

Abstract
Rationale: Rifapentine has been investigated at various doses, frequencies, and dosing algorithms, but clarity on the optimal dosing approach is lacking.
Objectives: To characterize rifapentine population pharmacokinetics, including autoinduction, and determine optimal dosing strategies for short-course rifapentine-based regimens for latent tuberculosis infection.
Methods: Rifapentine pharmacokinetic studies were identified though a systematic review of literature. Individual plasma concentrations were pooled, and nonlinear mixed-effects modeling was performed. A subset of data was reserved for external validation. Simulations were performed under various dosing conditions, including current weight-based methods; and alternative methods driven by identified covariates.
Measurements and Main Results: We identified nine clinical studies with a total of 863 participants with pharmacokinetic data (n = 4,301 plasma samples). Rifapentine population pharmacokinetics were described successfully with a one-compartment distribution model. Autoinduction of clearance was driven by rifapentine plasma concentrations. The maximum effect was a 72% increase in clearance and was reached after 21 days. Drug bioavailability decreased by 27% with HIV infection, decreased by 28% with fasting, and increased by 49% with a high-fat meal. Body weight was not a clinically relevant predictor of clearance. Pharmacokinetic simulations showed that current weight-based dosing leads to lower exposures in individuals with low weight, which can be overcome with flat dosing. In HIV-positive patients, 30% higher doses are required to match drug exposure in HIV-negative patients.
Conclusions: Weight-based dosing of rifapentine should be removed from clinical guidelines, and higher doses for HIV-positive patients should be considered to provide equivalent efficacy.
Keywords: tuberculosis, rifapentine, rifamycins, population pharmacokinetics, latent tuberculosis
At a Glance Commentary
Scientific Knowledge on the Subject
Rifapentine has become a principle component of novel short-course regimens for latent tuberculosis infection and a promising agent for treatment-shortening regimens for active disease. Evidence suggests that rifapentine induces its own elimination, but the implications for novel dosing strategies are not well understood. Furthermore, the evidence supporting the current weight-band dosing of rifapentine is lacking and requires further evaluation.
What This Study Adds to the Field
In this individual-participant data meta-analysis of rifapentine pharmacokinetics, we describe the population pharmacokinetics of rifapentine, including full characterization of the autoinduction profile. We find no evidence supporting weight-band dosing of rifapentine and thus recommend all individuals receive the same dose, with the exception of HIV-positive individuals, who would benefit from higher doses. This model will serve as a valuable tool for predicting drug exposure and determining optimal rifapentine doses for future clinical trials and in clinical practice.
The World Health Organization (WHO) estimates that 23% of the world’s population has latent tuberculosis infection (LTBI) and is at risk of developing active disease (1). Standard treatment for LTBI has historically been 9 months of daily isoniazid, for which patient compliance is poor and hepatotoxicity is a concern (2, 3). Recently, novel rifapentine-based regimens have demonstrated efficacy in preventing tuberculosis disease with much shorter treatment durations (4, 5). In addition, these regimens have shown equal or better safety profiles and higher patient compliance. The first regimen was 3 months of once-weekly rifapentine plus isoniazid (3HP) (4); it received U.S. Food and Drug Administration approval in 2014 and is now recommended by the CDC and the WHO for individuals with LTBI (6–8). An ultra–short-course regimen, 1 month of daily isoniazid plus rifapentine (1HP), has also shown efficacy, safety, and improved compliance in patients with HIV infection who are at high risk of developing tuberculosis disease (5); 1HP inclusion in WHO guidelines is under review (9).
Rifapentine has high antimycobacterial activity and a long elimination half-life of 15 hours that makes it an attractive candidate for treatment-shortening regimens (6, 10, 11). However, unlike in LTBI, it is still unknown if rifapentine will be effective in short-course regimens for active drug-sensitive tuberculosis disease (DS-TB). The only completed phase 3 clinical trial (RIFAQUIN [Rifapentine and a Quinolone in the Treatment of Pulmonary Tuberculosis]) failed to demonstrate noninferiority of intermittent rifapentine regimens in patients with DS-TB compared with the 6-month standard of care (12).
Robust characterization of rifapentine pharmacokinetics is required to determine optimal dosing strategies for new short-course regimens and for special populations. Current rifapentine-based regimens for LTBI use weight-band dosing (6, 8). However, these recommendations are not based on pharmacokinetic evidence; rather, they are drawn from the historical mg/kg doses used in rifampin-based therapy. The influence of body weight on rifapentine clearance remains inconclusive, as current studies have reported conflicting findings (13, 14). Meal type, dose amount, HIV status, race, and age may also impact rifapentine concentration (14–18). In addition, repeated twice-weekly dosing and daily administration results in lower rifapentine exposures over time, suggesting that rifapentine induces its own metabolism (19, 20).
Several pharmacokinetic studies have been conducted with varying rifapentine dosages (up to 20 mg/kg daily), frequencies (once weekly to twice daily), and methods (weight-based or flat dose) (19–22). Our aim here was to perform an individual-participant data meta-analysis and pool individual pharmacokinetic data from all relevant clinical studies in various populations (healthy volunteers and patients with LTBI and/or DS-TB with or without HIV infection). The goals were 1) to characterize rifapentine population pharmacokinetics, including the time course of autoinduction and relevant covariates that may have a significant clinical impact on rifapentine exposures and clinical efficacy, and 2) to derive dosing recommendations to inform optimal current and future use of rifapentine in tuberculosis infection and disease.
Methods
Clinical Studies
Rifapentine pharmacokinetic studies were identified through a literature search in PubMed with the terms “rifapentine” AND (“study” OR “trial”) from January 1, 1980, to December 31, 2015, according to Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines (23). Additional studies were identified through author collaborations. Corresponding authors of the study were invited to contribute data if the studies were prospective; if multiple-dose, pharmacokinetic measurements were available and validated; and if covariates of interest were documented (e.g., HIV status, meal type, and weight). All studies included in the analysis received ethical approval by their local ethical review boards.
Population Pharmacokinetic Analysis
Identified studies were split into an analysis cohort for structural model development and a validation cohort for external validation. We sought to conserve one-third of drug-concentration data for the validation cohort and to match dosing schedules and covariates (e.g., HIV) between cohorts when possible. Rifapentine plasma concentrations (Cp values) were natural log transformed and analyzed using nonlinear mixed-effects modeling with NONMEM (Nonlinear Mixed-Effects Modelling) version 7.41 (ICON Development Solutions). Pharmacokinetic data without an associated dosing record were excluded.
Population pharmacokinetic model building followed standard procedures by first characterizing the base structural model (24). To describe rifapentine autoinduction, a semimechanistic enzyme turnover model was used (25). Known covariate effects (i.e., HIV, meal type, and dose) were incorporated into the structural model. Additional covariate effects such as weight, age, race, body mass index, and sex were identified through a stepwise procedure with forward selection (P < 0.05) and backward elimination (P < 0.01). Final inclusion of covariates was based on statistical significance, scientific plausibility, and clinical relevance defined as a ≥20% change in the parameter estimate (26). Model development was guided by graphical assessment of goodness-of-fit plots, condition number, and the likelihood ratio test. Simulation-based diagnostics (e.g., visual predictive checks [VPCs]) were used for model validation. For detailed model-building procedures, see the online supplement.
Software
R software (version 3.4.2; R Foundation for Statistical Computing) was used for all data management, analyses, and graphical visualization. The xpose (version 0.4.4) and vpc (version 1.0.1) packages were used for visual diagnostics. Nonparametric bootstrap and covariate modeling were performed with Perl-speaks-NONMEM (version 4.7.0).
Dosing Simulations
Simulations were performed with the final model to 1) predict the autoinduction process with different doses and dosing schedules, 2) assess the impact of clinically relevant patient factors (e.g., HIV and weight) on rifapentine exposure, and 3) propose pragmatic dosing for rifapentine-containing LTBI regimens. Pharmacokinetic profiles were evaluated by different drivers of pharmacodynamics, including time above the minimum inhibitory concentration (MIC), area under the concentration–time curve (AUC), AUC/MIC ratio, maximum concentration (Cmax), and Cmax/MIC ratio, with MIC set to 0.06 mg/L (27). For 1HP and 3HP simulations, we predicted rifapentine exposure by following current weight-band dosing (1HP, 300 mg [<35 kg], 450 mg [35–45 kg], or 600 mg [>45 kg] daily; 3HP, 750 mg [<50 kg] and 900 mg [≥50 kg] once weekly) (4, 5). Alternative dosing methods were explored on the basis of identified covariates. All simulations were performed under low-fat meal conditions (the referent, where relative bioavailability = 1), given label recommendations.
Univariate Analysis of Month 2 Culture Conversion
Microbiological outcome data (i.e., liquid and solid culture data) were acquired from two phase 2 clinical studies: TBTC-29 (Tuberculosis Trials Consortium Study 29) and TBTC-29X (22, 28). Participant body weight and rifapentine AUC were evaluated as predictors of Month 2 culture conversion by logistic regression. Body weight was categorized as <50 kg or ≥50 kg, consistent with the weight-band dosing strategy used in these studies. AUC was categorized at the median AUC.
Results
Clinical Studies
We identified nine clinical studies with rifapentine pharmacokinetic data for the pooled analysis (Figure 1), including phase 3 (n = 2), phase 2 (n = 4), and phase 1 (n = 3) studies (12, 14, 19–21, 28–31). Overall, 863 subjects were included: 84 healthy volunteers, 702 patients with DS-TB, and 77 persons treated for LTBI. The analysis cohort included 360 subjects (n = 3,273 samples) from five studies. The validation cohort included 503 subjects (n = 1,115 samples) from four studies. Participant and trial characteristics are shown in Table 1. The analysis and validation cohorts were similar in design and participant characteristics. Overall, the median age was 34 years, the median weight was 59 kg, 31% were men, and 9% were HIV-positive. There was a wide range of rifapentine doses, dosing frequencies, and diets that were tested across studies (Table 1).
Figure 1.

Preferred Reporting Items for Systematic Reviews and Meta-analyses flow diagram.
Table 1.
Baseline Characteristics of the Study Participants in the Pooled Data Sets
| Trial* (Ref) | Rifapentine Regimen | N Individuals (N Samples) | Age (yr) | Weight (kg) | Female Sex | HIV-Positive |
|---|---|---|---|---|---|---|
| Analysis cohort | ||||||
| 06–0050 (19) | 900 mg thrice weekly with low-fat meal | 14 (269) | 41 (24–64) | 76 (50–97) | 3 (21.4) | — |
| Phase 1 | ||||||
| HV | ||||||
| PM | ||||||
| RIFAQUIN (12) | 900 mg twice weekly or 1,200 mg once weekly with high-fat meal | 241 (846) | 32 (18–80) | 56 (38–78) | 88 (36.5) | 46 (19.1) |
| Phase 3 | ||||||
| DS-TB | ||||||
| PM | ||||||
| TBTC-29B (14) | 5–20 mg/kg once daily with low-fat meal | 26 (504) | 47 (24–60) | 82 (60–99) | 5 (19.2) | — |
| Phase 1 | ||||||
| HV | ||||||
| Rifapentine, Mdz | ||||||
| TBTC-25 (29) | 600, 900, or 1,200 mg once weekly on empty stomach | 35 (357) | 44 (18–68) | 65 (46–110) | 12 (34.3) | — |
| Phase 2 | ||||||
| DS-TB | ||||||
| PH | ||||||
| ACTG-A5311 (21) | 10 mg/kg twice daily or 15 or 20 mg/kg once daily with low- or high-fat meal | 44 (1,210) | 35 (20–59) | 82 (60–99) | 12 (27.3) | — |
| Phase 1 | ||||||
| HV | ||||||
| Rifapentine | ||||||
| Validation cohort | ||||||
| TBTC-29X (28) | 10, 15, or 20 mg/kg once daily with high-fat meal | 225 (713) | 30 (18–70) | 55 (40–83) | 66 (29.3) | 19 (8.4) |
| Phase 2 | ||||||
| DS-TB | ||||||
| PHZE | ||||||
| TBTC-26 (30) | 900 mg once weekly with food | 77 (77) | 40 (19–63) | 81 (49–169) | 37 (48.1) | — |
| Phase 3 | ||||||
| LTBI | ||||||
| PH | ||||||
| TBTC-29 (22) | 10 mg/kg 5 d per week on empty stomach | 158 (158) | 36 (18–86) | 60 (40–101) | 46 (29.1) | 16 (10.1) |
| Phase 2 | ||||||
| DS-TB | ||||||
| PHZE | ||||||
| RioMar (31) | 7.5 mg/kg once daily with food | 43 (167) | NR | 58 (45–83) | NR | — |
| Phase 2 | ||||||
| DS-TB | ||||||
| PHMZ |
Definition of abbreviations: DS-TB = drug-sensitive tuberculosis disease; HV = healthy volunteers; LTBI = latent tuberculosis infection; Mdz = midazolam, only administered in some of the study participants; NR = not recorded; PH = rifapentine and isoniazid; PHMZ = rifapentine, isoniazid, moxifloxacin, and pyrazinamide; PHZE = rifapentine, isoniazid, pyrazinamide, and ethambutol; PM = rifapentine plus moxifloxacin; Ref = reference number; RIFAQUIN = Rifapentine and a Quinolone in the Treatment of Pulmonary Tuberculosis; RioMar = Rifapentine Plus Moxifloxacin-based Regimen for Treatment of Pulmonary Tuberculosis; TBTC = Tuberculosis Trials Consortium Study.
Data are expressed as median (range) or number (percentage) unless otherwise specified.
A description of each trial is provided, including study phase, population, and drug regimen.
Pharmacokinetic-Enzyme Model
The final rifapentine pharmacokinetic-enzyme model is shown in Figure 2, and final parameter estimates are shown in Table 2. All pharmacokinetic parameters were well estimated, with low relative SEs. Rifapentine apparent clearance was estimated to be 1.11 L/h in the typical adult and increased up to 1.92 L/h (173%) over time as a result of autoinduction. The induction process was described using an indirect-response, semimechanistic enzyme-turnover model (Figure 2). The effect of rifapentine drug concentration on enzyme production was described through a sigmoid maximum effect (Emax) relationship: effect = (Emax × Cpγ)/(EC50γ + Cpγ), where EC50 is the rifapentine Cp when half Emax is observed, and γ represents the steepness of the relationship. The maximum autoinduction effect is expected at the steady-state concentrations achieved with daily doses of 300 mg or more, and clearance stabilizes by Day 21 of therapy, assuming five half-lives to a steady state (Figure 3).
Figure 2.

Final rifapentine pharmacokinetic-enzyme model. The number of transit compartments was estimated using the relationship kTR = (N + 1)/MTT. The ka was assumed to equal the kTR. Rifapentine autoinduction was modeled with an enzyme turnover model, in which the EFF of rifapentine concentration in the central compartment increased the kENZ, thereby increasing the ENZ. Rifapentine CL increased as a result of increased ENZ. The F increased (+) or decreased (−) as indicated. CL = clearance; CLm = metabolite clearance; EFF = effect; ENZ = enzyme pool; F = fraction of drug absorbed or relative availability; ka = absorption rate constant; kENZ = enzyme production rate; kTR = transit-rate constant; MTT = mean transit time; V = apparent volume of distribution; Vm = metabolite volume of distribution.
Table 2.
Final Parameter Estimates for the Rifapentine Population Pharmacokinetic Model
| Parameter | Population Estimate |
Interindividual Variability |
||
|---|---|---|---|---|
| Value (%RSE) | 95% CI* | %CV (%RSE) | 95% CI* | |
| CL/F, L/h | 1.11 (1.92) | 0.952–1.48 | 24.3 (9.34) | 12.8–28.0 |
| V/F, L | 36.7 (1.99) | 28.5–40.9 | 17.6 (17.7) | 10.5–24.0 |
| MTT, h | 1.94 (2.97) | 1.83–2.04 | — | — |
| NN | 2.15 (5.44) | 1.66–2.70 | — | — |
| Bioavailability | 100 fixed | — | 29.8 (10.8) | 21.5–34.6 |
| Fixed effects on bioavailability† | ||||
| Dose | 0.0167 (5.30) | 0.00343–0.0287 | — | — |
| HIV infection | 0.729 (6.26) | 0.584–0.815 | — | — |
| High-fat meal | 1.49 (3.05) | 1.37–1.64 | — | — |
| Fasting | 0.731 (5.51) | 0.546–0.776 | — | — |
| kENZ, h−1‡§ | 0.00587 (32.1) | 0.00291–0.0135 | — | — |
| Emax, %‡ | 73.0 (25.2) | 51.0–116 | — | — |
| EC50, mg/L‡ | 4.27 (39.8) | 1.80–6.57 | — | — |
| γ | 10 fixed | — | — | — |
| Residual error of rifapentine | 0.577 (4.13) | 0.573–0.699 | — | — |
| CLm/fm, L/h | 3.11 (12.2) | 1.89–6.26 | 40.0 (6.69) | 34.2–44.6 |
| Vm/fm, L | 2.15 (7.07) | 1.67–3.15 | — | — |
| fm,dose‖ | 0.0185 (3.56) | 0.0004–0.0266 | — | — |
| HIV effect on CLm/fm | 1.36 (9.85) | — | — | — |
| Residual error of metabolite | 0.631 (5.59) | 0.560–0.695 | — | — |
Definition of abbreviations: CI = confidence interval; CL/F = apparent clearance; CLm/fm = apparent metabolite clearance; CV = coefficient of variation; EC50 = concentration at which effect is 50% of Emax; Emax = maximum effect; F = bioavailability; fm = fraction metabolized; fm,dose = dose-dependent reduction in fm; γ = steepness for Emax equation; kENZ = enzyme production rate; MTT = mean transit time; NN = number of transit compartments; RSE = relative SE; V/F = apparent volume of distribution; Vm/fm = apparent metabolite volume of distribution.
CIs were based on 926 (out of 1,000) successful bootstrap runs for rifapentine model and 999 (out of 1,000) successful bootstrap runs for metabolite model.
Fixed effects on F were relative to HIV-negative individuals receiving 300 mg of rifapentine with a low-fat meal, where F = 1 for each reference condition. Relative bioavailability is calculated as F = Fdose × Fhiv × Fhigh-fat × Ffasting, where Fdose is the relative reduction in bioavailability per 100 mg above 300 mg [equal to 1 − estimate × (dose/100 mg)], Fhiv is the relative bioavailability in HIV-positive individuals, Fhigh-fat is the relative bioavailability with a high-fat meal (vs. low-fat meal), and Ffasting is the relative bioavailability with fasting (vs. low-fat meal).
Autoinduction parameters were estimated on the basis of the analysis data set alone.
Translates to an enzyme turnover half-life of 118 hours.
The fm is a function of dose, where fm = 1 − fm,dose × (dose/100 mg).
Figure 3.
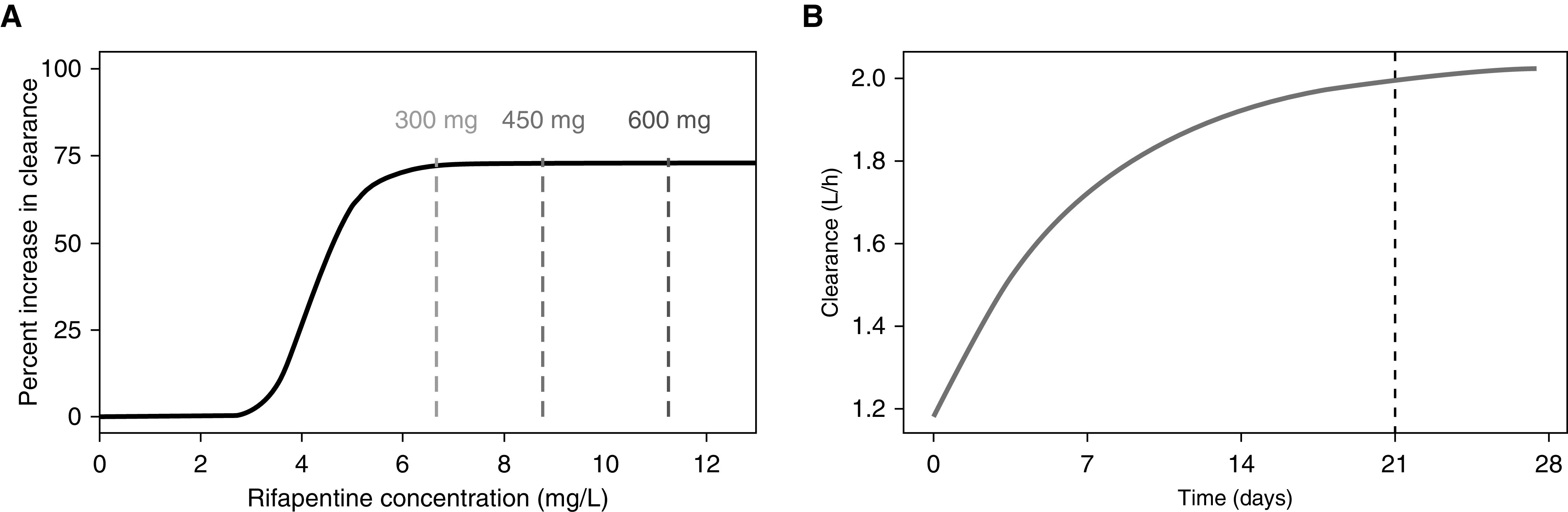
Rifapentine autoinduction profile. (A) The sigmoid relationship between rifapentine concentration and autoinduction is shown with the solid black line. Dashed lines represent the average concentration at a steady state of daily therapy with 300, 450, and 600 mg of rifapentine in a typical HIV-negative individual. (B) Rifapentine induction over time after daily administration of 600 mg. The black dashed line represents the time at which the induction process reaches a steady state.
Rifapentine Model Evaluation and Validation
The VPC of the basic structural model (built with analysis cohort data alone) shows that the model predicted the analysis cohort raw data well: the median, 5th, and 95th percentiles of raw data fell within or near the percentiles of model-predicted concentrations for all time points (Figure 4A). Furthermore, we show that model-predicted concentrations matched the raw data of an external data set (i.e., the validation cohort, which was not used in model development; Figure 4B).
Figure 4.
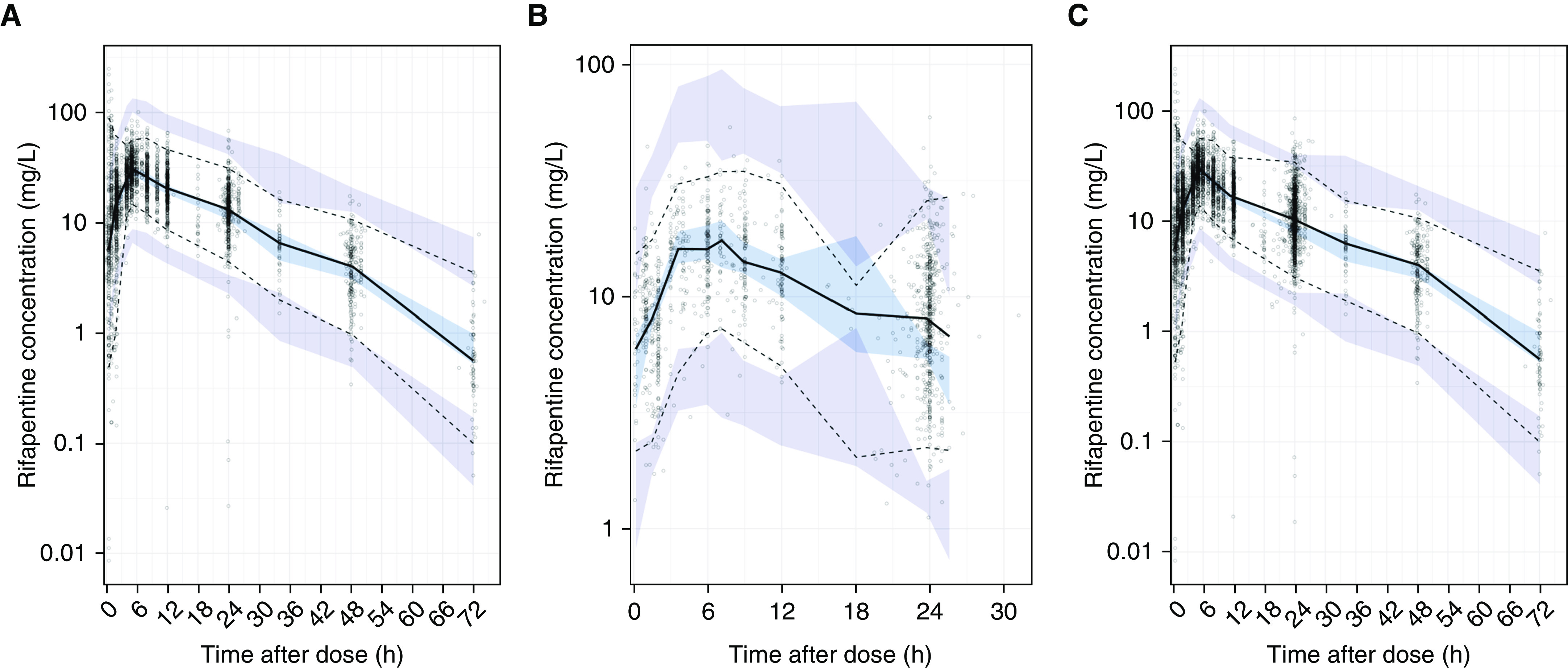
Validation of the structural rifapentine population pharmacokinetic model. Prediction-corrected visual predictive check of base model with (A) analysis data set, (B) validation data set, and (C) combined data set. Panels show the model predictions (shaded areas) compared with observed or raw rifapentine concentrations (dots). Model predictions were based on the base structural model, built from the analysis data set alone. The 5th (dashed line), 50th (solid line), and 95th (dashed line) percentiles of the observed raw data are overlaid onto the 95% confidence intervals of model-predicted concentrations at the 50th (light blue) as well as 5th and 95th (purple) percentiles, obtained from 500 simulations of each respective data set.
After model validation, data from both cohorts were pooled, and parameters were reestimated. VPCs of the final pharmacokinetic model for rifapentine and its metaboliteare shown in the online supplement. The final model predicted rifapentine (see Figure E2 in the online supplement) and metabolite (Figure E3) concentrations well for all studies.
Impact of Covariates on Rifapentine Pharmacokinetics
Rifapentine bioavailability was strongly (P < 0.001) influenced by HIV status, food, and dose, with clinically relevant effect sizes. The relative effects on bioavailability of HIV-positive status (vs. HIV-negative), high-fat meal or fasting condition (vs. low-fat meal), and dose per every 100 mg above 300 mg (the referent) are shown in Table 2. Body weight was related to rifapentine clearance (P < 0.001), with a 0.1-L/h (9%) increase in clearance for each 10-kg increase in weight (Figure 5). However, weight explained only 2.9% of the interindividual variability in clearance, and the effect size did not meet our criteria for clinical relevance. Furthermore, the majority of statistical significance was from a few influential individuals over 90 kg in weight (see online supplement). Allometrically scaling clearance did not provide any additional improvement over the linear relationship, and the functions were nearly identical at relevant weight ranges (40–100 kg). Therefore, the only covariates included in the final model were HIV, food, and dose.
Figure 5.
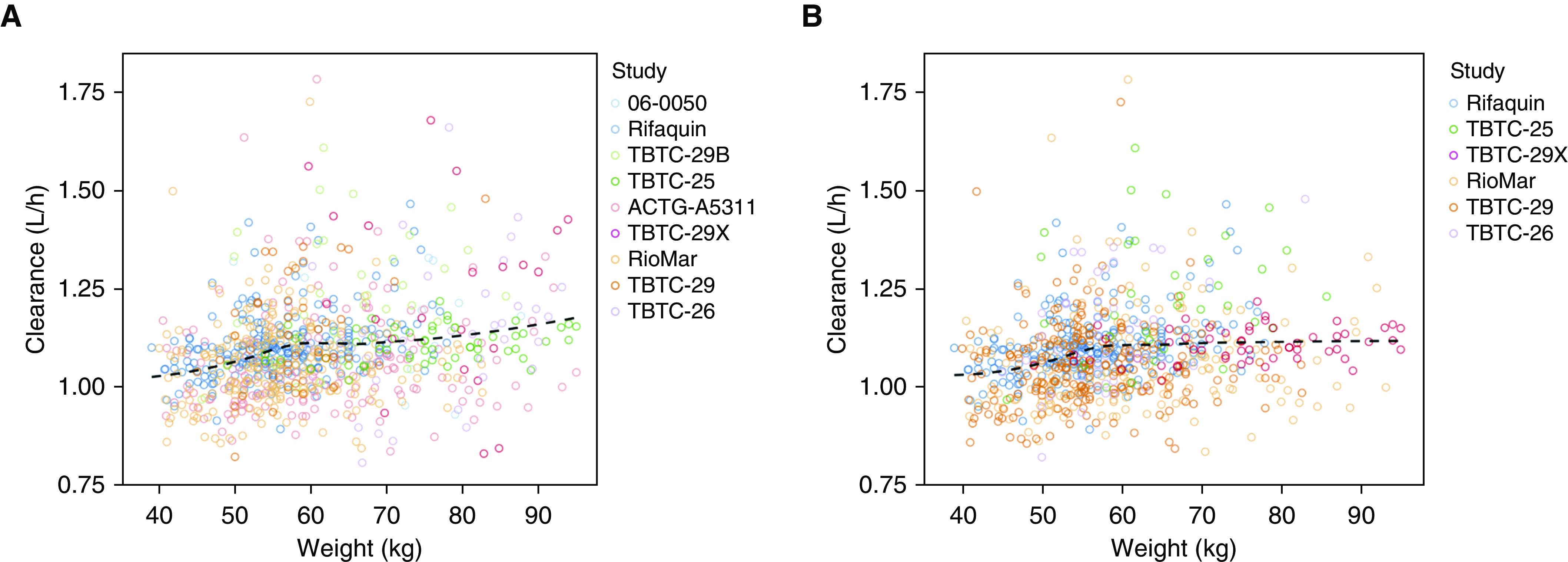
Relationship between weight and rifapentine clearance. The relationship was assessed for (A) all subjects and (B) only patients with drug-sensitive tuberculosis disease or latent tuberculosis infection with final model-parameter estimates. The dashed line represents local regression curve. ACTG = AIDS Clinical Trials Group; Rifaquin = Rifapentine and a Quinolone in the Treatment of Pulmonary Tuberculosis; RioMar = Rifapentine Plus Moxifloxacin-based Regimen for Treatment of Pulmonary Tuberculosis; TBTC = Tuberculosis Trials Consortium Study.
Rifapentine Simulations of Different Dosing Schedules
The effect of dose and dosing frequency on rifapentine pharmacokinetics is shown in Figure 6. With intermittent dosing, autoinduction was minimal to moderate and clearance increased slightly with larger doses (see online supplement). With daily dosing, maximum induction was achieved with doses of 300 mg or more. All dosing schedules were able to maintain concentrations above the MIC during the dosing interval, except for once-weekly schedules, in which concentrations fell below the MIC just before the next dose (Figure 6B). Cmax/MIC and AUC/MIC ratios were highest with daily dosing, because of drug accumulation, and increased with increasing dose (Table E2).
Figure 6.
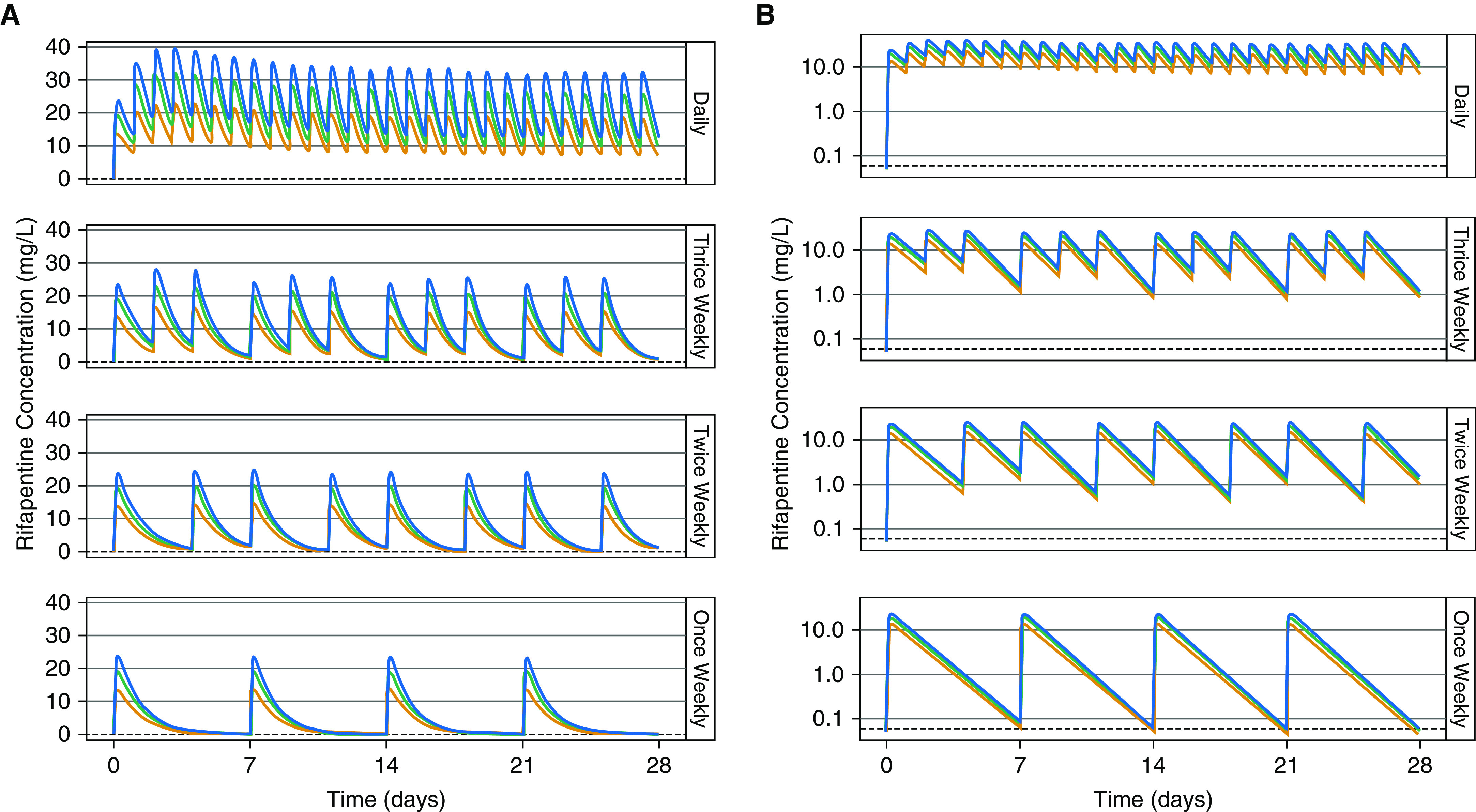
Effect of dose and dosing frequency on rifapentine exposure. (A) Rifapentine concentration over time and (B) concentration over time in log scale in a typical individual without HIV infection following once-daily, thrice-weekly, twice-weekly, and once-weekly administration of 600 mg (orange), 900 mg (green), or 1,200 mg (blue). The black dashed lines show the minimum inhibitory concentration (equal to 0.06 mg/L).
Rifapentine Simulations for 1HP and 3HP Therapy
We simulated rifapentine drug concentrations under the 1HP and 3HP regimens for LTBI in both HIV-positive and HIV-negative adults. The typical HIV-positive patient had lower drug concentrations than the typical HIV-negative patient when given the same dose because of decreased rifapentine bioavailability (Figure 7). Lower drug concentrations were also predicted in individuals with low weight who followed the current weight-band dosing (Figure 7). Removing weight bands and administering the same flat dose to all individuals would result in equal exposures across weights; however, it did not equalize exposures by HIV status (Figure 8). With a stratified regimen, in which HIV-positive individuals received ∼30% higher doses, similar exposures were expected by HIV status and weight for both 1HP and 3HP (Figure 8).
Figure 7.
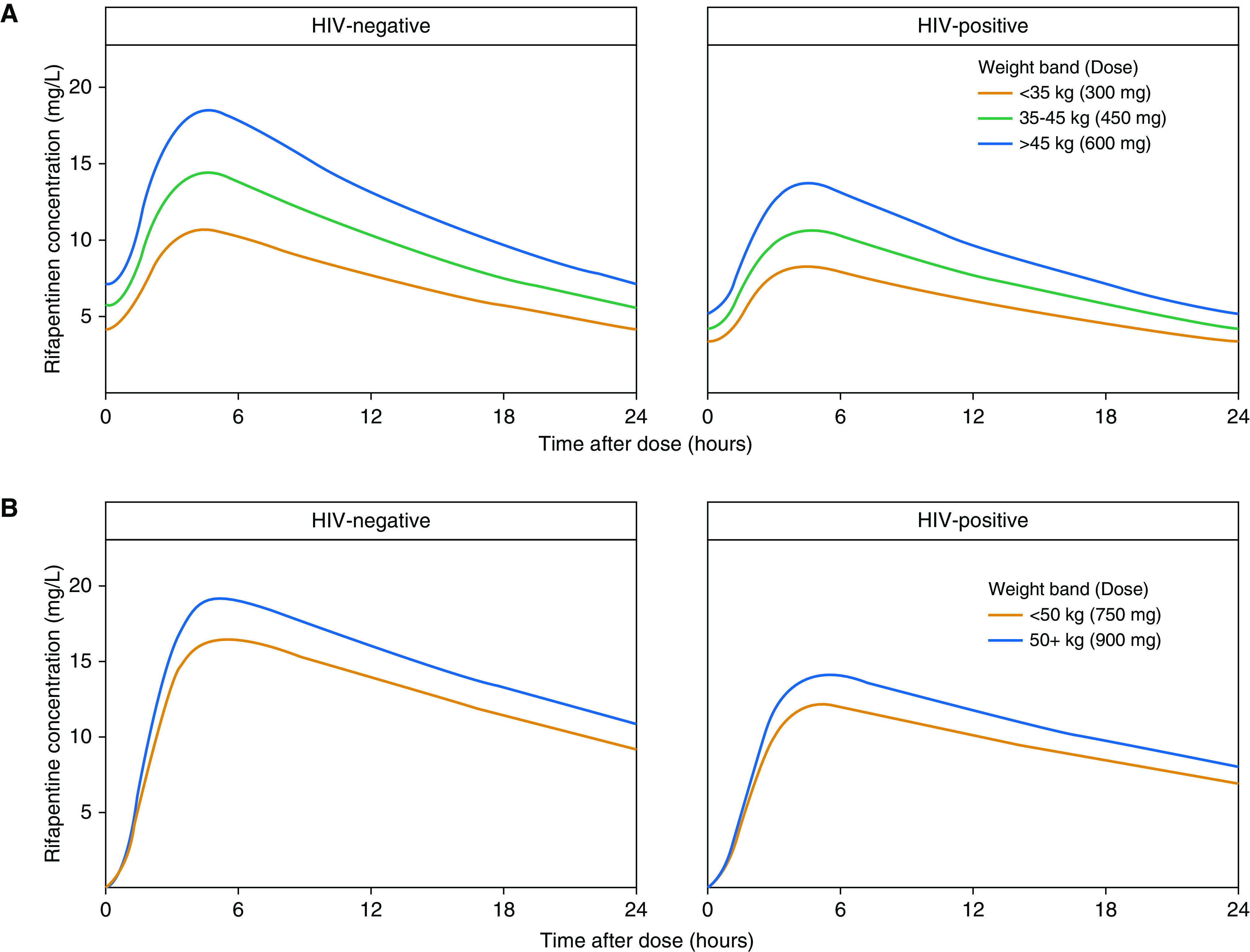
Pharmacokinetic profiles of rifapentine following (A) a regimen of daily isoniazid-rifapentine for 1 month and (B) a regimen of once-weekly rifapentine-isoniazid for 3 months. Concentration–time profiles over 24 hours are shown for the typical adult by HIV status on (A) Day 21 of therapy, to reflect steady-state concentrations, and (B) after the first dose, as no accumulation occurs with weekly dosing.
Figure 8.
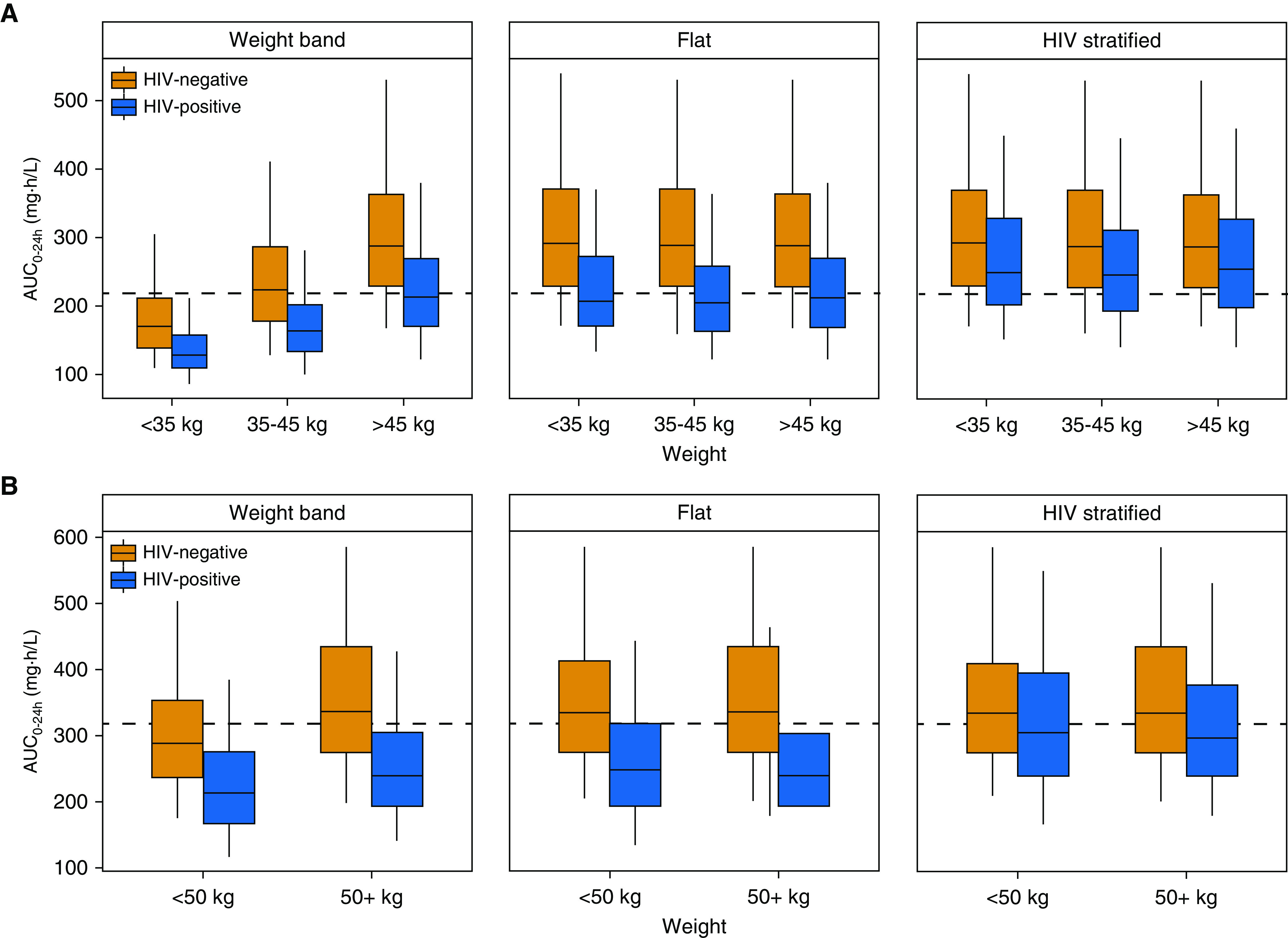
Predicted rifapentine exposures with different dosing methods for (A) a regimen of daily isoniazid-rifapentine for 1 month (1HP) and (B) a regimen of once-weekly rifapentine-isoniazid for 3 months (3HP). Profiles for drug-exposure area under the concentration–time curve over 24 hours (AUC0–24h) are based on 500 simulations. (A) 1HP predictions reflect steady-state exposures to account for autoinduction. “Weight-band” rifapentine doses were 300 mg for <35 kg, 450 mg for 35–45 kg, and 600 mg for >45 kg, as currently recommended for 1HP. The “flat” approach prescribed 600 mg to all individuals, and the “HIV-stratified” approach increased the dose in HIV-positive individuals to 750 mg. (B) 3HP doses were 750 mg for <50 kg and 900 mg for ≥50 kg for the weight-band approach, as currently recommended. The flat approach prescribed 900 mg to all individuals, and the HIV-stratified approach increased the dose in HIV-positive individuals to 1,200 mg. Dashed lines represent (B) the median AUC0–24h (317 mg · h/L) observed in patients treated with 3HP in the PREVENT-TB trial (i.e., TBTC-26 [Tuberculosis Trials Consortium Study 26]) and (A) the median predicted AUC0–24h in HIV-positive patients with 600 mg daily (219 mg · h/L).
Univariate Analysis of Month 2 Culture Conversion
A total of 363 individuals treated with 10 mg/kg of rifapentine had phase 2 microbiological data available. Univariate logistic regression results for Month 2 culture conversion of liquid media are shown in Figure 9. Month 2 culture conversion was less likely in individuals who had a lower rifapentine AUC (odds ratio, 0.49) and in those who weighed <50 kg (odds ratio, 0.60).
Figure 9.

Predictors of Month 2 culture conversion. Data were acquired from two phase 2 clinical studies (TBTC-29 [Tuberculosis Trials Consortium Study 29] and TBTC-29X), in which participants received 10 mg/kg of rifapentine daily. Odds ratios are from univariate analysis. AUC = area under the concentration–time curve; CI = confidence interval.
Discussion
In this study, we used a pooled individual-data approach with an external validation to describe rifapentine population pharmacokinetics in a large cohort of subjects. This analysis included nine clinical studies with a wide range of rifapentine doses and scheduling frequencies, allowing for successful characterization of rifapentine autoinduction with respect to drug concentration. It represents the largest analysis of rifapentine population pharmacokinetics to date. Our results establish several findings that may help guide rifapentine dosing strategies: 1) pharmacokinetic data do not support dosing rifapentine by body weight; 2) HIV-positive individuals require at least 30% higher doses to achieve equal drug exposures to HIV-negative persons; and 3) rifapentine autoinduction is strongly influenced by dosing frequency rather than by dose amount.
Since rifapentine’s approval, several studies have shown evidence of rifapentine inducing its own elimination, but none have characterized autoinduction with respect to rifapentine concentration (14, 16, 17, 19, 20). Previously published models have described rifapentine autoinduction empirically with time-varying clearance models (14, 17) or reduced bioavailability (16). Although these approaches are adequate for describing data, they have limited utility in clinical settings and for dose determination in new clinical trials. In our analysis, we used a semimechanistic turnover model in which rifapentine concentration was the driver of autoinduction (25). This method is advantageous in that it allows for predicting the magnitude of autoinduction in different rifapentine regimens of various doses and frequencies, including those that have not yet been tested in a clinical trial.
Rifapentine autoinduction is strongly influenced by dosing frequency. Simulated pharmacokinetic profiles showed increasing Cmax and AUC in the first week of therapy with daily dosing because of drug accumulation but decreased thereafter as a result of clearance induction. This effect was most prominent with daily dosing, moderate dosing thrice weekly, and minimal dosing with less frequency. These findings are in agreement with previous reports from noncompartmental analyses (20, 30, 32). Dose amount had little effect on the magnitude of autoinduction (∼10% higher clearance with 1,200 mg vs. 600 mg), regardless of dosing frequency. A dose effect on rifapentine autoinduction has been described previously (17, 19). In our model, nonproportional increases in drug exposure with increasing dose were described through a reduction in bioavailability, which is consistent with saturable absorption (14). Still, as the induction process is a function of rifapentine Cp in our model, any additional dose effects on clearance would be captured. Although full autoinduction is predicted with daily dosing, drug accumulation was also high, leading to superior Cmax/MIC and AUC/MIC ratios compared with less frequent dosing. This confirms that daily dosing has the highest potential for concentration-dependent killing of Mycobacterium tuberculosis. Furthermore, this work is an important contribution to the understanding of the rifapentine dose–exposure relationship, especially in the context of DS-TB, in which daily dosing is likely required (15).
Currently, body weight is the only dose-determining factor for rifapentine, which was not supported in our analysis. In three previously described population pharmacokinetic models, weight did not influence rifapentine pharmacokinetics (14, 15, 17). Furthermore, Savic and colleagues (15) supported flat dosing of rifapentine, which was later implemented in a phase 3 clinical trial for DS-TB (TBTC-31, clinicaltrials.gov identifier NCT 02410772). Contrarily, Langdon and colleagues (13) reported a change in rifapentine clearance by 0.5 L/h for each 10 kg of body weight in a small cohort of 46 patients. However, their model did not incorporate dose-dependent absorption (i.e., reduced bioavailability with increased dose), which likely would reduce the estimated weight effect on clearance because the study dosed by weight, and clearance and bioavailability are indirectly linked with oral dosing (13). Francis and colleagues (16) allometrically scaled clearance by fat-free mass. The model’s application to rifapentine dosing, which is based on total body weight, was not described. Our study is the largest population pharmacokinetic study to date, with over 800 patients and healthy volunteers. Although a small weight effect was observed (<10% change in clearance for each 10 kg in body weight), it does not justify a 150-mg (∼30%) change in dose, as currently recommended in LTBI dosing guidelines. Weight and patient population appeared to be correlated in our data set (i.e., patients with DS-TB weighed less on average); therefore, we investigated the weight effect in healthy volunteers, individuals with LTBI, and patients with DS-TB separately. The weight effect was comparable and remained clinically irrelevant. We conclude that weight is not a clinically relevant predictor of rifapentine clearance and that weight-based dosing should not be recommended.
Simulations of the 1HP and 3HP regimens showed lower rifapentine exposures in individuals with low weight who receive lower doses with current weight-band dosing. This ultimately puts the smallest, most vulnerable individuals at risk of underexposure and, consequently, treatment failure (33, 34). A univariate analysis of phase 2 culture data from two DS-TB studies showed that Month 2 culture conversion was less likely in individuals with low weight and in those with low rifapentine exposure. Although the pharmacokinetic–pharmacodynamic relationships in LTBI have not been established, rifamycins show concentration-dependent killing of M. tuberculosis, and rifapentine AUC is a strong predictor of Month 2 culture conversion (15, 35). Flat dosing of rifapentine (e.g., prescribing the same dose to all adults) ensures equal rifapentine exposure in adult patients of all sizes and thus allows an equal chance for a successful outcome. Moreover, flat dosing simplifies the regimen in adults and encourages coformulation of rifapentine and isoniazid into a fixed-dose combination tablet, reducing pill burden and simplifying the regimen even further.
Dose discrimination may be warranted by HIV status. HIV-positive persons have 27% lower rifapentine bioavailability, resulting in lower exposures than HIV-negative adults. Reduced bioavailability of rifamycins with HIV infection has been reported previously (15, 17) and has been attributed to malabsorption (36–38). Although antiretroviral drugs may also explain decreases in rifamycin concentration, the HIV-positive participants in our analysis did not receive antiretroviral therapy (12, 22, 28). Given that rifapentine’s main metabolite has activity against M. tuberculosis, we also looked at metabolite concentrations by HIV status. It appeared that HIV-positive individuals had lower exposures of both rifapentine and its metabolite, confirming the need for higher doses in HIV-positive patients. Increasing the 3HP dosage to 1,200 mg once weekly in HIV-positive patients results in exposures similar to those of 900 mg once weekly in HIV-negative patients. Likewise, 750 mg daily in HIV-positive adults is similar to 600 mg daily in HIV-negative adults for the 1HP regimen. Although 1HP at 600 mg daily was effective in preventing tuberculosis disease in HIV-positive individuals (5), this may reflect the minimum effective dose, and higher doses may provide better protection.
The proposed dosing recommendations are limited by the lack of established pharmacokinetic targets in LTBI. We proposed doses that would match median exposures after the standard doses tested in clinical trials with demonstrated efficacy. Given that the development of tuberculosis was rare in those studies, these pharmacokinetic targets are reasonable, and we would expect the proposed doses to result in efficacy similar to that observed in clinical trials. The pharmacokinetic target for the 1HP regimen reflects the median predicted exposure in a typical HIV-positive adult receiving 600 mg daily and may be on the low end. Pharmacokinetic data from the BRIEF-TB (Brief Rifapentine-Isoniazid Evaluation for Tuberculosis Prevention) trial, and future trials are urgently needed to confirm pharmacokinetic thresholds for 1HP. In addition, one study showed higher rifapentine bioavailability in Asians compared with Africans, which could impact the dose requirement (15). This finding could not be confirmed in our study because TBTC-29X was the only study contributing a substantial Asian population. Furthermore, investigation of race effects on rifapentine pharmacokinetics is required.
Our systematic review included all relevant studies published before 2016. Only one pharmacokinetic study was identified in more recent literature and would not have met our inclusion criteria because of nonstandardized meal administration (16). Thus, our model represents the most up-to-date analysis of rifapentine pharmacokinetics. Of note, the analysis includes only one study in LTBI participants. To date, these remain the only pharmacokinetic data in this population. Furthermore, there is no evidence to suggest pharmacokinetics would differ by disease state, so we do not expect this to impact the generalizability of our work to LTBI treatment.
In conclusion, rifapentine exhibits autoinduction that is strongly influenced by dosing frequency. Weight was not a clinically relevant predictor of rifapentine clearance; thus, dosing should not be based on an individual’s weight. In fact, weight-based dosing results in substantially lower drug concentrations that could ultimately compromise treatment efficacy. If stratified dosing is to be implemented, it should be done on the basis of HIV status to ensure that HIV-positive individuals are adequately exposed to drug. Lastly, as rifapentine use becomes more widespread in tuberculosis treatment and prevention, this model can serve as a useful tool in clinical practice and in clinical trial design for dose determination and exposure prediction.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank all of the study participants in each of the clinical studies for their significant contributions. They also thank the networks for sharing the data with them, including the TBTC, AIDS Clinical Trials Group, RIFAQUIN Study Team, and the International Consortium for Trials of Chemotherapy Agents in Tuberculosis.
Footnotes
Supported by the NIH/National Institute of Allergy and Infectious Diseases grant R01AI135124-01A (to R.M.S.).
Author Contributions: All authors contributed to the intellectual content of the manuscript and approved the manuscript version submitted for publication. Data was acquired by S.E.D., A.J., K.E.D., M.W., and H.M.M. Data analysis was performed by J.E.H., K.K.R., and R.M.S. J.E.H., K.K.R., and R.M.S. made substantial contributions to the study concept and design.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.201912-2489OC on May 15, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.World Health Organization. Global tuberculosis report 2018. Geneva, Switzerland: WHO Press; 2018. [Google Scholar]
- 2.American Thoracic Society; Centers for Disease Control and Prevention. Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a Joint Statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement as it relates to infants and children were endorsed by the American Academy of Pediatrics (AAP), August 1999. Am J Respir Crit Care Med. 2000;161:S221–S247. doi: 10.1164/ajrccm.161.supplement_3.ats600. [DOI] [PubMed] [Google Scholar]
- 3.Horsburgh CR, Jr, Goldberg S, Bethel J, Chen S, Colson PW, Hirsch-Moverman Y, et al. Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401–409. doi: 10.1378/chest.09-0394. [DOI] [PubMed] [Google Scholar]
- 4.Sterling TR, Villarino ME, Borisov AS, Shang N, Gordin F, Bliven-Sizemore E, et al. TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155–2166. doi: 10.1056/NEJMoa1104875. [DOI] [PubMed] [Google Scholar]
- 5.Swindells S, Ramchandani R, Gupta A, Benson CA, Leon-Cruz J, Mwelase N, et al. BRIEF TB/A5279 Study Team. One month of rifapentine plus isoniazid to prevent HIV-related tuberculosis. N Engl J Med. 2019;380:1001–1011. doi: 10.1056/NEJMoa1806808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prifitin (rifapentine) tablets. Product label. Silver Spring, MD: U.S. Food and Drug Administration; 2014. [accessed 2019 Sep 11]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021024s011lbl.pdf. [Google Scholar]
- 7.Centers for Disease Control and Prevention. Atlanta, GA: Centers for Disease Control and Prevention; 2016. Treatment regimens for latent TB infection (LTBI) [accessed 2019 Sep 11]. Available from: https://www.cdc.gov/tb/topic/treatment/ltbi.htm. [Google Scholar]
- 8.World Health Organization. Guidelines for treatment of tuberculosis. Geneva, Switzerland: WHO Press; 2010. [Google Scholar]
- 9.WHO Global TB Programme. Geneva, Switzerland: WHO Press; 2019. Latent tuberculosis infection: updated and consolidated guidelines for programmatic management. Background document on the 2019 revision. [Google Scholar]
- 10.Heifets LB, Lindholm-Levy PJ, Flory MA. Bactericidal activity in vitro of various rifamycins against Mycobacterium avium and Mycobacterium tuberculosis. Am Rev Respir Dis. 1990;141:626–630. doi: 10.1164/ajrccm/141.3.626. [DOI] [PubMed] [Google Scholar]
- 11.Sirgel FA, Fourie PB, Donald PR, Padayatchi N, Rustomjee R, Levin J, et al. The early bactericidal activities of rifampin and rifapentine in pulmonary tuberculosis. Am J Respir Crit Care Med. 2005;172:128–135. doi: 10.1164/rccm.200411-1557OC. [DOI] [PubMed] [Google Scholar]
- 12.Jindani A, Harrison TS, Nunn AJ, Phillips PP, Churchyard GJ, Charalambous S, et al. RIFAQUIN Trial Team. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 2014;371:1599–1608. doi: 10.1056/NEJMoa1314210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langdon G, Wilkins J, McFadyen L, McIlleron H, Smith P, Simonsson US. Population pharmacokinetics of rifapentine and its primary desacetyl metabolite in South African tuberculosis patients. Antimicrob Agents Chemother. 2005;49:4429–4436. doi: 10.1128/AAC.49.11.4429-4436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savic RM, Lu Y, Bliven-Sizemore E, Weiner M, Nuermberger E, Burman W, et al. Population pharmacokinetics of rifapentine and desacetyl rifapentine in healthy volunteers: nonlinearities in clearance and bioavailability. Antimicrob Agents Chemother. 2014;58:3035–3042. doi: 10.1128/AAC.01918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savic RM, Weiner M, MacKenzie WR, Engle M, Whitworth WC, Johnson JL, et al. Tuberculosis Trials Consortium of the Centers for Disease Control and Prevention. Defining the optimal dose of rifapentine for pulmonary tuberculosis: exposure-response relations from two phase II clinical trials. Clin Pharmacol Ther. 2017;102:321–331. doi: 10.1002/cpt.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francis J, Zvada SP, Denti P, Hatherill M, Charalambous S, Mungofa S, et al. A population pharmacokinetic analysis shows that arylacetamide deacetylase (AADAC) gene polymorphism and HIV infection affect the exposure of rifapentine. Antimicrob Agents Chemother. 2019;63:e01964-18. doi: 10.1128/AAC.01964-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zvada SP, Van Der Walt JS, Smith PJ, Fourie PB, Roscigno G, Mitchison D, et al. Effects of four different meal types on the population pharmacokinetics of single-dose rifapentine in healthy male volunteers. Antimicrob Agents Chemother. 2010;54:3390–3394. doi: 10.1128/AAC.00345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keung AC, Owens RC, Jr, Eller MG, Weir SJ, Nicolau DP, Nightingale CH. Pharmacokinetics of rifapentine in subjects seropositive for the human immunodeficiency virus: a phase I study. Antimicrob Agents Chemother. 1999;43:1230–1233. doi: 10.1128/aac.43.5.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dooley K, Flexner C, Hackman J, Peloquin CA, Nuermberger E, Chaisson RE, et al. Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob Agents Chemother. 2008;52:4037–4042. doi: 10.1128/AAC.00554-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dooley KE, Bliven-Sizemore EE, Weiner M, Lu Y, Nuermberger EL, Hubbard WC, et al. Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clin Pharmacol Ther. 2012;91:881–888. doi: 10.1038/clpt.2011.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dooley KE, Savic RM, Park JG, Cramer Y, Hafner R, Hogg E, et al. ACTG A5311 Study Team. Novel dosing strategies increase exposures of the potent antituberculosis drug rifapentine but are poorly tolerated in healthy volunteers. Antimicrob Agents Chemother. 2015;59:3399–3405. doi: 10.1128/AAC.05128-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorman SE, Goldberg S, Stout JE, Muzanyi G, Johnson JL, Weiner M, et al. Tuberculosis Trials Consortium. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J Infect Dis. 2012;206:1030–1040. doi: 10.1093/infdis/jis461. [DOI] [PubMed] [Google Scholar]
- 23.Moher D, Liberati A, Tetzlaff J, Altman DG PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151:264–269, W64. doi: 10.7326/0003-4819-151-4-200908180-00135. [DOI] [PubMed] [Google Scholar]
- 24.Byon W, Smith MK, Chan P, Tortorici MA, Riley S, Dai H, et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometrics Syst Pharmacol. 2013;2:e51. doi: 10.1038/psp.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smythe W, Khandelwal A, Merle C, Rustomjee R, Gninafon M, Bocar Lo M, et al. A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob Agents Chemother. 2012;56:2091–2098. doi: 10.1128/AAC.05792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development-part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2:e38. doi: 10.1038/psp.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alfarisi O, Alghamdi WA, Al-Shaer MH, Dooley KE, Peloquin CA. Rifampin vs. rifapentine: what is the preferred rifamycin for tuberculosis? Expert Rev Clin Pharmacol. 2017;10:1027–1036. doi: 10.1080/17512433.2017.1366311. [DOI] [PubMed] [Google Scholar]
- 28.Dorman SE, Savic RM, Goldberg S, Stout JE, Schluger N, Muzanyi G, et al. Tuberculosis Trials Consortium. Daily rifapentine for treatment of pulmonary tuberculosis: a randomized, dose-ranging trial. Am J Respir Crit Care Med. 2015;191:333–343. doi: 10.1164/rccm.201410-1843OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner M, Bock N, Peloquin CA, Burman WJ, Khan A, Vernon A, et al. Tuberculosis Trials Consortium. Pharmacokinetics of rifapentine at 600, 900, and 1,200 mg during once-weekly tuberculosis therapy. Am J Respir Crit Care Med. 2004;169:1191–1197. doi: 10.1164/rccm.200311-1612OC. [DOI] [PubMed] [Google Scholar]
- 30.Weiner M, Savic RM, Kenzie WR, Wing D, Peloquin CA, Engle M, et al. Tuberculosis Trials Consortium PREVENT TB Pharmacokinetic Group. Rifapentine pharmacokinetics and tolerability in children and adults treated once weekly with rifapentine and isoniazid for latent tuberculosis infection. J Pediatric Infect Dis Soc. 2014;3:132–145. doi: 10.1093/jpids/pit077. [DOI] [PubMed] [Google Scholar]
- 31.Conde MB, Mello FC, Duarte RS, Cavalcante SC, Rolla V, Dalcolmo M, et al. A phase 2 randomized trial of a rifapentine plus moxifloxacin-based regimen for treatment of pulmonary tuberculosis. PLoS One. 2016;11:e0154778. doi: 10.1371/journal.pone.0154778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keung A, Reith K, Eller MG, McKenzie KA, Cheng L, Weir SJ. Enzyme induction observed in healthy volunteers after repeated administration of rifapentine and its lack of effect on steady-state rifapentine pharmacokinetics: part I. Int J Tuberc Lung Dis. 1999;3:426–436. [PubMed] [Google Scholar]
- 33.Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis. 2013;208:1464–1473. doi: 10.1093/infdis/jit352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiner M, Benator D, Burman W, Peloquin CA, Khan A, Vernon A, et al. Tuberculosis Trials Consortium. Association between acquired rifamycin resistance and the pharmacokinetics of rifabutin and isoniazid among patients with HIV and tuberculosis. Clin Infect Dis. 2005;40:1481–1491. doi: 10.1086/429321. [DOI] [PubMed] [Google Scholar]
- 35.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother. 2007;51:3781–3788. doi: 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gengiah TN, Botha JH, Soowamber D, Naidoo K, Abdool Karim SS. Low rifampicin concentrations in tuberculosis patients with HIV infection. J Infect Dev Ctries. 2014;8:987–993. doi: 10.3855/jidc.4696. [DOI] [PubMed] [Google Scholar]
- 37.Gurumurthy P, Ramachandran G, Hemanth Kumar AK, Rajasekaran S, Padmapriyadarsini C, Swaminathan S, et al. Malabsorption of rifampin and isoniazid in HIV-infected patients with and without tuberculosis. Clin Infect Dis. 2004;38:280–283. doi: 10.1086/380795. [DOI] [PubMed] [Google Scholar]
- 38.Jeremiah K, Denti P, Chigutsa E, Faurholt-Jepsen D, PrayGod G, Range N, et al. Nutritional supplementation increases rifampin exposure among tuberculosis patients coinfected with HIV. Antimicrob Agents Chemother. 2014;58:3468–3474. doi: 10.1128/AAC.02307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.