 snRNA and nuclear extract derived U1 snRNA
To verify that the structure-probing protocol allows for the identification of U1 snRNA regions that interact with U1-specific proteins, we compared the SHAPE reactivity of in vitro transcribed U1 snRNA (in vitro U1 snRNA) with the reactivity of U1 snRNA present in nuclear extract (nuclear extract U1 snRNA) (Fig. 3A lane 1 vs. 3, B,C). Loop regions of stem–loop I and II, which are the known interaction sites of U1-70K and U1-A, should be more reactive for in vitro U1 snRNA when compared to nuclear extract U1 snRNA, which is bound by U1-specific proteins (Kondo et al. 2015). In agreement with these expectations, stem–loop I nucleotides U27–G38 of nuclear extract U1 snRNA show no SHAPE reactivity, whereas many of the same nucleotides of in vitro U1 snRNA display increased reactivity (Fig. 3A–C).
FIGURE 3.

SHAPE reactivity comparison of in vitro transcribed U1 snRNA and U1 snRNA from HeLa cell nuclear extract. (A) The two enlarged segments of the gel show regions of U1 snRNA that display different SHAPE reactivities between the conditions tested. The nucleotide sequences on the left correspond to the SHAPE reactivity of the nucleotide. The binding sites for U1-70K and U1-A are indicated on the right. IVT refers to in vitro transcribed U1 snRNA, NE to nuclear extract U1 snRNA. NAI indicates probing with SHAPE chemistry, DMSO are control lanes. (B) Reactivity plots of in vitro transcribed U1 snRNA and nuclear extract U1 snRNA. Black bars (0–0.3) signify background reactivity, orange bars (0.3–0.7) signify intermediate reactivity, and red bars (≥0.7) indicate highly reactive nucleotides. The y-axis represents the normalized SHAPE reactivity, the x-axis indicates the nucleotide position from 5′ to 3′ in the U1 snRNA sequence. Loop nucleotides for stem–loop I and II are indicated in addition to nucleotides that display different SHAPE reactivities between the IVT and NE condition. The reactive nucleotides for stem–loop III are also indicated. (C) Summary of SHAPE reactivity for in vitro U1 snRNA (top) and nuclear extract U1 snRNA (bottom) plotted onto the secondary structure of U1 snRNA. Loop definitions and protein binding sites are as indicated. Green boxes denote the binding site for Sm proteins.
Based on a published high-resolution substructure of U1 snRNA bound by the RRMs of U1-70K and U1-A, the first 7 loop nucleotides of stem–loop II are known to be ordered by the RNA recognition motif (RRM) of U1-A (Kondo et al. 2015). In agreement with U1-A binding, we observe reduced SHAPE reactivity for nucleotides G68 and C69 when comparing nuclear extract U1 snRNA with in vitro transcribed U1 snRNA and increased reactivity for C73 and C74 as they are shown to be single stranded and not interacting with the RRM of U1-A (Fig. 3A–C; Kondo et al. 2015). Interestingly, U66 is the only nucleotide that is reactive in the first 7 loop nucleotides of stem–loop II in nuclear extract U1 snRNA. This observation is plausible when considering the crystal structure, because the 2′ hydroxyl of U66 participates in the coordination of a water molecule between the amide carbonyl oxygen of leucine 49 and N3 of G68, which may activate it toward SHAPE electrophiles (Kondo et al. 2015).
Nucleotides U60, U62, and C86 display slightly elevated SHAPE reactivity in nuclear extract U1 snRNA compared to in vitro U1 snRNA (Fig. 3B,C). Several considerations could provide an explanation for these observations. It is possible that the bulged nucleotide preceding U60 adopts a unique conformation that enhances the SHAPE reactivity of its immediately downstream nucleotide. Nucleotide C86 is predicted to be bulged out and it is possible that protein interactions stabilize a conformation that also enhances its SHAPE reactivity in nuclear extract. Nucleotides C86 and U60 are adjacent to strong SHAPE-independent stops in the nuclear extract lanes. Thus, it is also plausible that the quantitation of nucleotides C86 and U60 is artificially influenced by these SHAPE-independent stops.
Loop nucleotides of stem–loop III, which are predicted to be single stranded and unbound by proteins across all conditions, are highly reactive toward 2-methylnicotinic acid imidazolide (NAI). The results obtained by SHAPE structure-probing are consistent with the crystallographic data for protein-associated U1 snRNA and they demonstrate that the use of SHAPE electrophiles, such as NAI (Spitale et al. 2013), can be used in conjunction with our pull-down approach to detect changes in the physical state of U1 snRNA.
Comparison between nuclear extract U1 snRNA versus enriched U1 snRNA
To determine whether the interaction of U1 snRNP with a 5′ splice site changes the structure or dynamics of U1 snRNA, we compared the SHAPE reactivity of enriched U1 snRNAs, defined as those bound to substrate RNAs, to nuclear extract U1 snRNA, defined as those unbound to RNA. The majority of U1 snRNPs in nuclear extract are assumed to be in an unbound state, with a minority of U1 snRNPs bound to genuine 5′ splice sites and other noncanonical sequences. The SHAPE analysis of nucleotides 26–108 showed minimal differences between nuclear extract U1 snRNA and enriched U1 snRNAs, suggestive of minimal changes in the protein bound core of U1 snRNA upon 5′ splice site binding (Fig. 4: compare lanes 5, 7, 9, 11, and 13 to lane 17, Fig. 5 nuclear extract compared to enriched conditions). As stated above, we observe that nucleotides U60, U62, and C86 are reactive in nuclear extract U1 snRNA. Interestingly, however, these nucleotides do not display SHAPE reactivity in U1 snRNA bound to the 5′ splice site sequence (Fig. 5, compare nuclear extract to enriched conditions). The loss of SHAPE reactivity for U60, U62 and C86 may therefore be an indication of 5′ splice site binding. Despite these small differences in SHAPE reactivity upon 5′ splice site binding, the overall lack of differences is largely consistent with expectations given that U1 snRNA is a highly structured RNA that forms its secondary structure even in the absence of U1 specific proteins. Therefore, 5′ splice site binding is not expected to significantly change the overall structure of U1 snRNA. We conclude that the core of U1 snRNA maintains a near uniform SHAPE reactivity regardless of its interactions with pre-mRNAs.
FIGURE 4.
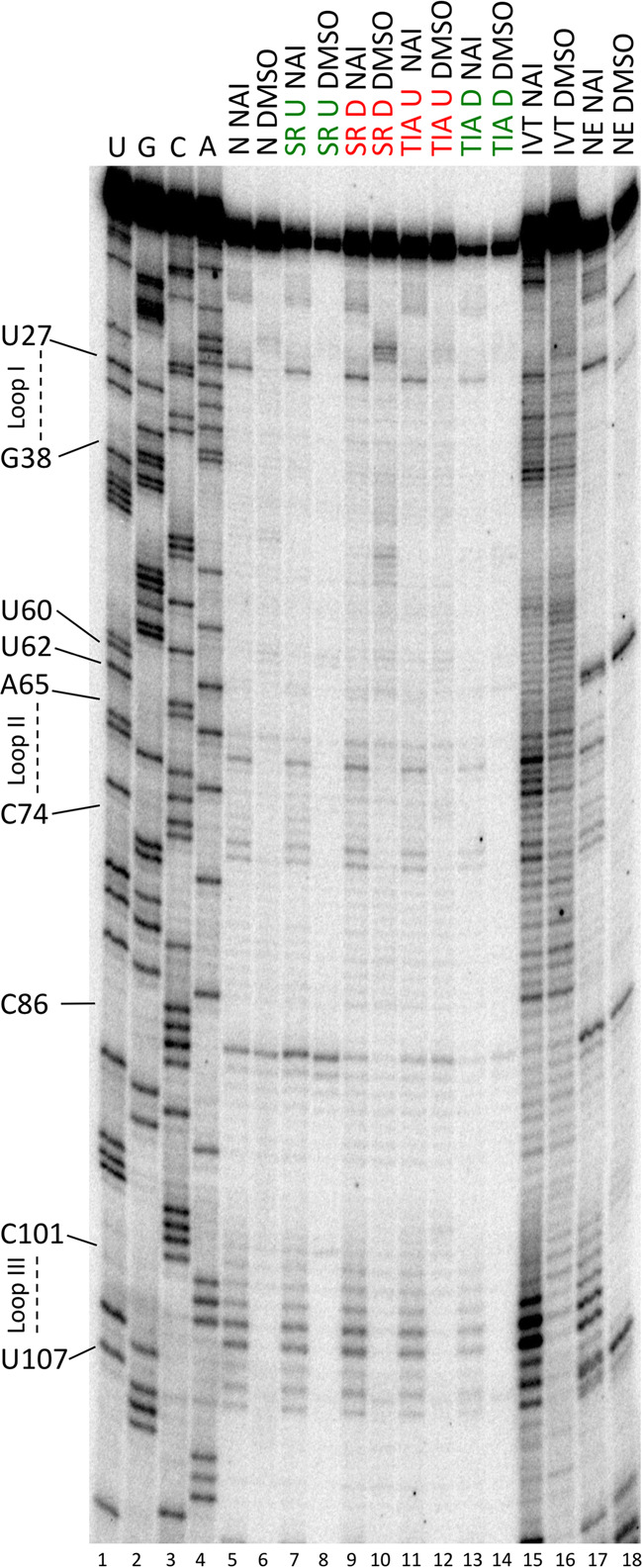
SHAPE reactivity of enriched U1 snRNAs bound to five different substrate RNAs, in vitro transcribed U1 snRNA and nuclear extract U1 snRNA. SHAPE chemistry performed on enriched U1 snRNPs. Loop I, II, and III regions are indicated on the left in addition to nucleotides that display differential SHAPE reactivities. N refers to neutral RNA sequence, SR U to SRSF7 bound upstream, SR D to SRSF7 bound downstream, TIA U to TIA-1 bound upstream, TIA D to TIA-1 bound downstream, IVT to in vitro transcribed U1 snRNA and NE to nuclear extract U1 snRNA. NAI indicates probing with SHAPE chemistry, DMSO are control lanes. Lanes 1–4 are sequencing lanes. The SHAPE stops correspond to the immediately upstream nucleotide in the sequencing lanes.
FIGURE 5.

Reactivity plots of enriched U1 snRNAs and nuclear extract U1 snRNA. Black bars (0–0.3) signify background reactivity, orange bars (0.3–0.7) signify intermediate reactivity, and red bars (≥0.7) indicate highly reactive nucleotides. The y-axis represents the normalized SHAPE reactivity and the x-axis indicates the nucleotide position from 5′ to 3′ in the U1 snRNA sequence. Loop nucleotides for stem–loops I and II are indicated in addition to nucleotides that display different SHAPE reactivities between enriched conditions and the NE condition. The reactive nucleotides for stem–loop III are also indicated.
We also compared the 5′ splice site binding region of U1 snRNA, defined as nucleotides 2–11, between nuclear extract U1 snRNA and enriched U1 snRNAs. We identified a 6 nt junction motif (5′-UACCUG-3′), which is more reactive to NAI in nuclear extract U1 snRNA compared to U1 snRNAs bound adjacent to activators (Fig. 6). This result shows that the SHAPE reactivity of the junction motif correlates with enhanced 5′ splice site binding.
FIGURE 6.
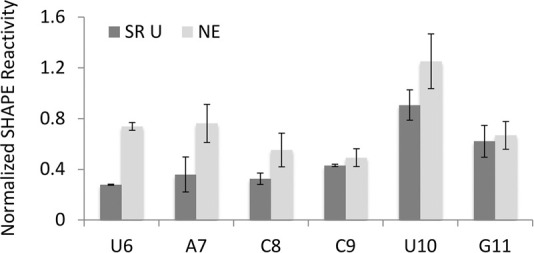
SHAPE reactivity of U1 snRNA junction motif (U6 to G11) upon RNA binding. The SHAPE reactivity of U1 snRNA enriched on substrate SR U is compared with the SHAPE reactivity of nuclear extract U1 snRNA (NE), which is not bound to substrate RNA. The y-axis represents the normalized SHAPE reactivity and the x-axis indicates the nucleotide position from 5′ to 3′ in the U1 snRNA sequence.
Impact of splicing activation and repression on SHAPE reactivity of U1 snRNA
A detailed analysis of the SHAPE reactivities of U1 snRNP bound to different substrate RNAs (activated versus repressed) was performed (Fig. 4, compare lanes 5, 7, 9, 11, and 13; Fig. 5, compare between enriched conditions). The core of U1 snRNA (nucleotides 26–108) has a uniform SHAPE reactivity regardless of whether it is in an activated or repressed state, suggesting that U1 snRNA structural changes that promote spliceosomal assembly are not mediated through this region. However, the junction motif is consistently observed to be less reactive toward NAI when splicing regulators are bound at activating positions (Fig. 7A–C). This result suggests that SRSF7 and TIA-1 promote more rigid base-pairing between U1 snRNA and the 5′ splice site when bound at activating positions (Fig. 7A–C; Supplemental Table 1). The increased SHAPE reactivity of the junction motif when splicing regulatory proteins are bound at repressive positions suggests that repressor/U1 snRNP interactions interfere with efficient base-pairing between the 5′ splice site and U1 snRNA. Interestingly, the average SHAPE reactivity of U1 snRNA bound to a neutral control substrate RNA lies between those observed for the activated and repressed substrates, suggesting that neutral sequences adopt an intermediate base-pairing state (Fig. 7C).
FIGURE 7.
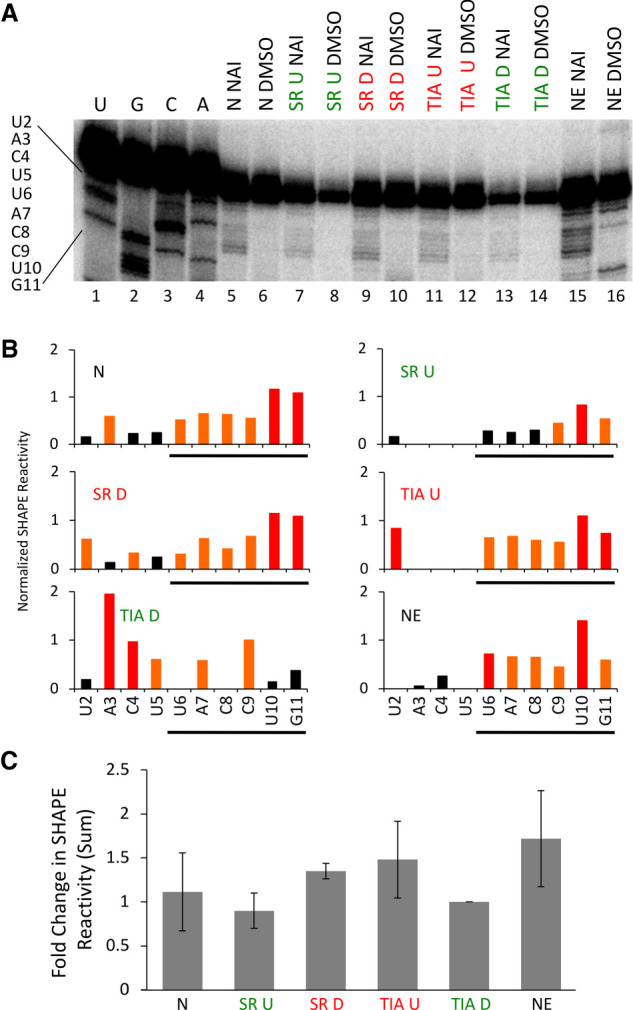
SHAPE reactivity of the 5′ end of U1 snRNA. (A) The U1 snRNA sequence is indicated on the left. N refers to neutral RNA sequence, SR U to SRSF7 bound upstream, SR D to SRSF7 bound downstream, TIA U to TIA-1 bound upstream, TIA D to TIA-1 bound downstream, IVT to in vitro transcribed U1 snRNA and NE to nuclear extract U1 snRNA. NAI indicates probing with SHAPE chemistry, DMSO are control lanes. Lanes 1–4 are sequencing lanes. (B) Representative reactivity plots of nucleotides 2–11 plotted for the data depicted in A. Nucleotides 2–11 represent the U1 snRNA sequence and the underlined nucleotides correspond to the junction motif (U6 to G11). The y-axis denotes the normalized SHAPE reactivity and the x-axis defines the nucleotide position in the direction of 5′ to 3′. (C) Fold change in junction motif SHAPE reactivity. The graph shows the average sum of SHAPE reactivities for the junction motif normalized to the TIA D condition across three biological replicates. Supplemental Table 1 details statistical significance between conditions. The y-axis indicates the fold change of the junction motif SHAPE reactivities and the x-axis defines the source of U1 snRNA.
To determine whether the observed differences in SHAPE reactivity are due to a lack of base-pairing between U1 snRNA and the 5′ splice site in repressed conditions or differences in base-pairing efficiency, we carried out psoralen crosslinking experiments of U1 snRNPs to activated or repressed substrates (Fig. 8A,B). The results show that U1 snRNA base-pairs with the 5′ splice site in all substrate configurations, however at varying levels of crosslinking efficiency. Activated substrate RNAs crosslink to U1 snRNA more efficiently than repressed or the neutral substrate RNAs (Fig. 8A, compare lanes 3 and 9 to lanes 1,5 and 7, Fig. 8B; Supplemental Table 2). One notable anomaly is the increased crosslinking efficiency of the SRSF7 bound downstream (SR D) substrate relative to the neutral substrate (N). Overall, the crosslinking results converge with the SHAPE structure-probing data, providing additional evidence that activators and repressors modulate the efficiency of 5′ splice site base-pairing interactions. U1 snRNP binds the 5′ splice site in various contexts, but splicing activators increase base-pairing, whereas strong splicing repressors such as TIA-U decrease base-pairing.
FIGURE 8.
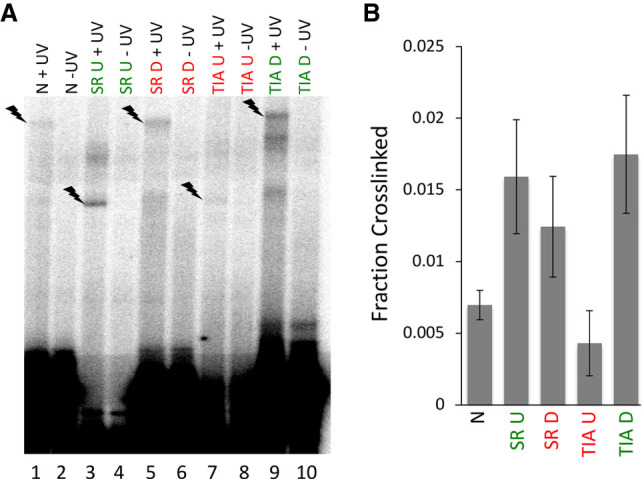
U1 snRNA psoralen crosslinking efficiency to the 5′ splice site of substrate RNAs. (A) Representative psoralen crosslinking gel. +UV lanes were irradiated with 365 nm light, −UV lanes were not irradiated. The lightning bolts indicate U1 snRNA-substrate RNA crosslinks. (B) Quantitation of the psoralen crosslinking efficiency. The graph indicates the average fraction of each substrate RNA that was crosslinked to U1 snRNA across three biological replicates. Supplemental Table 2 details statistical significance between conditions.
DISCUSSION
SR proteins and hnRNPs have dual and antagonistic activities depending on where they bind relative to a regulated 5′ splice site. During splicing repression, U1 snRNP is recruited to the pre-mRNA and forms E-complex, however the formation of higher order complexes is severely hindered (Erkelenz et al. 2013). U1 snRNP is the only stably associated snRNP at E-complex (Hong et al. 1997; Das et al. 2000). Here, we carried out a thorough investigation of the U1 snRNA secondary structure in different contexts of splicing regulation. Importantly, we show that the junction motif of U1 snRNA is more reactive for U1 snRNPs not engaged in RNA binding and repressed U1 snRNPs when compared to activated U1 snRNPs bound to RNA (Fig. 6). While this may seem intuitive, it highlights two important points. First, the enriched SHAPE method is sensitive enough to detect changes in base-pairing efficiency, such as splice site activation by splicing regulatory proteins. Secondly, the method used herein can be a useful tool for researchers probing dynamic RNAs or RNPs. While the use of cell permeable structure probes such as DMS, NAI and NAz is extremely useful and permits RNA structure-probing in native environments, RNPs such as the spliceosome are known to be highly dynamic (Spitale et al. 2013; Rouskin et al. 2014; Zubradt et al. 2017; Feng et al. 2018). In specific instances it may be essential to probe an RNP in one unique context. For example, this study focuses on U1 snRNP's functional role in the context of pre-mRNA splicing and not on U1 snRNP's functional role in the context of inhibiting premature polyadenylation (Gunderson et al. 1998; Kaida et al. 2010). SHAPE carried out on whole cells would not be able to distinguish between U1 snRNAs that are bound to canonical 5′ splice sites, those that are preventing pre-mature polyadenylation or those that are not bound to RNA. It would also be unable to distinguish between U1 snRNPs in activated versus repressed states. As a result, an average SHAPE reactivity would be collected for in cell SHAPE.
Our findings also add to previous U1 snRNP structure-probing studies, which include chemical and enzymatic probing of deproteinized U1 snRNA, hydroxyl radical probing of U1 snRNP in E and A complex and SHAPE chemistry performed on in vitro transcribed U1 snRNA as a validation method for the SHAPE probe FAI-N3 (Krol et al. 1990; Dönmez et al. 2007; Chan et al. 2018). The structure-probing performed with FAI-N3 is largely consistent with our SHAPE data. However, because FAI-N3 can undergo a click chemistry-mediated biotin-streptavidin enrichment step, it has a higher signal to noise ratio and shows all nucleotides within loop regions to be reactive toward SHAPE electrophiles (Chan et al. 2018). The hydroxyl radical probing data shows that U1 snRNA does not undergo any major structural rearrangements that lead to differences in solvent accessibility, from E to A complex (Dönmez et al. 2007). We also observe that the core of U1 snRNP maintains a near uniform SHAPE reactivity when not associated with RNA or when bound to different substrate RNAs that mimic activated or repressed states. Both of these observations suggest that U1 snRNP maintains a uniform structure and protein composition in the early spliceosome.
The most striking differences in SHAPE reactivity that correlate with splicing activation and repression were detected when we compared SHAPE profiles of the U1 snRNA 5′ splice site binding region, in particular the intron–exon spanning junction motif (Fig. 7A–C). These results provide evidence that splicing activation promotes base-pairing and strong splicing repression, such as the proven repressor TIA-1 bound exonically, interferes with base-pairing between the junction motif and the 5′ splice site pre-mRNA nucleotides. This proposal is supported by psoralen crosslinking (Fig. 8A,B) and consistent with functional splicing data for these RNAs (Erkelenz et al. 2013).
It is plausible that the conformation of the junction motif acts as a switch where increased base-pairing with the 5′ splice site promotes splice site pairing, while U1 snRNAs with less base-paired junction motifs cannot promote splice site pairing. The fact that the junction motif shows increased SHAPE reactivity and decreased psoralen crosslinking when both SRSF7 and TIA-1 are bound at repressive positions further suggests that its base-pairing state can be modulated by inputs from both faces of U1 snRNP (Fig. 1A).
The “bind and activate” mode of 5′ splice site selection could be a safeguard mechanism to differentiate between U1 snRNPs functional roles in spliceosomal recruitment and prevention of premature polyadenylation. It is possible that U1 snRNP binds the pre-mRNA in an inactive conformation that is allosterically activated by splicing regulators bound at activating positions. Such a mechanism may be necessary because U1 snRNP is known to not only interact with genuine 5′ splice sites, but also with cryptic 5′ splice sites that frequently inhibit premature polyadenylation (Kaida et al. 2010). Thus, enhanced 5′ splice site base-pairing with U1 snRNP may act as an allosteric switch for spliceosomal assembly. U1 snRNP binding to a 5′ splice site triggers E-complex formation regardless of where splicing regulatory proteins are bound (Erkelenz et al. 2013). However, locking in base-pairing between U1 snRNA and the 5′ splice site occurs only in the presence of splicing activators. This activated conformation may then allow U1 snRNP to engage in cross intron activities, which include U1 snRNA stem–loop IV-mediated contacts with the U2 snRNP component SF3A1 during A complex formation (Sharma et al. 2014).
The proposed allosteric mechanism for the initiation of splice site pairing does not rule out any of the other potential mechanisms of position-dependent splicing activation/repression. The conformation of U1 snRNA could be regulated through U1- specific proteins such as U1-C, which is known to enhance 5′ splice site binding (Kondo et al. 2015). It is possible that TIA-1, which has been demonstrated to contact U1-C and recruit U1 snRNP to the 5′ splice site when bound intronically, can disrupt U1 snRNP's structural integrity when bound exonically (Förch et al. 2002). A similar mode of action could be envisioned for SRSF7, which is likely to mediate its effect on the junction motif through U1-70K. Lastly, the recruitment of additional proteins cannot be ruled out. It is possible that splicing regulators recruit different proteins to the two different faces of U1 snRNP depending on where they bind the pre-mRNA.
We have developed a targeted SHAPE methodology, which allows for probing U1 snRNP in the context of position-dependent splicing activation or repression. This method can be adapted to probe other snRNPs/spliceosomal complexes and possibly other dynamic RNPs. Importantly, we have demonstrated that the SHAPE reactivity of the U1 snRNA junction motif is altered during splicing activation and repression. These results provide molecular insights into the mechanism of position-dependent splicing activation and repression.
MATERIALS AND METHODS
In vitro transcription of substrate RNAs
Sequences for substrate RNAs were simultaneously linearized and amplified using standard PCR in 50 µL reactions. Subsequently 40 µL of PCR product was transcribed in vitro using the RiboMax RNA transcription kit (Promega). DNase treatment was performed according to the RiboMax protocol and RNAs were purified with the RNA Clean and Concentrator Kit (Zymo Research). A Nanodrop 2000 instrument was used to obtain RNA concentration values.
U1 snRNP enrichment approach
U1 snRNP was enriched through a pull-down assay using various substrate RNAs that contained verified binding sites for SRSF7 or TIA-1 at activating or repressing positions (Fig. 1B). Enriched U1 snRNPs within the context of activated or repressed E-complex were then probed using NAI, a SHAPE reagent (Spitale et al. 2013). SHAPE electrophiles acylate the 2′OH of RNA nucleotides based on their flexibility and dynamics (Wilkinson et al. 2006; McGinnis et al. 2012). Unstructured regions of RNA are more likely to be modified by SHAPE reagents, which cause reverse transcription to terminate one nucleotide before the acylated nucleotide. To detect structural changes within U1 snRNA including stem–loop IV, an LNA modified primer was designed to bind the 3′ end of U1 snRNA stem–loop IV. SHAPE reactivities of stem–loop IV were poor due to structure stops. Despite the poor SHAPE reactivities obtained for stem–loop IV, the LNA primer provided excellent reverse transcription (RT) signal and high-resolution data for the rest of U1 snRNA. Dideoxy sequencing was performed on enriched U1 snRNAs and nuclear extract U1 snRNA to confirm that the reverse transcription signal obtained from RNAs was generated from U1 snRNA (Supplemental Fig. 1).
Oxidation of substrate RNAs and RNA affinity pull-down
An amount of 300 picomoles of each substrate RNA was incubated with 5 mM NaIO4 (sodium [meta]periodate) and 0.1 M NaOAc (sodium acetate) for 1 h in the dark. Substrate RNAs were subsequently precipitated with 100% ethanol (EtOH). Adipic acid dihydrazide agarose beads were prepared by washing 125 µL of beads per reaction four times with two volumes of 0.1 M NaOAc. After the final wash, the bead volume was adjusted to 250 µL beads per reaction and 250 µL of beads were added to each substrate RNA pellet. Beads were subsequently incubated at 4°C on a rotator overnight. Beads were washed two times with 1 mL 2 M NaCl and three times with Buffer D (20 mM HEPES-KOH [pH 7.6], 20%, glycerol, 0.1 M KCl, 0.2 mM EDTA, and 0.5 mM DTT). 200 µL of 30% nuclear extract diluted with Buffer D containing 1 mM MgCl2 was added to each sample and incubated on a rotator for 15 min at 30°C. All samples were subsequently washed five times with 1 mL Buffer D containing 1 mM MgCl2.
RNA acylation and isolation
After the final wash, 5 µL of 1 M NAI or DMSO was added to 45 µL of evenly mixed beads and samples were incubated at 30°C for 15 min. After incubation with NAI, 150 µL of H2O was added followed by the addition of 200 µL of formamide. Samples were mixed by pipetting and then heated at 95°C for 1 min. RNA was separated from beads using 0.45 µm cellulose acetate Corning Costar SpinX columns, spun at 6000 rpm for 8 min. Final volumes were adjusted to 600 µL with H2O and subsequently 600 µL of premixed phenol:chloroform:isoamyl alcohol (25:24:1) pH 6.6 (Ambion) was added. The aqueous phase was extracted and washed with chloroform. For nuclear extract U1 snRNA, 5 µL of 1 M NAI or DMSO was added to 45 µL of 30% nuclear extract and incubated for 15 min at 30°C. Samples were directly phenol:chloroform extracted as described above. For in vitro U1 snRNA, U1 snRNA was folded and probed as described by Spitale et al. (2013). In vitro U1 snRNA was phenol:chloroform extracted as described above. All samples were precipitated with 300 mM NaOAc, 3 volumes of 100% EtOH and 2 µL of GlycoBlue (Invitrogen). All samples were washed twice with 70% EtOH and resuspended in 5 µL of water.
Reverse transcription
Four picomoles of a gene specific LNA modified primer were added to each reaction. Primers were 5′ end phosphorylated with T4 polynucleatide kinase (New England Biolabs [NEB]) using standard procedures from NEB and yielded 9000–10,000 CPM/µL. To anneal the primer, samples were heated to 95°C for 2 min and then cooled to 50°C. A master mix containing dNTPs, DTT, water (to 20 µL), 5× FS Buffer (250 mM Tris-HCl [pH 8.3], 375 mM KCl, 15 mM MgCl2) and Superscript III Reverse Transcriptase (Invitrogen) was added according to the superscript III protocol. For dideoxy (dd) sequencing samples, 2 µL of 10 mM ddNTPs (TriLink Biotechnologies) were added to the RNA and primer mixture before annealing. All samples were reverse transcribed for 50 min at 50°C. The RNA template was hydrolyzed with addition of 2 µL (one tenth volume) of 4 M NaOH and incubation at 95°C for 5 min. Samples were ethanol precipitated as previously described and washed two times as previously described. Samples were resuspended in 95% formamide, 18 mM EDTA, 0.025% xylene cyanol and bromophenol blue.
Gel electrophoresis, visualization, and quantification
Samples were run on 10% polyacrylamide (19:1 acrylamide:bisacrylamide) 7 M urea gels, see reference for detailed gel casting protocol (Movassat et al. 2017). Gels were prerun at 20 watts for 1 h, followed by 50 watts for 4–6 h depending on the desired resolution. Gels were dried for 1 h under vacuum at 80°C and exposed to a phosphor screen overnight. Images were obtained using GE Typhoon imager and peaks were integrated using SAFA software (Das et al. 2005). Results from junction motif analysis were independently verified using ImageJ software (Schneider et al. 2012). For the reactivity plots, the counts at each position in the NAI lane were subtracted by counts in the corresponding DMSO control lane. The counts at each nucleotide position were then divided by the average of the top 92%–98% most reactive nucleotides within that lane to obtain reactivity plots. The value obtained for each nucleotide was then averaged across three biological replicates. For the fold change of the junction motif (Fig. 7C) the SHAPE reactivity of the junction motif was normalized to the SHAPE reactivity of nucleotide U22, which stays constant across all conditions. Normalized SHAPE reactivities were divided by the TIA D SHAPE reactivity to obtain a fold change. Fold changes were averaged across three biological replicates. One tailed t-tests were performed in a pairwise manner to determine statistical significance. Comparisons to the crystal structure reference PDB code=4PKD. Each experiment was performed in triplicate.
Western blot assay
Protein samples from RNA pull-down experiments were separated on precast 12% Tris-Glycine SDS-PAGE gels (Bio-Rad) at 125 V for 50–60 min at room temperature. Gels were transferred to methanol activated Immun-Blot PVDF membrane (Bio-Rad) using Tris-Glycine transfer buffer at 60 V, 2 h, 4°C, with an ice pack and stirring of transfer buffer. The membrane was blocked for 1 h, at room temperature with 5% nonfat milk in tris-buffered saline with Tween-20 (TBS-T), followed by an overnight incubation with primary antibody at 4°C. Primary antibody was diluted in 2.5% nonfat milk in TBS-T. Primary antibody conditions in this work were the following: U1-70K at 1:100 (Santa Cruz Biotechnology, clone C-18 or Millipore mouse monoclonal, clone 9C4.1), U1-C at 1:200 (Sigma, rat monoclonal, clone 4H12), hnRNP A1 at 1:250 (Santa Cruz Biotechnology, mouse monoclonal, clone 9H10), SRSF7 at 1:1000 (MBL, rabbit polyclonal), and TIA-1 at 1:100 (Santa Cruz Biotechnology, goat polyclonal, clone C-20). The membrane was washed with TBS-T prior to incubation with the secondary antibody. Secondary antibodies were diluted in 2.5% nonfat milk in TBS-T and incubated for 1 h at room temperature. The membrane was washed further with TBS-T and then developed for 1 min using SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). Blots were imaged on a GE Typhoon imaging station.
Psoralen crosslinking
An amount of 300 picomoles of the five different substrate RNAs were dephosphorylated using NEB Quick Dephosphorylation Kit in 20 µL reactions. Reactions contained 20 units NEB quick CIP (calf intestinal phosphatase), 1× NEB CutSmart Buffer (50 mM postassium acetate, 20 mM Tris-acetate, 10 mM Magnesium acetate, 100 µg/mL BSA, pH 7.9) and were incubated at 37°C for 2 h. The CIP enzyme was heat inactivated at 80°C for 5 min. Substrate RNAs were 5′ end labeled with the addition of 20 units NEB T4 PNK (polynucleotide kinase), 2 µL gamma-ATP, and DTT(final concentration 5 mM). Kinase reactions were incubated at 37°C for 3 h and heat inactivated at 65°C for 20 min. Substrate RNAs were purified using ZYMO RNA Clean and Concentrator kits, resuspended in 25 µL and yielded a specific activity of ∼600,000 cpm/µL. Crosslinking reactions were carried out using 1 µL of radiolabled substrate RNA, 30% nuclear extract and mirrored the conditions used in SHAPE experiments. Each reaction contained 20 µg/mL 4′-aminomethyltrioxsalen (AMT) and was exposed to 365 nm UV for 10 min or kept in the dark as described by Tarn and Steitz (1994). Samples were then digested with proteinase K, phenol chloroform extracted and resolved on 6% denaturing polyacrylamide gels as described previously (Movassat et al. 2017). Crosslinking efficiencies were obtained using ImageJ. Briefly, a box was drawn around each band. The intensity in the +UV lane was subtracted by the intensity in the −UV lane and the resulting value was divided by the intensity of the band corresponding to the full-length substrate RNA in the +UV lane. The crosslinking efficiencies were averaged across three biological replicates and two tailed t-tests were performed in a pairwise manner to determine statistical significance.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We would like to thank Robert Spitale, Dalen Chan, and Chao Feng for helpful conversations and the gift of NAI reagent. We are grateful to Andrej Lupták and Luiz Passalacqua for helpful conversations. This work was supported by grants from the National Institutes of Health (NIH) (R01 GM062287 and R01 GM110244 to K.J.H.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.075135.120.
REFERENCES
- Buratti E, Baralle FE. 2004. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol 24: 10505–10514. 10.1128/MCB.24.24.10505-10514.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Beasley S, Zhen Y, Spitale RC. 2018. Facile synthesis and evaluation of a dual-functioning furoyl probe for in-cell SHAPE. Bioorg Med Chem Lett 28: 601–605. 10.1016/j.bmcl.2018.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou N-T, Shankarling G, Lynch KW. 2013. hnRNP L and hnRNP A1 induce extended U1 snRNA interactions with an exon to repress spliceosome assembly. Mol Cell 49: 972–982. 10.1016/j.molcel.2012.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Zhou Z, Reed R. 2000. Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol Cell 5: 779–787. 10.1016/S1097-2765(00)80318-4 [DOI] [PubMed] [Google Scholar]
- Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. 2005. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11: 344–354. 10.1261/rna.7214405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dönmez G, Hartmuth K, Kastner B, Will CL, Lührmann R. 2007. The 5′ end of U2 snRNA is in close proximity to U1 and functional sites of the pre-mRNA in early spliceosomal complexes. Mol Cell 25: 399–411. 10.1016/j.molcel.2006.12.019 [DOI] [PubMed] [Google Scholar]
- Erkelenz S, Mueller WF, Evans MS, Busch A, Schöneweis K, Hertel KJ, Schaal H. 2013. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 19: 96–102. 10.1261/rna.037044.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Chan D, Joseph J, Muuronen M, Coldren WH, Dai N, Corrêa IR, Furche F, Hadad CM, Spitale RC. 2018. Light-activated chemical probing of nucleobase solvent accessibility inside cells. Nat Chem Biol 14: 276–283. 10.1038/nchembio.2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förch P, Puig O, Martínez C, Séraphin B, Valcárcel J. 2002. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J 21: 6882–6892. 10.1093/emboj/cdf668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Ares M. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. 10.1038/nrg3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson SI, Polycarpou-Schwarz M, Mattaj IW. 1998. U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol Cell 1: 255–264. 10.1016/S1097-2765(00)80026-X [DOI] [PubMed] [Google Scholar]
- Hertel KJ. 2008. Combinatorial control of exon recognition. J Biol Chem 283: 1211–1215. 10.1074/jbc.R700035200 [DOI] [PubMed] [Google Scholar]
- Hertel KJ, Maniatis T. 1998. The function of multisite splicing enhancers. Mol Cell 1: 449–455. 10.1016/S1097-2765(00)80045-3 [DOI] [PubMed] [Google Scholar]
- Hong W, Bennett M, Xiao Y, Feld Kramer R, Wang C, Reed R. 1997. Association of U2 snRNP with the spliceosomal complex E. Nucleic Acids Res 25: 354–361. 10.1093/nar/25.2.354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. 2010. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468: 664–668. 10.1038/nature09479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamma H, Portman DS, Dreyfuss G. 1995. Cell type-specific expression of hnRNP proteins. Exp Cell Res 221: 187–196. 10.1006/excr.1995.1366 [DOI] [PubMed] [Google Scholar]
- Kondo Y, Oubridge C, van Roon A-MM, Nagai K. 2015. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. Elife 4: e04986 10.7554/eLife.04986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol A, Westhof E, Bach M, Lührmann R, Ebel JP, Carbon P. 1990. Solution structure of human U1 snRNA. Derivation of a possible three-dimensional model. Nucleic Acids Res 18: 3803–3811. 10.1093/nar/18.13.3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SR, Hertel KJ. 2004. Commitment to splice site pairing coincides with A complex formation. Mol Cell 15: 477–483. 10.1016/j.molcel.2004.06.025 [DOI] [PubMed] [Google Scholar]
- Matera AG, Wang Z. 2014. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 15: 108–121. 10.1038/nrm3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis JL, Dunkle JA, Cate JHD, Weeks KM. 2012. The mechanisms of RNA SHAPE chemistry. J Am Chem Soc 134: 6617–6624. 10.1021/ja2104075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Graveley BR. 2011. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev 21: 373–379. 10.1016/j.gde.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Query CC, Sharp PA. 1993. Splicing of precursors to mRNAs by the spliceosome. Cold Spring Harb Monograph Series 24: 303. [Google Scholar]
- Movassat M, Shenasa H, Hertel KJ. 2017. Preparation of splicing competent nuclear extract from mammalian cells and in vitro pre-mRNA splicing assay. Methods Mol Biol 1648: 11–26. 10.1007/978-1-4939-7204-3_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Pan Z, Zhang Z, Lin L, Xing Y. 2018. The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet 102: 11–26. 10.1016/j.ajhg.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705. 10.1038/nature12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Maris C, Allain FH-T, Black DL. 2011. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell 41: 579–588. 10.1016/j.molcel.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Wongpalee SP, Vashisht A, Wohlschlegel JA, Black DL. 2014. Stem-loop 4 of U1 snRNA is essential for splicing and interacts with the U2 snRNP-specific SF3A1 protein during spliceosome assembly. Genes Dev 28: 2518–2531. 10.1101/gad.248625.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenasa H, Hertel KJ. 2019. Combinatorial regulation of alternative splicing. Biochim Biophys Acta Gene Regul Mech 1862: 194392 10.1016/j.bbagrm.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Hertel KJ. 2008. Conserved RNA secondary structures promote alternative splicing. RNA 14: 1463–1469. 10.1261/rna.1069408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi E-A, Busch A, Hertel KJ. 2011. Efficient internal exon recognition depends on near equal contributions from the 3′ and 5′ splice sites. Nucleic Acids Res 39: 8928–8937. 10.1093/nar/gkr481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. 2013. RNA SHAPE analysis in living cells. Nat Chem Biol 9: 18–20. 10.1038/nchembio.1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. 1994. SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev 8: 2704–2717. 10.1101/gad.8.22.2704 [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- Wang G-S, Cooper TA. 2007. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8: 749–761. 10.1038/nrg2164 [DOI] [PubMed] [Google Scholar]
- Warf MB, Berglund JA. 2010. Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem Sci 35: 169–178. 10.1016/j.tibs.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, Weeks KM. 2006. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1: 1610–1616. 10.1038/nprot.2006.249 [DOI] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR. 1999. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 402: 832–835. 10.1038/45590 [DOI] [PubMed] [Google Scholar]
- Zahler AM, Neugebauer KM, Lane WS, Roth MB. 1993. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science 260: 219–222. 10.1126/science.8385799 [DOI] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, Rouskin S. 2017. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat Methods 14: 75–82. 10.1038/nmeth.4057 [DOI] [PMC free article] [PubMed] [Google Scholar]