

Abstract
Biological nitrogen fixation is catalyzed by the enzyme nitrogenase, which facilitates the cleavage of the relatively inert triple bond of N2. Nitrogenase is most commonly associated with the molybdenum-iron cofactor called FeMoco or the M-cluster, and it has been the subject of extensive structural and spectroscopic characterization over the past 60 years. In the late 1980’s and early 1990’s, two ‘alternative nitrogenase’ systems were discovered, isolated, and were found to incorporate V or Fe in place of the Mo. These systems are regulated by separate gene clusters, however, there is a high degree of structural and functional similarity between each nitrogenase. Limited studies with the V- and Fe-nitrogenases initially demonstrated that these enzymes were analogously active as the Mo-nitrogenase, but more recent investigations have found capabilities that are unique to the alternative systems. In this review, we will discuss the reactivity, biosynthetic and mechanistic proposals for the alternative nitrogenases as well as their electronic and structural properties in comparison to the well-characterized Mo-dependent system. Studies over the past 10 years have been particularly fruitful, though, key aspects about V- and Fe-nitrogenases remain unexplored.
Graphical Abstract
1. Introduction
Nitrogen fixation occurs in biology through the activity or enzymes known as nitrogenase. The enzyme takes N2 from the atmosphere or other sources, and at ambient temperature and pressure can cleave the strong triple N2 bond to generate two equivalents of ammonia, as seen in a simplified reaction 1:
| (1) |
However, as will be discussed in Section 5, the stoichiometry and mechanism of this enzymatic process is more complex. Nitrogenases play a critical role in the global nitrogen cycle and provide bioavailable nitrogen for organisms to form the fundamental building blocks of life – amino acids and nucleobases. In diazotrophic organisms that can express nitrogenase, there are three different variants: Mo-dependent, V-dependent and Fe-only nitrogenases. These three systems are closely related as they are likely products of genetic duplication and evolutionary divergence as opposed to the simple substitution of the transition metal. The Mo-dependent systems have been studied more extensively than the others, however, the focus of this review will be on the mechanism and reactivity of the Mo-independent systems. Inherently there is less characterization reported for the V- and Fe-only nitrogenases due to the relatively recent isolation, so information will be discussed within the context of what is available for the alternative systems and will not constitute an exhaustive description of Mo-nitrogenase.
2. Genetic Distribution, Regulation and Evolution of Mo-Independent Nitrogenases
Within the last decade, genetic sequencing efforts have expanded the genome data bank, and this has allowed for the identification of an array of novel nitrogen fixing organisms. Bioinformatics, in combination with a better understanding of nitrogenase assembly, was used to identify 149 diazotrophs in 2012 after a comprehensive genome search using a minimum gene set nifHDKENB;1 this number increased to 359 organisms from a similar study conducted in 2019.2 In Archaea, nitrogenase is restricted to the Euryarchaeota phylum whereas in Bacteria, nitrogenase can be found in 13 different phyla; primarily in Proteobacteria, Firmicutes, Cyanobacteria and Bacteroidetes.1–3 The sources of these organisms were shown to be far more diverse than previously understood. Beyond the conventional soil dwellers in agricultural sites,1,3 nitrogen fixing organisms have been found in a variety of environments ranging from marine, coastal and freshwater sediments,4 to waste and sewage stacks,5 the termite midgut6 and in human pathogens.3 The number of organisms identified with putative alternative nitrogenases also increased proportionately with the number of available genomes,7,8 indicating a more widespread taxonomic distribution of alternative nitrogenases than considered previously. One study by Morel and co-workers estimated that alternative nitrogenases constitute 14–21% of the total nitrogenase activity, and are major participants of diazotrophic activity in almost a quarter of environments where biological nitrogen fixation is detected.3
The increasing prevalence of alternative nitrogenases identified in genomic databases highlights a potential for ecological importance and a role in the global nitrogen cycle, which raises the question of how alternative nitrogenase expression may be regulated. Most organisms that fix N2 only encode for Mo-nitrogenase (nif), but organisms that encode the alternative nitrogenases (vnf and/or anf) also encode for Mo-nitrogenase.1,9–12 Several prokaryotes, namely Azotobacter vinelandii, Methanosarcina acetivorans, and Rhodopseudomonas palustris, have the genes for all three nitrogenases,13 however, some diazotrophs only encode Mo- and Fe-only nitrogenases (Rhodobacter species),14 while others only encode Mo- and V-nitrogenases (Anabaena species).15 In A. vinelandii, the putative regulatory pathway involves the master regulator genes nifA, vnfA and anfA, which are responsible for the expression of Mo-, V- and Fe-nitrogenase encoding genes, respectively.16 In the presence of Mo, the gene product NifA turns on the expression of the nif-encoding genes while vnfA and anfA are both transcriptionally repressed. In the presence of V and absence of Mo, VnfA is expressed and it transcriptionally suppresses the nif- and anf- encoding nitrogenase genes. Lastly, in the absence of both Mo and V, the anfA gene is activated to turn on the synthesis of Fe-nitrogenase.17 There is certain degree of overlap between these nitrogenase systems given that neither the vnf nor the anf gene clusters contain the entire set of necessary biosynthetic genes. For one example, in A. vinelandii the biosynthesis of all nitrogenases require the action of NifU, NifS and NifB.18 Interestingly, transcriptome analysis also revealed that many of the genes required for V-nitrogenase assembly are also turned on when the organism was expressing Fe-nitrogenase.18 The NifA-mediated regulation framework seems to be employed by other diazotrophic proteobacteria, albeit with some modifications. For instance, in Rhodobacter capsulatus, NifA is believed to be necessary for the synthesis of Fe-only nitrogenase, although this is facilitated through a NifA-dependent factor known as RpoN.19 Alternatively, there are instances where the nifA gene does not appear have any encoded homologs, with many examples found in diazotrophic cyanobacteria. In one example, in Anabaena variabilis, nitrogenase expression is controlled by an interplay of a number of environmental factors, and ultimately through regulation of the nifB promoter.20 More recent evidence has revealed a more complex picture of alternative nitrogenase usage. Sequencing results taken from lichen and termite hindgut samples indicated a small or inconsistent correlation between alternative nitrogenase expression with trace metal availability.6,21,22 Even in the well-established system of A. vinelandii, it is known that “hybrid” nitrogenases, such as the V-dependent protein containing the Mo-dependent cofactor, can be produced under specific growth conditions (see Section 4.6).23 Thus, there are still many questions that remain concerning the gene regulation of alternative nitrogenase, and addressing them will become increasingly important for the future.
Due to improvements in sequencing techniques and the widespread availability of genetic information, the potential evolution of nitrogenase has been reassessed. Based on the availability of fixed nitrogen, Fe, and Mo in the ancient oceans, an initial proposal posited that the alternative nitrogenases predated Mo-nitrogenase.10,24 However, recent phylogenetic studies point to the existence of a proto-nitrogenase species that was a common ancestor of all three known nitrogenases, and it was suggested to contain a precursor cofactor that lacked Mo.2,12,25 Peters, Boyd and co-workers have argued that the structural genes of the modern Mo-nitrogenase, namely nifHDK, were the first to evolve from the proto-nitrogenase genes, potentially through gene duplication of nifD and differentiation events.17,25 Subsequently, it was proposed that the nifDK genes might have been duplicated in order to give rise to cofactor biosynthetic genes nifEN, thereby allowing for the development of a fully functional Mo-nitrogenase.2 Based on a concatenated phylogenetic lineage of the nifHDK genes, it was further suggested that the structural genes of V-nitrogenase, i.e. vnfHDGK, were derived from nifHDK while structural genes of Fe-only-nitrogenase, anfHDGK, were subsequently evolved from the vnf genes.25 This notion is consistent with the fact that the assembly of the V- and Fe-only nitrogenase cofactors rely on nif-encoded genes, particularly nifUS and nifB as well as nifEN for organisms other than A. vinelandii and R. palustris which instead encode for vnfEN.11,26 This proto-nitrogenase proposal was also compared with studies of ancient sedimentary rock from marine and fluvial sources that investigated the nitrogen isotope ratios of the encapsulated biomass from that time period.27,28 Nitrogenases are known to produce fixed nitrogen that disfavors the heavier 15N isotope relative to the natural isotopic abundance of N2. This can represented by the nitrogen fractionation where (δ15N = [(15N/14N)sample/ (15N/14N)standard] − 1), and Mo-nitrogenase can show δ15N values between −1% to −4% whereas the alternative nitrogenases favor 14N products more strongly with δ15N values between −6% and −8%.27–29 The sediments tested were between 3.2 and 2.75 billion years old, and in that time frame, the observed fractionation was δ15N ~ 0%, which is more consistent with Mo-nitrogenase than the Mo-independent variants.28 Compared to the phylogenetic analysis, the isotope experiments support the notion that Mo-nitrogenase predates the alternative systems. However, the isotope fractionation of a proto-nitrogenase cannot be accurately known, but it has been suggested that such a nitrogenase ancestor would be promiscuous, inefficient, and likely unable to discriminate against the heavy nitrogen isotope.17 Additionally, there is no clear time point of when such a proto-nitrogenase would have started to incorporate Mo. A detailed picture for the evolution of nitrogenase proteins has begun to emerge, but it will be interesting to see how this changes as further investigation unveils new nitrogen fixing species and probes the biochemistry the nitrogenase proteins from these sources.
2.1. Assembly of Mo- and V-Nitrogenases
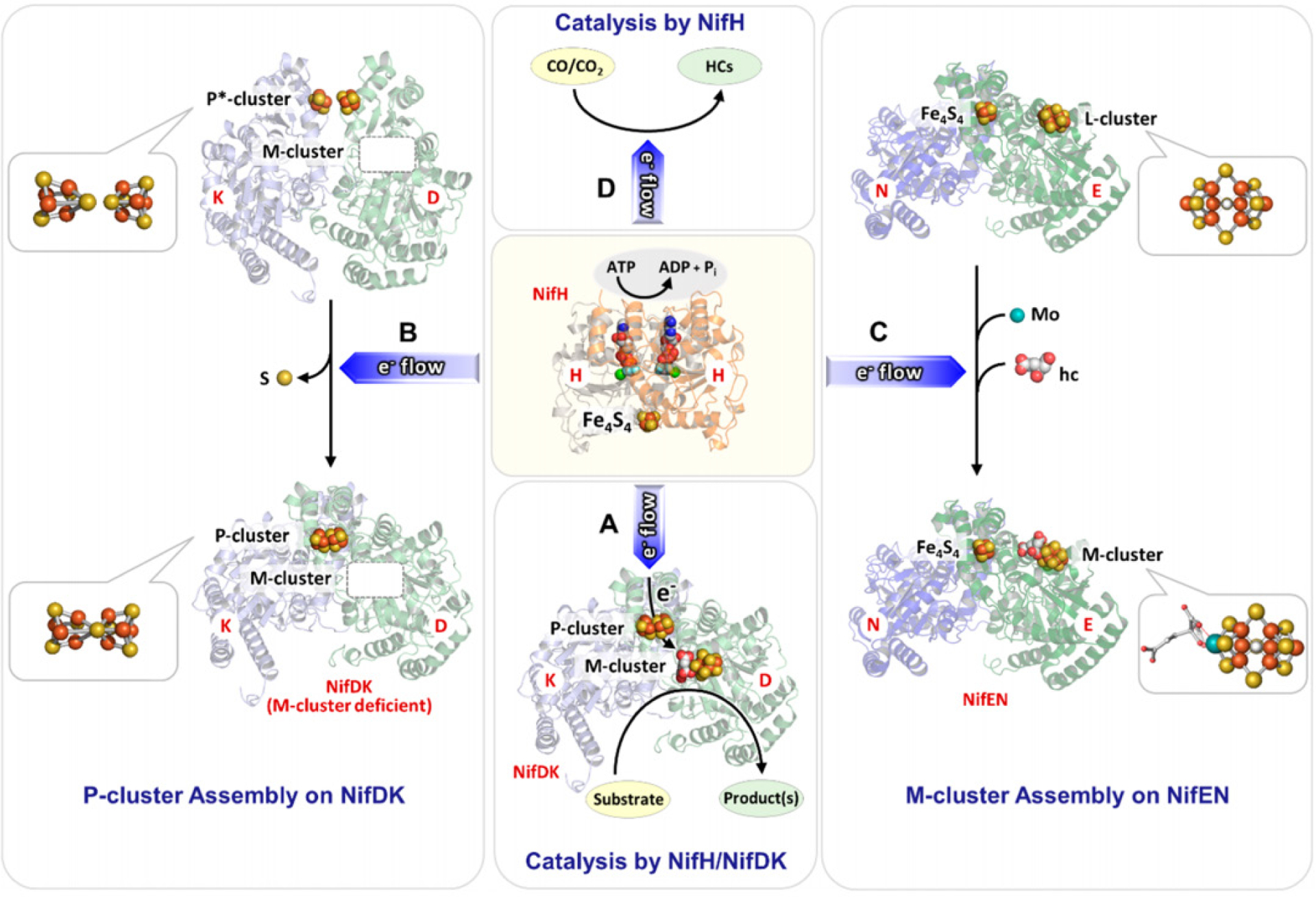
Extensive characterization efforts in the past two decades have provided valuable insight into the biochemistry and assembly of the catalytic component of Mo-nitrogenase, called the MoFe protein or NifDK. NifDK is a heterotetrameric protein that contains two cofactors – the [Fe8S7] P-cluster and the [MoFe7S9C-(R)-homocitrate] M-cluster (also called FeMoco). For A. vinelandii, it is understood that the synthesis of the P- and M-clusters of NifDK require a minimum set of proteins that include NifU, NifS, NifB, NifEN, NifV and NifZ, in addition to the structural gene products NifH and NifDK.30 The functions of these nif gene products have been studied in great detail and are briefly summarized in Table 1. Overall, the P-cluster of the MoFe protein was determined to be synthesized in situ, i.e. on the NifDK polypeptide scaffold, and the M-cluster was synthesized ex situ, i.e. outside of the NifDK protein.30–32 In the former case, a biosynthetic precursor of the P-cluster, denoted as the P*-cluster, was identified as a pair of [Fe4S4]-like clusters. The conversion of this P*-cluster to the fully matured P-cluster was described as the fusion of the two [Fe4S4] clusters, with the loss of a sulfur atom. The process also involves the action of NifZ and NifH in an ATP- and electron-dependent manner (Figure 1).33 The catalytic component of V-nitrogenase, called VFe protein or VnfDGK, has a similar composition as the Mo-dependent system, with a P-cluster and a V-containing analog of the M-cluster called the V-cluster. Interestingly, a significant degree of similarity was observed between the P*-cluster in the pre-matured MoFe protein and the matured P-cluster in V-nitrogenase based upon electron paramagnetic resonance (EPR) and X-ray absorption spectroscopic (XAS) analyses (see Section 3).34 The synthesis of the M-cluster involves a series of transformative steps, starting from a pair of [Fe4S4] clusters (denoted as the K-cluster). The K-cluster is then converted into a [Fe8S9C] core (denoted as the L-cluster), and finally Mo and R-homocitrate are incorporated to yield the matured M-cluster (Figure 1).30–33,35–37 It has been suggested that the M- and V-clusters share the same biosynthetic pathway until the formation of the L-cluster, and after this point the pathways diverge as either Mo or V is inserted along with the organic ligand R-homocitrate.31,32
Table 1.
Description of Relevant Gene Products from the nif, vnf and anf Operonsa
| gene | gene product label | known function |
|---|---|---|
| nifHb | NifH, dinitrogenase reductase, component 2, reductase component, γ subunit, Fe protein | Mediation of ATP-dependent electron transfer during catalytic turnover, facilitates formation of P-cluster on NifDK, facilitates conversion of L- to M-cluster on NifEN via insertion of Mo and R-homocitrate |
| nifD,Kb | NifDK, dinitrogenase, component 1, catalytic component, MoFe protein | Facilitates chemical transformation of N2 to NH3 at the active cofactor site |
| nifL | - | serves as a negative transcription regulator of nif genes |
| nifA | - | serves as a positive transcription regulator of nif genes |
| nifF | NifF, flavodoxin | involved in the transfer of electrons to nitrogenase during catalysis and/or assembly |
| nifS | NifS, cysteine desulfurase | transfers sulfur to NifU for the assembly of small FeS clusters |
| nifU | NifU | serves as a scaffold for the assembly of small FeS clusters, which can then be used for M- and P-cluster assembly |
| nifBb | NifB | mediates the radical SAM-dependent insertion of carbon concomitant with the formation of a Mo/homocitrate-free precursor of the M-cluster |
| nifE,Nb | NifEN | serves as a scaffold for the maturation of the M-cluster; a structural/functional homolog of NifDK |
| nifV | NifV, homocitrate synthase | synthesizes R-homocitrate for M-cluster assembly |
| nifZ | NifZ | serves as a key factor in the stepwise maturation of P-clusters in NifDK, possibly through a chaperone-like function |
| nifM | NifM, peptidyl-proyl cis-trans isomerase | involved in the maturation of Fe protein |
| nifX | NifX | proposed intermediate carrier in M-cluster assembly |
| nifY | NifY | proposed intermediate carrier in M-cluster assembly |
| nafY | NafY | proposed intermediate carrier in M-cluster assembly |
| nifW | NifW | putative role in protecting nitrogenase from oxygen damage |
| nifO | NifO | function unknown, although it resembles thioredoxin |
| nifT | NifT | Unknown function |
| vnfH | VnfH, dinitrogenase reductase, component 2, reductase component, γ subunit, Fe protein | Mediation of ATP-dependent electron transfer during catalytic turnover, likely participates in biosynthesis of VnfDGK cofactors by analogy to nif system but this has not been shown |
| vnfD,G,K | VnfDGK, dinitrogenase, component 1, catalytic component, VFe protein | Facilitates chemical transformation of N2 to NH3 and CO/CO2 into hydrocarbons at the active cofactor site |
| vnfE,N | VnfEN | Putative involvement in V-cluster biosynthesis by analogy to NifEN |
| vnfA | - | serves as a positive transcription regulator of vnf genes, also correlated to expression of anf genes |
| vnfX | VnfX | Unknown function |
| vnfY | VnfY | Unknown function |
| vnfO | VnfO | Unknown function |
| vnfU | VnfU | Unknown function |
| anfH | AnfH, dinitrogenase reductase, component 2, reductase component, γ subunit, Fe protein | Mediation of ATP-dependent electron transfer during catalytic turnover, may participate in biosynthesis of AnfDGK cofactors but has not been shown |
| anfD,G,K | AnfDGK, dinitrogenase, component 1, catalytic component FeFe protein | Facilitates chemical transformation of N2 to NH3 at the active cofactor site |
| anfA | - | serves as a positive transcription regulator of anf genes |
| anfO | AnfO | Unknown function |
| anfR | AnfR | Unknown function |
| anfU | AnfU | Unknown function |
Figure 1.
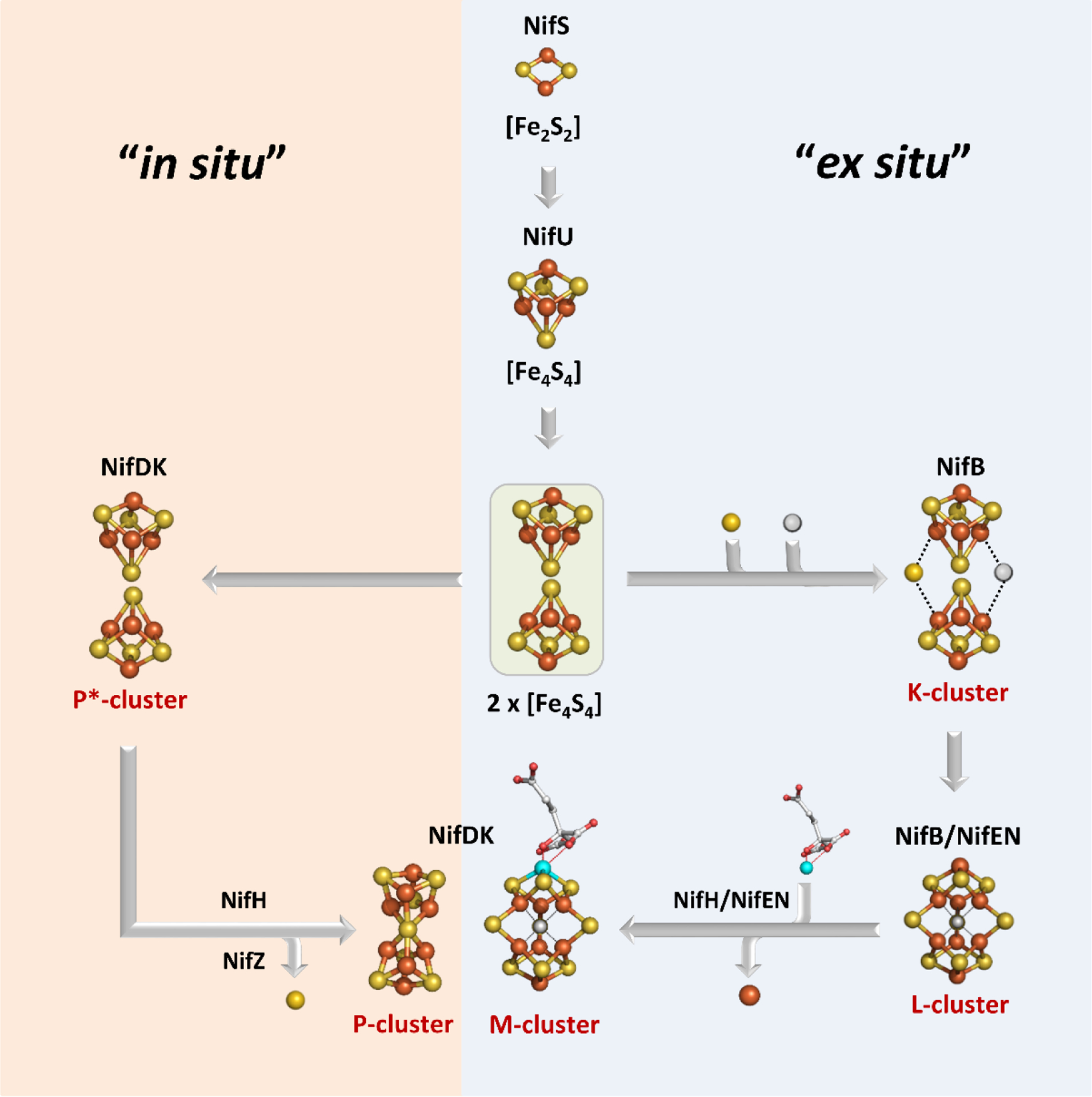
Flow diagram for the assembly of nitrogenase cofactors. The orange “in situ” pathway represents the assembly of the P-cluster on NifDK, whereas the blue “ex situ” block shows the assembly of the M-cluster. Assembly proteins are labeled in black; cluster species are labeled in red. Atoms are represented as ball-and-stick models and colored as follows: Fe, orange; S, yellow; C, grey; O, red; Mo, teal. Adapted with permission from ref 32. Copyright 2014 American Chemical Society.
While the topic of Mo-nitrogenase assembly has been covered in a number of reviews, there is currently little understanding of the assembly process for the alternative nitrogenases aside from indirect association to what is known about the Mo-dependent system. In light of this, a brief synopsis will be outlined in this portion of the review with a focus on the biosynthesis of the M-, V- and Fe-clusters. The intention is to emphasize the potential commonalities shared by the biosynthetic pathways of these analogous nitrogenase cofactors.
The biosynthesis of the M-cluster starts with the assembly of [Fe4S4] units, composed of mobilized iron and sulfur (Figure 1).38,39 In A. vinelandii the NifU and NifS gene products accomplish this task,18 however, not all diazotrophs encode nifUS but alternative systems can be used. In the genome of Paenibacillus sp. WLY78, there is no encoded nifUS, but several genes exist that can facilitate the same function, including a complete suf (sufCBSUD) operon, and partial suf (sufABC) and isc (iscR and fdx) systems.17,40 Additionally, it has been demonstrated that plasmids carrying the iscSUA and hscABfdx genes that encode for [Fe4S4] cluster assembly proteins can be used to heterologously express structural nitrogenase proteins from methanogenic organisms in Escherichia coli.41 These FeS biosynthetic building blocks are then loaded onto NifB, the radical S-adenosyl-L-methionine (SAM)-dependent enzyme that serves as the site where the precursor 8Fe core (L-cluster) is generated. On NifB, two types of FeS clusters were found as predicted by sequence analysis.42 The first is a [Fe4S4] cluster that is bound to a radical SAM-motif, referred to as the SAM cluster,43 and the second type is actually a pair of [Fe4S4]-like clusters, known as the K-cluster,43 that serves as building blocks for the L-cluster (Figure 1). When the NifB protein is reduced using dithionite (S2O42−), these two sets of metal clusters give rise to a composite EPR signal with g-values at 2.02, 1.95, and 1.90.43 Upon addition of SAM, this signal is greatly diminished, and is accompanied by the appearance of a signal at g = 1.94 associated with the L-cluster.44,45 This observation pointed to a mechanism where the two [Fe4S4] components of the K-cluster fused into the 8Fe L-cluster through the action of radical SAM cleavage. Indeed, radiolabel tracing has provided strong evidence for this proposal, as a 14C-labeled L-cluster can be generated through the addition of [methyl-14C] SAM to NifB.46,47 The carbon labeling along with the detection of the reaction product S-adenosyl-L-homocysteine (SAH), was strongly indicative of a SN2-type methyl transfer reaction. This is a common reactivity among SAM enzymes,48 and implied that the SAM cluster transferred carbon to the substrate K-cluster, which gave rise to the central carbide of L-cluster. The reaction of SAM with NifB (Figure 2) also yielded 5’-deoxyadensine (5’dA), indicating involvement of a 5’-deoxyadensyl radical (5’dA•). Deuterium labeling demonstrated that the radical species was involved in the hydrogen abstraction of the methyl group that was transferred to the K-cluster.46 Furthermore, methanethiol (CH3SH) was also detected upon acid quenching of the reaction, demonstrating that the SAM-derived methyl group is transferred to an acid-labile sulfur atom instead of an iron atom associated with the K-cluster.49 More recently, the site-directed mutagenesis of NifB has led to the identification of the so-called K2 cluster, one 4Fe half of K-cluster that is coordinated by Cys264, Cys274 and Cys277, as being the site of methyl transfer.50 The other half of K-cluster, designated as the K1 cluster, is coordinated by Cys30, Cys63 and Cys129, and was studied using advanced pulse EPR techniques and a NifB variant carrying only the K1 cluster.50 It was demonstrated that a histidine ligand from the protein might be coordinating to the K1 module and this coordination was lost upon the conversion of K- to L cluster.50 Further analysis of NifB is currently hindered by a lack of a reported crystal structure.
Figure 2.
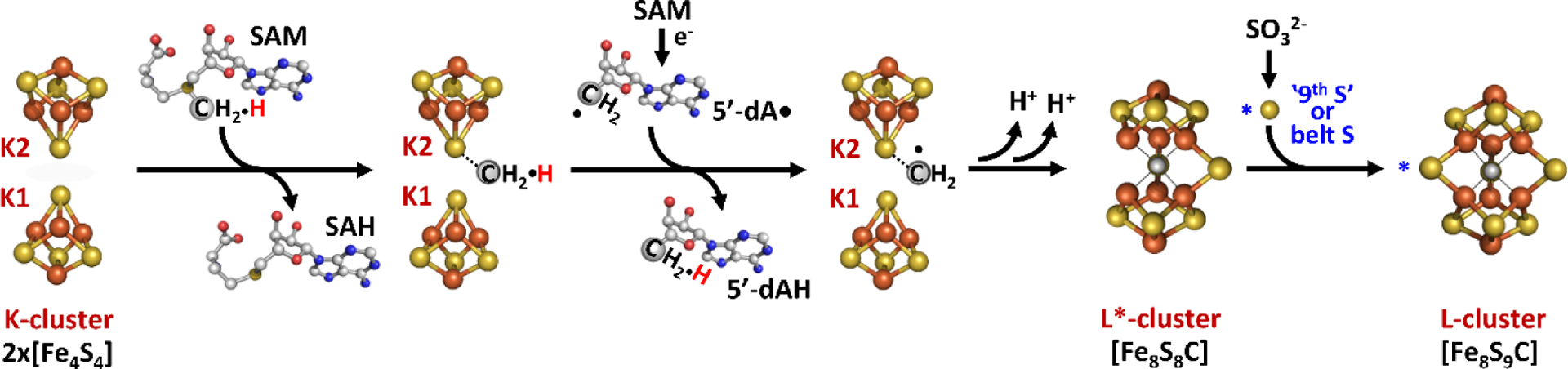
Proposed model for L-cluster assembly on NifB. Cluster species are represented as ball-and-stick models with N atoms as colored in blue and the other atoms colored as described in Figure 1. Adapted with permission from ref 51. Copyright 2018 Springer Nature.
Taken together, a mechanistic model emerges where the conversion of the K-cluster begins with the SN2-methyl transfer reaction from the first molecule of SAM to the K2 cluster, followed by hydrogen atom abstraction of the K2-bound methyl group by 5’dA• to generate a substrate bound methylene radical (Figure 2). This would then trigger a core rearrangement, possibly facilitated by the His ligand on K1, and the two K-cluster modules would fuse to form an [Fe8S8C] core. Biochemical and spectroscopic characterization of this core, designated the L*-cluster, suggested that it might have very similar topology compared to the L-cluster, with the so-called “9th-sulfur” atom missing.51 XAS analysis detected a slightly more open conformation of L*-cluster compared to L-cluster, and collectively, this was interpreted to mean that the missing 9th sulfur atom on L*-cluster was from one of the three ‘belt’ μ2-sulfide positions.52 Surprisingly, the source of the 9th-sulfur atom is not derived from SAM or NifS as previously understood,53,54 but rather from sulfite (SO32−) in solution.51 35S tracing experiments conclusively showed the sulfur incorporation into L-cluster from sulfite by incubating 35SO32− with NifB and SAM.51 Sulfite is one of the three central hubs of prokaryotic sulfur metabolism,55 the other two being sulfate (SO42−) and sulfide (S2−), and this intriguing finding offers a potential link between nitrogenase cofactor biosynthesis and cellular sulfur metabolism. Another interesting connection can also be drawn to the potential involvement of a bridging μ2 sulfur atom in catalysis. As observed in Mo- and V-nitrogenase, the S2B atom that bridges between Fe2 and Fe6 in M- and V-clusters was displaced by incoming inhibitor or substrate-like ligand (See Section 3 and 5). The mechanism of this process remains unclear, but it could bear similarities with the sulfur incorporation process observed for the L*-cluster.
In Mo-nitrogenase, the L-cluster is transferred from NifB to NifEN after its formation. NifE and NifN are paralogs of NifD and NifK, and as mentioned above, NifEN is a scaffold necessary for nitrogenase assembly. On NifEN, maturation of the L- to M-cluster occurs through the participation of NifH, which mobilizes both molybdate (MoO42−) and R-homocitrate, subsequently interacting with NifEN in order to insert these components into the L-cluster.56,57 This process in vitro requires the hydrolysis of ATP and a high concentration of dithionite as reductant.57 The role of NifH as Mo/homocitrate insertase was suggested by Mo K-edge XAS analysis, which pointed to a change in the oxidation state and the coordination environment of the NifH-bound Mo.58 EPR analysis also revealed perturbations to the [Fe4S4] cluster of NifH as Mo/homocitrate were loaded onto the protein.58 Additionally, Mo/homocitrate-loaded NifH could be separately purified and then reused as a reagent for the conversion of L- to M-cluster. After Mo and homocitrate insertion, NifEN undergoes a conformational rearrangement in which the newly matured M-cluster becomes less solvent exposed.59 In vitro experiments demonstrated that the M-cluster containing NifEN can then form a direct protein-protein complex together with apo-NifDK (i.e. M-cluster deficient yet P-cluster intact NifDK).60 In this complex, the M-cluster is directly transferred and inserted into the NifDK protein through a positively charged funnel of amino acid residues that also allows for the facile reconstitution of apo-NifDK with isolated M-cluster (or V-cluster, see Section 4).61 The ferrying of the M-cluster between NifEN and NifDK is made possible because NifEN lacks a number of key residues that either provide a covalent ligand for the cluster or sterically enclose the M-cluster within a positively charged insertion channel, analogous to that found in NifDK.33,36,62
In the V-nitrogenase system from A. vinelandii, cofactor assembly diverges from the Mo-dependent pathway as the NifB-bound L-cluster is likely transferred onto VnfEN instead of NifEN (Figure 3). Based on the high sequence homology between NifEN and VnfEN due to a gene duplication event, as well as the homology between NifH and VnfH, a similar mechanism of L-cluster maturation could be proposed in the alternative nitrogenase systems to make V- and Fe-cluster on VnfEN. This is substantiated by the fact that the residues ligating to the L- and M-clusters on NifEN are all conserved in VnfEN.31 Moreover, the expression of nifV, coded for homocitrate synthetase, has been observed when A. vinelandii cultures were grown under V- and Fe-nitrogenase expressing conditions, lending further support for this proposal.18,38 In addition, it has been shown that attempts to insert V and R-homocitrate into the L-cluster using NifEN and NifH have led to a cluster species with low V occupancy and limited ability to reconstitute and activate apo-NifDK.63 This observation emphasizes the optimization of NifEN/NifH and VnfEN/VnfH for the insertion of Mo and V atoms, respectively. In contrast, the combination of VnfEN/AnfH appears to be responsible for the in vivo biosynthesis of Fe cluster. In A. vinelandii deletion of the vnfEN gene was previously shown to eliminate the synthesis of functional V- and Fe-nitrogenases,64 and transcriptome analysis also demonstrated that the vnfEN gene is highly overexpressed (as much as 50-fold from baseline) under nitrogen fixing conditions in the absence of Mo-nitrogenase.18 By analogy to the Mo-nitrogenase system, V- or Fe-cluster could be delivered directly from VnfEN to its target via protein-protein interactions (Figure 3). If this hypothesis holds true, it would also imply that VnfEN might be able to dock to both VnfDGK and AnfDGK to deliver the V- and Fe-clusters respectively. This is conceivable, considering the homology between NifEN and VnfEN, but also between NifDK, VnfDKG and AnfDKG.38 Significantly, a number of residues that have been suggested to be important for the M-cluster insertion in NifD are either observed or substituted to similar residues in VnfD as well as in AnfD.38 This provides a rudimentary framework for future investigations of the alternative nitrogenase assembly.
Figure 3.
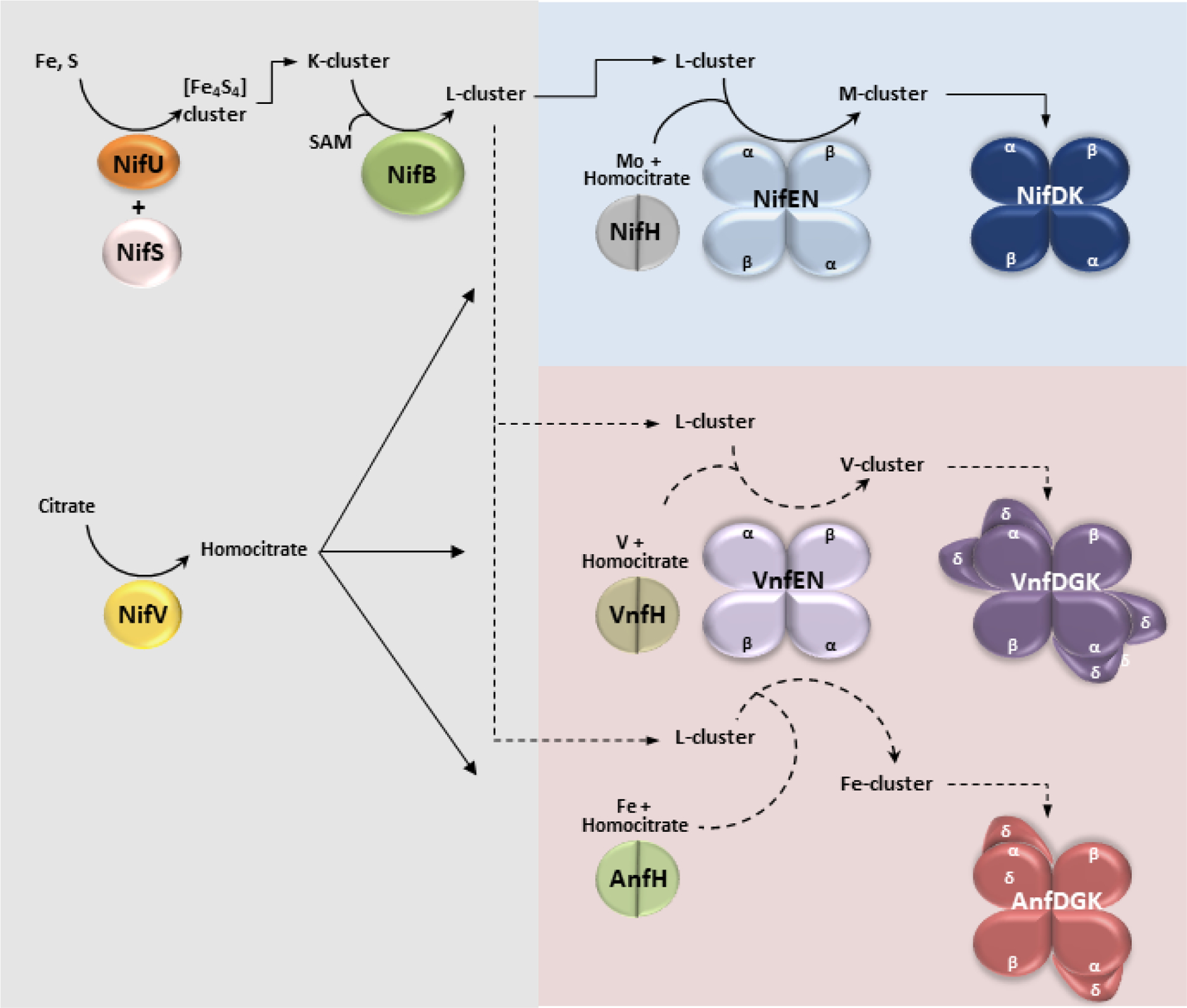
Proposed pathways for M-, V- and Fe-cluster biosynthesis. Area shaded in gray represents the common portion of the pathway, including the proteins NifU, NifS, NifV and NifB. Area shaded in blue represents the M-cluster specific maturation pathway, and the red shaded area depicts the V- and Fe-cluster specific maturation pathways. Experimentally verified steps are indicated by solid arrows whereas putative assignments are indicated by dotted arrows. See the main text for detailed descriptions.
3. Structure and Properties of Nitrogenase Proteins
A key to understanding nitrogenase has come from the structural and spectroscopic analysis of the two protein components and the transition metal cofactors housed therein.65,66 In 1992, Rees and co-workers solved the crystal structures of both the catalytic (NifDK)67,68 and reductase (NifH) components69 of the Mo-dependent nitrogenase from A. vinelandii. These structures, along with the structure of the NifH:NifDK complex70,71 provided necessary three-dimensional depictions of the enzyme in various states. This allowed for the contextualization of much of the spectroscopic and biochemical analysis that had been accumulated to that point. While the extent of this characterization for the Mo-dependent system is vast, relatively little is available for the Mo-independent nitrogenases in comparison. Initial structural information came in 1987 and 1988 from XAS analysis of the VFe proteins from A. vinelandii and Azotobacter chroococcum using the unique V atom as a spectroscopic probe,72,73 but a crystal structure would not be solved until 2017 when Sippel and Einsle reported the structure of the Av VFe protein.74 The following year, the structure of the Fe protein from the V-dependent system, VnfH, was also reported by Einsle and co-workers.75 Crystal structures from the Fe-only nitrogenase have not yet been reported, though in 2002, Fe K-edge XAS and 57Fe Mössbauer spectroscopy were used to analyze the cofactors of the FeFe protein from R. capsulatus, and this study suggested there is structural homology to the Mo- and V-nitrogenases.76
In general, the proteins from the different nitrogenase systems are broadly similar and share common structural features. For the reductase proteins, each contain a [Fe4S4]-cluster and nucleotide binding sites that facilitate the obligate electron transfer coupled to ATP hydrolysis during catalysis.77 On the other hand, the catalytic proteins contain two different cluster species; one is the [MFe7S9C-(R)-homocitrate] (M = Mo, V, Fe) cluster called the Mo-, V-, or Fe-cluster for the respective nitrogenase system and is the active site of substrate reduction, whereas the other cluster is the [Fe8S7] P-cluster, responsible for the transfer of electrons between the reductase and the active site.65,66 Each of these clusters also have spectroscopic properties that have been used to generate structural models prior to the availability of crystallographic evidence. Additionally, the V- and Fe-only nitrogenases also contain an additional cofactorless protein subunit that Mo-nitrogenase lacks. This section will cover the structural similarities and differences between the nitrogenase systems, and as the focus of this review is on the alternative nitrogenases, details will be briefly summarized for the Mo-dependent variant.
3.1. The Characterization of the Reductase Components – NifH, VnfH and AnfH
The reductase proteins of nitrogenase, also referred to as the Fe protein, component 2, or by the gene product (NifH, VnfH, AnfH for Mo-, V-, and Fe-only nitrogenases, respectively), are critical for both substrate turnover and for the biosynthesis of the clusters (Figure 4).77 The best characterized protein, Av NifH, has been recognized for three primary functions: (1) Mo and homocitrate insertase for the maturation of the precursor L-cluster to the M-cluster;58 (2) reductase that carries out P-cluster synthesis on the catalytic NifDK protein;34,79–82 (3) obligate electron transfer partner to NifDK for substrate turnover with concomitant hydrolysis of two ATP molecules per electron.83 Additionally, NifH and VnfH were also found to be capable of the interconversion of CO2 to CO, a reactivity that will be discussed in Section 4.7. While the Fe proteins all have been shown to behave as reductases for catalysis as in function 3, functions 1 and 2 for VnfH and AnfH have yet to be demonstrated experimentally. However, there is a high degree of structural similarity between the Fe proteins despite differences in sequence. Although, VnfH is more similar to NifH than is AnfH (91% and 61% homology, respectively),35 so there is an underlying assumption that each protein will function analogously. This allows for the cautious application of insights gained from one system (Mo-dependent) to the others.
Figure 4.
An overview of the functions of the Fe protein, as exemplified by NifH. Atoms are represented by ball-and-stick models with coloration as described in Figure 2. Reproduced with permission from ref 78. Copyright 2019 American Chemical Society.
3.1.1. Crystal Structures of NifH and VnfH
The Mo-nitrogenase reductase component, NifH, is encoded by the nifH gene and forms a homodimer (γ2) approximately 60 kDa in size (Figure 5A).69 The protein contains a [Fe4S4] cluster positioned on a 2-fold rotation axis at the subunit interface, bound by the Cys97 and Cys132 residues from each subunit. The reductase from V-nitrogenase, VnfH, is similarly sized (~60 kDa) and analogously structured, with Cys98 and Cys133 from each monomer providing a binding site for the [Fe4S4] cluster (Figure 5B).75 NifH and VnfH also bind nucleotides, one molecule to each subunit, in a Walker’s motif A protein fold,84 found between residues 9 and 16 (or residues 11 to 17 in VnfH).69,75 In the 1992 structure of NifH, an ADP molecule was modeled in with partial occupancy in this position, though subsequent reports yielded a structure with MgADP bound to Av NifH,85 as well as a structure with MgATP bound to a Av NifH variant that is unable to hydrolyze ATP.86 In contrast, the crystal structure of VnfH has only been reported with MgADP bound, and this overlays well with the analogous MgADP-bound NifH structure.75 The structures of the nucleotide-bound or free NifH proteins are rather similar, and only show minor conformational changes between the nucleotide-bound and unbound states.77 This finding initially conflicted with earlier biochemical and spectroscopic studies that implied there should be changes to the protein and/or [Fe4S4] cluster upon addition of nucleotide. The EPR spectra of Av NifH (and Av VnfH) become broadened in the presence of nucleotide,66,87 indicating a connection between the [Fe4S4] cluster and the coordination of the nucleotide ~20 Å away from the cluster. Chelation agents such has bathophenathrolinedisulfonate or 2,2’-bipyridine are limited or unable to extract Fe from the [Fe4S4] cluster of NifH, but in the presence of MgATP the Fe is rapidly removed.88–90 This indicated that in the presence of nucleotide, the cluster becomes more solvent exposed. Additionally, structural perturbations were measured for NifH using small angle X-ray scattering (SAXS) which showed an observable change to the protein conformation in the presence of MgATP compared to the MgADP-bound or nucleotide-free species.91 Collectively, these observations are consistent with a more dynamic structure of NifH (and by analogy, VnfH and AnfH) that can selectively expose the [Fe4S4] cluster to solvent in the presence of nucleotide.
Figure 5.
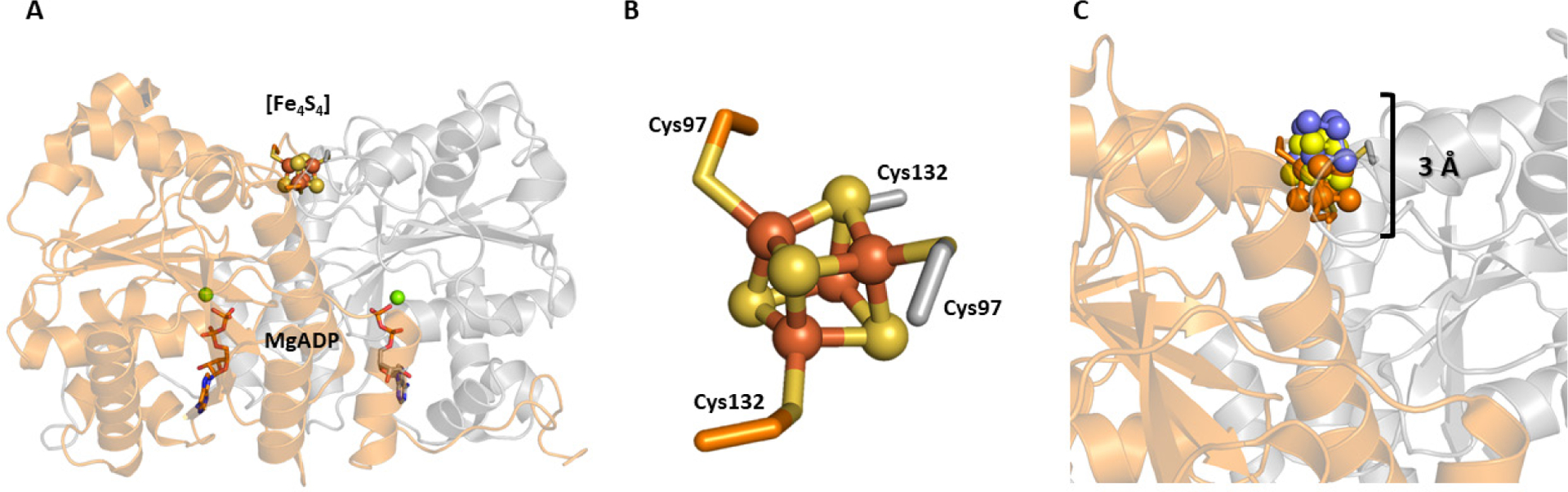
Crystal structure of NifH and the effect of nucleotide binding on [Fe4S4] position. A: Crystal structure of the MgADP-bound Av NifH (PDB ID 1FP6) with each subunit of the homodimer (γ2) colored differently (orange and grey). B: [Fe4S4] cluster of NifH. C: Overlay of [Fe4S4] clusters of the nucleotide bound (ATP analog in blue, ADP in yellow) and nucleotide free forms of NifH.
In 199770 and later in 200571 structures of the NifH:NifDK complex were reported that helped clarify the discrepancy between the solution state and solid state characterizations of NifH. The two nitrogenase components form a 2:1 complex, with one NifH unit binding to each αβ heterodimer of NifDK. Additionally, this complex had been crystallized with nucleotide-free, MgADP-bound and non-hydrolysable MgATP analog-bound (MgADP•AlF4 and MgAMPPCP) NifH proteins. If the Fe protein portions of the crystal structures are compared and overlaid (Figure 5C), the overall structures of the polypeptide are rather similar, but there is a ~3 Å displacement of the cluster between the nucleotide-bound and nucleotide-free states of the protein, with the MgATP analog-bound variant being the most exposed.71,77 This observation is in agreement with the solution studies, and likely reflects the structural changes that may be operative in the solution state. Unfortunately, structures of the VnfH:VnfDGK complex or of any Fe-only nitrogenase proteins are not yet available, but these species would likely be structurally similar to the partners in the Mo-dependent system.
3.1.2. Solution and Spectroscopic Properties of Fe Proteins
The [Fe4S4] cluster of the Fe protein can support three oxidations states, [Fe4S4]0, [Fe4S4]1+ , and [Fe4S4]2+, which is relatively uncommon for FeS proteins, as they generally support the two redox states required for one electron transfers.77,92 During catalytic turnover, the Fe protein is proposed to cycle between the reduced [Fe4S4]1+ and oxidized [Fe4S4]2+ states, and the reduction of the Fe protein is driven in vivo by a physiological reductant such as flavodoxin or ferredoxin.93–97 During in vitro studies, the ubiquitous chemical reductant dithionite (S2O42−) stabilizes the [Fe4S4]1+ state, whereas redox active dyes such as viologens or indigodisulfonate (IDS) can be used to oxidize the protein to the [Fe4S4]2+ state.66 The [Fe4S4]0 state is the so called all-ferrous “super-reduced” state first reported in Av NifH by Watt and Reddy,98 but the nature and relevance of this species is still controversial, and this will be discussed below.
The redox properties of the Fe proteins have been studied through potentiometric titrations, as opposed to other electrochemical methods. The midpoint potential (Em) for the [Fe4S4]2+/1+ redox couple in Av NifH was found to be approximately −300 mV versus the standard hydrogen electrode (SHE) at pH 8 in an Ar atmosphere.99,100 The potentials for NifH and VnfH from A. vinelandii were also determined in a CO2 atmosphere and found to be −301 mV and −346 mV versus SHE, respectively.87 When nucleotides are bound to the Fe proteins, the midpoint potentials decrease by ≥100 mV, consistent with the biochemical characterization described in Section 3.1.1. In an atmosphere of argon, potential values of Em = −430 mV and −440 mV versus SHE were found for the MgATP- and MgADP-bound species of Av NifH, respectively.99,100 The midpoint potential for the [Fe4S4]2+/1+ couple for both NifH and VnfH from A. chroococcum with MgADP bound had also been determined, with values of −450 mV and −463 mV versus the normal hydrogen electrode (NHE).101 This showed that the potentials for both Av and Ac NifH are similar to each other. Interestingly, the Em values shift by approximately +30 mV for MgADP-bound Av NifH and Av VnfH (compared to Ac VnfH) in an atmosphere of CO2 (−405 mV and −430 mV versus SHE, respectively).87 This observation could be consistent with CO2 interacting with the [Fe4S4] clusters of the Fe proteins, as these have been shown to facilitate the reversible conversion of CO2 into CO (See Section 4.7).
EPR and Mössbauer spectroscopies have also been invaluable techniques for the study of the Fe proteins, as each cluster oxidation state has associated signals that can be used to understand the electronic properties of the protein. In all cases, the fully oxidized [Fe4S4]2+ state of the Fe protein is EPR silent, and so with Av NifH, Mössbauer spectroscopy was employed to determine that the species was diamagnetic with an S = 0 ground spin state.102 The [Fe4S4]1+ state, the resting state of the cluster in the presence of dithionite, was also studied using parallel EPR and Mössbauer experiments. Av NifH was reported with a mixture of two spin states (Figure 6) in roughly equal proportions, one S = ½ species and one S = 3/2 species (Table 2). The S = ½ species had a rhombic signal with g-values of 2.05, 1.94, and 1.88, referred to as the “g = 1.94 signal,” and the signal was consistent with spectra obtained from other [Fe4S4]1+ species.102,103 On the other hand, the S = 3/2 species had an axial signal with g = 5.8, 5.15, and this spin state is unique to the nitrogenase Fe proteins.102,103 The ratio of the mixture could be modulated through additives such as glycerol (Figure 6), which pushed the mixture to primarily S = ½, or urea, which shifts the spectra to the opposite composition.102 Similar EPR signals have also been reported for VnfH from A. vinelandii and from the methanogenic archaeon M. acetivorans, as well as Av AnfH (Table 2).35,87,104 Nucleotide binding to the Fe proteins broaden the line shapes of both sets of signals, but this does not strongly change the spin state mixture of the protein-bound cluster.87,103–105
Figure 6.
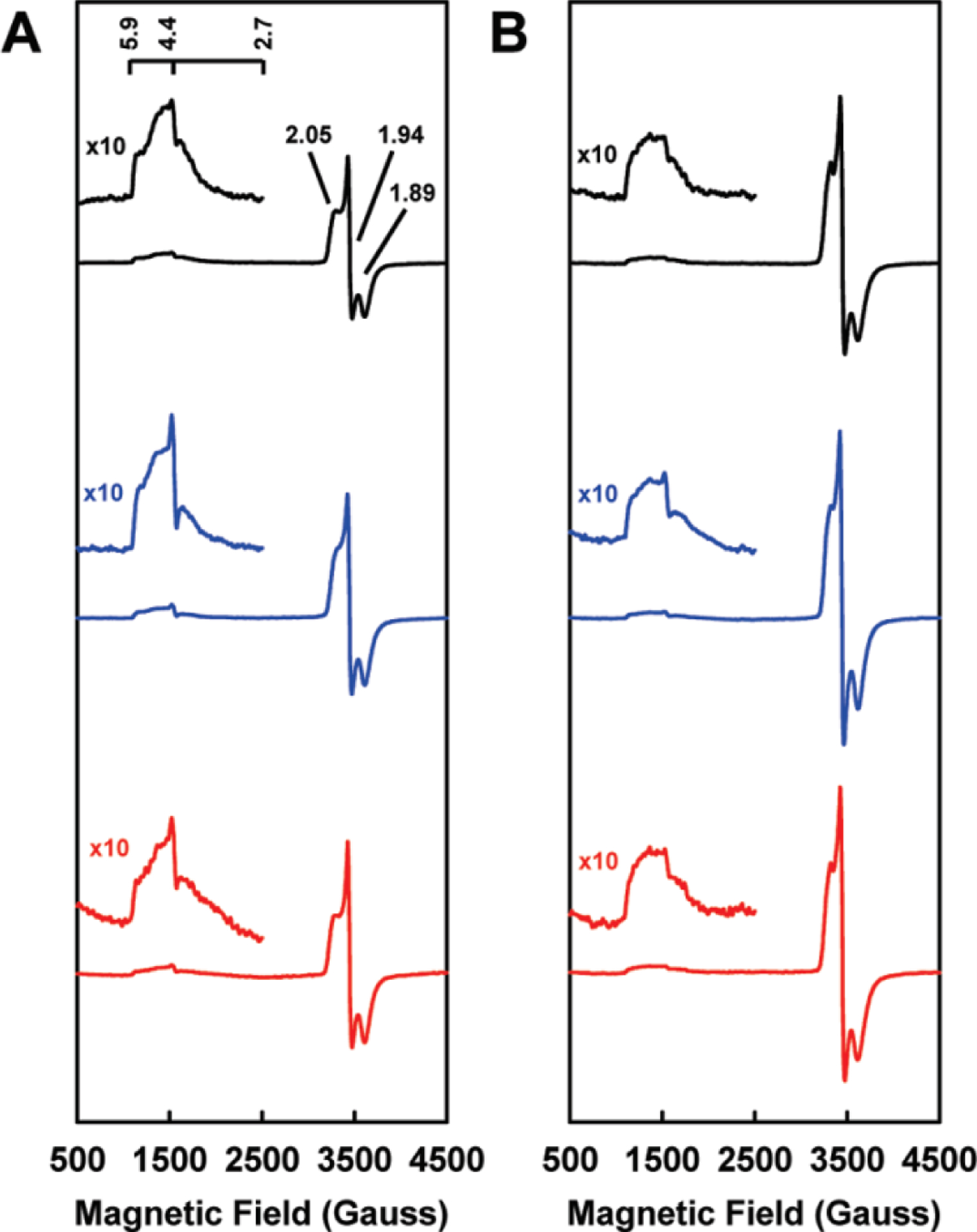
EPR spectra of Fe proteins in the dithionite-reduced form (NifH = black, VnfH = blue, AnfH = red) in the absence (A) or presence (B) of 50% glycerol. Reproduced with permission from ref 35. Copyright 2011 American Chemical Society.
Table 2.
EPR features of the Fe protein components from A. vinelandiia
| Protein | Oxidation state | Nucleotide | Spin State (S) | g-values | refs |
|---|---|---|---|---|---|
| NifH | [Fe4S4]2+ | - | 0 | - | 102 |
| [Fe4S4]1+ | - | ½ | 2.05, 1.94, 1.88 | 102 | |
| 3/2 | 5.8, 5.15 | ||||
| [Fe4S4]1+ | ADP | ½ | 2.02, 1.92 | 105 | |
| 3/2 | 4.8 | ||||
| [Fe4S4]1+ | ATP | ½ | 2.02, 1.92 | 103,105 | |
| 3/2 | 4.8 | ||||
| [Fe4S4]0 | - | 0 | - | 98,106 | |
| [Fe4S4]0 | - | 4 | 16.4 | 108,112 | |
| VnfHb | [Fe4S4]2+ | - | 0 | - | 87 |
| [Fe4S4]1+ | - | ½ | 2.05, 1.94, 1.89 | 35,87 | |
| 3/2 | 5.9, 4.4 | ||||
| [Fe4S4]1+ | ADP | ½ | 2.02, 1.92 | 104 | |
| 3/2 | Unresolved | ||||
| [Fe4S4]1+ | ATP | ½ | 2.02, 1.92 | 104 | |
| 3/2 | unresolved | ||||
| [Fe4S4]0 | - | 4c | 16.4 | 87 | |
| AnfH | [Fe4S4]1+ | ½ | 2.05, 1.94, 1.89 | 35 | |
| 3/2 | 5.9, 4.4 | ||||
X-band EPR spectra collected at temperatures between 2 and 12 K.
The Ma VnfH data from ref104 is similar to the Av VnfH data shown.
This spin state assignment has not been made from Mössbauer or other analysis, but by analogy to the NifH system.
The controversial “super-reduced” state, at the time of discovery, represented the first [Fe4S4]-containing species with all of the Fe centers having the 2+ oxidation state;98 VnfH from A. vinelandii and from M. acetivorans were subsequently shown to support the [Fe4S4]0 state as well.87,104 The physiological relevance of the super-reduced Fe protein is not clear, but Watt has proposed the 2-electron [Fe4S4]2+/0 couple to be operative in nitrogenase. This would allow for concomitant hydrolysis of only two molecules of ATP (compared to four ATP molecules), and NifH could then function more efficiently than as a 1-electron agent.106 However, the experiments discussed below by Burgess, Farmer and co-workers are not in agreement with this proposal.107 The generation of these species has been reported using several reductants; methyl viologen and flavodoxin hydroquinone yield a brown colored protein, whereas Ti(III)citrate, Cr(II)EDTA (EDTA = ethylenediaminetetraacetic acid) or a Eu(II)-reductant yield Fe protein that has a red-pink color in contrast to the typical brown hues associated with other FeS proteins.87,98,104,106,108,109 The [Fe4S4]1+/0 redox couple has been assessed for Av NifH, though, there are discrepancies in the reported midpoint potentials depending on the method of measurement. In the initial 1994 report by Watt, reduced methyl viologen was used as a titrant and an Em value of −460 mV versus NHE was determined.98 Subsequently in 2006, Watt reported that the all-ferrous Fe protein could also be generated using the flavodoxin hydroquinone from A. vinelandii (Em = −515 mV versus NHE), resulting in the same species that is generated from methyl viologen.106 In contrast, Burgess, Farmer and co-workers in 2002 used the more reducing Cr(II)EDTA (−1 V versus NHE at pH 8) as the redox titrant, and determined the Em for the all-ferrous redox couple to be −790 mV versus NHE.107 Additionally, they reported that the use of reduced methyl viologen or the hydroquinone form of flavodoxin II resulted in no observable reduction of the [Fe4S4]1+ state to all-ferrous NifH.107 The super-reduced state of Av NifH has also been analyzed in detail using a variety of spectroscopic and computational methodologies.98,106,108,110–112 The [Fe4S4]0 species reported by Watt and co-workers was found to have an EPR silent S = 0 ground state, measured using the Evans method but not Mössbauer spectroscopy.98,106 The super-reduced Av NifH generated using Ti(III)citrate is the best characterized, with an observed g = 16.4 signal in the parallel mode EPR spectrum,108,112 consistent with an S = 4 ground state established from Mössbauer spectroscopy and density functional theory (DFT) calculations.108,110–112 The Av and Ma VnfH proteins treated with Eu(II) reductants also show a similar EPR signal, but lack Mössbauer characterization.87,104 However, the Cr(II)EDTA reduced Av NifH protein has only been studied using electrochemical methods and UV-visible absorption spectroscopy, but has similar absorption features as the Ti(III)citrate-reduced Fe protein.107
This set of seemingly contradictory results involving the [Fe4S4]0 state of the Fe protein does not lend itself to clear interpretation. The best characterized example is the Ti(III)citrate-reduced Av NifH protein, but this species lacks a measured midpoint potential. Electrochemical measurements were carried out for the Cr(II)EDTA-reduced Fe protein, and this species has similar UV-vis absorption features as the Ti(III)citrate-reduced protein (red-pink color) but lacks EPR or Mössbauer spectroscopic characterizations.107 Additionally, the Av and Ma VnfH proteins treated with Eu(II) also share a red-pink color as well as similar EPR signals as the Ti(III)citrate-reduced species, but lack a measured midpoint potential or study by Mössbauer spectroscopy.87,104 In a sense, the proteins produced under these conditions could all reflect the same species, but without analogous characterization for every condition, it is difficult to determine with certainty. This set of [Fe4S4]0 Fe proteins also differs from the diamagnetic methyl viologen/flavodoxin hydroquinone-reduced protein that has a brown color and an Em value that is 330 mV more positive than the Cr(II)-reduced species.106,107 One interpretation is that the all-ferrous Fe protein can exist in two different spin states, one that is capable of functioning under physiological conditions (S = 0) and one that cannot (S = 4) because of the potentials required to generate the species. The possibility of a spin state mixture is not unprecedented; it has been well established that the [Fe4S4]1+ state of NifH is a mixture of S = ½ and S = 3/2 states, the composition of which is highly dependent on the solvent and chemical additives.102,103 However, Watt and co-workers claim to generate the S = 0 state while Burgess, Farmer and co-workers stated that reduction of the Fe protein with Watt’s reported reductants was not possible. It is also unclear how such a spin state change would drastically affect the measured redox potentials. Another interpretation is that one of the forms of the [Fe4S4]0 state is an artifact, produced from adventitious binding of reductant molecules or other components of the solution. Further still, it is possible that neither form of the all-ferrous state of the Fe protein is relevant for the physiological function of nitrogenase. There is simply not enough information to clearly validate these interpretations, so additional characterization of the [Fe4S4]0 state of the Fe proteins remains necessary.
As discussed previously, the crystal structures of the free NifH proteins in various states had very similar physical metrics despite changes in the biochemical behavior that suggested otherwise (See Section 3.1.1). The structure of Av NifH with the [Fe4S4]0 state has also been reported,113 but similar to the other structures, the bonding metrics of the cluster do not vary from the more oxidized species discussed above.77 This difference could be due to experimental limitations, such as oxidation reactions that occur during the crystallization process or radiation damage to the protein during data collection, but could also could reflect accurate bond metrics. One method to further assess the solution state structural properties of the Fe protein [Fe4S4] clusters is the use of X-ray absorption spectroscopy.35,114 The Fe K-edge XAS analysis was reported for all three Fe proteins from A. vinelandii in the dithionite-reduced [Fe4S4]1+ state.35 The extended X-ray absorption fine structure (EXAFS) region of the data can be used to obtain absorber-scatterer distances from the Fe atoms in the protein, and gain information about the structural configuration. Largely, the structural metrics from EXAFS were fit similarly for NifH, VnfH and AnfH, with 4 Fe–S distances at ~2.30 Å, 1 short Fe---Fe distance at ~2.50 Å and 2 longer Fe---Fe scatterers at ~2.70 Å.35 Additionally, NifH benefitted from the inclusion of a third Fe---Fe distance at 2.62 Å, distinguishing it from the other two proteins. These structural metrics differ slightly from the distances reported for the NifH and VnfH crystal structures (PDB codes 1G5P and 6Q93, respectively).75,113 In both crystal structures, the Fe–S distances range from 2.3 to 2.4 Å which is in agreement with the EXAFS derived data, but the Fe---Fe distances for the NifH structure range between 2.6 and 2.7 Å, whereas the VnfH structure has distances that range between 2.7 and 2.8 Å. There is not an Fe---Fe length in either crystal structure consistent with the short ~2.50-Å pathway from the XAS model, but the longer 2.70-Å distance agrees with the crystallographically determined metrics. Overall, the combined X-ray absorption and X-ray diffraction data suggests that the clusters in both the crystal and in solution have similar structures and continues to support that the [Fe4S4] cluster remains in similar configuration in all three of the Fe proteins. It is also important to note that the MgADP molecule bound to VnfH in the crystal structure could be the source of the perturbation relative to the NifH structure, but additional data would be needed to explore this facet further.
3.2. The Properties of Mo-Nitrogenase
The Mo-nitrogenase from A. vinelandii is the best characterized nitrogenase system, though the catalytic proteins have also been isolated from A. chroococcum, Klebsiella pneumoniae, Clostridium pasteurianum, and R. capsulatus.66,115 The nifD (α subunit) and nifK (β subunit) genes encode for a heterotetrameric α2β2 protein, NifDK, that is ~220 kDa in size.65 NifDK contains two different complex metalloclusters in each αβ-dimer that are essential for electron transfer and substrate turnover, designated as the P- and M-clusters (Figure 7).67,116,117 The P-cluster is a [Fe8S7] cofactor that facilitates electron transfer from NifH to the M-cluster during catalysis, and the cluster is positioned at the α/β interface of NifDK ~10 Å below the surface of the protein.67,118 In the resting state, the P-cluster appears as two [Fe4S3] cubane cluster units that share a common vertex, a μ6-sulfide, and is additionally ligated by two Cys residues (α-Cys88, β-Cys95), each in a bridging mode. A total of six cysteine residues, three from each subunit, (α-Cys62, α-Cys88, α-Cys154, β-Cys70, β-Cys95, and β-Cys153) coordinate to the P-cluster. The M-cluster (also known as FeMoco) is a [MoFe7S9C-(R)-homocitrate] cofactor that is responsible for substrate reduction. This cluster is buried below the surface of the protein in each of the α subunits of NifDK and is located ~19 Å from the P-cluster (Figure 7). The M-cluster appears as an asymmetric combination of two partial cubane units, [Fe4S3] and [MoFe3S3], with a μ6-insterstitial carbide as a shared vertex, and is additionally bridged by three “belt” μ2-sulfide ligands (Figure 8C).67,116,117,119 The Mo atom is further coordinated to (R)-homocitrate by the 2-hydroxy and 2-carboxy groups of the organic acid. NifDK binds to the M-cluster though two amino acid residues; α-Cys275 at the Fe-capped end and α-His442 at the Mo-capped end.32 As mentioned in Section 3.1.1, NifDK can also form a 2:1 NifH:NifDK complex that places the [Fe4S4] cluster of the Fe protein between ~18 and 24 Å away from the P-cluster depending on if a nucleotide is bound (Figure 7).71
Figure 7.
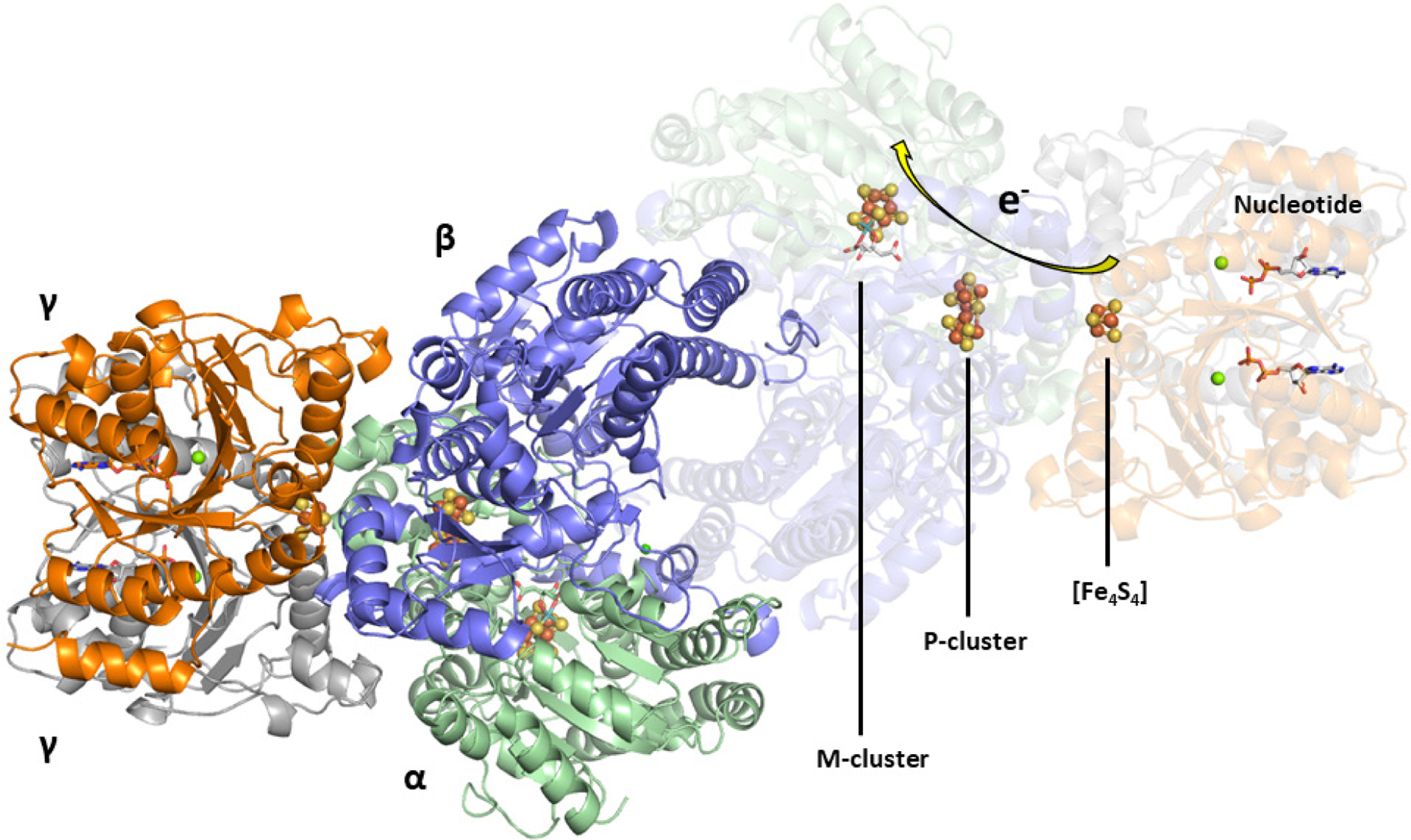
The crystal structure of the NifH:NifDK complex with [Fe4S4], P- and M-clusters shown (PDB ID 1N2C). The arrow shows the proposed flow of electrons through the enzyme. The coloring of the protein: NifD (green), NifK (blue), NifH (orange and grey). The clusters are represented as ball-and-stick models with the same coloration as described in Figure 2.
Figure 8.
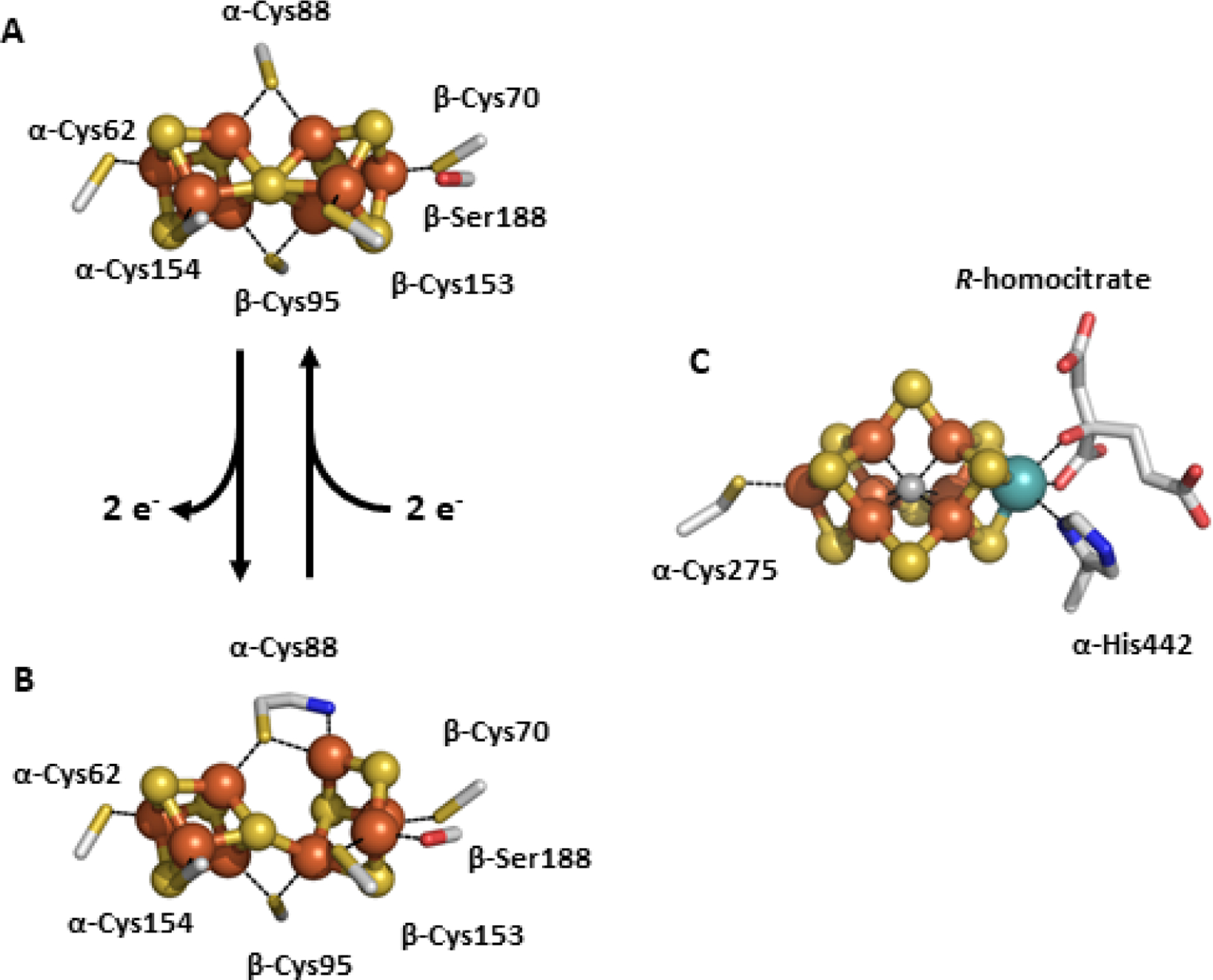
The P- and M-clusters of NifDK. The P-clusters can undergo a conversion from the all-ferrous PN state (A) via 2-electron oxidation to the POX state (B). This opens the cluster and causes the cleavage of two Fe–S bonds from the central μ6-sulfide and Fe centers bind to the backbone of α-Cys88 and β-Ser188. The M-cluster is bound in NifD to α-Cys275 and α-His442. The atoms represented as ball-and-stick models, and are colored as follows: Fe, orange; S, yellow; C, grey; O, red; N, blue (PDB IDs: 1MIN and 3MIN).
3.2.1. P-cluster from NifDK
There are three recognized reversible states of the P-cluster in Mo-nitrogenase; the reduced PN state, the one-electron-oxidized P1+ state and the two-electron-oxidized POX state. The cluster can be further oxidized but it is not clear if there is physiological relevance due to observed degradation of the cluster.66 The resting state of the P-cluster is the PN state, and this is obtained under dithionite reducing conditions. Mössbauer and EPR analysis indicates that all 8 of the Fe atoms in the PN state are ferrous, and result in a diamagnetic S = 0 spin state.120,121 The structure of this P-cluster species is described in the section above as a fusion of two cubanes incorporating the μ6-sulfide, a feature that is unique to the PN-cluster and not observed in any other biological system. The one-electron-oxidized P1+ state has been studied by EPR and magnetic circular dichroism (MCD) spectroscopies, and this analysis assigns the cluster as a mixture of S = ½ and S = 5/2 species, with g-values of 2.06, 1.95, 1.81 for the S = ½ system and two sets of g-values (A,B) for the 5/2 system, gA = 6.7, 5.3 and gB = 7.3.120,122 The structure of P1+ has not been definitively observed through X-ray diffraction methods, but Lawson and co-workers reported a structure of NifDK from K. pneumoniae with evidence that a P-cluster species consistent with the P1+ may have a conformation somewhere between the PN and POX states.123 However, MCD analysis of the Av NifDK protein is consistent with an electronic description of the P1+ state as two connected [Fe4S4]1+ clusters.122 The oxidation of PN to POX was shown to have a midpoint potential of −307 mV versus NHE for the two electron event,120,124 which is a more positive potential than for nucleotide-bound NifH (−430 mV versus SHE).100 Subsequent analysis by EPR and Mössbauer spectroscopies assign an S = 3 or 4 spin state to POX, with a sharp signal observed at g = 11.9.120,125 It was also observed that POX has a structure that undergoes drastic rearrangement compared to the PN state (Figure 8, A and B).126 Two of the Fe atoms from one cubanoid half of the P-cluster break the Fe–S bonds from the central μ6-sulfide and each form a new bond; one Fe center binds to the O atom of α-Ser188 and the other to the backbone N atom of α-Cys88. Despite the rearrangement, the observed changes are reversible, though there is no clear consensus about which P-cluster states are operative during substrate turnover.66
While it may be unclear which redox states of the P-cluster are functional during substrate reduction, one proposal has been put forth by Seefeldt, Hoffman and co-workers called the ‘deficit-spending electron transfer’model.83,127 If the normal flow of electrons during catalysis starts with the Fe protein, then transfers to the M-cluster through the P-cluster, the deficit-spending model begins with the PN cluster slowly transferring one electron to the MN state of the M-cluster (the deficit), and then the P-cluster in the P1+ state is quickly re-reduced by the Fe protein, regenerating PN (repaying the deficit).83 The logic is that the PN cluster is composed of all ferrous iron centers,120,121 and generating Fe centers in the 1+ oxidation state is not favored in biological FeS clusters, therefore, the P-cluster must first transfer an electron before it can receive an electron.83 This was supported by experiments showing that the P-cluster was redox active during substrate reduction,128 and that electron transfer from the Fe protein to the MoFe protein is conformationally gated, thus, when NifH and NifDK form a complex, gated electron transfer is somehow allowed to occur.127,129 The deficit-spending electron transfer model is certainly intriguing, however, analogous experiments have not been reported for either the V- or Fe-only nitrogenases, so it is unclear if the model holds for the alternative systems. Further investigation will be necessary to better understand this process across all nitrogenase proteins.
3.2.2. M-cluster from NifDK
The M-cluster is an interesting metallocofactor in biology for many reasons other than the capability to facilitate N2 reduction. The cluster is asymmetric, with one Fe-capped end and one Mo-capped end, employing a chiral R-homocitrate ligand to bind to the heterometal, and unlike the P-cluster, the M-cluster is only coordinated to the protein through two amino acid residues despite the high nuclearity (Figure 8C).68,117 In the initial stages, the resolution of the protein crystal structures was not sufficient to see all of the atoms of the M-cluster, so it was unclear if there was a central atom or what the identity of that atom was.68,118 Subsequent improvements of the resolution allowed for the identification of an interstitial atom,117 and a 1.0 Å resolution crystal structure in combination with X-ray emission spectroscopy (XES) identified the atom as a carbon.116,119 Ribbe, Hu and co-workers were able to definitively confirm that the central atom of the M-cluster was a carbon derived from SAM.46 Another notable feature is that the cofactor is synthesized on proteins other than NifDK, and is only transferred to the apo-protein in the final step of the assembly (see Section 2.1).32 The M-cluster can also be selectively extracted from NifDK in a destructive process discussed in Section 4.5, but the isolated cofactor has never been crystallized independently from the protein.
The catalytic cofactor of NifDK has a characteristic rhombic EPR signature, with intense signals at g = 4.3, 3.7 and 2.0 that are associated with an S = 3/2 spin system.66,103 This state (MN) is the most commonly observed, as it is stabilized in the presence of dithionite, though the metallocluster can be readily oxidized by one electron to an S = 0 diamagnetic state (MOX).120,124 The EPR spectrum of the isolated M-cluster still maintains the S = 3/2 spin state, but with broader, shifted resonances (g = 4.6, 3.3, 2.0) indicative of a different ligand environment.130,131 The redox behavior of Mo-nitrogenase is challenging to study because of the multiple redox states that both the P- and M-clusters can support, however, the signature EPR signal for the M-cluster can be monitored during potentiometric titrations with chemical redox mediators to obtain a midpoint potential. O’Donnell and Smith were able to determine the Em for the MN to MOX redox couple in Av and Ac NifDK, with values of −42 mV versus NHE for both proteins.132 The equivalent midpoint potential reported for Rc NifDK was also found in the same range (Em ~ −50 mV versus SHE).133 The Em for the one-electron reduced M-cluster, MR, has also been measured to be −465 mV versus NHE, but this could only be obtained in the presence of NifH.134–136 Interestingly, the isolated M-cluster has a much more negative potential than when bound to the protein, with reported values for the MOX/MN couple between −320 and −270 mV versus SHE. For a more specific example, the isolated M-cluster in the solvent N-methylformamide (NMF) was found to have potentials for the MOX/MN and MN/MR redox events at −320 mV and −1.00 V versus NHE, respectively.137–139 The dramatic change observed in the reduction potentials for the M-cluster bound to and free from the nitrogenase protein demonstrates that the protein environment plays a significant role in tuning the properties of the M-cluster.
In a recent study by Minteer and co-workers, the electrochemical properties of the MoFe protein were also studied by immobilizing the protein on a pyrene-modified hydrogel film that allows the protein to be attached to an electrode directly as opposed to requiring redox mediators in solution.140 Square wave voltammetry was employed and the MoFe protein was found to have two redox features, one at −230 mV (E½A) and the other at −590 mV (E½B) versus NHE. Protein variants with single-point mutations at the P-cluster and M-cluster sites, as well as apo-NifDK, were used to correlate the redox features to a metallocofactor. This resulted in E½A and E½B being assigned to the PN/P1+ and MN/MR couples of the P- and M-clusters, respectively. Additionally, the V- and Fe-only nitrogenase proteins were also studied, and assignment by analogy resulted in the P-cluster in both alternative systems being the same as in Mo-nitrogenase, but the V- and Fe-clusters were respectively assigned to potentials of −380 mV and −400 mV versus NHE.140 The E½A potential for the MoFe protein initially appears similar to the Em = −307 mV value reported by Hagen and co-workers,120 but what is important to note is that E½A is assigned to a one-electron event, PN/P1+, whereas the previous midpoint potential from Hagen was assigned to a two-electron event PN/POX. It is also interesting that the P-cluster potential would remain invariant across all three nitrogenases, considering that the P-cluster of V-nitrogenase does not appear to have identical properties as the P-cluster of Mo-nitrogenase (discussed in Section 3.3.2). Additionally, E½B = −590 mV for the M-cluster i ~200 mV more negative than the analogous redox couple for either the V- or Fe-clusters. These differences are not clearly discussed in the report by Minteer,140 though, the alternative nitrogenases are not particularly stable (see Sections 3.3 and 3.4 for details), so immobilization of the proteins in a film for the electrochemical experiments may substantially affect the observed properties. There was also no characterization of the nitrogenase proteins reported to assess the properties of the cofactor before or after immobilization, so it may also be possible that the P-, M-, V- and Fe-clusters were adversely affected by this process. Additional experiments will be necessary to rectify the observed differences between solution state and immobilized nitrogenase variants.
3.3. The Characterization of V-Nitrogenase
While the Mo-dependent nitrogenase is preferentially expressed in most organisms, the alternative nitrogenases provide important backup systems for when molybdenum is not readily available. As will be discussed in Section 4, the effectiveness of the alternative systems with respect to dinitrogen fixation ranks Mo > V > Fe-only, with a bias in V- and Fe-only nitrogenases towards proton reduction to H2. To date, the V-nitrogenase protein (VnfDGK, or VFe protein) has only been isolated from only two organisms, A. chroococcum and A. vinelandii,141,142 but vnf genes have been observed in a wide range of other species. This section will describe the characterization of these V-nitrogenases, including the recently reported crystal structures of Av V-nitrogenase and the relevant comparisons to Mo-nitrogenase.
The catalytic component of V-nitrogenase, VnfDGK or VFe protein, is encoded by the vnfDGK genes, and the VnfD (α, ~53.8 kDa) and VnfK (β, ~53 kDa) gene products have a ~30% conserved sequence identity as compared to the NifD and NifK proteins.143 V-nitrogenase additionally has a VnfG subunit (δ, ~13.3 kDa) that Mo-nitrogenase lacks. The exact function of the additional subunit is still unknown,144,145 but it has been proposed to be involved in the transfer of the V-cluster to the apo-enzyme based on sequence similarity to identified cofactor chaperones from A. vinelandii and K. pneumoniae.146,147 The VnfDGK forms a α2β2 hetrotetrameric core analogous to NifDK, but the amount of the VnfG subunit is variable depending on the conditions of protein purification (Figure 9). Preparations of the Ac VFe protein were consistent with an α2β2δ2 formulation with an Mr = 239600 Da.141 On the other hand, in the initial report of the Av VFe protein by Hales and co-workers, only an α2β2 species was observed,142 but later preparations resolved αβ2(δ) and α2β2(δ) variants with a variable amounts of VnfG, but the Mr ~ 240 kDa based on a heterohexameric α2β2δ2 formulation.148 Ribbe, Hu and Lee later reported a histidine-affinity-tagged version of the Av VFe protein that was consistent with a heterooctameric α2β2δ4 formulation (Figure 9) having Mr ~ 270 kDa, demonstrating the variability and sensitivity of the VnfDGK protein compared to NifDK.149 Then in 2017, the crystal structure of VnfDGK was reported by Sippel and Einsle showing an α2β2δ2 formulation74, which supports that α2β2δ4 and/or α2β2δ2 could be active in solution.
Figure 9.
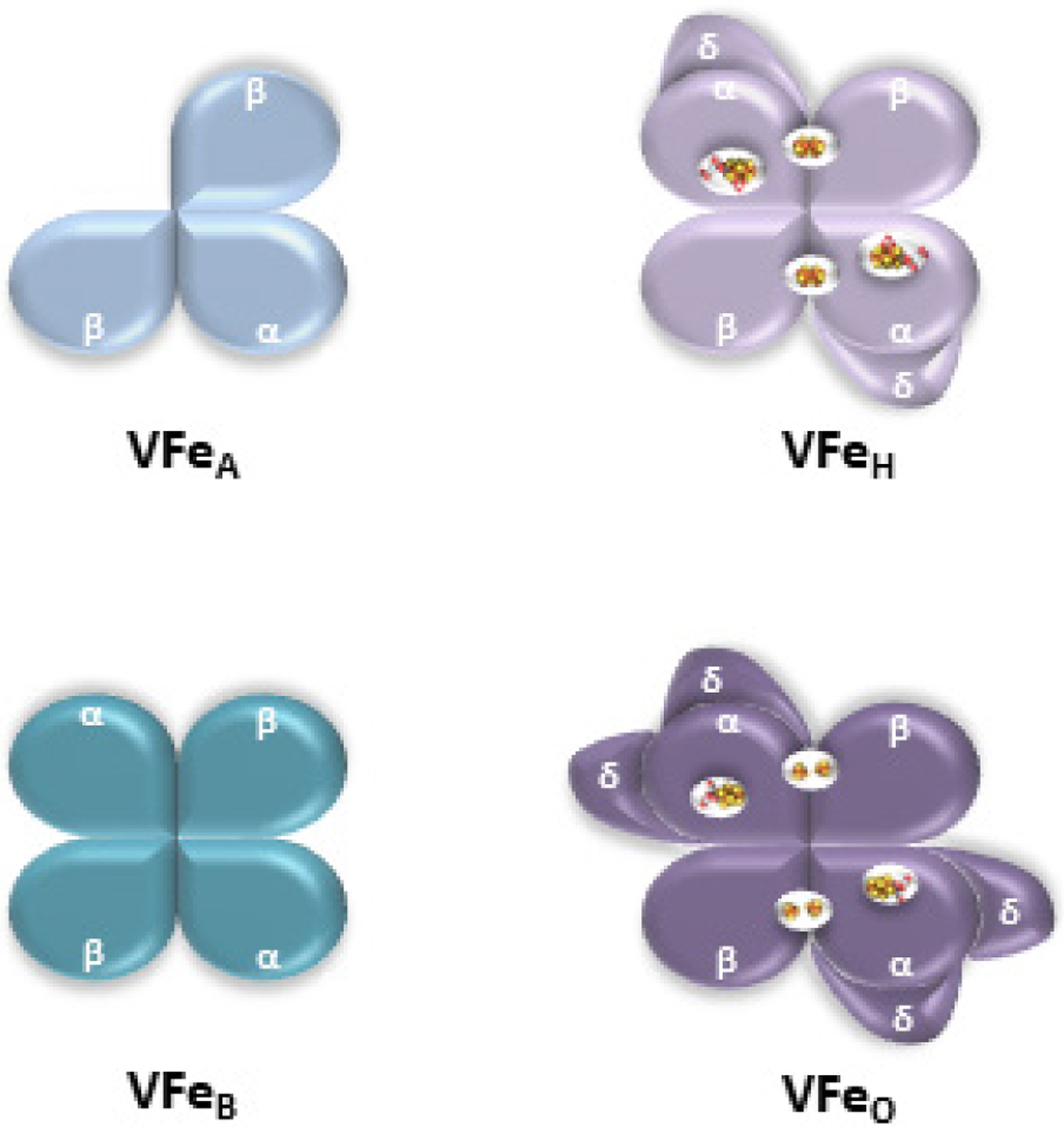
Summary of different conformations of the VFe protein that have been reported. VFeA = αβ2(δ), VFeB = α2β2(δ), the hexameric VFe protein (VFeH) = α2β2δ2, the octameric VFe protein (VFeO) = α2β2δ4. Information known of the metal cofactors were incorporated into VFeH and VFeO. See the main text for details.
3.3.1. Crystal Structures of VnfDGK
VnfDGK has a structure that is very similar to that for NifDK but with some key differences (Figure 10).74 The VnfD and VnfK subunits form the pair of VnfDK dimers consistent with the same overall α2β2 core as NifDK, but VnfG is a globular 113-amino acid protein with a fold composed of 4 α-helices that bind exclusively to the VnfD subunit. This raises a possible concern that the 2 VnfG subunits (or 4 subunits in solution) might interfere with the binding of VnfH to the catalytic component.149 While the VnfH:VnfDGK structure has not yet been reported, overlaying the structures of NifH:NifDK complexes with various nucleotide-bound states on the VnfDGK crystal structure showed that the VnfG protein is sufficiently removed such that there would not be interference, however, it is unclear where additional VnfG units may bind.74
Figure 10.
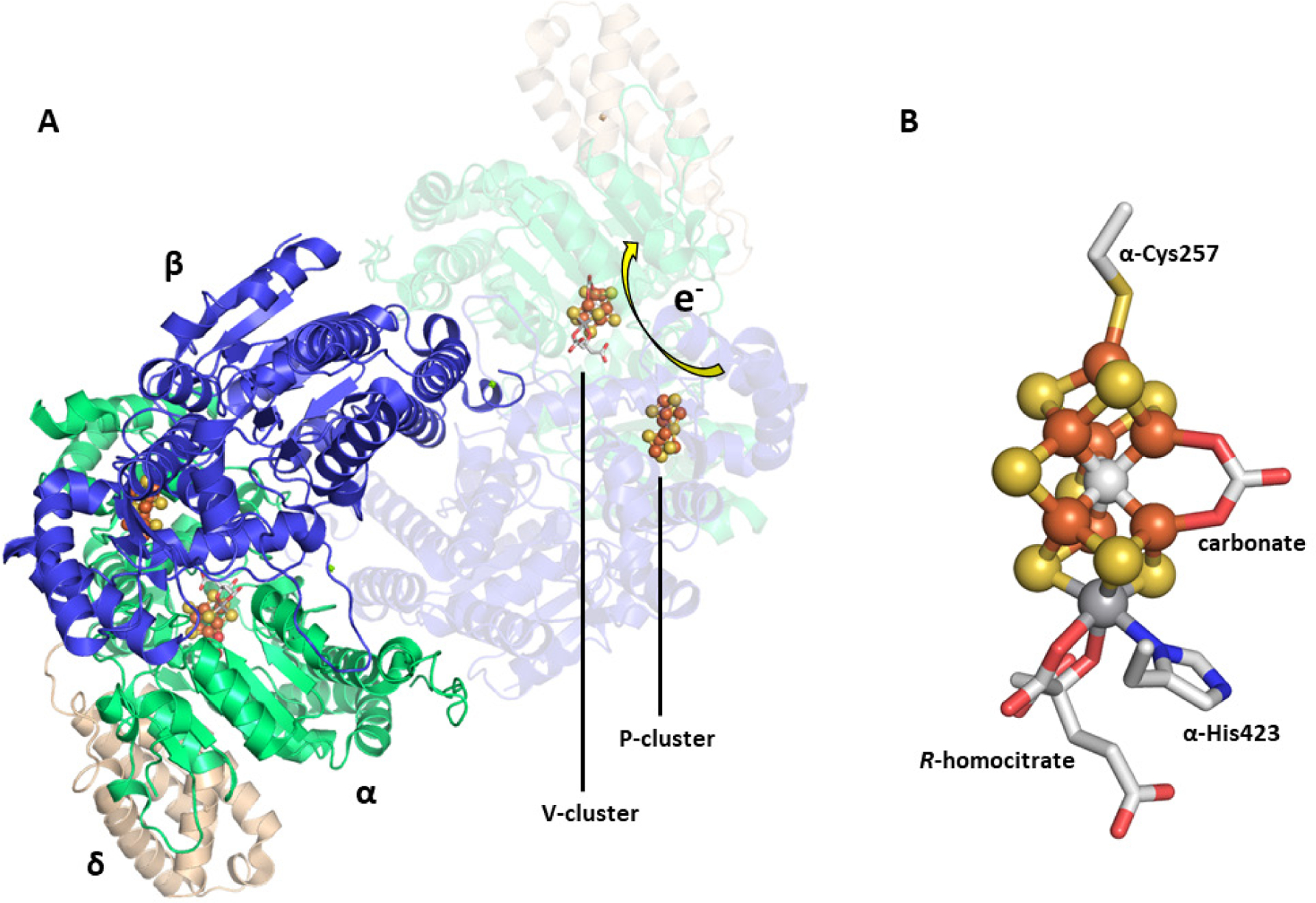
The crystal structure of Av VnfDGK (A) and the carbonate-containing V-cluster (B). The protein is colored as follows: VnfD (green), VnfG (tan), VnfK (blue). The atoms of the V-cluster are shown as a ball-and-stick model, with the carbonate, R-homocitrate and protein residues shown as sticks. Atomic coloring: Fe, orange; S, yellow; V, dark grey; C, light grey; N, blue; O, red (PDB ID 5N6Y).
Like NifDK, VnfDGK houses two types of metalloclusters necessary for electron transfer and substrate turnover, similarly designated as the P-cluster and V-cluster (Figure 10A). Crystallographically, the VnfDGK structure reveals that the P-cluster appears primarily as the PN cluster does in NifDK, with the [Fe8S7] cofactor positioned between the VnfD and VnfK subunits via six cysteine residues (α-Cys49, α-Cys75, α-Cys138, β-Cys31, β-Cys56, and β-Cys115) with a central μ6-sulfide atom.74 However, additional electron density consistent with one of the Fe atoms (Fe6) moving closer to β-Ser153 was observed. This is somewhat analogous to the changes observed for the POX state of NifDK where the equivalent Fe6 and Fe5 break from the central sulfide atom and bind to a serine (α-Ser188) and the backbone of a cysteine residue (α-Cys88).126 In the VnfDGK structure only one of these changes occurs, and based on this, the authors assign the partially occupied state of the P-cluster to the one-electron-oxidized P1+ state.74 However, EPR or other magnetic measurements were not reported for the V-nitrogenase protein used, to corroborate this assignment. The V-cluster reported in the structure is formulated as a [VFe7S8C(CO32−)(R-homocitrate)] cluster that is buried in the α subunit near the P-cluster, and is anchored to the protein by two residues, α-Cys257 and α-His423, on the Fe- and V-capped ends, respectively (Figure 10B). While the overall structure of the V-cluster core is similar to that of M-cluster from NifDK, there are several observations that should be noted. While the V atom occupies the same place as the Mo center, including coordination by His and R-homocitrate ligands, the average V–Fe distance of 2.77 Å to the closest three Fe centers in the V-cluster is slightly longer than the analogous 2.69-Å Mo–Fe distance in the M-cluster.74,116 This causes the V-cluster to appear slightly elongated relative to the M-cluster. The largest difference between the M- and V-clusters is the replacement of one of the μ2-sulfide ligands (S3A) by a CO32− moiety in the V-cluster.74 Substituting other molecules such as nitrate (NO3−) or acetate (CH3COO−) during refinement did not result in adequate agreement with the electron density, but an independent confirmation of the carbonate ligand has not been reported. There is currently no definitive function for, or source of the carbonate ligand identified in the crystal structure.
In 2018, Einsle and co-workers reported a second crystal structure of VnfDGK with a reaction intermediate purportedly bound to the V-cluster, achieved through the limited use of dithionite as reductant.150 The structure of the protein fold as well as the P- and V-clusters were largely unchanged from the previous structure, except for the loss of another μ2-sulfide ligand (S2B) and its replacement by a light atom (X) with Fe–X distances of 2.01 Å (Figure 11). Electron density was observed ~7 Å away from the S2B position on the V-cluster, near the α-Gln176 residue, that was best fit with a hydrosulfide ion (HS−, Figure 11).150 This location was then proposed to be the ‘holding pocket’ for the displaced S2B sulfur atom. The same α-Gln176 residue also becomes reoriented relative to the previous structure, such that the side chain amide O atom is 2.55 Å from the X atom and 2.84 Å from the nearby α-His180 residue, implying the formation of hydrogen bonding interactions (Figure 11). The equivalent residues in Mo-nitrogenase (α-Gln191, α-His195 in A. vinelandii) have been shown to play important roles in substrate reduction,151–154 though the equivalent investigations have not been carried out using V-nitrogenase. Based on this information, the X atom was tentatively assigned to a nitrene (HN−) species, and a hydroxide (HO−) was ruled out based on the assumption that water binding to the V-cluster after the release of S2B would make water a competitive inhibitor of N2 reduction.150 Additionally, the authors note that the Fe–XH bond distances of ~2.0 Å were in line with Fe2–(μ-NH) diiron model complexes. However, the synthetic complex cited has metrics that are incongruent with those from the VnfDGK structure. Peters and co-workers reported an Fe(II)–(μ-NH)(μ-H)–Fe(II) complex supported by phosphine ligands that features Fe–NH bond distances of 1.826 and 1.790 Å, an Fe---Fe separation of 2.659 Å, with an Fe–N–Fe angle of 94.7°.155 The equivalent measurements from VnfDGK are 2.0 Å for Fe–X, 2.6 Å for Fe---Fe and 82° for the Fe–X–Fe angle.150 While hydrogen bonding can cause perturbations to bond lengths, a ~0.2 Å elongation is not reasonable and would be inconsistent with assignment to a nitrene species based on the synthetic model cited. Additionally, quantum mechanics / molecular mechanics (QM/MM) calculations carried out by Bjornsson and co-workers favor the X ligand as a hydroxide species (HO−), citing better agreement between the bonding metrics and hydrogen bonding interactions.156 It is clear that further investigation of the identity of the light atom will be necessary to address the situation.
Figure 11.
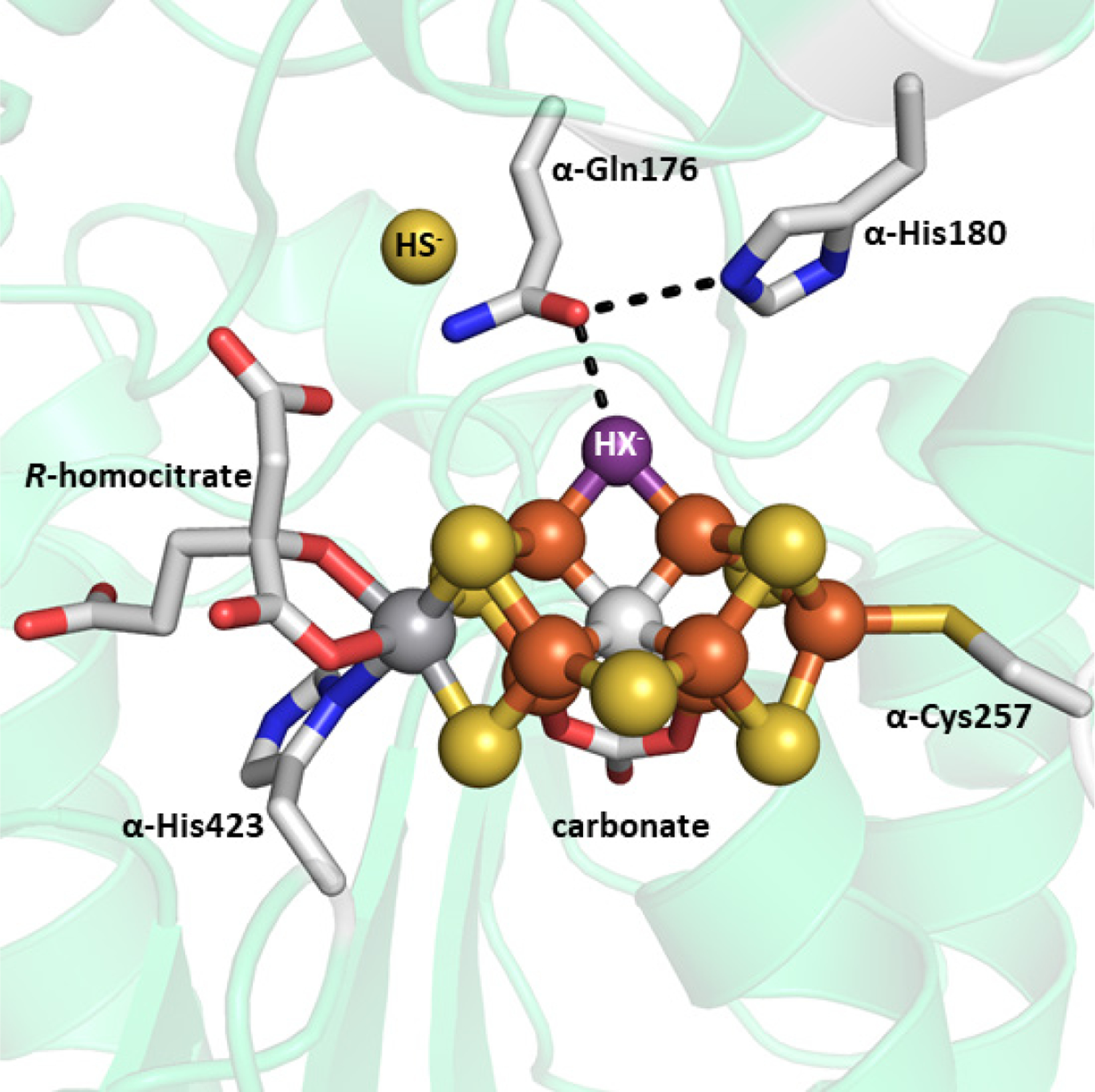
Crystal structure of a putative reaction intermediate bound to Av VnfDGK (PDB ID 6FEA). The cluster atoms are shown as ball-and-stick models, the exogenous HS− molecule is shown as a sphere, and the remaining atoms are shown as stick models. HX− ligand (purple) is shown with hydrogen bonding interaction (black dashed line) to α-Gln176, which is itself interacting with α-His180. The coloring is as described in Figure 10.
3.3.2. P- and V-clusters from VnfDGK
Prior to the report of the V-nitrogenase crystal structure, the identity and composition of the metallocofactors in the protein was derived from a combination of EPR, Mössbauer, MCD and XAS spectroscopic analyses. The study of the clusters on the protein is made challenging due to several overlapping paramagnetic signals in addition to the VnfDGK composition issues described earlier. Initial reports of the EPR spectra of dithionite reduced forms of the Ac and Av VFe proteins had an intense S = ½ signal in the high field region with g = 2.04, 1.93, and a series of resonances in the low field region between g = 3.0–6.0 (Table 3, Figure 12 A and B).141,142 In the spectrum of the Ac VFe protein, the specific g-values found were 5.60, 4.35, 3.77. These were assigned to a mixture of S = 3/2 species affiliated with the V-cluster based on analogy to the M-cluster, but with an order of magnitude lower intensity than in the MoFe protein.141 The S = ½ species observed for VnfDGK was believed to be an irrelevant contamination. This assignment was, in part, supported by the EPR spectrum of V-cluster extracted from the Ac VFe protein, where g = 4.5, 3.6 resonances were observed, albeit in a very noisy spectrum.157 In addition, the POX state for the Ac VFe protein was observed with a g = 12 signal in parallel mode EPR and was assigned to an S = 4 system, while the intensity of the S = ½ signal was not found to be correlated to the specific activity of the protein.158
Table 3.
Selected EPR signals and g-values associated with the metallocofactors of the alternative nitrogenases
| Species | Redox Statea | Spin State (S) | g-values | ref |
|---|---|---|---|---|
| Av MoFe | MR | 0 | - | 103 |
| MN | 3/2 | 4.3, 3.7, 2.0 | ||
| MOX | 0 | - | 120,124 | |
| PN | 0 | - | 120,121 | |
| P1+ | 120, 122 | |||
| 5/2 | A = 6.7, 5.3; B = 7.3 | |||
| POX | 3 or 4 | 11.9 | 120, 125 | |
| Av V-MoFe | V-cluster | 168 | ||
| ½ | 1.93 | |||
| Av M-cluster | MN | 3/2 | 4.6, 3.3, 2.0 | 130 |
| Ac VFe | VN | 3/2 | 5.60, 4.35, 3.77 | 141 |
| PN | 0 | - | 158 | |
| POX | 3 or 4 | 12 | 158 | |
| ? | ½ | 2.04, 1.93 | 141 | |
| Av VFe | VN | 3/2 | 5.80, 5.40 | 160 |
| ? | ½ | 2.04, 1.93 | ||
| Av VFeBR | VN | 3/2 | 5.71, 5.42 | 162 |
| PN | 0 | - | ||
| ? | ½ | 2.04, 1.93 | ||
| Fe(III) | 5/2 | 4.34 (sharp) | ||
| Av VFeBN | VN | 3/2 | 5.68, 5.45 | |
| p1+ | 5/2 | 6.67, 5.3, 4.3 (broad) | ||
| ? | ½ | 2.04, 1.93 | ||
| Fe(III) | 5/2 | 4.34 (sharp) | ||
| Av VFeBOX | POX | 3 | 11.5 (⊥)b, 12.8 (‖)c | |
| Av VFeON | VN | 3/2 | 5.50, 4.32, 3.77 | 149 |
| P1+ | 5/2 | 6.68 | ||
| P-cluster | ½ | 2.03, 1.92 | ||
| Av VFeOOX | POX | 3 or 4 | ~12 (weak) | |
| Av M-VFe | MN | 3/2 | 4.65, 3.49 | 163 |
| P-cluster? | ½ | 2.04, 1.93 | ||
| Ac V-cluster | VN | 3/2 | 4.5, 3.6, 2.0 | 157 |
| Av V-cluster | VN | 3/2 | 5.55, 3.25, 2.00 | 168 |
| Rc FeFe | FeR | ½ | 1.96, 1.92, 1.77 | |
| FeN | 0 | - | ||
| FeOX | ½ | 2.27, 2.06 | 133 | |
| PN | 0 | - | ||
| P3+ | ½ | 2.00, 1.98, 1.96 | ||
| Av L-clusterd | LN | ½ | 1.97, 1.83 | 45 |
Relates to the assigned redox state of the cluster, N = dithionite reduced form of the cluster, OX = an oxidized cluster, ? = not been assigned, Fe(III) = adventitious high-spin iron, P-cluster = has not been assigned to a specific state of the cluster.
observed in perpendicular mode EPR.
observed in parallel mode EPR.
The [Fe8S9C] cluster that serves as the precursor to the M-cluster, and likely the V- and Fe-clusters as well.
Figure 12.
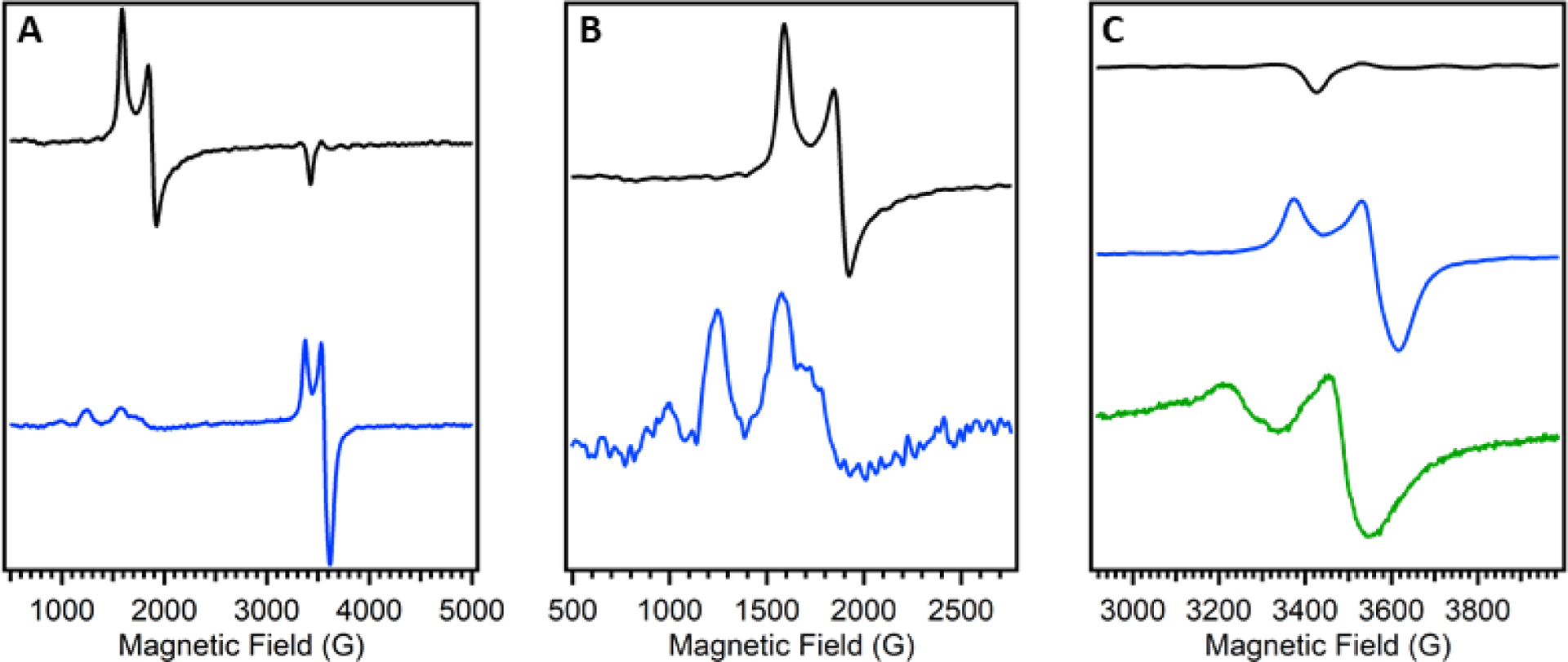
Representative EPR spectra of Av NifDK (black) and Av VnfDGK (blue). A: Spectrum of the full range of data. B: Spectrum of the low field region, Av VnfDGK data has been scaled for visibility. C: Spectrum of the high field region, additionally the apo-VFe protein (Av ΔnifB VnfDGK, green) is included. Data for the MoFe and VFe proteins was reported in ref 149 and data for apo-VFe protein was reported in ref 34. Note that the spectra shown in the figure were not recorded under identical conditions.
The properties of Av VnfDGK have more extensively been explored and these results are summarized in Table 3.148,159–161 The EPR spectrum of the Av VFe protein at low field showed signals at g = 5.80, 5.40 that demonstrated temperature-dependent behavior, and they were assigned to the ground state and an excited state of the same S = 3/2 system, respectively.160 The S = 3/2 signal was spin quantified to one spin per V atom, consistent with assignment to V-cluster, and the S = ½ signal was determined to be a minor component with 0.2 spins per V atom per protein. There was also an additional signal around g = 4.3 that was thought to be from adventitious Fe(III) bound to the protein. MCD analysis established that one of the cluster species in the dithionite reduced state of VnfDGK was an S = 3/2 system that becomes diamagnetic when it undergoes one-electron oxidation.159 The other cluster was diamagnetic in the dithionite reduced state, then becomes a paramagnetic S = 5/2 system upon oxidation. Further, Mössbauer spectroscopy was employed to provide oxidation state information about the clusters.161 In the as-isolated form, the VFe protein contained 5–10% of clusters in an oxidized state, so additional samples were prepared with limiting reductase (1:10 molar ratio of VnfH:VnfDGK) which served to reduce the oxidized species to simplify the analysis. The VnfH-reduced VFe protein (VFeR) was consistent with 52% of the Fe centers being assigned to P-clusters in the all-ferrous PN state, analogous to MoFe protein, and 48% of the Fe was associated with the V-cluster, with a small amount of adventitiously bound Fe covering the difference. It was then suggested that both the S = 3/2 and ½ EPR signals should be associated with the V-cluster, as the PN clusters would be EPR silent. Together, these results were interpreted to mean that (1) the P-clusters were in the diamagnetic PN state when reduced by VnfH, (2) the dithionite-reduced VFe protein contained some amount of an oxidized P-cluster, and further oxidation of the VFe protein clearly produced a paramagnetic S = 5/2 system, (3) the V-cluster was associated with the S = 3/2 and ½ signals in the resting dithionite reduced state (VN), but the spin quantification of these spin systems by EPR did not agree.160
After the discovery that Av VnfDGK could be isolated in two forms, αβ2 (VFeA) and α2β2 (VFeB), the spectroscopic data was reanalyzed and further explored because previous samples of VnfDGK were recognized as mixtures of the two forms (Table 3).148,162,163 The VFeB variant is more active, with VFeA having ~75% of the activity of the former, so VFeB will be the primary focus. In the as-isolated dithionite reduced form (VFeBN), the EPR spectrum shows g-values of 6.67, 5.68, 5.45 as well as a broad resonance at 4.3 at low field, and the g = 2.04, 1.93 S = ½ signal at high field.162 The V-cluster was assigned to the g = 5.68, 5.45 signals as part of an S = 3/2 system, in line with previous observations, but the identity of the remaining g-values was unclear.160 The protein could be reduced further using substoichiometric VnfH (VFeBR), and this showed loss of the g = 6.67 resonance and the broad signal at g = 4.3, while the S = 3/2 signals shift to g = 5.71, 5.42 (Table 3, Figure 13A).162 Subtraction of these spectra (VFeBN–VFeBR), revealed the same g = 6.67, 4.3 signals from the spectrum of VFeBN as well as a new derivative signal at g = 5.3. Tittsworth and Hales previously reported similar signals (g = 6.67, 5.3) for the Av MoFe protein that were assigned to the one-electron-oxidized S = 5/2 P1+ state of the P-cluster, so those observed in VFeB were designated analogously (Table 3).162,164 Oxidation of VFeBR by 1.5 equivalents of thionine caused g = 6.67, 4.3 signals to reappear (Figure 13B), and an additional 1.5 eq. increased the intensities of both signals similarly (Figure 13C), so the broad g = 4.3 was also assigned to the P1+ state, but it was unclear which spin system was the origin of the signal.162 Oxidation by a total of 6 eq. (VFeBOX) produced a new broad signal at g = 11.5 in perpendicular mode EPR, and at g = 12.8 in parallel mode. This compares reasonably well to the two-electron oxidized POX state of the P-cluster in the MoFe protein (sharp signal at g = 11.8 in parallel mode), and so was assigned to an S = 3 POX species (Table 3).120,125 Further oxidation of VFeBOX eliminates all signals other than the sharp g = 4.34 feature (Figure 13D), which increases, and was assigned to adventitious high-spin Fe(III), likely from degradation of the P- and V-clusters.162
Figure 13.
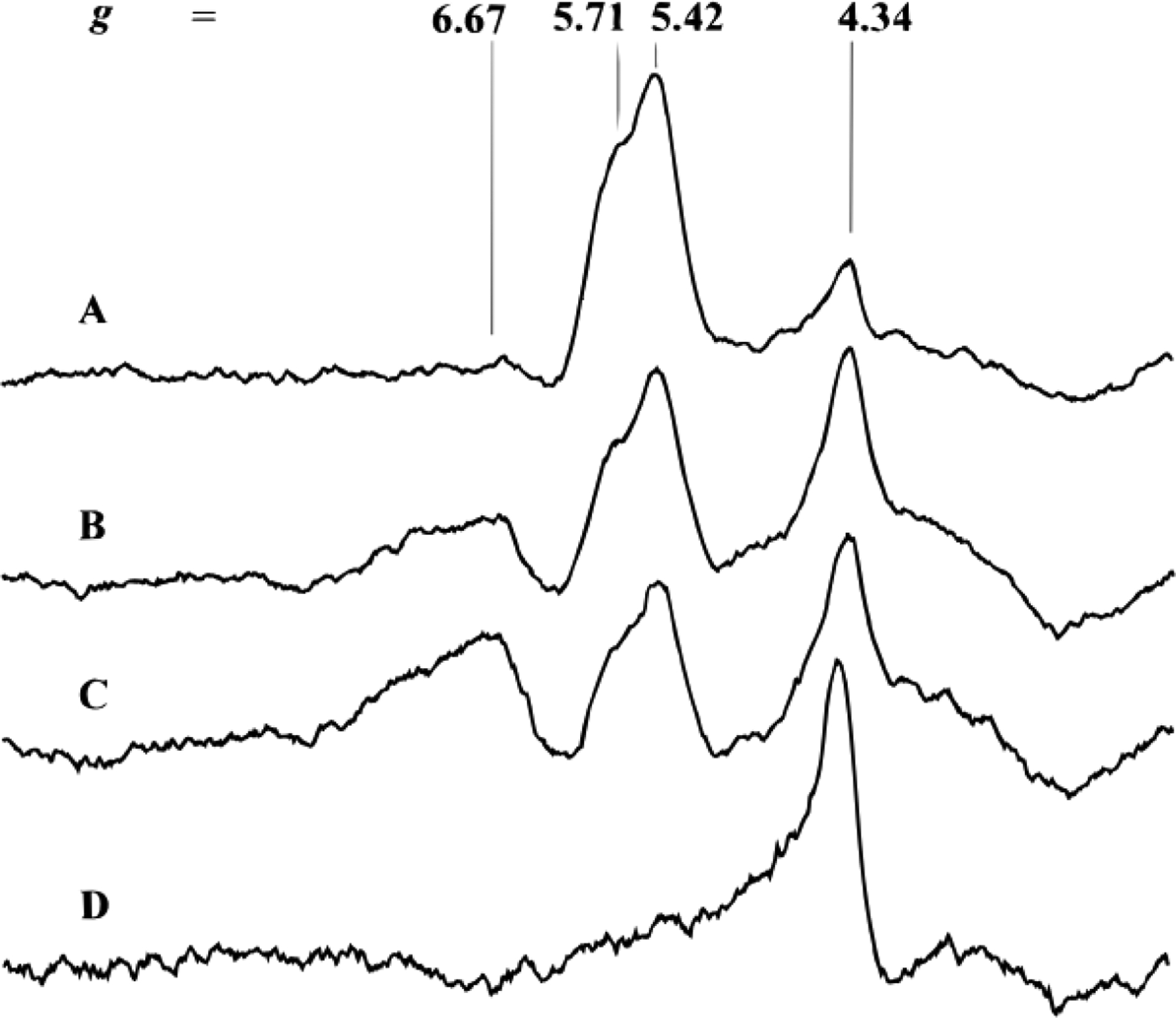
EPR spectra of the low field region of the thionine-titrated Av VFeB protein. A: VnfH-reduced VFeBR; B: VFeBR oxidized by 1.5 equivalents of thionine; C: VFeBR oxidized by 3 equivalents; D: VFeBR oxidized by 6 equivalents (VFeBOX). Reproduced with permission from ref 162. Copyright 1996 American Chemical Society.
The remaining high field signals in the EPR spectra, however, were more complicated to analyze. The authors reported that upon reduction of VFeBN to VFeBR, the S = ½ signal would disappear, but only on occasion.162 The signal could be recovered with 1 eq of oxidant if it had disappeared (VFeBR → VFeBN), but if it remained, it would disappear with the addition of 1 eq of oxidant. This bizarre behavior suggested that the signal was only a minor component of the system, but the identity was unclear. Hales and co-workers also reported a hybrid VFe protein generated by adding isolated M-cluster to cell extracts of an A. vinelandii strain expressing an apo-VFe protein that lacked the V-cluster (Figure 14).163 The resultant protein that was purified contained the M-cluster instead of the V-cluster (M-VFe). EPR spectroscopy showed that the prominent S = 3/2 signal for the M-cluster on NifDK (g = 4.32, 3.68, 2.01, Figure 14A) was shifted and drastically broadened in the dithionite reduced M-VFe sample (g = 4.65, 3.49, Figure 14B). Additionally, the S = ½ region yielded the same g = 2.04, 1.93 signal for both the VFe protein and M-VFe (Figure 14 B and C). This suggested that the S = ½ system was not a property of the V-cluster, but was instead inherent to the VFe protein, lending assignment to a P-cluster signal. However, this was still inconsistent with the Mössbauer analysis that showed the P-cluster was primarily composed of S = 0 PN clusters.161
Figure 14.
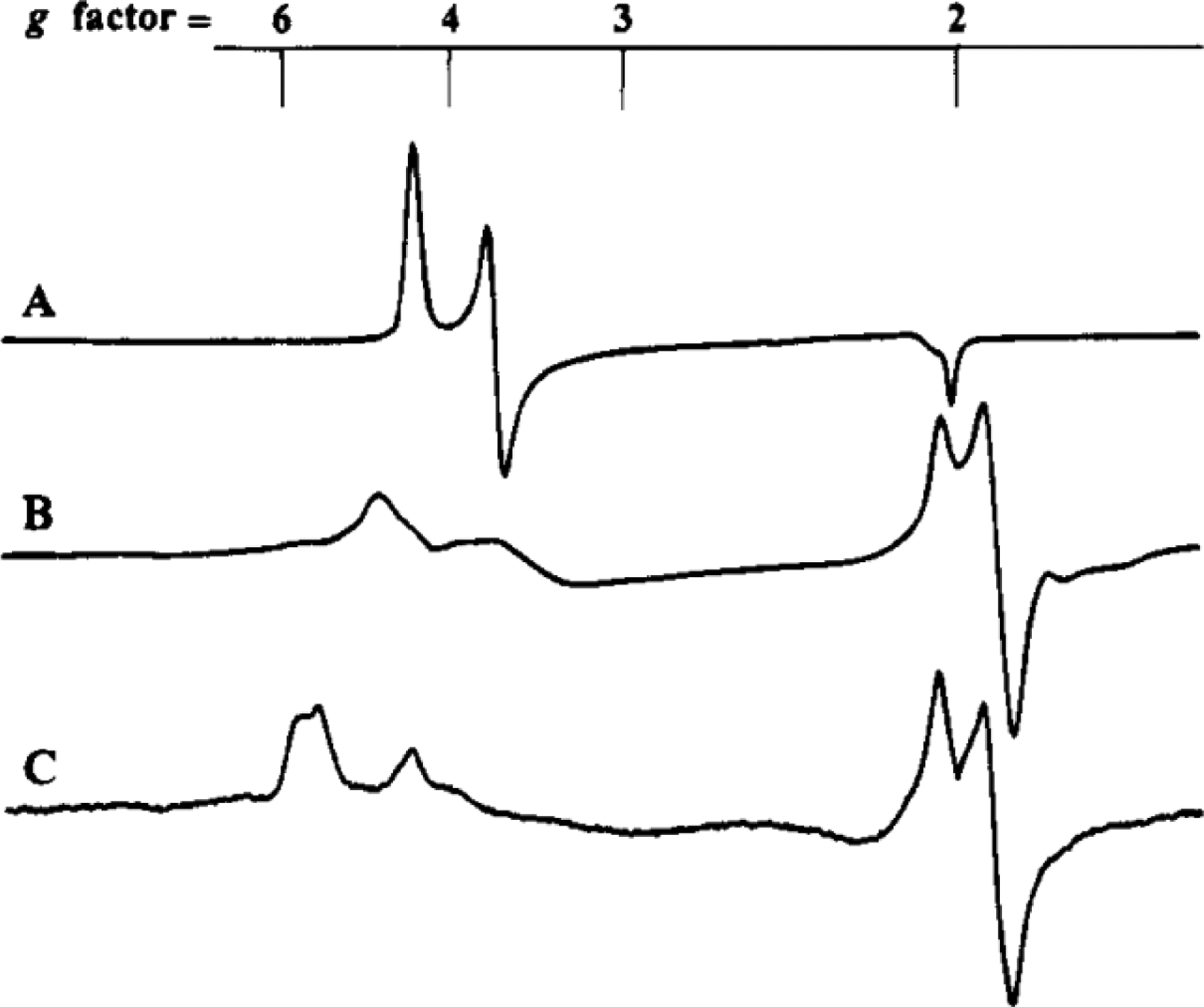
EPR spectra of the Av MoFe protein (A), Av M-VFe protein (B), and Av VFe protein (C) in the dithionite reduced state. Reproduced with permission from ref 163. Copyright 1994 American Chemical Society.
Ribbe, Hu and Lee also reported EPR studies of Av VnfDGK, but used a protein purified with a His-affinity tag and a heterooctameric composition of α2β2δ4 (VFeO) in contrast to VFeB (Figure 9).149 This VFeO protein is highly active (see Section 4) and as such, provided a good opportunity to study the spectroscopic properties of V-nitrogenase. The dithionite reduced form, VFeON, shows an EPR spectrum with resonances at g = 6.68, 5.50, 4.32, 3.77, 2.03, 1.92, which are similar to those seen for the Ac VFe protein and Av VFeBN (Table 3).141,160,162 The g = 6.68 signal was assigned to an S = 5/2 species, likely associated with the P1+ state of the P-cluster, analogous to the assignment for Av VFeB.162 VFeO could also be oxidized (VFeOOX), and this species showed a very minor signal at g ~ 12 that was previously assigned to the POX state of the P-cluster in VFeOOX.162 The g-values at 5.50, 4.32, 3.77 were attributed to S = 3/2 signals, and when Av VFeO was put under substrate turnover conditions with C2H2, N2 or Ar atmospheres, the S = 3/2 signals diminished, supporting that those signals were associated with the V-cluster.149 However, there was an additional observation that the g = 5.50 resonance had a different temperature dependence than the g = 4.32, 3.77 signals. This was interpreted to mean that each set of signals corresponded to different S = 3/2 species, similar to VFeB where the g = 5.68 signal was assigned to the S = 3/2 V-cluster and the broad signal at g ~ 4.3 was likely associated with a P1+-cluster but without a clear spin assignment.162
The S = ½ signal was also found to behave differently for VFeO than in previous reports.149 The intensity of the S = ½ EPR features appeared to increase with increasing specific activity, similar to the S = 3/2 signal, and in stark contrast to Ac VnfDGK.141,158 Additionally, when VFeO was under catalytic turnover conditions, the g = 2.03, 1.92 signal was attenuated. These observations suggested that the S = ½ signal is related to the activity of the protein and not just an adventitious species, and further, was attributed to the P-cluster (Table 3).149 This is in agreement with S = ½ signals observed for the M-VFe protein by Hales and co-workers.163 Additionally, an isolated apo-VnfDGK protein (ΔnifB background, lacking V-cluster but with P-clusters present) was also studied by Ribbe and co-workers using EPR and X-ray absorption spectroscopies.34 The protein was purified as a trimeric αβ2 species ~160 kDa in size, similar to the V-cluster replete VFeA.148 The EPR spectrum of the apo-VFe protein shows a similar S = ½ feature to the holo-VFe proteins, with g-values at 2.08, 1.89 (Figure 12C, green trace), which supports the association of the signal with the P-cluster.34 Additionally, it was found that unlike the P-clusters on the MoFe protein, the apo-VFe protein was unable to be oxidized to the POX state, which suggested that the P-cluster on VnfDGK may not look like the ‘normal’ P-clusters of NifDK. Further, the XAS data showed a feature in the Fourier transform at R + Δ ~ 2.4 Å composed of primarily Fe---Fe contributions associated with a [Fe4S4]-like species, and this feature is completely absent in the analogous apo-MoFe protein with typical P-clusters (Figure 15, red versus black trace).34,81 To further complicate matters, Sippel and Einsle report the EPR of the Av VFe protein used to obtain a crystal structure that showed the same S = ½ signal, but the P-clusters in the structure are modeled similarly to those in MoFe.150 It seems that it may be possible for the P-clusters on the VFe protein to take on conformations similar to those found in MoFe protein under certain conditions. However, the solution state characterization is not entirely consistent with a well-behaved P-cluster and suggests that an alternative conformation may be operative in V-nitrogenase. Additional work will be necessary to further elucidate the structure of the P-cluster.
Figure 15.
Fe K-edge k3-weighted EXAFS (A) and Fourier transforms of the data (B) for a series of apo-nitrogenase proteins lacking the catalytic cofactor (M- or V-cluster) but containing a P-cluster variant. ΔnifB NifDK with normal P-cluster (black), ΔnifH NifDK in the presence (blue) or absence (green) of NifH with P-cluster variants, and ΔnifB VnfDGK (red) with P-cluster. Adapted with permission from ref 34. Copyright 2005 National Academy of Sciences.
The structures of the metallocofactors of the V-nitrogenases from A. chroococcum73,157,165,166 and A. vinelandii72,167–170 had primarily been explored using X-ray absorption spectroscopy before the recent appearance of crystal structures. Smith and co-workers first reported the V K-edge XAS data of V-nitrogenase from A. chroococcum in 1987, and a parallel study with better resolution of the Av VFe protein followed in 1988 by Cramer and co-workers (Figure 16).72,73 Both proteins were fit similarly with an octahedral environment around the V-center from the cluster, with V–S of 2.32 and 2.34 Å, V–O of 2.14 and 2.13 Å, and V---Fe distances of 2.74 and 2.76 Å, for the Ac and Av VFe proteins, respectively. This compared well to the analogous Mo K-edge experiments for the Mo-nitrogenase of C. pasteurianum and K. pneumoniae, though the Mo---Fe distance was slightly shorter at 2.69 Å.72 The V K-edge XAS of the VnfH-reduced (VFeR) and thionine-oxidized forms of Ac VFe protein were also investigated.165 The X-ray absorption near edge structure (XANES) for the VFeR, dithionite reduced VFeN, and thionine-oxidized (VFeOX) forms were largely similar, with identical K-edge energies (5466 eV) and similar pre-edge intensities, though the data for VFeR was of poor quality and prevented additional analysis. The oxidation state of the V atom was estimated to be between V(II) and V(IV).73,165 The EXAFS of the oxidized form was essentially identical to the dithionite reduced form, with 3 V–S distances at 2.34 Å, 3 V–O at 2.13 Å and 3 V---Fe at 2.74 Å, indicating that with respect to the V center, there is no large-scale rearrangement of the cluster upon oxidation. The Fe K-edge XAS data for Av VFeN and Av VFeOX were also reported and compared to a series of FeS model complexes to assist with structural determination.167 These experiments were further complicated because the Fe from both the P- and V-clusters contribute to the average signal observed in the XAS experiment. The EXAFS fits of VFeN and VFeOX were very similar, with distances for Fe–S at ~2.30 Å, and for Fe---Fe scatterers at 2.67 and 3.76 Å. The Av MoFe protein was also analyzed in parallel, and the distances fit for both MoFe and VFe proteins were in agreement, supporting that the M- and V-clusters have similar structure.167 Interestingly, Fe---Mo scatterers were necessary to obtain the best fit of the Mo-nitrogenase EXAFS data, but an analogous Fe---V was not required. This is likely because V and Fe have similar scattering characteristics, and so any V-specific interactions are averaged with Fe signals. Additionally, XES experiments in combination with DFT calculations were carried out on the VFe protein from A. vinelandii to establish the existence of a central carbon atom, similar to what was observed in the MoFe protein.119,169,315 These studies identified that there was indeed a carbide present in the V-cluster similar to the M-cluster, though analogous biochemical confirmation though the use of isotopically labeled carbon has not yet been reported.
Figure 16.
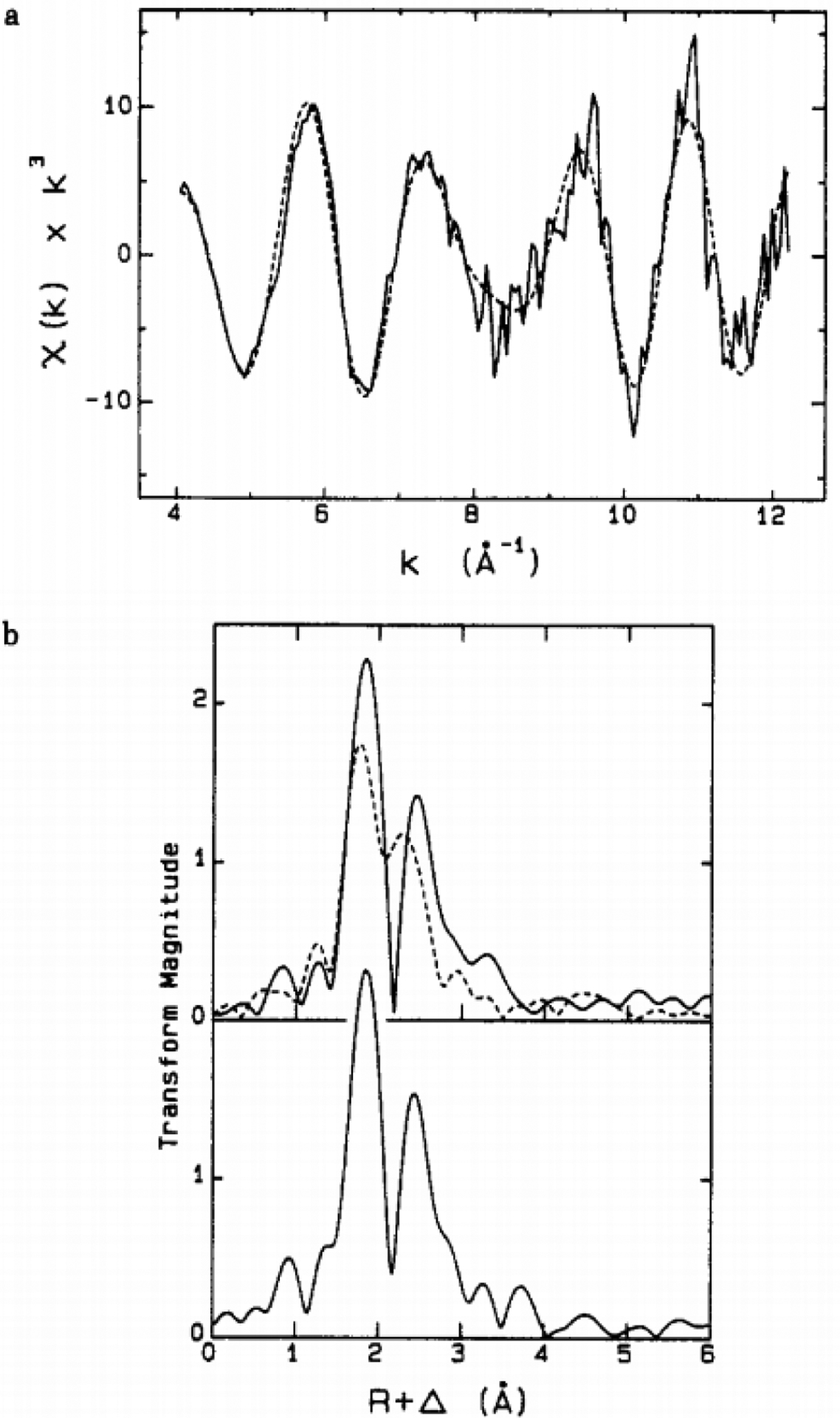
The V K-edge EXAFS data from Av VnfDGK. a: the best fit (dotted) of the k3-weighted EXAFS data (solid) for Av VnfDGK. b: Fourier transforms of the EXAFS data for Av VnfDGK compared to Kp NifDK (top, black vs dashed, respectively) and to the model complex [Me4N][VFe3S4Cl3(DMF)3]•2DMF (bottom, solid). Reproduced with permission from ref 72. Copyright 1988 American Chemical Society.
The extracted V-cluster has also been studied from V-nitrogenase from both A. chroococcum and A. vinelandii.157,166,168 Smith and co-workers first demonstrated the extraction of V-cluster from the Ac VFe protein in 1988, and were able to use the cluster to reconstitute apo-MoFe protein.157 However, the reconstitution of the protein with the V-cluster only conferred a partial restoration of activity to the protein, leaving the resulting V-MoFe protein unable to reduce N2. As mentioned previously, the V-cluster extracted from the Ac VFe protein also showed weak EPR signals at g = 4.5, 3.6, 2.0, and while these resonances are similar to those found for the isolated M-cluster (g = 4.6, 3.3, 2.0), the spectrum of the isolated V-cluster was rather noisy.130 Then in 2010, Ribbe and coworkers also reported the extracted V-cluster, but the cluster came from A. vinelandii.168 The isolated Av V-cluster was capable of restoring activity to apo-MoFe protein, including the ability to fix nitrogen. Characterization by EPR spectroscopy showed g-values for the extracted V-cluster at 5.55, 3.25, 2.00, though the g = 3.25 resonance is very broad, which differs from the extracted Ac V-cluster signals (Table 3, Figure 17A).157,168 The g = 5.55 signal from the isolated Av V-cluster compares well to the S = 3/2 g = 5.5–5.7 resonance from the spectrum of the VFe protein, assigned to the V-cluster (Figure 17A, black versus blue trace). In addition, the isolated Av V-cluster showed a much less intense signal at g = 2.00, which is not likely to substantially contribute to the S = ½ signal in the protein. The EPR spectrum of the V-MoFe protein showed resonances at g = 5.50, 4.32, 3.72 that are broader than the analogous region for the wild-type VFe protein, and also displayed much weaker g-values at 2.01, 1.93 (Figure 17B, black versus red trace). This supports the notion that the protein environment plays a large role in tuning the properties of the nitrogenase cofactors. Both the isolated V-clusters from A. chroococcum and A. vinelandii have also been characterized by Fe K-edge XAS.166,168 The EXAFS analysis for each cluster is similar, showing a best fit consisting of ~3 Fe–S scatterers at 2.23 Å, ~2.5 Fe---Fe distances at 2.63 Å, 1 Fe---Fe/V scatterer at 2.90 Å and a longer Fe---Fe scatterer at 3.69 Å. Additionally, the isolated Av V-cluster was fit with Fe–O and Fe---C scatterers from bound solvent molecules.168 These fits are quite comparable to the Fe K-edge data obtained for the Av V-nitrogenase holo-protein, consistent with a similar structure,167 though the data does not give an indication of the presence or absence of the carbonate (CO32−) ligand that is observed in the crystal structure.74
Figure 17.
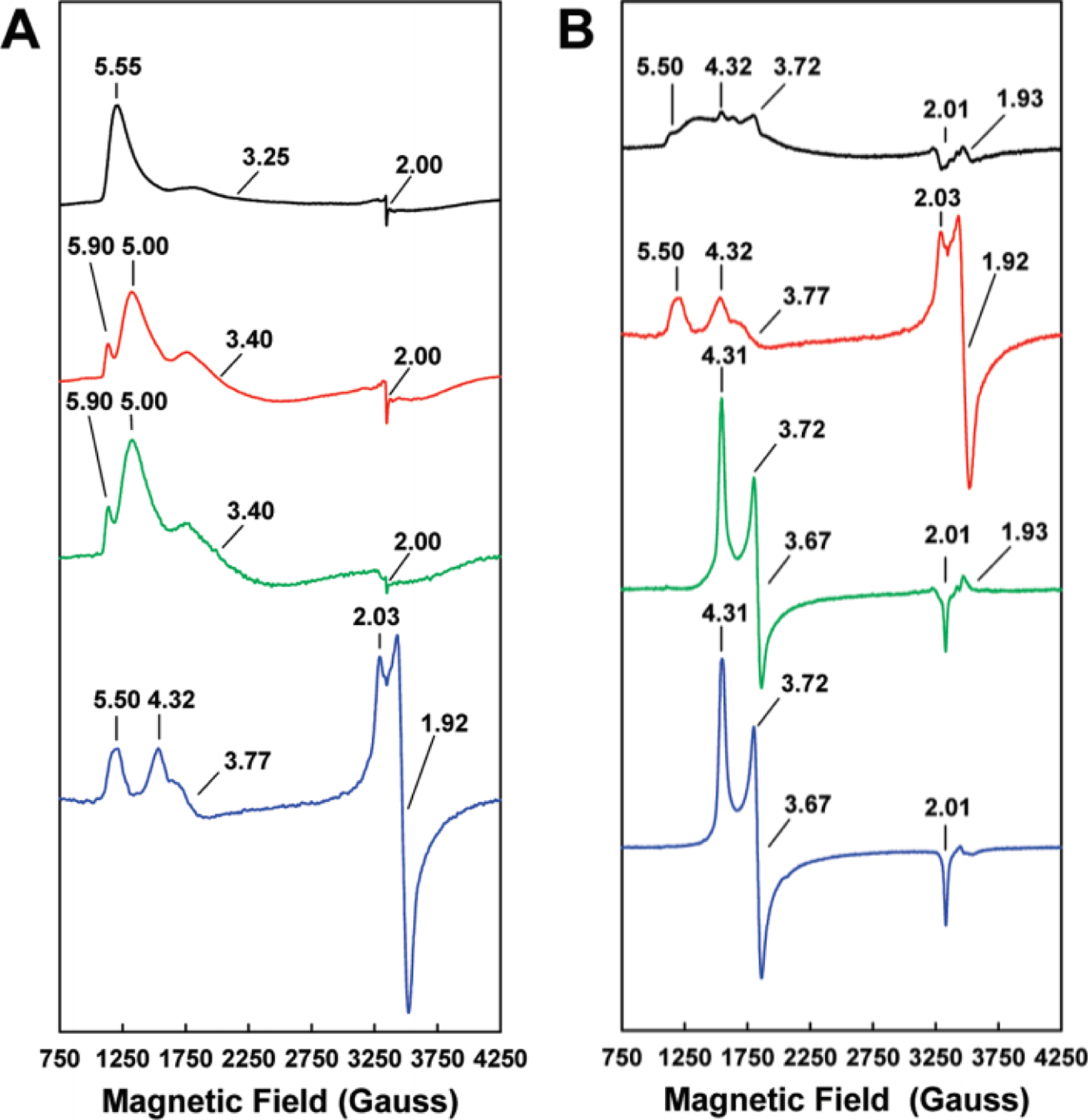
EPR spectra of the isolated Av V-cluster (A) and apo-MoFe protein from A. vinelandii, reconstituted with V- or M-cluster (B). A: EPR spectra of V-cluster in NMF (black), in NMF plus 10 mM thiophenol (red), in NMF plus 1,4-benzenedithiol (green) or in the wild-type VFe protein (blue). B: EPR spectra of V-cluster reconstituted apo-MoFe protein (V-MoFe, black), wild-type VFe protein (red), M-cluster reconstituted apo-MoFe protein (green) and wild-type MoFe protein (blue). Adapted with permission from ref 168. Copyright 2010 American Chemical Society.
More recently, DeBeer, Kovacs and co-workers170 used a combination of DFT calculations, high-energy resolution fluorescence detected (HERFD) XAS, as well as non-resonant XES techniques to further probe the electronic properties of the isolated Av V-cluster. The calculations suggested that the oxidation state of the V atom in the V-cluster was V(III), which is similar to the assignment made of Mo(III) in the M-cluster using a similar methodology.170–172 Interestingly, the subsequent X-ray analysis also suggested that the Fe atoms of the V-cluster were in a more reduced state compared to the M-cluster counterparts.170 This observation seems to correlate with the measured midpoint potentials of −414 mV and −270 mV versus SHE for the isolated V- and M-clusters, respectively,139 with the more negative potential relating to a seemingly more reduced V-cluster. Further, the iron-heterometal bonding interactions were found to be weaker for the V-cluster compared to the M-cluster, which would align with the elongated V---Fe distances observed by XAS of the protein, cofactor and crystallographically characterized V-cluster relative to the Mo-dependent counterpart.170
3.4. The Characterization of Fe-only Nitrogenase
The Fe-only nitrogenase catalytic protein (AnfDGK or FeFe protein) is encoded by the anfDGK genes, and has been expressed in bacterial strains that delete or inactivate the genes for Mo- and V-nitrogenases, or the cells are grown in the presence of tungstate (WO42−) to repress the expression of Mo-nitrogenase (when the organism lacks genes for VnfDGK).173–177 The AnfDGK protein has been isolated from A. vinelandii, R. capsulatus, Rhodospirillum rubrum, and R. palustris, but the determination of the subunit composition was fraught with issues similar to those observed for V-nitrogenase.178,179 There were initially two compositions reported for Av AnfDGK, an α2β2 variant analogous to the Mo-nitrogenase, and an αβ2 species similar to that observed for the Av VFeA protein.66,148 Subsequent studies resulted in a composition for FeFe protein from A. vinelandii and R. capsulatus consistent with a heterohexameric α2β2δ2 formulation and a size of Mr = ~250 kDa (AnfD ~ 59 kDa, AnfK ~ 51 kDa, AnfG ~ 14 kDa).115,180 To date, there is no reported crystal structure of AnfDGK, though, one might anticipate that the protein would retain a structure analogous to those of the Mo- and V-dependent systems based on the similarity between the three systems. AnfDGK has similar subunit size and composition to V- and Mo-nitrogenases (noting that Mo-nitrogenase lacks a NifG subunit), and when the V- and Mo-nitrogenase crystal structures are overlaid, there is modest overlap, with a root-mean-squared deviation of 1.97 Å for all atoms.74 However, these factors alone are not sufficient to predict structure, so additional comparison will need to wait for the crystallography of AnfDGK.
Fe-nitrogenase has limited characterization compared to the other nitrogenase species discussed, however, spectroscopic analysis of the catalytic protein from R. capsulatus by EPR, XAS and Mössbauer spectroscopies provided insight into the structure and properties of the cofactors.76,115,133 In the presence of dithionite, the EPR spectrum of the Rc FeFe protein (FeFeN) is featureless, indicating that both the Fe- and P-clusters have a diamagnetic S = 0 spin state.115 This deviates from the observations in the MoFe protein where the dithionite-reduced state (MN) of the protein has a characteristic S = 3/2 signal (g = 4.29, 3.67, 2.01 for Rc MoFe protein) assigned to the M-cluster (Table 3). The Mössbauer spectrum of the 57Fe-enriched FeFe protein demonstrated that the Fe-cluster in the resting state (FeN) was consistent with 8 total Fe centers, 4 Fe(II) and 4 Fe(III), while the P-cluster was composed of 8 ferrous centers, consistent with the PN assignment from Mo-nitrogenase.76 However, there are some caveats associated with the Mössbauer analysis. The P- and Fe-clusters were not separately labeled with 57Fe like has been done for the MoFe protein,181,182 so the contributions from both clusters appeared in the spectra of AnfDGK. The Mössbauer analysis of the VFe protein also suffers from this uniform 57Fe labeling, and similar methodology is employed for its analysis.161 As such, for the V- and Fe-only nitrogenases an assumption was made that the P-clusters of the alternative systems were spectroscopically analogous to those in NifDK. This was based on the similarity of the fit parameters for the Mössbauer features associated with the P-cluster in NifDK, but also as a means to reduce the number of free variables required for least-squares fitting of the data.76,161 The remaining V/Fe-cluster contributions were fit, and isomer shifts as well as quadrupole splittings were extracted that compare well to those of the M-cluster.161,181 For AnfDGK, the oxidation state assignment for the Fe sites of the Fe-cluster was then based on the observed diamagnetism in the FeN state, which could be achieved with equal amounts of ferrous and ferric Fe sites.76 More recently, X-ray spectroscopy, DFT calculations, and crystallographic methods have been used to reassess the oxidation states of the Fe sites in the M-cluster due to the controversial assignment of the Mo center to be in the 3+ rather than the previously assumed 4+ state.171,183–187 While these efforts have raised questions about the previous Mössbauer analysis, there have been no studies published in parallel for the alternative systems, so it is unclear how the oxidation state assignments would differ, if at all.
The FeFe protein can also be subjected to turnover conditions, and with a component ratio of 1:10 AnfH:AnfDGK a new rhombic S = ½ signal appeared in the EPR spectrum with resonances at g = 1.96, 1.92, 1.77, which were assigned to the one-electron-reduced form of the Fe-cluster (FeR).133 Under the same conditions, the MoFe protein yielded a featureless EPR spectrum, further differentiating the Fe-only nitrogenase. Attempts to oxidize the Fe- and P-clusters were also carried out using potentiometric titrations to correlate the EPR signals to the redox behavior. Two S = ½ species were observed (Figure 18, spectrum 3 and 4), one species had a narrow signal with g = 2.00, 1.98, 1.96 and an Em = −80 mV versus SHE, and the other signal was broad with g = 2.27, 2.06 and an Em ~ +80 mV versus SHE (Table 3).133 The narrow signal compared well to the three-electron-oxidized P3+ species observed for the Av MoFe protein, so was assigned analogously for the Rc FeFe protein.120 The broad signal was tentatively assigned to the one-electron-oxidized state of the Fe-cluster (FeFeOX).133 Surprisingly, there was no evidence of the P1+ or POX states of the P-cluster, as both produce EPR active signals in Mo-nitrogenase (see Section 3.2.1).
Figure 18.
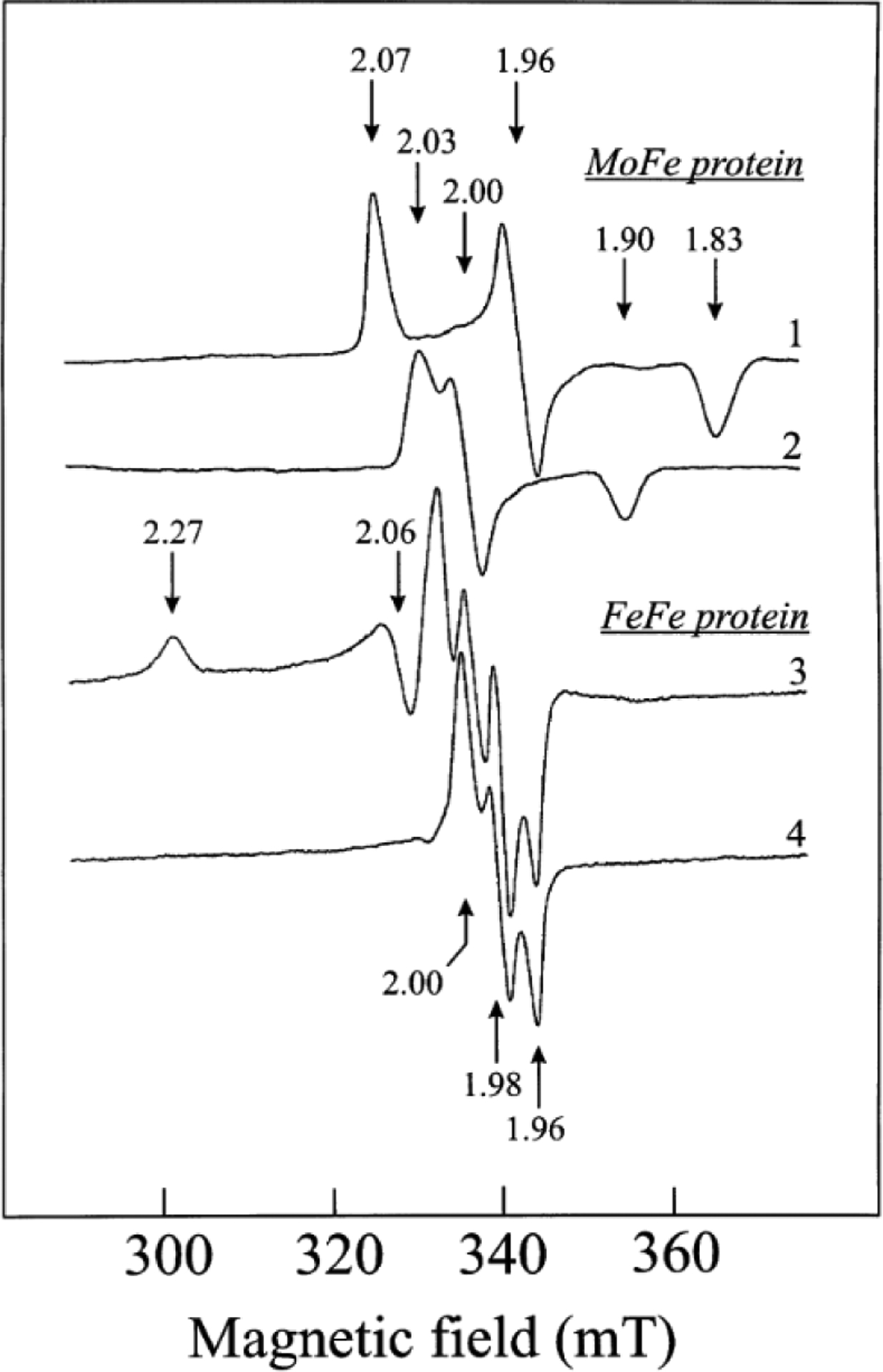
The EPR spectra of the Rc MoFe protein (1, 2) and the Rc FeFe protein (3, 4). The Rc MoFe protein oxidized with 2 mM (spectrum 1) and 4 mM (spectrum 2) K3[Fe(CN)6]. The Rc FeFe protein oxidized with 2.5 mM K3[Fe(CN)6] and measured at 16 K (spectrum 3) or 10 K (spectrum 4). Reproduced with permission from ref 133. Copyright 2002 John Wiley and Sons.
The structural information available about AnfDGK and its metallocofactors is limited to the Fe K-edge XAS analysis that provided characterization of the metal clusters housed in the protein.76 The Rc FeFe protein was analyzed, but XAS provides average information, so the absorber-scatterer pairs that were fit reflect contributions from both the P- and Fe-clusters in the protein. Other methods of analysis have been used for the MoFe protein that involve obtaining XAS data on the holo-protein (P- and M-clusters) and the apo-protein (P-cluster only) then performing a weighted subtraction to characterize the M-cluster contributions alone,44 but this type of study has not yet been extended to the FeFe protein. Despite the logistically challenging circumstances, the EXAFS analysis of Rc AnfDGK provided data that appeared similar to analogously collected Rc NifDK, both in the position and general intensity of the Fourier transform features and k3-weighted data.76 Importantly, an Fe---Fe separation of 3.68 Å was a necessary component for the best fit of the data, and this feature is diagnostic in the M-cluster of trigonal prismatic Fe sites, assigned to Fe---Fe distances between the 6 core Fe centers of the cluster (Figure 19). The 3.68-Å distance paired with the other fitting parameters allowed for the proposal that the Fe-cluster is structurally analogous to the M-cluster. This also agrees with the XAS characterization of the precursor L-cluster, as this species should be effectively the same as the Fe-cluster, absent the R-homocitrate ligand.45,56,59
Figure 19.
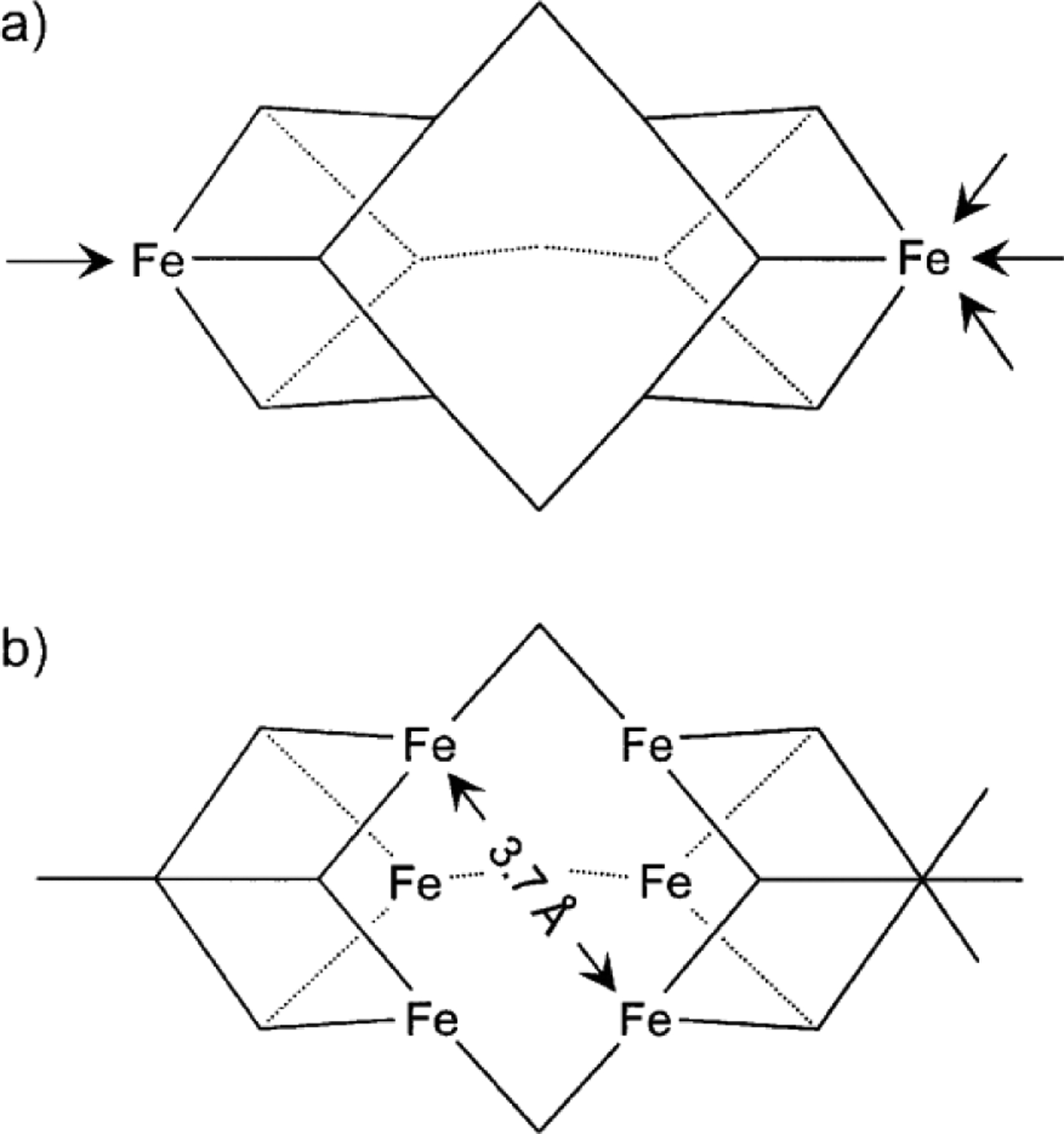
Structural elements of the Fe-cluster from the Rc FeFe protein based on spectroscopic analysis, the terminal Fe-capped ends of the cluster (a) and the trigonal prismatic Fe-atom arrangement of the cofactor from EXAFS analysis (b). Reproduced with permission from ref 76. Copyright 2001 Springer Nature.
4. Reactivity of the Alternative Nitrogenases
The reactivity of nitrogenase is intriguing, as it is the only biological system capable of completely cleaving the strong triple bond of N2 to generate ammonia under ambient temperature and pressure. It is a multi-electron, multi-proton transfer process that requires nucleotide-dependent electron transduction from the reductase component (Fe protein) to the catalytic component (MFe protein; M = Mo, V or Fe), where the P- and M/V/Fe-clusters facilitate transfer to the substrate.66,83,101,188,189 Proton translocation and substrate hydrogenation are also key aspects of nitrogenase that have been explored through biochemical,190–192 crystallographic,193 and theoretical studies.194–196 However, the data is primarily from Mo-nitrogenase and has yet to be replicated to the same degree, if at all, in the V- or Fe-only nitrogenases. Thus, relevant detail will be described as it relates to the alternative systems but will not constitute an exhaustive review of the MoFe protein.
N2 is reduced using 8 H+ and 8 e− equivalents in Mo-nitrogenase, even though N≡N bond scission requires 6 H+/e− equivalents (reaction 1). This is because one equivalent of H2 is generated as part of the mechanism for N2, a phenomenon that is described in more detail in Section 5. In the absence of N2 or any other substrate, nitrogenase continues to generate H2 from protons (reaction 2) concurrent with ATP hydrolysis, demonstrating hydrogenase activity.66,197
| (2) |
Over time, other gaseous substrates with triple bonds, such as acetylene (C2H2) and carbon monoxide (CO) have been investigated as N2 analogs with differing behavior. Subsequent study expanded the substrate scope to include nitrile compounds such as hydrogen cyanide (HCN), nitrogen-containing species such as azide (N3−), nitrite (NO2−) and nitric oxide (NO), unsaturated cyclic compounds (cyclopropene, diazirine) and alkyne species with terminal triple bonds such as propyne (HC≡C–CH3) and propargyl alcohol (HC≡C–CH2–OH).66,197–200 These substrates are reactive to varying degrees, some of which mildly or strongly inhibit the enzyme. Most of these compounds have no real physiological relevance but are useful for understanding the reductive capability of nitrogenase. An overall summary of the substrate reactivity with respect to the alternative nitrogenases is shown in Figure 20.
Figure 20.
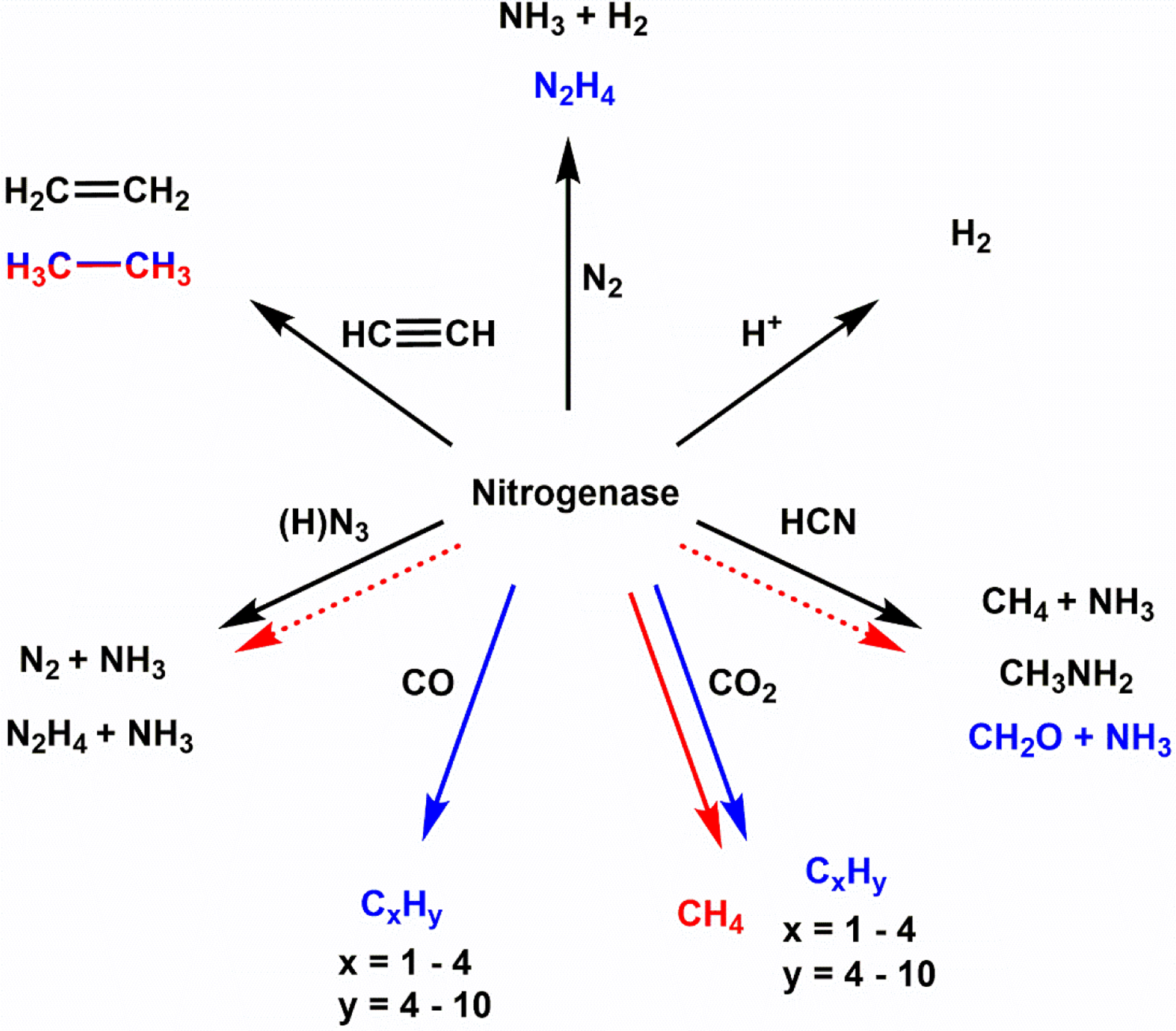
Overall summary of the reactions catalyzed by the alternative nitrogenases. Black lines and products represent the capabilities of all nitrogenases, blue and red lines and products represent the capabilities of V/Fe-nitrogenases, respectively. The dotted red lines reflect that Fe-nitrogenase may be able to carry out the reaction, but it has not been shown experimentally.
The following section will cover the reactivity of the alternative nitrogenases, including the individual catalytic and reductase components, and how they compare to the well-characterized Mo-dependent system. The reactions of V- and Fe-only nitrogenases have been explored with the ‘standard’ scope of nitrogenase substrates, N2, H+ and C2H2, the ‘alternative’ substrates azide and cyanide, as well as the ‘newly-discovered’ substrates carbon monoxide and carbon dioxide. The reductase proteins NifH and VnfH are also independently reactive, and have been recently shown to facilitate the reversible conversion of CO to CO2 in addition to their electron transfer capabilities.
4.1. Technical Considerations for the Study of Nitrogenase Reactivity
An important concept for the study of nitrogenase and the associated reactivity is called the electron flux or electron flow, which is the availability of electrons during a reaction. The reduction of protons to H2 in the absence of any other substrate provides a direct measure of the electron flux in nitrogenase, and as there is no other substrate known to increase H2 production, the proton reduction activity reflects a maximum turnover rate for the system.198 Along with this, Watt and Burns showed that the total electron flux through nitrogenase is not sensitive to the substrate,201 so the total electron equivalents found in substrate reduction products should be equivalent to H2 production in the absence of substrate, in general, for a well behaved system. For example, V-nitrogenase can reduce acetylene to ethylene (2-electron product) and ethane (4-electron product) but will also generate H2 (2-electron product) in the acetylene atmosphere. The rate of electron consumption with respect to C2H4, C2H6 and H2 formed from this reaction should roughly equal H2 production in an Ar atmosphere, and for this reason, the concept has also been referred to as the electron balance. There are examples of inhibitors, like cyanide (CN−), that reduce the total electron flow through the system (i.e. CN− binding decreases all substrate reduction including of H+ under Ar, but CO binding arrests substrate reduction, but H+ under Ar is unchanged) though this is not as common. The electron flux can also be controlled through the amount of reductase component that is used during catalytic turnover. The nucleotide-loaded Fe protein needs to dock with the MFe component to form a complex before an electron can be transferred and ATP can be hydrolyzed, so increasing the molar ratio of Fe protein to MFe protein can allow for an increased rate of electron consumption, and thus a higher electron flux. The component ratio can affect the activity of the enzyme, but each species of nitrogenase and each substrate may require different ratios to fine tune the system. Typical ‘high-flux’ ratios are 20:1 or 30:1, but Fe protein can be at saturating concentrations at ~8:1 under certain conditions,202 and ‘low-flux’ would constitute ratios at ~1:1 or smaller.
Another important aspect to the reactivity is the quality of the proteins that are used in assays. Acquisition of the individual nitrogenase component proteins is nontrivial, and the method of purification can potentially affect the activity. Both the MFe and Fe proteins contain FeS clusters, each of which is sensitive to O2, so all of the purification is necessarily done under anaerobic conditions.202 Nitrogenase proteins do not overexpress particularly well in the organism, and so hundreds of grams of cells are required to yield protein on a scale of a few hundred milligrams, and up to a gram. Initial reactivity studies often began with cell extracts, the soluble fraction of the cell lysate, which contain the target nitrogenase proteins with many other proteins from the cell for this reason. Whole cell assays, with acetylene as a substrate, have also been used as a diagnostic tool for identifying nitrogen fixation capability in bacteria, as obtaining isolated protein is time intensive and the direct measure of NH4+ is challenging.203,204 Purification can take several days in some cases, but longer when affinity tags are not used. The alternative nitrogenase proteins are also rather sensitive, and can produce material with variable subunit composition, complicating characterization.148,175,178
4.2. Standard Nitrogenase Substrate Reduction Activity Towards Dinitrogen, Protons and Acetylene.
Nitrogenase reactivity has been extensively studied, but the proteins utilized do not always originate from the same organisms. The Mo-nitrogenase from A. vinelandii (Av) is one of the best characterized, but the MoFe protein from K. pneumoniae (Kp), C. pasteurianum (Cp), A. chroococcum (Ac), and R. capsulatus (Rc) have also been reported. However, alternative nitrogenase proteins have been isolated and studied from a smaller selection of organisms. V-nitrogenase has only been isolated from A. vinelandii142 and A. chroococcum,141 while the Fe-only variant has been isolated from A. vinelandii,175,176 R. capsulatus,115 R. rubrum (Rr),173 and R. palustris (Rp).177 Generally, each subset of nitrogenase proteins reacts in similar ways to proteins from other organisms, though the same experiments or same analysis is not always available. Table 4 reflects a selection of reported specific activities for the reaction of various nitrogenase proteins with the physiological substrates N2 and H+, as well as C2H2.
Table 4.
Specific Activities for Standard Nitrogenase Substrate Assaysa
| Products | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sp. | Protein | NH3gg (N2) |
H2gg (N2) |
C2H4b (C2H2) |
C2H6b (C2H2) |
H2b (C2H2) |
H2c (Ar) |
N2Hgg (N2) |
Protein Ratiod |
Ref |
| NifDK | ||||||||||
| Ac | MoFee | 436±32 | 296±4.9 | ~2100f | - | - | 1541f | - | 4:1 | 205–207 |
| Av | MoFeg | 520 | - | 2000 | - | 303 | 2200 | - | ~10:1f | 142,175 |
| Av | MoFe | 78h | - | 1003±93 | - | - | - | - | 60:1 | 150,208 |
| Av | M-MoFei,j | 715±82 | 218±13 | 1393±101 | <0.3 | - | 1481±110 | - | 30:1 | 168,222 |
| Av | V-MoFei,j | 166±17 | 174±10 | 366±27 | 6±1 | - | 363±39 | - | 30:1 | 168,222 |
| Rc | MoFek | 235 | 210 | 1200 | - | 170l | 1300 | - | 40:1 | 115 |
| VnfDGK | ||||||||||
| Ac | VFem | 259±8h (177n) |
375±18h | - | - | - | 1516 | 1.25n | 3:1 | 205,207 |
| Ac | VFem | 265 | - | 426 | 8.5 | - | 1094 | - | 10:1 | 217 |
| Ac | VFem | 350 | 928 | 608 | 15 | 998 | 1348 | - | 20:1 | 141 |
| Av | VFeo | 330 | - | 220 | - | - | 1400 | - | ~10:1f | 142 |
| Av | VFep | ~400dd | - | - | - | - | - | - | 20:1 | 220 |
| Av | VFep | - | - | 454±21 | 4.93±0.64 | - | - | - | 60:1 | 208 |
| Av | VFep | 19 | “similar range to ref 208” | 4:1 | 150 | |||||
| Av | VFep | - | - | 11.0±1 | 0.258±0.05 | - | - | - | 60:1 | 221 |
| Av | VFei,q | 458 | 792 | 560 | 17 | - | 1730 | - | 30:1 | 149 |
| Av | M-VFei,r | 20±3ee | 52±3ee | 39±1ee | 3.6±0.1ee | 61±3ee | 104±7.8ee | - | 30:1 | 23 |
| Av | M-VFei,s | 15±1 | 188±23 | 66±4 | - | - | 183±11 | - | 30:1 | 34 |
| AnfDGK | ||||||||||
| Av | FeFet | 181±5 | 599±57 | 306±3 | - | 484±3 | 1085±41 | - | 30:1 | 180 |
| Av | FeFeu | 110 | 220 | 58 | 26 | 226 | 350 | - | 20:1 | 176 |
| Av | FeFev (3Slow) |
38 | 213 | 28 | - | 202 | 253 | - | 10:1 | 175 |
| Av | FeFev (3Fast) |
30 | 145 | 18 | - | 124 | 203 | - | 10:1 | 175 |
| Rc | FeFew | 190 | 1300 | 260 (345x) |
ND | 1800l | 2600 | - | 40:1 | 115,178 |
| Rr | FeFey | 3.6z | 67 | 6.5 | 0.22 | 63 | 71 | - | 1:1 | 173 |
| Rp | FeFei,aa | ~80bb | - | - | - | - | ~480bb | - | - | 177 |
| W-incorporation | ||||||||||
| Rc | W-MoFecc | NDhh | - | 1 | - | - | 102 | - | 2:1 | 223 |
| Av | W-MoFeff | 145 | 165 | 550 | - | 70 | 610 | - | ~10:1f | 224 |
Examples are selected for comparison of relevant Mo-dependent and Mo-independent systems. The protein labels with a dash indicate the metallocluster before the dash, and the polypeptide used after the dash (e.g. W-MoFe is the tungsten cluster inside of the MoFe protein). Specific activities are reported with units of nmol product × min−1 × (mg protein)−1. Temperature of assays reported at 30 °C. Gas in parenthesis reflects the substrate conditions used for the assay.
assays reported in 10% C2H2, 90% Ar atmosphere.
assays reported in 100% Ar atmosphere.
ratio of the reductase Fe protein to catalytic nitrogenase protein.
purified from a WO42− tolerant strain MCD50.
reported under saturating conditions of Fe protein.
purified from WT strain UW.
reduction assay uses 4:1 ratio of Fe protein:MFe protein.
histidine affinity tag.
apo-MoFe protein purified from ΔnifB deletion strain DJ1143. The appropriate isolated cofactor, M- or V-cluster are then used to reconstitute the protein.
purified from the WT strain B10S.
assay reported in 5% C2H2 atmosphere.
purified from ΔnifHDK deletion, WO42− tolerant strain MCD1155.
assay reported at 40 °C.
purified from ΔnifHDK deletion strain modified from WT UW.
purified from WT Lipman 1903 strain, after growing cells multiple times in liquid culture and agar plates with Na3VO4 media in lieu of Na2MoO4-containing media as described in ref 208.
purified from ΔnifHDK deletion, WO42− tolerant strain YM68A.
protein purified from ΔnifHDK deletion, WO42− tolerant strain YM68A. Cells grown after depletion of V salt from the fermenter, with subsequent inclusion of excess Mo salts.
apo-VnfDK protein was purified from ΔnifHDKB deletion, WO42− tolerant strain YM7A. The protein was then reconstituted with M-cluster, was found to have a αβ2 composition.
purified from ΔnifDK, ΔvnfK deletion, WO42− tolerant strain DJ1255.
purified from ΔnifHDK, ΔvnfDGK deletion, Mo-tolerant strain RP306.
purified from ΔnifHDK deletion, WO42− tolerant strain CA11.
purified from ΔnifHDK deletion strain modified from WT B10S.
assay reported in 100% C2H2 atmosphere.
purified from WT strain UR206 in the presence of NaWO4.
assay reported in 46% 15N2, 54% Ar atmosphere.
purified from ΔnifH, ΔvnfH deletion strain CGA7554.
converted from nmol product × nmol protein to nmol product × min−1 × (mg protein)−1.
purified from an anfA mutant of the WT strain B10S that inactivates Fe-only nitrogenase, cells grown with Na2WO4.
converted from nmol product × s−1 × (nmol protein)−1 at 1 atm N2 to nmol product × min−1 × (mg protein)−1.
activities reported with units of nmol product × min−1 × (nmol cofactor)−1, where nmol cofactor is determined by metal analysis of the protein.
purified from WO42− tolerant strain UW-LM2, after a series of growths under Mo-free conditions to deplete Mo-stores in the cell.
assay reported in 100% N2 atmosphere.
ND = not detected.
For Mo-nitrogenase, the proteins from A. vinelandii and A. chroococcum are similarly active, however, making a comparison of activity can be difficult because the molar ratio of Fe:MoFe proteins is not consistent for each set of experiments, even for the same protein. For instance, specific activities (units of nmol product × min−1 × (mg MFe protein)−1) for NH3 formation in Ac and Av MoFe proteins are 436 and 520, respectively, but the Ac experiments report a component ratio of 4:1, whereas the Av experiments state that Fe protein is present in saturating concentrations, with a likely ratio of ~10:1.142,205 At first glance, it appears that the Av MoFe protein is ~20% more active than the Ac MoFe protein, though with a higher electron flux they might be comparable. Proton and acetylene reduction for both species was reported with saturating amounts of Fe protein, and the Ac MoFe protein appears ~30% less active during proton turnover (1541 compared to 2200), but is comparable to the Av MoFe for the reaction with acetylene (~2000).175,206,207 Taking electron balance into account, in the Ac MoFe protein series, the N2 reduction products roughly equal H2 production under Ar (~3000 nmol electrons per minute) whereas C2H4 formation accounts for 4200 nmol of electrons per minute, and does not include H2 formed (was not reported, Table 4). This difference could reflect variable reaction conditions between the N2, H+ and C2H2 experiments, or could be the result of either changing protein ratios or batch-to-batch variation of the proteins used. To further illustrate this point, Einsle and co-workers reported ammonia and ethylene formation from reaction with the Av MoFe protein, and the specific activities were 78 and 1003, respectively.150,208 These values reflect a smaller percentage (15% and 50%) of the previously reported activities for the Av MoFe protein, but the H2 production is not reported under any conditions, so it is not possible to accurately assess the electron balance. The smaller values are not likely the result of low electron flux, as the component ratio is 60:1. The Rc MoFe protein appears to be genuinely less active than either Ac or Av MoFe proteins, even with a 40:1 molar ratio of reductase.115
The activities for various reports of V- and Fe-only nitrogenase are also summarized in Table 4. In general, there is a decrease in activity by the alternative systems across the standard scope of substrates compared to Mo-nitrogenase from the same organism. The Ac VFe protein is ~60% as active for N2 reduction as the Ac MoFe protein, and Av VnfDGK is ~75% as active as the respective Mo-system.142,149,205 The FeFe protein from A. vinelandii shows an even lower N2 reduction activity, 35% compared to MoFe protein, but the Rc FeFe protein maintains 80% of the specific activity of Rc NifDK (190 versus 235). However, the activities for Av and Rc AnfDGK are similar (180 and 190, respectively).115,176,180 Compared to MoFe protein, the alternative nitrogenases direct a larger fraction of electron equivalents towards H2 formation during N2 turnover (Table 4) with FeFe protein> VFe protein> MoFe protein, and this has been interpreted to mean there is a general inefficiency of VnfDGK and AnfDGK to facilitate the reduction of N2. The differential production of H2 during turnover is still an active area of investigation and has important mechanistic considerations that will be discussed further in Section 5. Proton reduction is also decreased in the alternative systems compared to the Mo-dependent nitrogenase, except in the case the Rc FeFe protein, which has an H2 formation rate two times that of the Rc MoFe protein (Table 4).115
Similar to NifDK, the VFe proteins from A. chroococcum and A. vinelandii are comparably active towards the standard substrates, but the electron flux conditions should be taken into account for the best comparisons. The Ac VFe protein fortunately has been reported with varying degrees of completeness using different ratios of Fe protein to VFe protein, and the general trend observed is an increase in activity with an increase of the ratio from ~4:1 to 20:1 (Table 4). Av VnfDGK has been shown to generate products with component ratios of 4:1 to 60:1, but the closest comparison is at 20:1 for Ac and 30:1 for Av.141,149 VnfDGK from A. chroococcum is ~25% less active for N2 turnover than A. vinelandii (specific activity of 350 compared to 458), but Ac VFe protein produces 15% more hydrogen during N2 turnover than Av VFe protein (928 versus 792). This suggests that the Av VFe protein is slightly more efficient at nitrogen fixation than the Ac VFe protein. The reduction of protons is also found with a 20% higher rate for the A. vinelandii protein (1730) compared to A. chroococcum (1348), supporting that the Av VFe protein is more active in general. Interestingly, Dilworth and Eady found that Ac VnfDGK was able to additionally turnover N2 and release hydrazine (N2H4) as a product with a specific activity of 1.25 nmol product × min−1 × (mg VFe protein)−1 at 40 °C using 3:1 ratio of reductase.207 Hydrazine was also found not to serve as a substrate for the VFe protein, which is in contrast to the Mo-systems that will turnover hydrazine but do not produce it, except when the protein is acid or base quenched.209–211 This activity has not been reported for the V-nitrogenase from A. vinelandii.
The comparison of Fe-only nitrogenase proteins also echoes similar reactivity trends towards the physiological substrates as the V-nitrogenase. The activity of the Av FeFe protein has been reported several times,175,176,180 but the highest activity comes from Seefeldt and co-workers using a protein component ratio of 30:1.180 The FeFe protein produces even more H2 during N2 turnover than the VFe protein, divertinĝ50% of the electron flux towards H2 and decreasing efficiency of the reaction, whereas the Av VFe and MoFe proteins employ 37% and ~20% of electrons towards proton reduction, respectively. AnfDGK from R. capsulatus favors H2 formation to an even higher degree, using 70% of the total electron flow under an N2 atmosphere, and consequently has a much higher specific activity for proton reduction in an Ar atmosphere than Av AnfDGK (2600 versus 1085). The Fe-only nitrogenases from R. palustris and R. rubrum are capable of 40% and 2% of the activity observed for N2 reduction by Av and Rc FeFe proteins, respectively, and are not particularly active towards proton reduction (Table 4), implying further optimization of conditions may be necessary.173,177 Though, the Rr FeFe protein was reported with a Fe protein to FeFe protein ratio of 1:1, reflecting low flux conditions, and a component ratio was not provided for the Rp FeFe protein at all.
Generally, the alternative nitrogenases are not particularly efficient at converting N2 to NH3 and are biased toward H2 formation as compared to the Mo-dependent system, but the non-physiological substrate acetylene reacts differently with the alternative systems. Acetylene is a good substrate for Mo-nitrogenase with apparent Km values reported between 0.3–2 kPa, and Mo-nitrogenase exclusively produces ethylene (C2H4) with a specific activity upwards of 2000 nmol product × min−1 × (mg MoFe protein)−1 following the general reaction 3.142,206,212,213
| (3) |
A small amount of H2 is produced during acetylene turnover, but the substrate strongly inhibits proton reduction such that >90% of electron flux is funneled towards ethylene formation.66,214 Further, as the concentration of acetylene is increased, H2 production can be completely stopped, whereas H2 is always produced during N2 turnover.215,216
Compared to the MoFe protein, acetylene is generally considered a poor substrate for the alternative nitrogenases. The VFe and FeFe proteins have a slightly lower affinity for the substrate, with apparent Km values of 6 kPa for Av and Ac VFe proteins179,217 and 14 kPa180 and 12.5 kPa115 for Av and Rc FeFe proteins, respectively. In contrast, the VFe and FeFe proteins achieve ~30% and ~20% of the specific activity for the formation of C2H4 relative to the Mo-dependent system (Table 4). Despite the lower activity, formation of C2H4 by VnfDGK (and extension AnfDGK) is proposed to follow a similar pathway as NifDK.217 Eady and co-workers found the transformation catalyzed by the Ac VFe protein to be highly stereospecific, producing [cis-2H2]C2H4 when 2H2O is used in the buffer, in line with observations of the Mo-dependent system.217 Unlike for MoFe protein, the alternative nitrogenases are less affected by the C2H2-induced inhibition of proton reduction, as only ~35% and ~15% of the electron flow is diverted to C2H4 formation by VFe and FeFe proteins, respectively, while the majority is converted into H2.115,217 Ethane can also be generated from acetylene, likely following reaction 4, but it is only observed in the alternative nitrogenases and equates to ≤ 5% of electron flux for ethylene formation.178,179
| (4) |
The exclusivity of ethane as a product has led to its use in the identification of nitrogen fixation activity by alternative nitrogenases in whole cell assays.13,14,218,219 It is not exactly clear how the reduction occurs, but free C2H4 is not a reaction intermediate. This implies there might be different mechanisms for the formation of C2H4 and C2H6, because pure C2H4 is not reduced to C2H6.217 The reaction also has different pH profiles for the VFe and FeFe proteins. Ac VnfDGK is able to maintain consistent ethane production across a pH range of 6.5 to 7.5, then the specific activity decreases above pH 7.5;217 this is in contrast to the Rc FeFe protein where product formation maximizes at ~pH 6.6 and steadily decreases at increased pH values.115
It is not understood why there is a difference in reactivity between the Mo-dependent and Mo-independent nitrogenases. One possible contributing factor for the V-nitrogenase protein is that it may be more sensitive than the Mo-dependent counterpart. Blanchard and Hales reported that during purification of the A. vinelandii VFe protein, two forms of the enzyme were isolated, one with an α2β2(δ) structure, and another slightly less active form with an αβ2(δ) composition where the amount of the δ subunit was variable.148 There were no isolated alternative forms of the A. chroococcum VFe protein reported, however, there were mentions of multiple unresolvable forms of V-nitrogenase with lower activities during the 6-day purification, but these less active fractions were discarded.141 The use of a histidine-affinity-tag on the A. vinelandii VFe protein substantially reduced the time and column runs required to obtain purified protein, and while there may be several factors involved, the His-tagged VFe protein currently produces the highest specific activities for substrate reduction for the system (Table 4).149
Within the literature, there are several indications that expression and purification of the VFe protein could play roles in the observed reactivity. The A. vinelandii VFe protein expressed and purified by Einsle and co-workers is interesting, as it came from a wild-type organism, unlike the gene deletion variants expressed by others.208 The resulting protein was selectively expressed through repeated growths under strictly Mo-free conditions in the presence of Na3VO4 as a vanadium source. VnfDGK was purified without reported alternate conformations, in contrast to the previous studies of non-tagged VnfDGK from A. chroococcum and A. vinelandii.141,148 Specific activities of 454 and 4.93 nmol product × min−1 × (mg VFe)−1 for the generation of C2H4 and C2H6, respectively, were indicative of alternative nitrogenase activity (Table 4). However, neither N2 or H+ reduction activities, nor metal analyses are reported.208 Subsequently, this protein variant was used to generate the first crystal structure of V-nitrogenase,74 as well as to trap a putative N2 reduction intermediate in crystallo,150 though, the specific activity for N2 reduction was reported with a value of 19, reflecting ~5% of the activity of untagged or tagged A. vinelandii VFe protein.142,149 Seefeldt and co-workers showed N2 reduction activity of ~400 with the VFe protein obtained using the same strain and purification protocols reported by Einsle, but acetylene and proton reduction activities are not presented in the study.220 Additionally, Minteer and co-workers221 also use the same methodology to obtain and study the VFe protein, and report specific activities of 11 and 0.26 for the formation of C2H4 and C2H6 from acetylene, reflecting ~3% of the activity seen by Einsle.208 So far, the field has not yet reached a general consensus with the standardized activity of Av VFe protein for C2H2 reduction.
For AnfDGK, it was initially unclear if the first reported protein from A. vinelandii had inherently low activity,175 or if minor Mo incorporation into the FeFe protein was responsible for the reactivity,176 implying that the Fe-only cofactor was not active. Like for the VFe protein, the FeFe proteins have also been reported to be generally unstable during purification, which may also have contributed to the attenuated activity.115,175 However, Müller and co-workers were able to purify AnfDGK from R. capsulatus using a faster protocol, demonstrating specific activities that were much higher than the contemporary A. vinelandii and R. rubrum variants, and additionally showed that Mo was not being incorporated into the FeFe protein.115,173,178 More recently, Seefeldt and co-workers180 reported the purification of A. vinelandii FeFe protein from a ΔnifDK, ΔvnfK deletion strain analogous to previous reports,175,176 but the activities were higher, similar to what was observed for Rc FeFe.115,178 The most active Av and Rc proteins show similar specific activities for N2 and C2H2 reduction (181 vs 190 and 306 vs 260, respectively) though the R. capsulatus FeFe protein produces between 2 to 4 times more H2 than the A. vinelandii FeFe protein (Table 4). Metal analysis of the A. vinelandii protein variant showed that Mo and V levels were at or below the detection limit, supporting that the Fe-only cofactor is indeed active.180 A FeFe protein from R. palustris has also been reported by Harwood and co-workers, and is the first Fe-only nitrogenase to implement a histidine-affinity-tag on the protein to assist with purification.177 The R. palustris FeFe protein appears to be mildly active in general; NH3 production is found with a specific activity of ~80 nmol product × min−1 × (mg FeFe)−1, reflecting 40% of the activity as compared to the Av or Rc proteins. A complete substrate study has not yet been shown, so it is unclear the reasons for the difference in activity.
4.3. Reactions of Nitrogenase with Azide and Cyanide
The so-called ‘standard’ nitrogenase substrates beyond dinitrogen are C2H2 and H+, and these have been successfully employed in biochemical assays due to their predictable gaseous reaction products that allow for facile detection.66,198 The acetylene to ethylene and proton to hydrogen reductions are 2e− and 2H+ processes that have a clear product; ethylene can also be further reduced by 2e−/2H+ to ethane by the alternative nitrogenases, as described earlier. Other non-gaseous substrates have also been explored for their reactivity with nitrogenase.66,198 The triatomic azide (N3−) is a logical extension of N2 as a substrate, being a multiply bonded nitrogen-containing species, and the same can be said for cyanide (CN−), as an isoelectronic alternative to dinitrogen. However, these substrates have more complex reduction pathways than the standard counterparts, and they can generate products that stay in solution which can be challenging to detect.
Azide was first shown to be a substrate for Mo-nitrogenase in 1967 by Schöllhorn and Burris, reporting the two-electron reduction using cell extracts from nitrogenase-expressing A. vinelandii and C. pasteurianum strains, that produced equal amounts of N2 and NH3 as shown in reaction 5.225
| (5) |
Subsequently, Dilworth and Thorneley, then later Burgess and co-workers, reinvestigated this reaction using purified protein components from A. vinelandii and K. pneumoniae, finding that ammonia was not generated with equal stoichiometry to dinitrogen, but in excess.226,227 This was interpreted to mean that there was more than one reaction pathway for azide reduction. To further rationalize the observation, extensive product analysis revealed that hydrazine, previously established as a substrate of Mo-nitrogenase,209 was also a product of the reaction. This led to the proposal that nitrogenase could facilitate two additional azide reduction reactions (reaction 6, 7).
| (6) |
| (7) |
While reactions 5–7 were consistent with the experimental observations, it was also possible that excess ammonia could be produced either by the additional reduction of hydrazine into ammonia, or through the reduction of the N2 produced from the 2-electron reaction of azide shown in reaction 5. To test for hydrazine, Dilworth and Thorneley used isotopically labeled 15N2H4 in azide reduction assays, and found that none of the resulting ammonia detected contained the labeled nitrogen, demonstrating that under those conditions hydrazine was not being further reduced.226 Burgess and co-workers were able to assess if the generated N2 could be reduced through native nitrogenase reaction by conducting the experiment using an atmosphere of 2H2 (D2).227 When D2 was added to the azide reduction assay, the amount of N2 produced increased, and a corresponding amount of NH3 decreased, while N2H4 was unaffected. The inhibition of NH3 production by D2 was consistent with the well-known inhibition of N2 reduction by H2(D2),228 and supported that N2 was the source of the excess ammonia. This H2 inhibition behavior will be discussed with more detail in Section 5.
A further complication arose with the analysis due to the speciation of the azide in solution. N3− is a weak base (pKa= 4.6)229 and nitrogenase is active between pH 7 and 8, so both azide and the conjugate hydrazoic acid (HN3) can exist in the reaction; however, the major component would be N3−. As a result, the pH-dependence of the reactions 5 and 6 were also explored, and it was found that N3− was the substrate for reaction 5 while HN3 was the substrate for reaction 6.226,227 Additionally, Mo-nitrogenase was found to have a much higher affinity for HN3 (Km = 12 μM) than N3− (Km = ~1–3 mM), so even with the low concentration of HN3 in solution, it could still serve as a substrate. It was also unclear if and how reaction 7 was participating during the assay.
V-nitrogenase from A. vinelandii was also investigated for a reaction with azide, and compared to Mo-nitrogenase from the same organism by Newton and co-workers.230 In general, the V-dependent enzyme generated the same set of products, including excess NH3 but was less active than the Mo-dependent counterpart, producing N2 and N2H4 at 8% and 33% of the rates for the MoFe protein, respectively (Table 5). It was also found that the VFe protein has a slightly higher affinity for HN3 (Km = ~4 μM) than what was reported for the MoFe protein.227 Interestingly, when the electron pairs required to generate the product are taken into account (e.g., N2H4 is produced from a 6-electron reduction, or 3 electron pairs), the rates of formation by the VFe protein for N2 and N2H4 are roughly equivalent, whereas N2 is formed at a rate 4 times higher than N2H4 for the MoFe protein.230 This was interpreted to mean that the VFe protein has no preference for the 2-electron (reaction 5) or the 6-electron (reaction 6) processes, while the MoFe protein preferentially carries out the 2-electron reduction. The source of the excess ammonia formation was also probed by carrying out the reaction in the absence or presence of an H2 atmosphere. The specific activities for NH3 and N2H4 formation of ~110 and 26 nmol product × min−1 × (mg protein)−1, respectively, were determined under both sets of conditions, implying that N2 generated from azide was not the source of excess ammonia in the V-dependent system, and it was proposed that reaction 7 was instead responsible.230 However, several things should be noted from the study. One point is that an analogous experiment to assess VnfDGK-catalyzed hydrazine reduction was not reported, nor if hydrazine could be generated from N2 formed through reaction 5. This is a salient point, as it has been shown that N2H4 is produced from N2, but not reduced by the VFe protein from A. chroococcum.207 Another point is that the V-dependent reductase, VnfH, was not used for the reported assays and instead NifH was employed, which could potentially contribute to the differences in observed results for the VFe protein. Further, Newton reports that they observe no change in azide reduction products in the presence or absence of H2 in the wild-type and variant Av Mo-nitrogenase systems, in contrast to the study by Burgess using the same enzyme.151,227,230 It is clear that additional investigation is required to clarify and better understand azide reduction by nitrogenase.
Table 5.
Specific Activities for the Reduction of Cyanide and Azide by Nitrogenasea
| Product | ||||||||
|---|---|---|---|---|---|---|---|---|
| Protein | Substrate | H2 (1)h |
N2 (1) |
CH3NH2 (2) |
N2H4 (3) |
CH4 (3) |
NH3 (3) |
ref |
| Av MoFe | KN3 | 1300 | 630 | 53 | 290b | 152 | ||
| Av VFe | KN3 | 470 | 51 | - | 18 | - | 54b | 230 |
| Av MoFe | HCNc,f | 270 | - | 29 | - | 50 | 50 | 151 |
| Av MoFe | HCNd,f | 500e | - | 70e | - | 190e | 250e | 233 |
| Av VFe | HCNc,f | 560 | - | 20 | - | 20 | 20 | 230 |
| Av VFe | HCNc,g | 260 | - | 27 | - | 17 | 22 | 230 |
The reactions are all reported at 30 °C in an Ar atmosphere with a 20:1 molar ratio of the reductase component to the catalytic component. Units are reported as nmol product × min−1 × (mg protein)−1, converted from nmol of electron pairs × min−1 × (mg protein)−1 from ref 230.
there are several potential routes that can generate NH3 from N3−, the activities were converted based on a 6-electron reduction.
substrate added as NaCN dissolved in HEPES buffer pH 7.4 with added HCl.
substrate added as NaCN dissolved in Tes-KOH buffer, adjusted to pH 7.3 with HCl.
reactions reported at pH 7.3 using 8:1 molar ratio of reductase component to catalytic component.
5 mM NaCN used.
50 mM NaCN used.
the number in parenthesis refers to the number of electron pairs required to generate the product.
Cyanide reduction by nitrogenase is equally complex, as the substrate is capable of forming several carbon-containing products in addition to ammonia. Hardy and Knight first showed that HCN could be reduced using Mo-nitrogenase containing cell extracts from A. vinelandii and C. pasteurianum.231 They observed methane (CH4), ammonia, and a small amount of another base that they proposed was methylamine (CH3NH2) by the following reactions:
| (8) |
| (9) |
Further, Hardy and Knight put forward the idea that cyanide was reduced in 2-electron steps in analogy to N2 fixation, starting from HCN to methyleneimine (CH2=NH) in the first step shown by reaction 10,
| (10) |
then subsequently to methylamine in the next 2-electron reduction, finally ending with the cleavage of the C–N bond to form methane and ammonia after a total addition of 6 electrons. However, methyleneimine was not detected in their experiments, and the methylamine product was proposed but the extra base could not be clearly identified.231 Kelly and co-workers were also able to show that cyanide reduction occurred using cell-free extracts from A. chroococcum, and formed a very small amount (< 0.1% of methane formed) of the C2 products ethylene and ethane.232
The use of cell extracts complicates analysis because of the plethora of additional proteins present in solution, so Burgess and co-workers used purified Mo-nitrogenase components from A. vinelandii to probe cyanide reduction.233 Similar to azide, cyanide is present as either the free base CN− or the conjugate acid HCN in solution; a pKa of 9.11 indicates that the major species under the reaction conditions is HCN.234 The pH-dependent reduction studies revealed that in the base form, CN− is a strong reversible inhibitor of electron flow through nitrogenase, but was not the substrate for the reaction.233 HCN was the substrate (Km = 4.5 mM), and could be reduced by 6 electrons as in reaction 8, or by 4 electrons as in reaction 9, and unsurprisingly produces H2 in parallel. The specific activities for the formation of CH4, NH3, CH3NH2 and H2 were found to be 190, 250, 70 and 500 nmol product × min−1 × (mg protein)−1, respectively (Table 5). However, NH3 was generated in excess relative to the amount of CH4 produced, which raised the possibility that either an unobserved 2-electron reduced CH2=NH or the 4-electron reduced CH3NH2 could be responsible. Methylamine was tested as a substrate for Mo-nitrogenase, and CH4 was not detected, indicating that the product could not be further reduced by the enzyme and was likely not a source of the NH3.233 This left a putative 2-electron reduced product as the source of excess ammonia, as methyleneimine would be susceptible to hydrolysis, forming formaldehyde (CH2O) and NH3 (reaction 11), though CH2O was not detected. It was also possible that C–C bond formation from coupling HCN molecules could generate ammonia, but ethylene and ethane were only observed after doubling the protein concentration and still only yielded 0.036% and 0.02% of the CH4 formed, invalidating them as a potential source of ammonia.
| (11) |
Newton and co-workers also investigated cyanide reduction by V-nitrogenase from A. vinelandii, with a specific goal to determine if formaldehyde could be produced and detected.230 There were no pH-dependence experiments done analogously to the Burgess studies with the MoFe protein,233 so the assumption was made that HCN was the substrate for the reaction. Subsequently, CN− was assigned as the inhibitor species in solution. Overall, the VFe protein was less active than the MoFe protein, but was determined to have a similar affinity for the substrate as the MoFe protein (Km = 1.9 mM), and experience less inhibition from CN− than for Mo-nitrogenase (Table 5).230 The product distribution was similar for the VFe and MoFe proteins, though the VFe protein produces a higher amount of CH3NH2 relative to CH4 than the MoFe protein, with CH3NH2:CH4 ratios of 0.66:1 and 0.39:1, respectively. The V-dependent system also generated excess NH3, so to assess if reactions 10 and 11 were operative, extensive effort went into the detection and identification of CH2O or a further reduced CH3OH as products. There were many problems that arose while conducting the analysis, but Newton and co-workers reported the detection and quantification of formaldehyde as a product of HCN reduction, but no methanol was observed, suggesting that the 2-electron reduced product, CH2=NH, is generated. However, the small amount of CH2O detected does not correspond to the excess ammonia observed, so further investigation is necessary to clarify the source of the additional NH3.230
4.4. Expanded Substrate Reactions for Alternative Nitrogenases
Carbon monoxide is a small gaseous molecule that is isoelectronic to dinitrogen, and therefore was of interest in the study of nitrogenase as a substrate analog.66 The strong triple bond of CO can be completely cleaved and converted into methane and water in a 6-electron reduction that is analogous to the basic reduction of N2 (reaction 1).
| (12) |
Carbon monoxide can also be used in coupling reactions to produce longer chain, complex hydrocarbons in a process known as Fischer-Tropsch synthesis, often employing H2 as a source of both protons and electrons.235 In a fundamental sense, coupling of two CO molecules requires 8 protons and electrons to form C2H4 as a product, shown in the following reaction:
| (13) |
With respect to nitrogenase, CO has been established as an inhibitor of substrate reactions with the exception of H+ reduction, and the Mo-dependent system has been extensively explored.66 However, recent discoveries over the past decade have revealed that CO can also behave as a substrate for the V-dependent nitrogenase, showing Fischer-Tropsch-like chemistry. These studies, as well as reactions with carbon dioxide, will be described in the following section.
Like in Mo-nitrogenase, the V-nitrogenases from Av and Ac as well as the Rc Fe-only nitrogenase, in the presence of CO, show complete inhibition of N2 reduction with increasing partial pressures of CO, but C2H2 reduction is affected for the alternative systems less than the Mo-dependent system.217,236 This apparent insensitivity to CO for the reaction has been interpreted to suggest that in some nitrogenases there are multiple acetylene and CO binding sites, each with different affinities for CO, but this is not supported for all of the alternative systems.149,179 Aside from the difference in acetylene reduction, it has been broadly considered that MoFe protein and the alternative nitrogenases react with CO in a similar manner, though careful analysis reveals differences between the systems that are quite intriguing.143 It was observed that for acetylene reduction by Av VnfDGK in the presence of CO, low electron flux (1:5, VnfH:VnfDGK) led to an increase in ethylene formation relative to higher flux (8:1) conditions, and further, ethane formation was enhanced at low pressures of CO.236 This seems to be a unique feature of the A. vinelandii system, as the VFe protein from A. chroococcum demonstrates more typical inhibition of acetylene reduction in the presence of CO.217 Rc AnfDGK has also been shown to feature analogous inhibition of substrate reduction as the MoFe and VFe proteins, particularly at lower partial pressures of CO.115 However, acetylene reduction by AnfDGK in the presence of high concentrations of CO produces an amount of ethane that exceeds ethylene production by ~80%, whereas in V-nitrogenase the two products formed are inhibited with similar behavior.174 As mentioned, hydrogen production by the MoFe and Ac VFe proteins is unsurprisingly stable in a CO environment,217 however, for Av V-nitrogenase it was reported that increasing the atmosphere from 0 to 100% CO (while balancing the pressure with Ar), resulted in a decrease in the specific activity for proton reduction of ~75%.149 It is uncertain why the Av protein has this inhibition pattern, but it has been proposed that there may be two separate mechanisms for H2 evolution, one of which is more sensitive to CO binding.149
It was also discovered that Av V-nitrogenase is capable of reducing CO to longer chain hydrocarbon products, forming C–C bonds (Table 6).237,238 For comparison, Av Mo-nitrogenase is minimally able to convert CO to hydrocarbons with specific activities for C2H4 and C2H6 of 0.025 and 0.013, compared to 32 and 1 for the VFe protein. These CO-based activities are much lower than the standard N2, C2H4, and H+ substrates, as a high degree of electron equivalents are diverted to H2 formation (specific activities of 1700 and 540 nmol H2 × (mg protein)−1 × min−1 for the MoFe and VFe proteins, respectively).238 The product distribution changes when 2H2O and deuterated Tris buffer are used to carry out these reactions.237,238 Greater yields of product are observed in the presence of deuterated solvent and C4 products are generated by NifDK, whereas similar products are observed for VnfDGK in proteo- and deutero-solvents (Table 6).237 These differences also show inverse kinetic isotope effects (KIE) for product formation from CO by the MoFe and VFe proteins, with average KIEs of ~0.1 and ~0.5, respectively. The KIE can be affected by many contributing factors, particularly from modified dynamics of the protein, so more work would be necessary to ascertain the meaning of these effects. Another observation was that the presence of H2 inhibits the formation of C2H4 from CO by ~10% at lower concentrations of H2 (~10% H2, 90% CO) and upwards of 25% at higher concentrations (67% H2, 33% CO).238 H2 is only known to inhibit N2 reduction for nitrogenases,66,217 so inhibition of CO reduction by H2 may indicate that there are similar mechanistic features between the CO and N2 reduction reactions.
Table 6.
Reported Specific Activities for CO and CO2 Reduction by Nitrogenasea
| Products | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sp. | Protein | H2 (CO)q |
CH4 (CO) |
C2H4 (CO) |
C2H6 (CO) |
C3H6 (CO) |
C3H8 (CO) |
C4H8 (CO) |
C4H10 (CO) |
Protein Ratioh |
ref |
| Av | MoFeb,c | ~1700000f | 25 [730] |
13 [110] |
2.9 [55] |
5.9 [63] |
- [6.8] |
- [7.4] |
10:1 | 237,238 | |
| Av | VFed,c | ~540000f | 510 [1200] |
32000 [35000] |
1000 [1500] |
43 [220] |
560 [590] |
2.1 [6.4] |
3.3 [9.8] |
10:1 | 237,238 |
| Av | M-MoFee,c | - | [100] | 26 [410] |
13 [71] |
3.0 [43] |
4.5 [31] |
- [6.6] |
- [7.0] |
30:1 | 222 |
| Av | V-MoFee,c | - | [81] | 33 [370] |
8.8 [93] |
3.4 [49] |
3.1 [21] |
- [6.5] |
- [10] |
30:1 | 222 |
| Av | M-VFeg,c | 42200p | - | 650p | 21p | 1.7p | 7.3p | - | 0.75p | 30:1 | 23 |
| Products | |||||||||||
| Sp. | Protein | CO (CO2)i |
CH4 (CO2) |
C2H4 (CO2) |
C2H6 (CO2) |
C3H6 (CO2) |
C3H8 (CO2) |
C4H8 (CO2) |
C4H10 (CO2) |
Protein Ratioh |
ref |
| Av | MoFeb,c | 3.3 [14] |
0.27 | 0.14 | - | - | - | - | - | 6:1 | 242 |
| Av | VFed,c | 4.3 [13] |
- [8.7] |
- [1.5] |
- [0.083] |
- | - | - | - | 6:1 | 242 |
| Av | VFed,c | 4.3 [5.5] |
0.92 [0.19] |
0.15 [0.34] |
0.15 [0.22] |
0.078 [0.21] |
0.054 [0.099] |
0.021 [0.070] |
0.021 [0.045] |
j | 243 |
| Av | VFek | - | ~35l | 25±5l | - | 42±7l | - | - | - | m | 221 |
| Rp | FeFen,c | - | ~10o | - | - | - | - | - | - | - | 177 |
Activities in [brackets] were run in 2H2O and deuterated buffer and correspond to the deuterated product. Values reported in units of 10−3 nmol product × min−1 × (mg protein)−1 except where indicated otherwise. Temperature of assays reported at 30 °C.
purified from strain YM13A with His-affinity-tagged MoFe.
Histidine affinity tag.
purified from ΔnifHDK deletion, WO42− tolerant strain YM68A.
apo-NifDK protein purified from ΔnifB deletion strain DJ1143. The appropriate cofactor, M- or V-cluster are then used to reconstitute the protein.
ratio of reductase to nitrogenase used is 30:1.
protein purified from ΔnifHDK deletion, WO42− tolerant strain YM68A. Cells grown after depletion of V salt from the fermenter, with subsequent inclusion of excess Mo salts.
ratio of the reductase Fe protein to catalytic nitrogenase protein.
NaHCO3 is the CO2 source in an Ar atmosphere.
no reductase protein used, chemical reductant only – EuIIDTPA (DTPA = diethylenetriamine pentaacetic acid).
purified from WT Lipman 1903 strain, after growing cells multiple times in liquid culture and agar plates with Na3VO4 media in lieu of Na2MoO4-containing media as described in ref 208.
reported values with units of 10−3 nmol product × (nmol protein)−1.
reaction driven electrocatalytically.
purified from ΔnifH, ΔvnfH deletion strain CGA7554.
assay reported 0.45 atm of CO2 (no counter gas specified – likely Ar).
activities reported with units of nmol product × min−1 × (nmol cofactor)−1, where nmol cofactor is determined by metal analysis of the protein.
100% atmosphere of CO.
CO has not only been shown to be a substrate during in vitro assays with purified enzyme, but A. vinelandii cells expressing VnfDGK (strain YM68A) are also competent for CO reduction.239 When the cell cultures have depleted the ammonium from the initial growth media, a CO atmosphere is added and the headspace of the reaction vessel is monitored over 8 hours. Ethylene, ethane and propane are observed with maximum hydrocarbon formation occurring in a 15% CO, 85% Ar atmosphere (specific activity ~6 nmol product × (mg protein)−1 × min−1; mg protein is based on average yield from cells), which reflect ~20% of the activity of isolated VnfDGK in a 100% CO atmosphere for the same products.237,238 Above 15% CO, the hydrocarbon yield drops; in a 37.5% CO atmosphere the specific activity for hydrocarbon formation is ~2.5.239 This in vivo activity is suggestive of vestigial metabolic ability of nitrogenase and may not simply be an adventitious feature.
Carbon dioxide is another carbon-containing small molecule in a more oxidized state than CO and can serve as a potential substrate for nitrogenase. CO2 has been shown by Seefeldt and co-workers to be reduced to CO and H2O (reaction 14) by the wild-type Av MoFe protein, and to CH4 and H2O (reaction 15) by a modified Av MoFe protein variant.240,241
| (14) |
| (15) |
These reactions proceed with specific activities of 0.8 and ~5 nmol product × (mg protein)−1 × min−1, respectively, though no CO2 reduction to CH4 was observed for the wild-type enzyme. This is markedly slower than reaction with the standard nitrogenase substrates, or with CO (Table 6). CO2 reduction was then revisited by Ribbe and co-workers using Av V-nitrogenase in addition to the Mo-dependent enzyme.242 Reaction of CO2 with the MoFe protein was able to produce CO, CH4 and C2H4 with specific activities of 3.3, 0.27 and 0.14 (units of ×10−3), where the VFe protein generated CO comparable with MoFe protein, but no other observed products. However, both enzymes saw an order of magnitude increase of CO formation in D2O, and the VFe protein also produced CD4, C2D4 and C2D6 with higher rates than observed for Mo-nitrogenase in H2O (Table 6). This means that CO production from CO2 has an inverse KIE for both nitrogenases, with similar activities, indicating that this reaction is carried out with a similar mechanism. However, the fact that use of H2O/D2O results in different profiles for C–C bond formation indicates that the MoFe and VFe proteins may undergo chain extension through different routes.242 The isolated Fe-only nitrogenase from R. palustris was also shown to react with CO2 to produce CH4 with a specific activity of ~10 × 10−3 nmol product × (mg protein)−1 × min−1, which is a higher activity than measured for the MoFe or VFe proteins, though longer chain hydrocarbon products or CO were not reported.177
CO2 reduction by alternative nitrogenases has been observed to have metabolic relevance in the native organism.177 Harwood and co-workers reported that a strain of the phototrophic R. palustris expressing V-nitrogenase produced a small, but detectible amount of methane from CO2, whereas a strain expressing the Fe-only nitrogenase produced roughly an order of magnitude more methane than V-nitrogenase, and methane formation was found to increase commensurate with light intensity. Isotopically labeled NaH13CO3 was incubated with the Fe-only nitrogenase strain as a source of CO2 and 13CH4 formation was measured, confirming a CO2-based chemical transformation. Additionally, the Fe-only nitrogenase expressing organisms R. rubrum, R. capsulatus and A. vinelandii were also tested for in vivo CO2 reduction activity.177 When the cells were grown under nitrogen-fixing conditions (no supplemented source of reduced nitrogen species), R. palustris, R. rubrum, and R. capsulatus produced 400–500 nmol CH4 × (mg total protein)−1 in the absence of supplemented Mo, but with a Mo source present, the Fe-nitrogenase proteins were repressed and the activity was absent. This indicated that the Fe-only nitrogenase was the reactive species. Similar behavior was observed with wild-type A. vinelandii, but CH4 production reflected only ~1% of the activity for the other organisms.177 This difference could indicate that the Av FeFe protein is not efficient at CO2 reduction, or it could be that the VFe protein is being preferentially expressed in the absence of Mo, despite the fact that VO43− is not added to the growth media.
The reduction of CO2 is a challenging 8 electron process (reaction 15), not dissimilar from N2 reduction, and in nitrogenase, electron delivery is intimately linked to the reductase Fe protein. Efforts from several groups have been made to understand how the Fe protein and associated electron delivery can change CO2 reduction. Ribbe and co-workers studied CO2 reduction using a Eu-based chemical reductant EuIIDTPA (DTPA = diethylenetriamine pentaacetic acid) in an ATP-independent reaction without Fe protein.243 VnfDGK from A. vinelandii was studied under these conditions, and C1 to C4 products were observed, with the primary products being CO and CH4 (4.3 and 0.92 × 10−3 nmol product × (mg protein)−1 × min−1, respectively). This reflects an improvement in the activity for the VFe protein, as products from CO2 were not observed in proteo-buffers in the presence of the reductase protein,242 but the VFe protein is still more active towards CO reduction.237,238 The CO2 reduction has also been carried out analogously in isotope labeled buffers, and KIEs have been reported for product formation. In comparison, for CO reduction under ATP-dependent conditions, all products had inverse KIE values,237 but in the ATP-independent system for the reduction of CO2, VnfDGK has an inverse KIE for all but methane, which has a more classical KIE value of ~4.8.243 This may indicate there could be a change in mechanism for methane formation from CO2 compared to CO reduction, as the latter has an inverse KIE of ~0.9. The Fe protein may also play a role in the generation of methane (or lack thereof, see Section 4.7). However, the introduction of the chemical reductant could have additional effects on the protein or the reaction, so detailed mechanistic experiments would be necessary to further interpret this phenomenon.
The electrocatalytic reduction of CO2 has been explored by Minteer and co-workers using the V-dependent nitrogenase from A. vinelandii in combination with derivatives of cobaltocene as electron meditators.221 The VFe protein was added to an electrolyte solution containing NaHCO3 as the CO2 source, and when potential was applied, CH4, C2H4 and C3H6 products were measured in roughly equal amounts (Table 6). Interestingly, CO is not observed in the analysis, in contrast to the ATP-dependent and ATP-independent systems reported by Ribbe where CO formation constitutes ≥75% of observed products.242,243 However, the rate of typical acetylene reduction reported by Minteer is rather low (Table 4), and may indicate that the VFe protein used was less active and therefore not reflective of the full product distribution.221 Seefeldt and co-workers also report the electrocatalytic, as well as ATP-driven, reduction of CO2 by the MoFe and FeFe proteins from A. vinelandii.244 It was shown that both proteins were able to generate formate (HCO2−), a 2-electron reduced product, using Fe protein or an electrode, though in the FeFe protein, a higher percentage of electron equivalents (31% vs 9% for the MoFe protein) go towards CO2 reduction instead of H2 production. If CO2 is primarily converted to formate, this set of results may be consistent with the in vivo observation of low CH4 production by Harwood for A. vinelandii,177 but further studies would be necessary to confirm this finding.
4.5. Activities of the Isolated Nitrogenase Cofactors
The alterative nitrogenase proteins have been studied and their enzymatic properties have been compared to the well-characterized Mo-dependent system. However, while each nitrogenase has conserved structural features and reactivity, it can often be challenging to untangle the effects of the protein from the cofactor when analyzing the Mo-, V- and Fe-only nitrogenases. To this end, the catalytic nitrogenase clusters can be extracted from the protein in order to explore their properties, as first demonstrated for Mo-nitrogenase by Shah and Brill in 1977.245 Over the years, several methodologies have been utilized to acquire cofactor,131,246 but the general outline involves the purification of large quantities (gram scale) of nitrogenase enzyme and carefully denaturing the protein using acids and organic solvent under strictly anaerobic conditions. The denatured protein can then be neutralized, pelleted by centrifugation and subsequently washed with organic solvent to prepare for cluster extraction. NMF (N-methylformamide), DMF (dimethylformamide) and MeCN (acetonitrile) in the presence of base have all been used to extract the target cofactor, though basified NMF has the highest success. Initial study of the isolated cofactor was done to obtain spectroscopic parameters associated with the M- and then later V-cluster directly, but also as a method to obtain active nitrogenase from the apo-enzyme.66,131
The M- and V-clusters from A. vinelandii, but not the Fe-cluster, have been isolated and had their catalytic abilities studied, but the precursor L-cluster lacking the R-homocitrate ligand has been investigated as a Fe-cluster analog.247–251 The substrate reduction activity of the M- and V-clusters with small molecules was first reported in 2012 by Ribbe and co-workers, using Tris-HCl buffer at pH 8 with EuIIDTPA as the reductant, in the presence of both CO and NaCN.247 The cofactors were shown to be stable in the aqueous media, as both the M- and V-clusters are able to reactivate the apo-(ΔnifB)-MoFe protein with similar proton reduction activities over the course of an hour, indicating that the cofactors are not decomposing. CO reduction generated a distribution of C1–C4 products with comparable activity for both the M- and V-clusters, verified through using isotopically labeled 13CO (Table 7).247,249 However, the total turnover number (TON; nmol product × (nmol cofactor)−1 × n, where n is the number of C for each product (CnHm)) for product formation is ~0.3 indicating a substoichiometric reaction. The reduction of CN− was slightly more successful, producing up to C6 hydrocarbons that were identified from 13CN− with the primary carbon product being C2H4 (~60% of hydrocarbon products formed). The total TON for carbon products from cyanide was reported as 17 for M-cluster and 16 for V-cluster, however, ammonia production had a slightly higher TON than total hydrocarbon formation, for both clusters.249 Later, the L-cluster was reported along with the M- and V-clusters using the same buffer system as well as reductant, and the L-cluster was found to be as stable, and react with CO and CN− similarly to the M- and V-clusters (Table 7).249 Reduction of CO yielded a total TON of 0.3, whereas the reaction with CN− reflected carbon-based TON of 20. There is a tendency for the L-cluster to form more methane from both CO and CN− than the other two clusters, and one suggested rationale for this difference was that the M- and V-clusters both had the organic acid ligand bound to the heterometal site, whereas L-cluster lacks the native ligand and therefore has an additional open reaction site.249 This was additionally supported through the use of Anderson-Schulz-Flory plots that are often used to describe product distributions in Fischer-Tropsch synthesis. For the M- and V-clusters these plots demonstrated that for the reduction of CO and CN−, the generation of methane deviates from the predicted behavior, whereas for the L-cluster the behavior does not. This indicated that the M- and V-clusters are somehow capable of facilitating the coupling of CHx groups to produce longer chain hydrocarbons which disfavors the release of methane.249
Table 7.
Activity Values for the Reactions of Isolated Nitrogenase Cofactors with Small Molecule Substratesa
| Products | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | Red. | Sol. | Sub. | CH4 | C2H4 | C2H6 | C3H6 | C3H8 | C4H8 | C4H10 | C5H10 | C5H12 | NH3/CO | Sumj | Ref |
| 0.081 | 0.10 | 0.042 | 0.054 | 0.018 | 0.030 | 0.010 | - | - | - | 0.34 | |||||
| 0.38 | 11 | 0.17 | 3.8 | 0.17 | 0.98 | 0.12 | 0.50 | 0.086 | 18/- | 17h | |||||
| 1.3 | 0.30 | 0.73 | 0.18 | 0.31 | 0.040 | 0.11 | - | - | - | 3.0 | |||||
| 7.2 | 3.2 | 2.7 | 0.55 | 0.68 | 0.16 | 0.21 | - | - | - | 15 | |||||
| 0.81 | 0.026 | 0.040 | 0.0021 | 0.0030 | - | - | - | - | −/0.55 | 1.4 | |||||
| 150 | 6.0 | 46 | 4.8 | 14 | 1.2 | 3.2 | 0.40 | 0.65 | - | 230 | |||||
| 580 | 84 | 110 | 66 | 39 | 21 | 10 | 2.8 | 2.2 | - | 910 | |||||
| 43 | 3.0 | 15 | 2.1 | 3.9 | 0.36 | 0.68 | - | - | - | 68 | |||||
| 0.055 | 0.092 | 0.030 | 0.039 | 0.016 | 0.019 | 0.0056 | - | - | - | 0.26 | |||||
| 0.76 | 9.6 | 0.25 | 3.3 | 0.13 | 1.2 | 0.11 | 0.62 | 0.056 | 17/- | 16h | |||||
| 1.2 | 0.33 | 0.58 | 0.14 | 0.25 | 0.036 | 0.10 | - | - | - | 2.7 | |||||
| 6.2 | 2.9 | 2.3 | 0.43 | 0.59 | 0.20 | 0.27 | - | - | - | 13 | |||||
| 1.2 | 0.034 | 0.058 | 0.0036 | 0.0039 | - | - | - | - | −/0.58 | 1.8 | |||||
| 250 | 4.6 | 39 | 2.5 | 8.3 | 0.57 | 1.4 | 0.23 | 0.069 | - | 300 | |||||
| 44 | 1.1 | 7.2 | 1.0 | 1.2 | 0.81 | 0.11 | - | - | - | 55 | |||||
| 0.12 | 0.070 | 0.024 | 0.042 | 0.017 | 0.021 | 0.0080 | - | - | - | 0.30 | |||||
| 3.401 | 7.5 | 1.6 | 3.2 | 0.61 | 1.3 | 0.36 | 0.80 | 0.22 | 23/- | 20h | |||||
| 2.4 | 0.25 | 1.0 | 0.20 | 0.45 | 0.068 | 0.096 | - | - | - | 4.5 | |||||
| 8.3 | 1.8 | 2.4 | 0.20 | 0.45 | 0.028 | 0.052 | - | - | - | 13 | |||||
| 1.52 | 0.042 | 0.080 | 0.0051 | 0.0069 | - | - | - | - | −/0.66 | 2.3 | |||||
| 110 | 4.4 | 38 | 4.2 | 13 | 0.88 | 2.4 | 0.15 | 0.45 | - | 180 | |||||
| 390 | 54 | 66 | 48 | 24 | 15 | 6.8 | 3.2 | 1.7 | - | 610 | |||||
| 21 | 0.80 | 5.6 | 0.60 | 1.5 | 0.16 | 0.44 | - | - | - | 30 | |||||
Activity numbers are reported in plain text for yield in units of nmol product × (nmol cofactor)−1, and corresponding turnover numbers are reported in bold with units of nmol product × (nmol cofactor)−1 × n, where n is the number of C for each product (CnHm) to reflect total C-substrate consumption.
100% CO atmosphere.
from NaCN.
DMF was buffered with 2,6-lutidinium triflate and triethylamine.
from tetrabutylammonium cyanide [Bu4N(CN)].
100% CO2 atmosphere.
DMF was buffered with triethylammonium tetrafluoroborate Et3NH(BF4) and triethylamine.
C6 products (C6H12, C6H14) observed for M-, V-, L-clusters. Yield (nmol product × (nmol cofactor)−1 [TON (nmol product × (nmol cofactor)−1 × n, where n is the number of C for each product)]. C6H12: M-cluster 0.051 [0.30], V-cluster 0.065 [0.39], L-cluster 0.067 [0.40]; C6H14: M-cluster 0.0051 [0.030], V-cluster 0.0042 [0.025], L-cluster 0.031 [0.19].
unpublished results.
Sum of yield and turnover values for C-based products.
The isolated cofactor chemistry was further explored by changing the solvent to dry DMF, introducing 2,6-lutidinium triflate and triethyl amine as a buffering system, and using the stronger samarium (II) iodide (SmI2) reductant to replace EuIIDTPA (E0’Sm = −1.55 V versus SCE in tetrahydrofuran compared to E0’Eu = −1.14 V at pH 8).248 The carbon-based TON for CO reduction by all three cofactors is increased by an order of magnitude with SmI2, having values of 3, 2.7, 4.5 from CO for the M-, V- and L-clusters, respectively. Cyanide reduction was mildly increased by use of the more reducing SmI2 (Table 7). Compared to the Eu-reductant experiments, all of the clusters tended to strongly favor formation of C1 (CH4) and C2 (C2H4, C2H6) products instead of chain elongation, though C4 products (C4H8, C4H10) are observed from gas chromatography – mass spectrometry (GC-MS) analysis and confirmed by 13C labeling.248 This tendency to form methane from CO differs from the protein-based activity of the MoFe protein, where C2 and C3 (C3H6, C3H8) products are observed, but not methane.237,238 Additionally, the stronger reductant system was able to facilitate reduction of CO2, which produces C1, C2 and C3 products with total TONs ~50% less than those for CO reduction, and favors formation of CO and CH4 (Table 7).248 By far the most abundant product formed in these isolated cofactor reactions is H2, with TON from reaction with CO and CO2 of ~5 for all three cofactors, but in the presence of CN− proton reduction is slightly higher at ~14 for M- and Fe-clusters and ~11 for the V-cluster. Although, there is nonzero H2 formation from the SmI2 directly, this reduction also happens to be the ‘standby’ reaction facilitated by the respective enzymes when other substrates are absent.66 Subsequent refinement of conditions, in addition to using triethylammonium tetrafluoroborate (Et3NH(BF4)) instead of 2,6-luitdinium triflate as the proton source for the organic buffer system, and SmI2 reductant allowed for further increased TON of substrate reduction for the M- and L-clusters.250,251 For the M-cluster, the carbon-based TON for CO, CN− and CO2 reduction increased to 230, 910 and 68, respectively, with longer chain hydrocarbons being observed up to C5 (C5H10, C5H12).251 CO2 reduction with the optimized conditions yielded up to C4 hydrocarbons (up from C3), with a compensating decrease in the percentage of C1 (CO, CH4) formation (97% vs 63% C1). The same reactions facilitated by the L-cluster reflect 70%, 80% and 40% of the total TON compared to M-cluster for CO, CN−, and CO2, respectively, though a similar shift from C1 to longer chain hydrocarbon products is observed (Table 7).250 The chemistry of the V-cluster with these improved conditions was also explored for CO and CO2, with TON of 300 and 55, respectively. The TON for CO is higher than for the M or L-clusters, though the values for CO2 reduction fall in between the other clusters (Table 7). Collectively, these experiments with isolated cofactor demonstrate that all three of the clusters studied react in broadly similar ways, but there are indeed some differences that seem to derive from the choice of heterometal as well as the organic ligand that binds to the cluster.
The isolated M-cluster has also been shown to reduce and couple formaldehyde and acetaldehyde to longer chain hydrocarbons using a Eu(II) reductant in buffered aqueous solution.252 These reactions cannot be analogously studied with the protein systems as aldehydes tend to denature or otherwise disrupt the protein structure. Compared to CO reduction, formaldehyde has a much higher total TON of 67 as compared to 1.5 for CO with the same product scope. Primarily, formaldehyde produces methane with C2 carbon products being the next most abundant, but minor amounts of C3 and C4 products are observed. Acetaldehyde on the other hand features a total turnover of 112 for substrate reduction, and is primarily reduced to ethane, followed by butylene, ethylene and butane. Additionally, it was observed that both aldehyde substrates can be coupled with C1 compounds CO and CN− to produce up to C4 carbon products, confirmed by isotope labeling studies.252 For the reaction of formaldehyde with CO, the total TON was 42, with methane as the primary product, though coupling to CN− is slightly more successful with TON of 64, and methane, ethylene and ethane are produced in roughly equal amounts. Acetaldehyde appears to be less reactive towards coupling with CO or CN− (TON of 84 for either), a ~70% of the total product is ethane, similar to the aldehyde alone, though C3 products are observed, demonstrating that coupling does occur.
4.6. Hybrid Nitrogenase Enzymes and their Reactivity
The activity of the alternative nitrogenases compared to Mo-nitrogenase raises questions about the origins of the differentiation in reactivity – is it derived from the heterometal of the cofactor or more to deal with the specific protein environment? As described above, the individual proteins and the isolated cofactors have been studied in an effort to understand their inherent properties.66,131 Primarily, isolated cofactors were used for the reactivation of apo-nitrogenase that lacks the catalytic cofactor, a protein expressed in a cell line with a deletion of the nifB gene.32 Reactivation was often accomplished first through lysis of the cells expressing ΔnifB nitrogenase, followed by an initial centrifugation of the lysate to remove cell debris. The resulting ‘cell extract’ then contained the target protein along with many others, and the mixture could be incubated with isolated cofactor to form holo-nitrogenase. Subsequent purification and substrate reduction assays would then be done to demonstrate activity.131 This reactivation could also be achieved through the initial isolation of the apo-enzyme, followed by a ‘clean’ incubation of only the protein and the isolated cofactor. In principle, there isn’t a restriction on which isolated cofactor can be incorporated into a given apo-nitrogenase. M-cluster could be incubated with the apo-VFe protein to produce a hybrid enzyme (M-VFe), or vice versa (V-MoFe). In this way, the native protein, isolated cofactor, and hybrid protein can all be analyzed to better understand the reactivity of nitrogenase. This strategy has been employed by several groups to further explore the properties that are inherent to both the protein framework (MoFe or VFe proteins) and the cofactor (M- or V-cluster).157,163,168,222 Additionally, a cell strain expressing a particular nitrogenase could be grown in the presence of an alternative heterometal in order to generate a hybrid in vivo that can be purified from the cell directly. The in vivo method has been used in the VFe system to incorporate M-cluster, and with the MoFe protein to incorporate tungsten into the catalytic cofactor.23,223,224 A collection of various hybrid nitrogenase enzymes has been summarized in Table 8.
Table 8.
Hybrid Enzymes and the Reported Reactivitya
| Products Observed | |||||||
|---|---|---|---|---|---|---|---|
| Protein (cell background) |
Hybridb | Methodc | NH3 | C2H4 | C2H6 | H2 | ref |
|
Kp NifDK (ΔnifB) |
V-MoFe | Apo-NifDK cell extract or isolated protein incubated with V-cluster | ND | + | + | + | 157 |
|
Av NifDK (ΔnifB) |
V-MoFe | Apo-NifDK cell extract incubated with V-cluster | NR | + | + | NR | 157 |
|
Av VnfDGK (ΔnifHDKB) |
M-VFe | Apo-VnfDGK cell extract incubated with M-cluster | + | + | + | + | 163 |
|
Av NifDK (ΔnifB) |
V-MoFe | Isolated apo-NifDK incubated with V-cluster | + | + | + | + | 168,222 |
|
Av VnfDGK (ΔnifHDKB) |
M-VFe | Isolated apo-VnfDGK incubated with M-cluster | + | + | NR | + | 34 |
|
Av VnfDGK (ΔnifHDK) |
M-VFe | Cells grown with excess Na2MoO4 (−Na3VO4) | + | + | + | + | 23 |
|
Av NifDK (W-resistant) |
W/Mo-MoFe | Cells grown with Na2WO4 (−Na2MoO4) | + | + | NR | + | 224 |
|
Rc NifDK (FeFe-suppressed) |
W-MoFe | Cells grown with Na2WO4 (−Na2MoO4) | ND | + | NR | + | 223 |
ND = not detected. NR = not reported. Plus (+) sign indicates that the activity is observed/reported.
the nomenclature of the hybrid protein is listed as ‘cluster-protein’ where cluster can be the M, V or W-clusters and the protein is NifDK (also called MoFe protein) or VnfDGK (also called VFe protein). Thus, V-MoFe refers to the hybrid protein that has the Mo nitrogenase polypeptide (NifDK) with the V nitrogenase cofactor (V-cluster).
the description for the method used to obtain the hybrid protein
After the isolation of V-nitrogenase from A. chroococcum and A. vinelandii, there was a great deal of interest in studying the V-containing analog of the M-cluster.141,142 One strategy carried out by Smith and co-workers involved the extraction of the V-cluster from the Ac VFe protein, followed by incubation of the cofactor with extracts of cells expressing the ΔnifB MoFe protein from K. pneumoniae and A. vinelandii.157 When the incubated extracts for both systems were assayed for C2H2 reduction, both demonstrated the formation of C2H4 and C2H6, showing that V-cluster had been incorporated. The ΔnifB Kp MoFe protein was also isolated, then incubated with V-cluster and the resultant protein showed minor but detectible activity with specific activity values of 2 (nmol product × min−1 × (mg protein)−1) for H+ reduction in an Ar atmosphere, as well as 1.1 and 0.09 for the formation of C2H4 and C2H6, respectively.157 Additionally, H2 production was only mildly hindered in a 10% acetylene atmosphere. This pattern of reactivity for V-MoFe is indicative of the wild-type VFe protein, but not wild-type MoFe protein, suggesting that the V-cluster is able to confer the activity. However, the Kp V-MoFe protein was reported to not be competent for N2 reduction to NH3, and the other substrate activities reflect only a small percentage of the wild-type VFe protein, indicating that there might be problems with either the protein or the cofactor.
Hales and co-workers were also able to generate a hybrid nitrogenase, but reversed the components from the Smith study, using an A. vinelandii ΔnifB VFe strain and isolated M-cluster to produce M-VFe.163 Cell extracts containing the cofactor-deficient VFe protein were incubated with isolated M-cluster from the Av MoFe protein, and the resultant M-VFe protein was isolated and characterized. In general, the hybrid reacts towards N2 with similar electron allocation as NifDK, with 70% of electrons going towards NH3 production and the remaining 30% going to H2 evolution. In comparison, wild-type VnfDGK has a 50:50 split between N2 and H+ reduction in a nitrogen atmosphere, and the Kp V-MoFe hybrid was not reported to react with N2 at all.157,163 The ability of the M-VFe protein to produce H2 in an argon atmosphere is hindered, demonstratinĝ40% of the activity of the wild-type Av VFe protein (Table 4), suggesting that there may be issues with proton delivery to the M-cluster site in V-nitrogenase. Additionally, C2H2 reduction by Av M-VFe has a product distribution more closely resembling the wild-type VFe protein with 35%, 10% and 55% of electrons going towards C2H4, C2H6 and H2 formation, respectively, whereas 95% of electrons goes toward C2H4 for the MoFe protein without observation of ethane.163 This seems to imply that the VFe protein is also able to confer the ability to produce ethane from acetylene, even with the M-cluster present.
The formation of hybrid enzymes was also explored by Ribbe, Hu and co-workers using histidine-tagged ΔnifB apo-MoFe/VFe proteins from A. vinelandii, reconstituted with isolated cofactor as well as protein expressed in the presence of an alternative metal.23,34,168,222 The isolated ΔnifB apo-MoFe protein was incubated with both the M- and V- clusters to compare the reactivity towards the standard nitrogenase assays (N2, C2H2 and H+ reduction).168 The protein reconstituted with M-cluster (M-MoFe) was able to recover most, but not all, of the enzymatic activity as compared to the wild-type MoFe protein (Table 4). This observation demonstrates that reconstituted nitrogenase is slightly less active than the wild-type enzyme, in accordance with previous reports.157,163 However, the affinity-tagged Av V-MoFe protein displays a greatly increased activity compared to Kp V-MoFe, including the reduction of N2 to NH3, and the formation of ethane from acetylene.168 The activity of Av V-MoFe reflects ~25% of the formation observed for Av M-MoFe for comparably reported products. This shows that purification using affinity tags helps to increase the reactivity of the protein, and further finds that the V-cluster can impart the acetylene reduction phenotype to the hybrid protein but is less active than the M-cluster containing variant. An affinity-tagged ΔnifB apo-VFe protein was analogously isolated and reconstituted with M-cluster (M-VFe).34 The hybrid was shown to have substrate reducing capability that reflected < 20% of the activity of the M-MoFe control, though, it was unclear if the V-nitrogenase acetylene reduction phenotype was observed as ethane formation was not reported. The decreased activity in this instance was attributed to the unstable ΔnifB apo-VFe protein with an αβ2 composition, notably without the δ subunit.34 It was later reported that M-VFe could also be generated in vivo by expressing the protein in the presence of excess Na2MoO4.23 The resultant in vivo hybrid had an α2β2(δ) composition, had the V-nitrogenase phenotype, producing ethylene and ethane from acetylene, and H2 formation was only mildly attenuated by acetylene. However, metal analysis indicates that only one M-cluster is incorporated per protein, likely leaving the second cofactor site empty, in agreement with the generally decreased activity relative to the native VFe protein.
The CO reduction capabilities for the heterologous V-MoFe and M-VFe hybrids were also investigated and compared to the wild-type enzymes.23,222 Both hybrid nitrogenases were capable of facilitating C–C bond formation from the reduction of CO; V-MoFe formed C2 and C3 products in proteo-solvent whereas M-VFe generated C2–C4 products under similar conditions (Table 6). The V-MoFe hybrid was found to yield an increased amount of hydrocarbon products in deutero-solvent, including C4 products, with inverse KIEs similar to the native MoFe system.222 Of particular note is that CD4 is observed with V-MoFe (and wild-type VFe protein) but not with the wild-type MoFe protein. This strongly suggested that while the presence of the V-cluster can subtly tune the reactivity of the enzyme, the polypeptide plays a larger role in determining product formation. In support of this notion, the in vivo M-VFe hybrid converted CO to C2H4 and C2H6 with higher specific activities (650 and 21 × 10−3 nmol product × min−1 × (nmol cofactor)−1 for C2H4 and C2H6, respectively) relative to V-MoFe (33 and 8.8 × 10−3 nmol product × min−1 × (mg protein)−1) and wild-type MoFe protein (25 and 13 × 10−3 nmol product × min−1 × (mg protein)−1), in line with C2 hydrocarbons being primary products of the native VFe protein.23
Attempts have also been made to generate hybrid enzymes through the incorporation of the non-native tungsten as a heterometal for the catalytic nitrogenase cofactor.223,224 Hales and Case grew a MoFe protein-expressing W-resistant A. vinelandii strain in the presence of WO42−, transferring the culture multiple times in an attempt to deplete the Mo stores of the organism as much as possible.224 The resulting MoFe protein was purified, and incorporation of one equivalent of both Mo and W into each heterotetramer was observed, consistent with one M-cluster and one W-cluster per protein. The hybrid W/Mo-MoFe protein was active towards substrate reduction but reached only ~30% of the activity of the native MoFe protein, so it was unclear if the W-cluster was active and if the reduction of activity simply reflected the lower M-cluster incorporation. Subsequently, Müller and co-workers developed a strain of R. capsulatus that inactivated the regulatory gene anfA responsible for the expression of the FeFe protein under Mo-depleted conditions.223 This strain was grown in Mo-free conditions, in the presence of WO42−, the MoFe protein that was expressed was consistent with the incorporation of one W-cluster per α2β2 heterotetramer, without the detection of substantial Mo. The Rc W-MoFe protein was then assayed with the standard substrate scope and was not particularly active towards C2H2 or H+, with specific activities of 1 and 102 nmol product × min−1 × (mg protein)−1, respectively (Table 4). Additionally, the hybrid was not active towards N2 reduction indicating that the W-cluster is generally inert for nitrogenase activity.
4.7. Substrate Reduction by the Reductase Components NifH and VnfH
As described in Section 3.1, nitrogenase is a two-component system comprised of the catalytic protein that contains the M-, V- or Fe-cluster, as well as the [Fe4S4]-containing reductase protein. In the Mo-dependent system, the reductase, NifH, has been shown to have three primary physiological functions; the protein is involved with insertion of Mo and homocitrate to convert the L-cluster to the M-cluster on NifEN, the biosynthesis P-cluster on NifDK, and the ATP-dependent electron transfer during nitrogenase catalysis.77 These functions have largely been unexplored for the reductase proteins from the alternative nitrogenase systems, VnfH and AnfH for the V- and Fe-only nitrogenases, respectively. Though it has been shown that Av NifH is capable of reducing VO43− to VO2+,253 the lack of a reported VnfEN, and various ΔnifZ and ΔvnfH VnfDGK/AnfDGK variants prevents the extensive characterization of biosynthetic reactivity. However, a seemingly adventitious reactivity of NifH and VnfH was discovered recently. It was found by Hu and co-workers that the Fe proteins could facilitate the conversion of CO2 to CO.87,254 This section will detail the observed activity of these reactions.
CO2 reduction by NifH and VnfH was studied in parallel by modifying the redox agent and the presence of a nucleotide.87 Using nucleotide-free conditions and sodium dithionite as the reductant, the two reductases generated CO with specific activities of 0.10 and 0.13 (10−3 nmol product × min−1 × (nmol protein)−1) for NifH and VnfH, respectively, and these values moderately increased in the presence of ATP (Table 9). For comparison, MoFe and VFe proteins can facilitate CO2 reduction to CO an order of magnitude faster than the reductase components alone (Table 6).242 The difference in reactivity for the Fe proteins in the presence or absence of nucleotide was attributed to the ~100 mV decrease in redox potential that is observed for the Fe proteins in the presence of nucleotide.87 To further investigate this phenomenon, dithionite was exchanged for the more reducing EuIIDTPA, and this resulted in specific activities for CO formation of ~3 × 10−3 nmol product × min−1 × (nmol protein)−1, which is similar to the rate for the catalytic nitrogenase proteins. Repeated additions of EuII reductant to the system continued to drive CO formation with near-linear increases of product. Unsurprisingly, in the presence of the oxidant indigo disulfonate (IDS), neither Fe protein could facilitate appreciable CO production, however, it was found that CO could be oxidized back to CO2 with much faster rates compared to CO2 reduction (Table 9).87 This established the nitrogenase Fe protein as a mimic of the enzyme CO dehydrogenase, which is the only other known system that can facilitate the reversible interconversion of CO2 and CO, albeit with much higher rates.
Table 9.
Substrate Reduction Activity Facilitated by NifH and VnfHa
| Product | ||||
|---|---|---|---|---|
| Protein | Redox Agent | Nucleotide | COb | CO2c |
| Av NifH | DT | - | 0.10 | - |
| DT | + | 0.15 | - | |
| IDS | - | 0.0056 | 200 | |
| EuIIDTPA | - | 3.2/7.7d | - | |
| EuIIDTPA | + | 7.6d | - | |
| Av VnfH | DT | - | 0.13 | - |
| DT | + | 0.17 | - | |
| IDS | - | 0.0083 | 190 | |
| EuIIDTPA | - | 3.2/7.8d | - | |
| EuIIDTPA | + | 7.6d | - | |
The data is reported from ref 87, all proteins are from A. vinelandii. Units reported as 10−3 nmol product × min−1 × (nmol protein)−1. All reactions reported at 30 °C. DT = sodium dithionite. IDS = indigo disulfonate.
assay reported in 100% CO2 atmosphere.
assay reported in 99.9% CO atmosphere.
CO production after multiple additions of EuII reductant; values reported with units of nmol CO × (nmol protein)−1.
Additionally, the in vivo reduction of CO2 was reported in A. vinelandii strains that express either the NifH or VnfH components without the paired catalytic component (NifDK and VnfDGK, respectively).87 In the presence of supplemented ammonia, which represses expression of Fe protein, effectively no CO was observed. However, when the ammonia was depleted, CO could be measured and was maximally produced in a 40% CO2, 60% Ar atmosphere with higher specific activities (~170 × 10−3 nmol product × min−1 × (nmol protein)−1) than observed in the in vitro assays (Table 9). These results indicate that there may be a native electron donor in A. vinelandii that permits the high degree of CO2 reduction carried out by the Fe proteins. It is unclear what physiological relevance this activity has, but the CO2 activity paired with the recent discovery that a homolog of NifH can generate longer chain hydrocarbons, further highlights the versatility of the Fe protein and warrants additional investigation.78,254
5. Mechanism of Nitrogenase
The multi-electron reduction of dinitrogen has been the focus of long-term multi-pronged research that utilizes biochemical, kinetic, spectroscopic, and structural biology approaches.66,143,197 This process is challenging because the individual components of nitrogenase have been studied, providing spectroscopic detail and physical properties. However, the study of substrate reduction requires both the catalytic and reductase components to function together, complicating the analysis. As described in Section 3, there are several metallocofactors housed between the two components of nitrogenase, a [Fe4S4] cluster on NifH and both the P- and M-clusters on the catalytic NifDK protein.32 Each of these clusters can contribute in spectroscopic studies, so it can be difficult to isolate the components from a single source. The crystallography of nitrogenase proteins has shed light on structural aspects of nitrogenase catalysis and has assisted the spectroscopic assignments, but the X-ray diffraction structures represent a snapshot of the solid-state protein and do not necessarily reflect the solution-state dynamics in the reaction mixture. To study the reactions of nitrogenase under physiological conditions requires a combination of rapid freeze-quench and stopped-flow methods to identify reaction intermediates and analyze the multi-step pathway.197 The bulk of the published kinetic and mechanistic work on substrate reduction has focused on N2 reduction through the characterization of the Mo-dependent nitrogenases from A. vinelandii66,197 and K. pneumoniae255–258 among others. Although, the kinetic and detailed spectroscopic studies have not all been conducted with one specific nitrogenase system. The mechanisms for the alternative V-dependent and Fe-only nitrogenases from A. vinelandii, A. chroococcum, and R. capsulatus have been studied alongside the Mo-dependent system,143,178 however, it wasn’t until recently that reports indicated that the alternative nitrogenases from A. vinelandii operate with similar mechanisms to the Mo-dependent system.220,259 The N2 reduction mechanism for Mo-dependent nitrogenase has been extensively detailed by Hoffman and co-workers in 2014,197 but this does not address the recent developments for the alternative nitrogenase systems. Below we provide a general summary of the mechanism proposed for N2 reduction, as well as other substrates with comparisons for the Mo-, V-, and Fe-only nitrogenases.
5.1. N2 Reduction Mechanism for Mo-nitrogenase
The most well described kinetic framework for N2 reduction by Mo-nitrogenase, including a mechanistic outline, involved the work of many research groups and resulted in the Lowe-Thorneley (LT) model (Figure 21).188,255–258 This model was later updated by Hoffman and co-workers,197 and subsequent additions have been made in recent years.220,259,260 This model described the kinetics of the chemical transformations that occur on NifH and NifDK from K. pneumoniae during nitrogenase catalysis, and the delivery of proton and electron equivalents to the catalytic component is tracked with an En notation, where n is the number of electrons and protons accumulated on one αβ-dimer of NifDK.188,197 This formalism does not distinguish the location of the electrons or protons, whether it be associated with the P-cluster or the M-cluster, simply that an electron and proton pair has been provided to the system. The electrons are transferred by NifH through the Fe protein cycle,77,261,262 so by analogy the LT model is also referred to as the MoFe protein cycle.188 In the literature, N2 reduction is sometimes referred to as the first reduction of N2 to a N2H2-level intermediate (can also be called N2 activation), whereas elsewhere it can be used as an umbrella term covering the complete triple bond scission into two equivalents of NH3. In this review, we ascribe N2 reduction to mean the entire process of N2 bond cleavage to produce ammonia.
Figure 21.
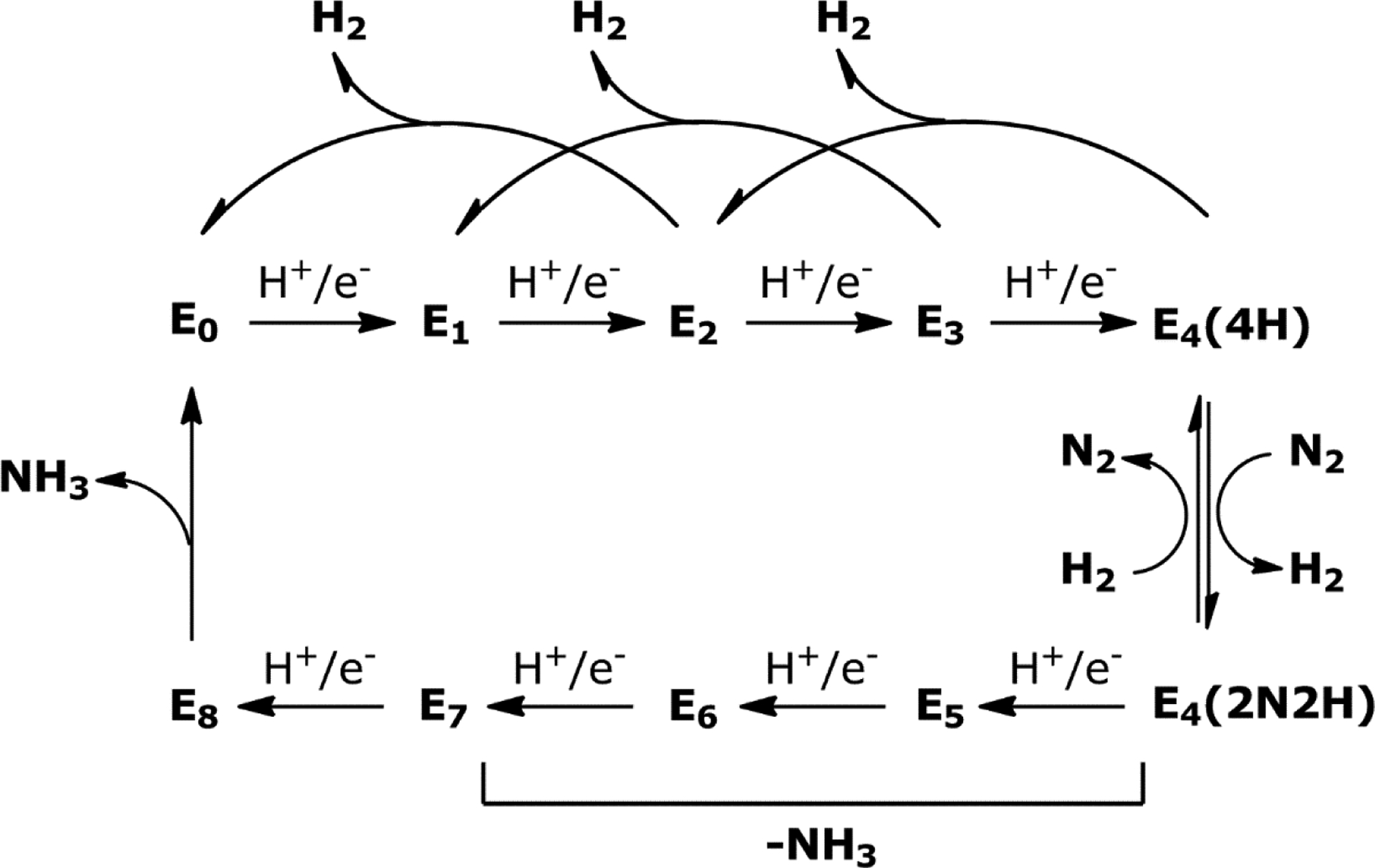
A modified Lowe-Thorneley scheme for N2 reduction in Mo-nitrogenase based on work from refs 188 and 197. The En notation refers to the n number of proton and electron equivalents loaded on to one αβ-dimer of NifDK. The resting state of the cycle is E0 and the release of H2 and NH3 during the cycle is indicated.
The first state, E0, represents the resting state of NifDK and is consistent with an M-cluster in an S = 3/2 state under dithionite reducing conditions.103 Much of the work on Mo-nitrogenase reported in the 1970’s and 80’s has involved the characterization of this state of the enzyme. The E0 state has an odd number of unpaired electrons, so the even numbered states of the LT model (n = 2, 4, 6, 8) will also have an odd number of unpaired electrons loaded on NifDK. This property allows for a convenient spectroscopic handle to study these species using EPR techniques, including pulsed methods like electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM), if the intermediate species can be appreciably accumulated.197 The odd numbered En states (n = 1, 3, 5, 7) will have an even number of unpaired electrons, which should either lead to a diamagnetic species that is EPR silent or to an integer spin (S = 1, 2, 3, etc) that would show EPR transitions.197 The E1 through E3 states occur before N2 reduction, while E4 is the active state where N2 is proposed to bind and where reduction begins. Further addition of protons and electrons to nitrogenase through the E5 – E8 states promotes the cleavage of the N2 bond, producing H2 and two equivalents of NH3, returning the cycle to the E0 state. An additional feature of the cycle is that the E2, E3, and E4 states can ‘relax’ by generation of H2, converting back to the E0 and E1 and E2 states, respectively (Figure 21), which represents non-productive hydrogen release that competes with N2 reduction.188,197
The initial LT model posited that N2 could bind to either the E3 or E4 states, but the E4 state was the point at which N2 reduction could occur.188 This proposal was based on kinetic measurements, as well as observations that with the A. vinelandii nitrogenase system (later for K. pneumoniae and C. pasteurianum as well263) HD gas could be produced from an atmosphere of D2 (2H2) and N2, and that TOH (T = 3H) is not appreciably formed in an atmosphere of T2 (3H2) and N2.188,210,263,264 These results suggested that H2 (or D2) can inhibit N2 reduction by reversing N2 binding (Figure 21), and this process is not a simple exchange with solvent but allows for the scrambling of the isotope label. The experiments also reinforced that one equivalent of H2 is produced by NifDK during N2 reduction.188,198 Subsequently, active site point mutants of residues that are nearby the M-cluster were generated for NifDK of A. vinelandii at α-His195 and α-Gln191 by Newton and co-workers,152 and these variant proteins were used to address the E3 versus E4 state for N2 binding. The relevant nitrogenase variants were inactive for N2 reduction, but in the presence of N2, the acetylene (C2H2) reduction activity was decreased. Activity could be restored through the addition of H2, implying that N2 binds to the M-cluster and can be knocked off by H2 (consistent with inhibition), but as the nitrogenase proteins in a N2/D2 atmosphere are not active for N2 reduction nor is HD produced, the E4 state was not likely being accessed.152 Based on these findings, Hoffman and co-workers argue that N2 must productively bind in the E4 state, and propose a reductive elimination mechanism for H2 production during N2 reduction to rationalize the generation of HD in a N2/D2 atmosphere.197 This mechanism was tested by carrying out C2H2 reduction assays in the presence of N2 and D2, and it was observed that deuterium was incorporated into the ethene (C2H4) product as either C2H2D2 or the more abundant C2H3D, supporting the proposed reductive elimination process.265
Consequently, the E4 state is inherently of interest within the LT scheme because it is a central point in the cycle that either produces H2, reverting to the E2 state, or can bind N2 on the M-cluster pushing the reaction cycle forward.188,197 As mentioned previously, when N2 binds to the E4 state (or E4(4H)), there is a concomitant loss of one equivalent of H2 from NifDK, leaving an E4(2N2H) species (Figure 21). However, it is not clear the reason why nitrogenase produces H2 as part of ammonia production, as 25% of the ATP (and therefore electrons) required for the reaction is funneled into this apparent byproduct. Recent DFT studies have suggested that the thermodynamically favorable formation of H2 is coupled to the less favorable binding and reduction of N2, such that the overall process is slightly favored or thermoneutral,266,267 though additional work is necessary to further explore this idea. To better understand the nature of the E4 state, Hoffman and co-workers have used freeze-quench spectroscopic experiments to trap E4 as well as other intermediate states.197 A single-point mutant (α-Val70→α-Ile70) of NifDK was identified that accumulated an intermediate, that was later assigned to be in the E4(4H) state.268,269 1,2H, 95Mo, and 57Fe ENDOR experiments demonstrated that the intermediate was consistent with the presence of bound hydride (H−) ligands that were interacting with the core Fe atoms of the M-cluster, but were not involved with the Mo center.270–273 This species, termed the “Janus intermediate,” was then proposed to have an M-cluster model with two μ2-hydride ligands bridged between two different sets of Fe atoms and two protons bound elsewhere on the cluster.197,273 It was later reported that spectroscopic features associated with the so-called Janus intermediate were observed in wild-type NifDK, suggesting that hydrides are not an artifact of the point-mutation, but are involved in the on-pathway mechanism of N2 reduction.260
The proposal of hydride intermediates as part of nitrogen fixation provides a framework for storing the proton and electron equivalents without accumulating charge on the metallocofactor directly. There is also established chemistry that involves hydride species both in biology, such as in the hydrogenase systems,274,275 as well as extensive examples in synthetic transition metal systems.276,277 However, it is still unclear how protons/electrons or hydride species are involved in the N2 bond scission, specifically. There are two general pathways that have been proposed for the hydrogenation of N2 – the distal and alternating pathways (Figure 22).66,197,278 In both cases, N2 is proposed to bind in an end-on mode to a metal center (M–N2) of the M-cluster and eventually merge at the formation of a terminal amido species (M–NH2). The final step of the mechanism is the delivery of one proton/electron equivalent to a terminal amido intermediate, releasing NH3 and regenerating the E0 state of NifDK. The two mechanisms differ in the specific sites of protonation of N2, the intermediates formed during the reaction, and which of the En states allow for NH3 to be released.
Figure 22.
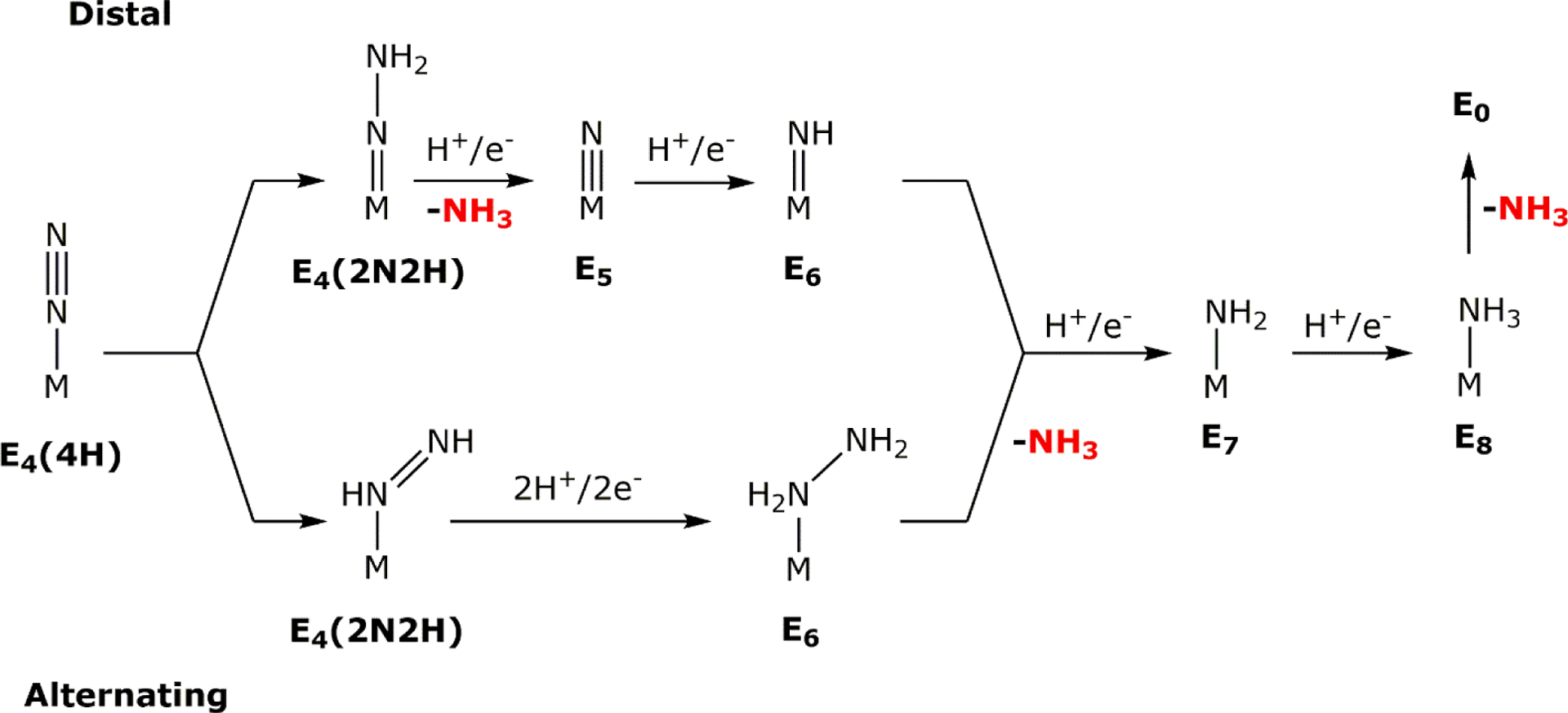
A mechanistic outline demonstrating a distal (top) and an alternating (bottom) pathway for the reduction of N2 by nitrogenase. The corresponding En state from the LT-model is indicated under each intermediate, along with the proposed release of NH3.
The distal pathway proposal is based on the chemistry of inorganic Mo complexes by Chatt279–282 and Schrock.283,284 In the first step, N2 binds to a metal center of the M-cluster in the E4(4H) state, and this promotes two of the proton/electron equivalents loaded on NifDK to add to the distal site of N2, forming a hydrazido (M=N–NH2) species, as well as H2. The next proton/electron addition (E5) also occurs at the distal nitrogen atom, releasing one equivalent of NH3 and generating a terminal metal nitrido (M≡N) species. The following two protonation and reduction steps (E6, E7) promote the formation of metal imido (M=NH) and amido (M–NH2) species, respectively, with the final addition (E8) generating and releasing the second equivalent of NH3. In contrast to the distal pathway, the alternating pathway of N2 cleavage is derived from studies with Fe complexes.285,286 The mechanism begins with N2 binding to the E4(4H) state, but one proton/electron equivalent is added to each nitrogen atom, forming a diazene-bound species (M–(H)N=NH) with the requisite loss of H2. The following two reduction/protonation steps (E5 and E6) form a hydrazine-type species (M–(H2)N–NH2), alternating the site of addition to the nitrogen atom. Protonation and reduction in the following step (E7) releases NH3, generating an amido-bound species that is subsequently liberated as the second equivalent of NH3 in the final step (E8).
There is currently no definitive consensus of the N2 reduction mechanism that is operative in nitrogenase, or if this reduction takes place at the Mo or Fe centers.197 Ammonia generation from N2 has been reported using synthetic Mo-based complexes, and this extensive and thorough body of work supported a Mo-centered distal pathway for the mechanism in nitrogenase.197,287 There is also a growing support for Fe-centered N2 reduction based on synthetic work with iron complexes,288–290 as well as from calculations and active site point mutants that modify the area adjacent to the Fe face of the M-cluster.200,278,291 However, there are several experimental observations that have been used to favor the alternating over the distal pathway. The first evidence of a reduced N2 intermediate was shown by Thorneley, Eady and Lowe by use of acid/base quenching techniques during turnover of the K. pneumoniae enzyme.211 Hydrazine (N2H4) was observed after quenching the reaction with acid or base, but the identity of the enzyme-bound intermediate was unclear. Subsequent studies with A. vinelandii and other variants demonstrated that nitrogenase could use hydrazine as a substrate to produce ammonia, showed that H2 did not inhibit hydrazine consumption, and that HD formation was not enhanced in the presence of hydrazine.198,210,292 Methyldiazene (CH3–N=NH) and diazene (HN=NH) were both found to be substrates for the Mo-nitrogenase from A. vinelandii, and additionally, diazene reduction was inhibited in the presence of H2.199,293,294 These results, together with spectroscopic measurements, are suggestive of the alternating mechanism, as both diazene and hydrazine are substrates for nitrogenase that are competent to produce ammonia, and the mechanistic observations of inhibition and HD formation are in line with other N2 reduction studies.197,198
5.2. N2 Reduction Mechanism by the Alternative Nitrogenases.
Compared to the Mo-dependent enzyme, the alternative nitrogenase systems have had far less characterization over the same time period, particularly with respect to the mechanism of N2 reduction.66,143 The two best characterized V-nitrogenase proteins come from A. chroococcum141 and A. vinelandii.142 As mentioned, the stoichiometry for the Mo-dependent N2 reduction has been established as:
| (16) |
where for each molecule of N2 that is reduced, only one equivalent of H2 is produced, and it is unclear why this 1:1 N2:H2 ratio occurs.66 However, analogous studies with the V-nitrogenase system from A. chroococcum295,296 establish a 1:3 stoichiometry for N2 reduction:
| (17) |
The source of this difference was also not clear but was largely understood to be an inherent inefficiency of the V-nitrogenase to reduce N2 compared to the Mo-dependent system, as the affinities of the Mo- and V-nitrogenases from A. chroococcum for the substrate N2295 and the electron transfer behavior from the respective reductase are similar.101,141 Additionally, the difference in reactivity was not believed to be the result of reaction conditions, as the differences were also observed during in vivo experiments with modified chemostat cultures of A. chroococcum297 and A. vinelandii.143,298 It was also observed that the V-nitrogenase from A. chroococcum produces a small amount hydrazine (~0.5% of the electron flux resulting in ammonia formation)207 as a product that increases with increasing temperature, and for Mo-nitrogenase, hydrazine is only observed when acid or base is used to quench the N2 reduction reaction.143,179,295 Intriguingly, hydrazine does not serve as a substrate for V-nitrogenase at low concentrations, but does so for Mo-nitrogenase.207,209,292 It was also reported that for the His-tagged A. vinelandii proteins, the well-known inhibitor of N2 reduction, CO, behaves differently for the Mo- and V-nitrogenases.149 In the presence of CO (atmosphere, 90% CO, 10% N2) NH3 formation was abolished in both systems, however, electron flux was diverted to H2 formation in the Mo-dependent system whereas H2 production and total electron flux was decreased by >50% in the V-dependent protein. This apparent loss of flux indicated that there may have been unidentified formation of products. Indeed, follow up studies (see Section 4.4) demonstrated that CO behaved as a substrate for V-nitrogenase, which facilitates C–C bond formation, further distinguishing the reactivity behaviors of the two nitrogenases.237,238
The Fe-only nitrogenase has been shown to follow similar reactivity trends as the V-nitrogenase system, demonstrating diminished reactivity compared to the Mo-dependent system, and also has little by way of mechanistic characterization.143,178 There are several Fe-only nitrogenase proteins that have been studied, from R. capsulatus,178 R. palustris177, R. rubrum,173 and A. vinelandii,175,176 with more variants continuing to be identified in biological nitrogen fixing systems. The N2:H2 ratio for Fe-only nitrogenase was established in the R. capsulatus system by Müller and co-workers as 1:9 (reaction 18), demonstrating that even more of the electron flux is dedicated to generate H2 during turnover than in V-nitrogenase.115 Thus, with respect to N2 reduction, the efficiency of the reaction was established as Mo > V > Fe-only.
| (18) |
However, the differences in the stoichiometry of the reaction between the nitrogenase variants raised several concerns about a unified mechanism for nitrogen fixation. Each variant had key mechanistic experiments carried out using nitrogenases from different species, and the analysis often was carried over between proteins without confirming that each behaved similarly. Why do the alternative nitrogenases produce more H2 during N2 reduction? If H2 production and alternative substrate interactions varied by nitrogenase, how could this be reconciled in a unified mechanism?
Advancements by Seefeldt and Hoffman in recent years suggest that the mechanisms of N2 reduction for Mo-, V- and Fe-only nitrogenases are similar.180,220,259 Protocols were used to acquire V- and Fe-only nitrogenase from A. vinelandii without the use of an affinity tag,180,208 similar to what had been described during the initial discoveries.142 However, the V-nitrogenase protein expressed by Hales was in a nifHDK deletion strain that later was found to produce two different conformers during purification – the standard α2β2(δ) V-nitrogenase as well as a less active αβ2(δ) form,142,148 whereas the protein used by Seefeldt came from a wild-type bacterial strain that did not have any genetic modifications and without reported irregularities in the protein.208,220 On the other hand, the Fe-only nitrogenase Seefeldt reported was derived from a strain with deletions of the nifHDK and vnfHDGK genes encoding the Mo- and V-nitrogenases, consistent with previous expression strategies.
These recent mechanistic studies focused primarily on the E4(4H) state and the ability of the enzymes to bind N2. Both A. vinelandii V- and Fe-only nitrogenases showed inhibition of N2 reduction in the presence of H2, a phenomenon that was observed for the V-nitrogenase of A. chroococcum and is well established for Mo-nitrogenase.66,180,217,220 Additionally, HD formation was measured in the presence of an N2/D2 atmosphere, in line with Mo-dependent experiments described in Section 5.1. These results were together interpreted to mean that N2 binds similarly to the respective nitrogenase cofactor, suggesting that the same E4(4H) ‘Janus intermediate’ was operative in all three systems.220 However, spectroscopic evidence of the E4(4H) species analogous to the Mo-dependent system has not yet been reported for either the V- or Fe-nitrogenases. Kinetic analysis was also carried out with V- and Fe-only nitrogenases to determine the ratio of N2:H2 to compare to the Mo system, such that all three enzymes come from the same organism.180,220 Extrapolation of kinetic fits relating to the N2:H2 ratio with respect to the pressure of N2 indicated that at high pressures all three systems would have a ratio of ~1, and would therefore reduce N2 following the stoichiometry of reaction 16. This was compared to an experiment with Mo-nitrogenase by Simpson and Burris that measured N2 reduction and H2 production at high (50 atm) pressures of N2, and this study showed that H2 production could not be abolished, and approached a N2:H2 ratio of 1.216 Analogous high pressure experiments were not conducted for the alternative nitrogenases due to the requirement of much higher pressures (>>50 atm) to carry out the same analysis, though there are no reported experimental values above 1 atm N2.180,220 The difference in apparent N2:H2 ratios was then contextualized with respect to the relative rates of N2 binding to E4(4H), relaxation of E4(4H) to yield H2, and formation of E4(2N2H) with concomitant decrease of the N2 bond order. The reported interpretation was that Mo-nitrogenase is able to produce E4(2N2H) faster than either of the alternative enzymes, so E4(4H) is less likely to unproductively relax to a lower En state to generate H2, thus, less H2 is observed.220 Extensive substrate analog studies have not been reported for all of these systems, so it is unclear based on this work how the alternative nitrogenases move through the higher En states as compared to Mo-nitrogenase.
Around the same time, Einsle and co-workers reported a crystal structure of V-nitrogenase from A. vinelandii with a putative reaction intermediate bound.150 In the structure there was electron density for a light atom (X) smaller than sulfur that apparently displaced the S2B bridging sulfide ligand from the V-cluster. There was also density for an HS− ion present ~7 Å away from the S2B site in a ‘holding pocket’ as well as a bridging carbonate (CO32−) ligand in the S3A position (see Figure 23 for labeling). Interactions from nearby residue α-Gln176 to the light X atom seemed to be indicative of a protonated species, however, protons cannot be directly observed from the diffraction experiments. These results together were used to assign the X atom as a nitrogen, and more specifically a μ-HN− nitrene group,150 although, this assignment is not strongly supported. As described in Section 3.3.1, the putative intermediate did not have metrics that compared well to the available model complex,155 there was no reported independent verification of the light atom identity, and subsequent calculations were highly suggestive of a μ-OH group.156 Regardless, based on the assignment of the X atom as N, a mechanistic proposal was described for N2 binding and reduction. The observation of the S2− loss was reported to be mechanistically important, as incipient hydride accumulation would force the sulfur ligand into the ‘holding pocket’ until returning later to kick out the final amido species in the E8 state, regenerating the E0 resting state. There is precedent in Mo-nitrogenase for the lability of the S2B ligand, as demonstrated in its replacement by both CO and Se2− in crystal structures from Rees,299,300 however, theoretical calculations of Mo-nitrogenase disfavor complete sulfide loss during catalysis.266,273,301–303 In addition, the release of S2B occurs after S3A has already been replaced by a carbonate, and the loss of two μ-sulfido ligands from the cluster had not been otherwise explored as a mechanistic possibility. Einsle further suggested that the two μ-hydride ligands bridge between Fe2 and Fe6 (equivalent to E4(4H) state) and the hydrides undergo reductive elimination to produce H2, leaving the V-cluster primed with 2 electrons and open coordination sites (termed E4*), allowing for N2 to bind to the cofactor and generate the E4(2N2H) state (Scheme 1B). This would allow for N2 to bind symmetrically between Fe2 and Fe6 in a μ1,1-N–NH2 bridging mode (Figure 23). The conversion between the E4(4H), E4* and E4(2N2H) states would also all be reversible.150 In this proposal, the crystallographically assigned HN− species would then be consistent with the E6 state following a distal-type reduction pathway (Figure 22). The authors also noted that N2 could alternatively bind to the E4* state between Fe2 and Fe6 in a μ-η2:η2 mode more similar to the μ-η2:η1 mode shown in Figure 23C to form diazene and subsequently hydrazine, as N2H4 is a minor product of V-nitrogenase as described earlier (Scheme 1B). Interestingly, there was no discussion of the purpose the carbonate ligand serves in catalysis or why it appears in V-nitrogenase when carbonate is nonessential for turnover by Mo-nitrogenase.
Figure 23.
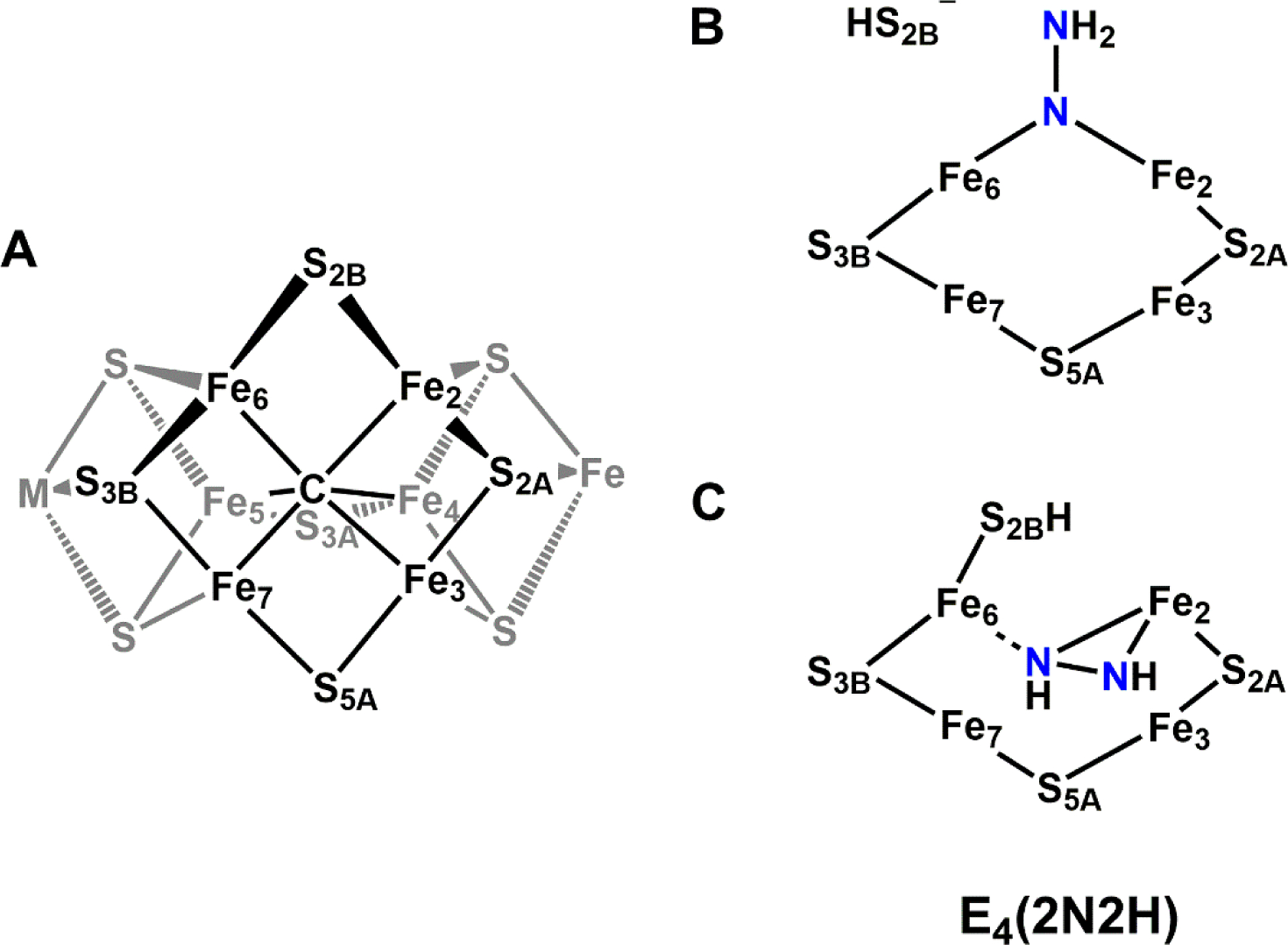
Proposed intermediates for the E4(2N2H) state of Mo/V-nitrogenase during N2 reduction. A: Depiction of the catalytic cofactor (M = Mo for M-cluster, V for V-cluster), where S3A is replaced by carbonate (CO32−) in the crystal structure of VFe. The catalytic surface of the M-cluster as identified by Hoffman and co-workers is colored black. B: Proposed E4 state by Einsle in ref 150 with a μ1,1-N–NH2 species. C: Lowest energy configuration of E4 state proposed by Raugei, Seefeldt and Hoffman in ref 266 with a μ-η2:η1-diazene-type species.
Scheme 1.
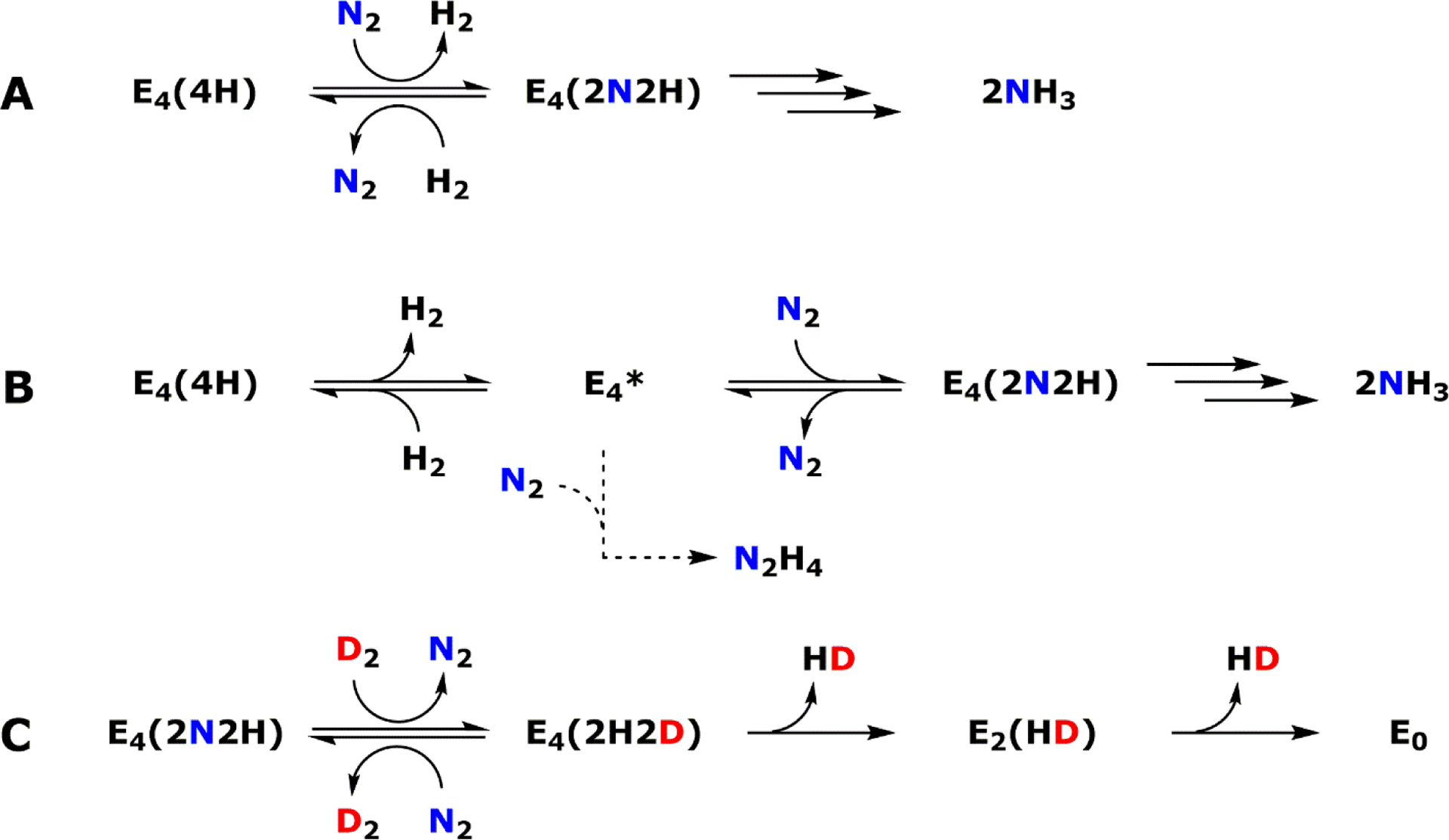
Simplified scheme of N2 binding for the proposed E4 state of the nitrogen reduction mechanism. A: The concerted N2 binding and H2 release from the LT model for Mo-nitrogenase, with multiple arrows representing the remaining steps required to produce NH3. B: The mechanism for N2 binding proposed for V-nitrogenase by Einsle and co-workers in ref 150. The multiple solid arrows represent the ‘on pathway’ reduction of N2 to NH3, and the dashed arrow reflects a proposed ‘off pathway’ method to produce N2H4. C: Depiction of D2 reacting with the E4(2N2H) state to produce a deuterium charged E4(4H) state (termed E4(2H2D)) and the subsequent relaxation as one means to produce HD.
If one assumes that the assignment of the X atom as N is confirmed, the result would seem to indicate that V-nitrogenase follows a different mechanism for N2 reduction than Mo-nitrogenase, or at minimum, there is more than one active N2 reduction pathway. One pathway would be analogous to the Mo-dependent system that produces ammonia, and V-nitrogenase would follow a distal-type reduction mechanism opposed to an alternating mechanism (Figure 22). The second pathway in the V-dependent system would bind N2 in a μ-η2:η2 mode to terminally produce hydrazine in agreement with experimental observations.207 The requirement of sulfur loss during catalysis is also an interesting idea, though validating biochemical investigations will be required to further understand this behavior. However, there are several issues with Einsle’s proposal for N2 binding and reduction by V-nitrogenase. The dubious identity of the X atom is problematic, but if to assess the proposal we assume this assignment is correct, the next issue stems from the existence of the E4* state. As discussed in the previous sections, H2 is an inhibitor of N2 reduction, and the conversion of the E4(4H) state to the E4(2N2H) state as shown in Scheme 1A requires N2 binding concomitant with H2 loss. Conversely, the presence of H2 reverses N2 binding and regenerates E4(4H) consistent with experimental observations of inhibition. Further, if nitrogenase is turned over in the presence of N2 and D2, 2 equivalents of HD are observed per N2, but no isotopically scrambled dihydrogen is observed in the absence of N2.188,210,263,264 In Einsle’s proposal, the E4* state is added, and that splits H2 release from the N2 binding step (Scheme 1, compare A to B). In the mechanism as described, it is less clear how H2 could serve as an inhibitor to N2 reduction, and more importantly, it would allow for the generation of HD if the E4(4H) species was exposed to D2 in the absence of N2. This is in opposition to the experimental observations that HD is only formed in the presence of D2 and N2. There is also no experimental evidence provided to support an alternate binding of N2 to the V-cluster that could result in a reaction pathway that generates N2H4, though its inclusion was described as ‘hypothetical.’ This proposal also differs from the studies described by Seefeldt and Hoffman that indicate all nitrogenases follow a similar mechanism, and favor retention of the S2B sulfide ligand.220,259,266,273 In addition, intermediate trapping studies with Mo-nitrogenase are supportive of an alternating type mechanism,197 though it is also important to note that the proposal by Seefeldt and Hoffman does not currently explain the observation that V-nitrogenase can produce hydrazine without turning it over as a substrate. It is clear that more work is needed to assess these differing proposals.
5.3. Mechanism of Nitrogenases for the Reduction of Carbon Monoxide
In addition to N2, nitrogenase has been known to reduce a variety of small molecules, as described in Section 4. Historically, Mo-nitrogenase was known to reduce substrates such as alkynes, nitriles, hydrogen cyanide, azide, cyclopropene and isonitriles, and these reactions have been summarized and discussed in detail in previous reviews.66,197,198,200 In the past two decades, exciting findings have established that V-nitrogenase can additionally moonlight as a CO reductase, with the ability to reduce and couple CO into hydrocarbons ranging from C1 to C4.237,238 In this context, there has been a renewed interest to study the reactions of CO, not only in the V-dependent system, but in Mo-nitrogenase as well. The objective of the following section is to present this development in light of the mechanistic insights that have been gained regarding these interesting reactions since their discovery.
5.3.1. The Binding and Reactivity of CO in Mo-Nitrogenase
It has long been established that CO behaves as an inhibitor of the Mo-nitrogenase, attenuating the reduction of all substrates except protons under conventional turnover conditions.66,215 The binding of CO had been observed by EPR in early studies, in which Mo-nitrogenase was subjected to turnover conditions with low electron flux.66 Two distinct signals were observed in the spectra; one at a low concentration of CO (at 8% CO, 92% Ar atmosphere, designated as the ‘lo-CO’ state), with g-values of 2.10, 1.98, 1.92, and the other at a high concentration of CO (at 50% CO, 50% Ar) atmosphere, designated as the ‘hi-CO’ state), with g-values of 2.17, 2.10, 2.05.213 13C and 57Fe ENDOR analysis of these signals confirmed that CO was bound to the Fe atom(s) of the M-cluster, and it was proposed that the lo-CO signal corresponded to a single molecule of CO bridging between two Fe atoms (Figure 24A), while the hi-CO signal corresponded to two CO molecules, each binding end-on to different Fe atoms (Figure 24B).304–306 These assignments were further supported and explored using Fourier Transform-IR (FT-IR) spectroscopy.307–309 In the Mo-nitrogenase from K. pneumoniae, the FT-IR spectrum of the lo-CO form had a single band at 1904 cm−1, while three additional bands corresponding to the hi-CO species were observed at 1906 cm−1, 1935.6 cm−1, and 1958 cm−1.307 The multiple signals observed in the hi-CO form of the enzyme indicated that CO was binding to more than one Fe atom, and the time-dependent analysis of these IR bands also suggested that the lo-CO form was primarily responsible for the inhibitory effect of CO on nitrogenase activity.307,308 The cryo-annealing (warming a sample to a higher temperature while remaining frozen) of EPR samples demonstrated that the lo-CO and hi-CO forms are inter-convertible, and real-time monitoring of this process using FT-IR revealed new spectroscopic features consistent with alternative CO binding modes that do not have an EPR signal.308,310,311 Additionally, both EPR and FT-IR studies indicated that the bound CO ligands could be photolyzed from the protein.308–311
Figure 24.
Possible CO binding configurations on the M- or V-clusters. A reflects a μ2-CO species bridged between two Fe centers of the catalytic cofactor, designated ‘lo-CO.’ This state was observed in both Mo- and V nitrogenase. B and C reflect potential ‘hi-CO’ species that were observed in Mo- and V-nitrogenase, respectively. B reflects two end-on-bound CO molecules (with uncertainty in the status of the bridging sulfide, as depicted with dashed lines), while C was proposed to be a combination of the μ2-CO moiety (i.e. ‘lo-CO’) in addition to an end-on bound CO (designated ‘extra CO’).
Perhaps the most exciting advance comes from the seminal work of the Rees group, in which the crystal structure of the CO bound MoFe protein from A. vinelandii was solved (Figure 25).299 To capture intermediates in the MoFe protein during turnover, Rees and co-workers developed an innovative procedure where substrate turnover was initiated under an atmosphere of CO, potentially producing CO-inhibited species. The MoFe protein was then re-isolated from the reaction mixture and crystallized within several hours. In the crystal structure, CO was found to bind in a μ2-bridging mode between Fe2 and Fe6 at both of the M-cluster sites in the heterotetramer, within close proximity to the catalytically important residues α-Val70 and α-His195.154,312,313 The former residue was previously indicated to be involved in CO binding, and single-point mutation of α-Val70 led to minor reducing activities towards CO.309,314 Surprisingly, the binding of CO in the structure was achieved by displacing the bridging sulfur atom (S2B) that was originally at that position; an unprecedented finding at the time. The replacement of S2B with a CO molecule causes the core geometry of the M-cluster to slightly distort from the known resting state structure, such that the molybdenum, the interstitial carbide, and Fe1 atoms no longer align along the 3-fold symmetry axis.299 Importantly, S2B was shown to reappear in the original position after the CO inhibited MoFe protein was once again subjected to turnover conditions, but in the absence of CO. This experiment demonstrated the interconversion of the resting and CO-inhibited states of the MoFe protein and thereby, the physiological relevance of the captured structural snapshot. A putative sulfur binding site was found ~22 Å away from the S2B position at the interface between the NifD and NifK subunits of NifDK, but it is unclear how the S2B sulfur would be able to reversibly migrate across such a long distance during activation and/or substrate turnover. This is in contrast to the ‘holding pocket’ observed in the crystal structure of VnfDGK, only ~7 Å away from the S2B position.150
Figure 25.
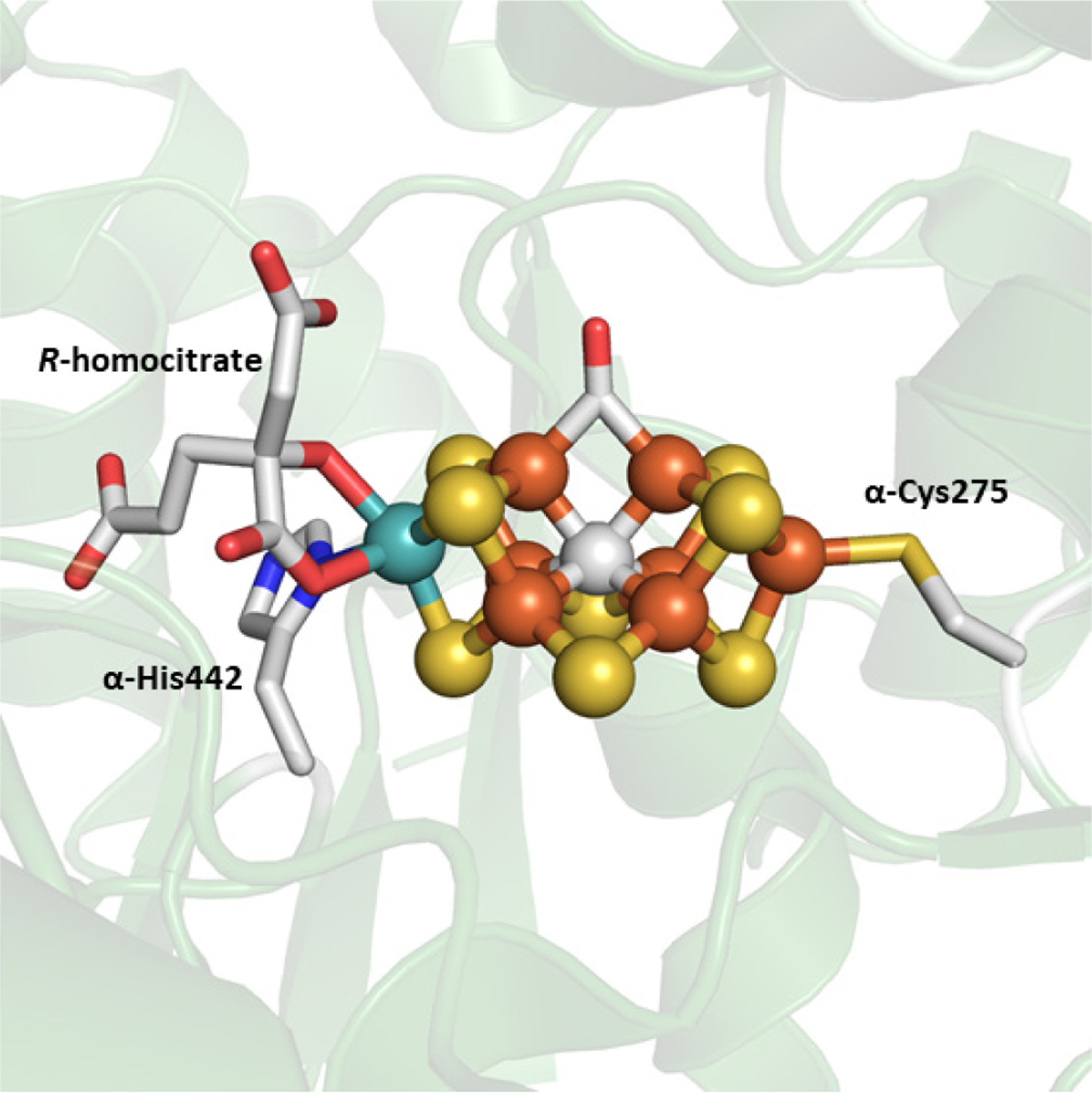
The crystal structure of Av NifDK with CO-bound to the M-cluster (PDB ID 4TKV). Coloration is the same as described in Figure 2.
While the CO-bound M-cluster observed in the crystal structure bore the same ligand-binding geometry as the lo-CO species proposed from EPR and ENDOR studies, it was unclear if the crystallographic and spectroscopic species were equivalent, due to notable differences in sample preparation. For example, the CO-bound MoFe protein from the crystal structure was generated under a saturating amount of CO (1 atm CO) with a high electron flux (excess Fe protein, high molar ratio), and the sample was allowed to sit idle for several hours under non-turnover conditions. In contrast, the lo-CO MoFe protein as described by EPR/ENDOR analysis was prepared using low concentrations of CO (0.08atm CO) with a low electron flux (excess MoFe protein, low molar ratio) and the samples were rapidly frozen. To resolve this ambiguity, an experiment was conducted to reproduce the sample using conditions reported by Rees and then analyzing it using EPR spectroscopy.315 A ‘lo-CO’ signal appeared in the spectra generated from this sample, and this demonstrated that the bound-CO species observed in the structure was indeed the same as the species proposed from EPR and ENDOR experiments. This finding suggested that the displacement of the belt sulfur atom was now a mechanistic possibility, a notion that had not previously been considered. One potential complication with this finding is in understanding how the loss of a μ2-sulfide ligand would affect the spectroscopic properties of the metallocofactor, if at all. In a recent study, the biochemical characterization of the L-cluster, the [Fe8S9C] biosynthetic precursor of the M-cluster, showed that the presence or absence of a bridging sulfur atom only minimally affects the EPR spectrum.51 Thus, it is conceivable that the spectroscopically derived lo-CO state might also lack a ‘belt’ sulfur ligand, a feature that is difficult to detect using EPR spectroscopy alone, and warrants further evaluation.
Based on the unprecedented CO-bound structure, Rees and co-workers argued for the catalytic relevance of the displacement of S2− from the S2B position during N2 and CO reduction.299 They further suggested that S2B might be protonated by the nearby α-His195, generating HS− as the leaving group compared to a dianionic sulfide. The release of the sulfur species would then open up a reactive diiron face of the cluster that can more easily bind substrate. This idea was later supported in the context of N2 reduction by calculations performed by Nørskov and co-workers, as well as experimental observations made by Einsle and co-workers, where a light atom assigned as HN−/HO− was observed in place of the bridging S2B sulfur atom in the crystal structure of the Av VFe protein.150,316 It is unclear whether the release of the bridging sulfur and subsequent binding of CO involves the “Janus” intermediate (See Section 5.1), though previous work seems to indicate that inhibition of CO might occur at less reduced states than E4.317 It is also uncertain if the binding of CO requires the reductive elimination of H2, analogous to N2 reduction. One interesting fact to consider is that CO does not inhibit proton reduction in Mo-nitrogenase.66 Since the binding of CO to the S2B position seems stable, the structural picture points to the possibility that hydrogen evolution, and perhaps hydride formation, might occur at a site different than the proposed Fe2 and Fe6 for the Janus intermediate, which would be consistent with the lack of inhibition of H2 formation by CO. Further investigation is required to elucidate these structure-function relationships.
5.3.2. The Binding and Reactivity of CO in V-Nitrogenase
Initially, little was known about CO-binding to V-nitrogenase, but early observations were made that CO inhibits C2H2 reduction less strongly when compared to Mo-nitrogenase.217,236 Later, it was discovered that unlike for Mo-nitrogenase, proton reduction by the V-nitrogenase from A. vinelandii could be inhibited by CO.149 In addition, the lo-CO and hi-CO signals from EPR studies were not initially reported for the V-dependent protein when it was subjected to turnover conditions in a CO atmosphere, analogous to those for Mo-nitrogenase.163 These findings could all be rationalized by the fact that CO is actually a substrate for V-nitrogenase, as demonstrated by Ribbe and co-workers in 2010.237,238 Since this discovery, efforts have been made to use DFT calculations to gain insight into the mechanism of CO reduction.318,319 While this early work did not have much by way of experimental calibration, it was recognized that reduction of CO likely occurs through sequential reduction and protonation of a bound CO moiety. In both the studies of Dance, and Nørskov and co-workers, the most energetically demanding step is the first reduction, where a cofactor bound CO (M–CO) species was converted to a metal-formyl (M–CHO) species. These theoretical calculations also suggested that a methylene moiety (H2C2−) was likely to be involved in the C–C coupling reaction for the formation of C2 and C3 products. Lastly, it was also noted that the mechanism of CO reduction to longer chain hydrocarbons may involve more steps than are proposed for N2 reduction. The challenges in the experimental characterization of CO reactions with the VFe protein are equally daunting, and in many ways mirror those that have been encountered in the mechanistic investigation of Mo-nitrogenase. The chief hurdle is the complex and uninterrupted electron transfer process during substrate turnover, such that the isolation and characterization of bound substrate or reaction intermediates were unattainable by conventional means. In the case of Mo-nitrogenase, this problem was overcome through site-directed mutagenesis near the active site, in combination with rapid freeze-quench spectroscopic techniques, as well as structural analysis of inhibitor- and analog-bound MoFe protein samples (See Section 5.3.1). Similar approaches have more recently been undertaken by the Ribbe and Hu groups to understand the mechanism of CO reduction by V-nitrogenase.
It was recognized by Ribbe and co-workers that when the V-nitrogenase of A. vinelandii was subjected to turnover conditions in a CO atmosphere, the protein displayed an EPR signal that is almost identical to the lo-CO signal that was observed in Mo-nitrogenase, albeit at much lower intensity.315 This finding was reasonable, considering that the reaction of V-nitrogenase with CO was slow (Table 6), so a small but still detectable spectroscopic feature can be observed. The result also agrees with predictions from DFT calculations that the initial reduction of a bound CO species would be rate determining, and therefore, has the highest likelihood to be trapped and studied.318,319 Importantly, this result sheds light on the reduction of CO by V-nitrogenase by comparison to the well-established framework of CO binding in Mo-nitrogenase, as described in the previous section. Consequently, it was proposed that CO may also bind to the V-cluster in a μ2-bridging conformation, analogous to that observed for the Mo-dependent system, as evidenced by an identical ‘lo-CO’ signal.299 To better characterize this species, strategies were devised to accumulate a CO-bound species by uncoupling the binding of CO to the VFe protein from further reaction. One strategy implemented was to subject the VFe protein to low redox potentials using the strong reductant EuII-DTPA (−1.14 V versus SHE) under a CO atmosphere, with non-turnover conditions.315 At 1 atm CO, a dramatic increase of the lo-CO signal with g-values at 2.09, 1.99 and 1.91, was observed (Figure 26A). CO release experiments revealed that one CO per VFe protein was trapped under these conditions. This result was consistent with one bridging CO molecule binding the V-cluster in only one of the V-cluster sites. Starting from similar conditions but increasing the pressure of CO to 2.6 atm, a new composite signal emerged in the EPR spectrum, that was assigned to the hi-CO form of V-nitrogenase (Figure 26B).320 The hi-CO state from the VFe protein is comprised of a lo-CO component similar to that observed for the MoFe protein, but when this species is subtracted from the spectrum, a residual component remains, which appears to have a more axial signal with g-values at 2.13 and 2.01 (Figure 26C). This residual signal was called the ‘extra-CO’ species, and CO release experiments identified that ~3–4 CO molecules per VFe protein were observed, a marked increase from the study of the lo-CO species from VnfDGK.315 A model for the hi-CO species was then proposed to have 2 CO molecules bound on each of the V-clusters in the protein.320 One of the CO ligands was consistent with the previously described μ2-bridging mode, while the ‘extra CO’ signal was tentatively assigned as a terminally bound CO molecule, adjacent to the bridging CO (Figure 24C). This assignment was speculative and required verification through in-depth spectroscopic analysis.321
Figure 26.
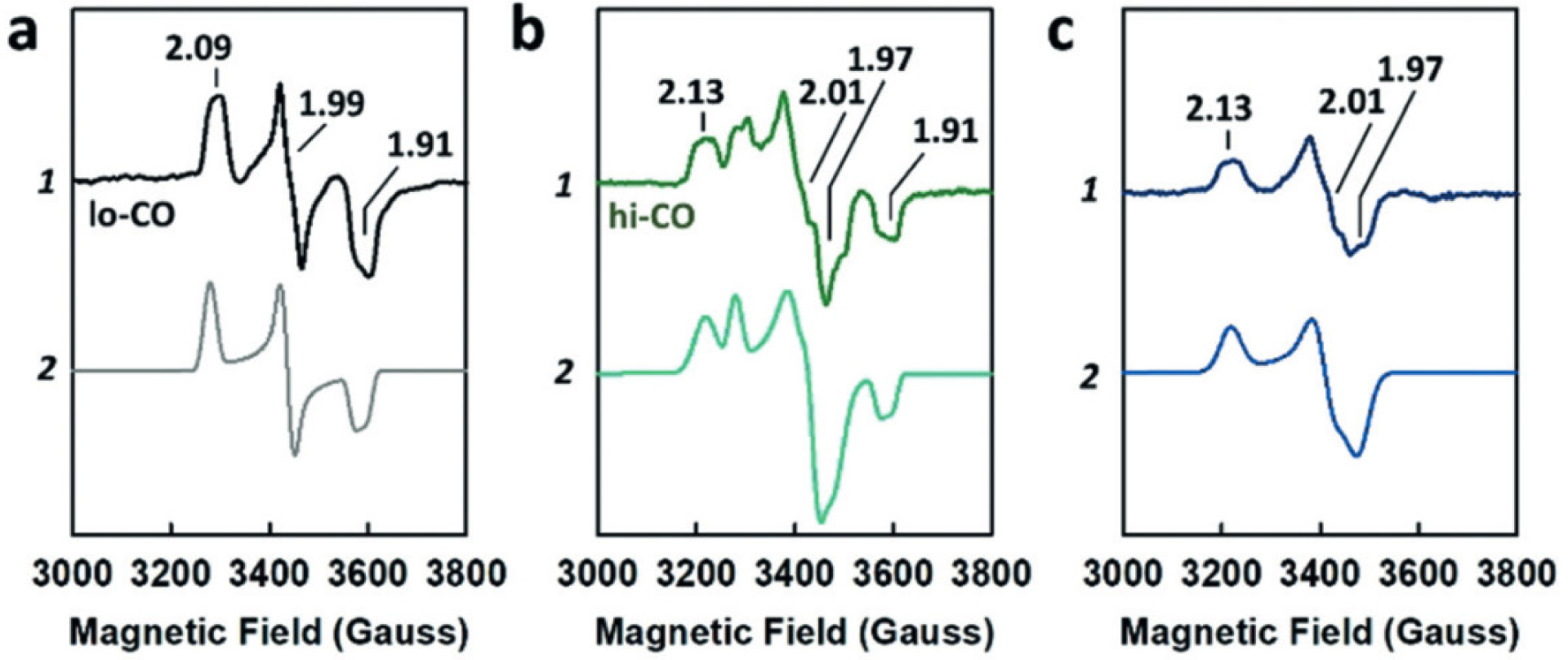
The EPR spectra (1) and respective simulation (2) for the Av VFe protein in the lo-CO (a) and hi-CO (b) states. The difference spectrum (c) of (hi-CO)–(lo-CO) uncovers a signal associated with the ‘extra-CO’ species. Adapted with permission from ref 320. Copyright 2018 John Wiley and Sons.
With the emergence of two distinct CO-bound species in the Av VFe protein, a question was raised regarding which of the species was competent for the reduction and coupling of CO into longer chain hydrocarbons. This notion had been previously considered for Mo-nitrogenase, as some kinetic and spectroscopic evidence suggested that the lo-CO species was responsible for the inhibitory effect on nitrogenase activity. On the other hand, the possibility of the hi-CO species with two CO molecules bound at the cofactor site seemed like a convenient starting point for C–C coupling. To address this question, Ribbe, Hu and co-workers tested the reactivity of the chemically generated lo-CO and hi-CO species of V-nitrogenase.320 When the purified lo-CO species was subjected to turnover conditions in an argon atmosphere, CH4 was detected. The origin of the generated CH4 was traced to the CO bound to the V-cluster using isotopically labeled 13CO. When the isolated 13C-labeled lo-CO VFe protein was reacted with additional equivalents of 12CO, ethylene and ethane with mixed carbon labels (12CH2=13CH2 and 12CH3−13CH3, respectively) were produced.315,320,321 This strongly implied that the putatively bridging lo-CO species had catalytic relevance in V-nitrogenase. In contrast, when the hi-CO VFe protein was subjected to turnover conditions in an Ar atmosphere, C2 products were not detected above the background. Treating the 13C labeled hi-CO species with additional 12CO only yielded mixed labeled ethylene and ethane (12CH2=13CH2 and 12CH3−13CH3), but not fully labeled products (12CH2=13CH2 and 13CH3–13CH3). These results suggest that the lo-CO, but not the hi-CO species, was the catalytically relevant form for hydrocarbon formation and C–C coupling.
Another line of evidence that further supports this conclusion came from an alternative approach in trapping the bound CO species using a “mismatched” V-nitrogenase system.321 In these studies, the A. vinelandii VFe protein (Av VnfDGK) was subjected to substrate turnover conditions in a CO atmosphere, but used a reductase protein from a different organism, M. acetivorans (Ma VnfH). This mismatched nitrogenase pair was previously shown to have much lower specific activity due to inefficient electron transfer.104 The reduced electron flux served to stall CO reduction and allow for the accumulation of CO-bound species. The hi-CO signal on the VFe protein was reproduced at 1 atm of CO pressure, whereas previously it required 2.6 atm of CO. This enabled the study of the competition of N2 and CO binding through the correlation between the N2 reduction activities and the intensities of lo-CO and hi-CO EPR signals.320,321 At 0.1 atm of N2 and increasing concentrations of CO (0.1, 0.5, 0.9 atm), ammonium production dropped drastically – losing most of the activity at 0.1 atm of CO. Coincidentally, the intensity of the lo-CO EPR signal increased concomitantly with the decrease in activity, gaining more than half of its maximum intensity at 0.1 atm CO. In contrast, the hi-CO EPR signal on the VnfDGK increases more slowly, remaining at < 20% of the maximum signal intensity even at 0.5 atm CO. This implied that N2 could be competing with the lo-CO species for reactive Fe sites on the V-cluster (but not the hi-CO species), likely at the S2B belt sulfide position.
Additional insights into the CO reduction pathway were gained from the novel aldehyde condensation reaction catalyzed by isolated M-cluster.252 Driven by EuII-DTPA, formaldehyde and acetaldehyde were found to be reduced to methane and ethene/ethane respectively (See Section 4.5).252 Interestingly, these aldehydes can be coupled to form C2 products from formaldehyde and C4 products from acetaldehyde. This suggests that an aldehyde-like species could be a potential intermediate for CO reduction. The connection between the CO- and aldehyde-reduction pathways were shown through cross coupling experiments, where 13C labeled formaldehyde and acetaldehyde were provided 12CO and/or 12CN−. The presence of mixed labeled products definitively showed that aldehydes can be coupled with CO and CN− and this is strongly indicative of reaction pathways that share a common coupling intermediate. This finding suggests that a direct coupling of two bound CO molecules, similar to the proposed hi-CO structure, is not likely to occur. The natural question that follows is, at which point of the reduction pathway does C–C coupling occur? To that end, isotope analysis using 2H labeling reveals that activation of formaldehyde leads to a metal bound hydroxymethyl moiety, which is supported by the finding that both substrate-derived hydrogen atoms (i.e. C2H2O) were retained in the product.252 This observation agreed with the proposed reactivity of the lo-CO species, as described above, and further indicates that the plausible candidates of coupling intermediates may either be metal-bound alcohol (M–ROH), allyl (M–RCH2) and (M–RCH3) alkyl species.
6. Summary and Perspectives
Nitrogenase is a complex two-component metalloprotein capable of facilitating the difficult chemical transformation of N2 into NH3 under standard temperature and pressure. This multi-electron and multi-proton reaction involves the concerted effort of three different FeS clusters, with the activity taking place at the Mo-, V-, or Fe-clusters for the Mo-, V-, and Fe-only nitrogenases, respectively. On the surface, it may seem that all the nitrogenase variants should behave in the same way, as they carry out identical reactions, but closer investigation reveals that this may not be the case.
In general, the alternative nitrogenases are less effective at nitrogen fixation as well as the catalytic turnover of other small molecule substrates (C2H2, HN3, HCN) in comparison to Mo-nitrogenase. There is a tendency to divert more reducing equivalents to the production of H2 during substrate reductions, as observed in the increased specific activities (Table 4) during turnover of N2 or C2H2. For N2, studies have demonstrated different stoichiometries for the concomitant generation of H2 during N2 fixation by each version of nitrogenase, pointing to different mechanisms for each nitrogenase variant. Additionally, V-nitrogenase was shown to produce hydrazine as a product, in contrast to the Mo system that can consume N2H4 as a substrate. The source of these differences has not been clearly identified, but kinetic analysis hints at a unified mechanism for all three systems that show different relative rates for the reactions. However, new structural evidence suggests there could be different mechanisms for N2 reduction by Mo and V-nitrogenases, so further work will help clarify these observations. On the other hand, V- and Fe-only nitrogenase have a weaker affinity for acetylene than does Mo-nitrogenase, and so are less capable of turnover. Uniquely, acetylene can be reduced to ethane by the alternative systems while this cannot be achieved with the Mo-dependent enzyme. This (along with other experiments) indicates that different binding sites for C2H2 may exist in the alternative proteins, and further supports an alternate mechanism for the turnover of C2H2 by these enzymes.
The alternative nitrogenases were also found capable of converting CO and CO2 into hydrocarbons, an activity that was not observed in the Mo-dependent enzyme. CO normally inhibits the reduction of N2 but not H2 in Mo-nitrogenase, but both N2 and H2 activities were attenuated for V-nitrogenase. This coincided with an increase in the formation of C1 to C4 hydrocarbon products, with C2 products (C2H4, C2H6) being favored. The reduction of CO was found to be a property of the VFe protein, as swapping the V-cluster for the M-cluster to make a hybrid enzyme (M-VFe protein) still allowed for hydrocarbon formation, albeit with lower rates than wild-type V-nitrogenase. Reconstituting the apo-MoFe protein with V-cluster (V-MoFe protein) did not confer increased CO-reducing capabilities, but it did allow for the reduction of acetylene to ethane, indicating that V-cluster itself carries some of the activity properties when in a protein scaffold. This contrasts with the activities of the isolated M- and V-clusters, which both react similarly towards small molecule substrates CO, CO2 and CN−. The Fe-only nitrogenase was also discovered to convert CO2 into methane, which was only observed for the V-nitrogenase in the presence of D2O. The unique properties of the alternative nitrogenases indicate that there may still be much to learn about the reactivity of nitrogenase.
While great effort has gone into the study of nitrogenase over the last two decades, the number of questions that still require answers seem to grow – partly due to the technical challenges of working with delicate nitrogenase proteins and partly due to the ever-increasing avenues of research. For instance, the proposed biosynthetic pathway for the metal cofactors of the VFe and FeFe proteins require experimental validation. In particular, the elucidation of the exact role of VnfEN will be crucial, as this protein is supposedly the central hub for the assembly for both the V- and Fe-clusters. Effort in this line is chiefly hindered by the isolation of intact VnfEN and other relevant protein components, but even with isolated protein, the analysis would be plagued by the lack of structural knowledge of the alternative nitrogenases. Most important would be the structure of the P- and Fe-clusters of the FeFe protein, as there is relatively little spectroscopic and structural characterization. Even for the more established V-nitrogenase, several of the structural features remain controversial, including the exact configuration of the P-cluster of the VFe protein, as well as the relevance of the carbonate ligand that sits in place of a sulfur in the V-cluster.
Furthermore, the intriguing discoveries of the CO and CO2 reducing abilities of V- and Fe-only nitrogenases seem to suggest that the alternative nitrogenases are more versatile small molecule reductases. This raises questions regarding the physiological relevance of these activities in the native environment, as well as in the evolution history of the enzyme. From a biochemical standpoint, there are ongoing efforts to try to address how much overlap, if any at all, there is between the mechanisms of CO/CO2 reduction and that of N2 reduction. One caveat here, as discussed earlier, is that there is no strong consensus whether the N2 reduction mechanisms are identical for the three nitrogenase systems. Additional verification of the experimental parameters must be carefully carried out in each individual system for the best basis of comparison. However, one thing that is certain is that the trapping of any bona fide substrate- or intermediate-bound nitrogenase species will be crucial in gaining further insights. To this end, the investigation of V-nitrogenase has already proven to be greatly useful. Hopefully, future research will yield even more exciting details as the century-old enigma known as the N2 reduction mechanism continues to be unraveled.
Acknowledgements
The authors were supported by NIH-NIGMS grant GM67626 (to M.W.R. and Y.H.), which funded research related to the assembly of nitrogenase. The authors were also supported by the Department of Energy grants DOE (BES) DE-SC0016510 (to Y.H. and M.W.R.) and DE-SC0014470 (to M.W.R. and Y.H.), which funded work related to the mechanistic investigation of ammonia formation by nitrogenase and hydrocarbon formation by nitrogenase hybrid systems, respectively. In addition, the authors were supported by NSF grants CHE-1904131 (to M.W.R. and Y.H.) and CHE-1651398 (to Y.H.) that supported work related to CO activation by nitrogenase and CO2 reduction by nitrogenase Fe proteins, respectively.
Biographies
Andrew J. Jasniewski is from West Allis, WI and received his B.S. degree (2011) from the University of Wisconsin—Madison working for Thomas Brunold on functional models of the Mn-dependent superoxide dismutase. He then moved to the University of Minnesota to study the structures and spectroscopy of nonheme diiron enzymes and related model complexes with Lawrence Que Jr., receiving his Ph.D. degree in 2017. He currently works at the University of California, Irvine with Markus Ribbe and Yilin Hu on the biochemistry and spectroscopy of nitrogenase.
Chi Chung Lee received a B.S. degree in Molecular Biology from the University of California, San Diego, and a Ph.D. degree in Molecular Biology and Biochemistry from the University of California, Irvine (UCI). He was a postdoctoral fellow at UCI for several years and is currently a Project Scientist at the same institute. During the last 13 years, his research interests have been centered on the assembly of nitrogenase cofactors as well as the mechanisms of substrate reduction.
Yilin Hu received a B.S. degree in Genetics from Fudan University, China, and a Ph.D. degree in Biochemistry from Loma Linda University. She was a postdoctoral fellow at the University of California, Irvine, and is currently Associate Professor at the same institute. During the last 17 years, she has been focused on studies related to nitrogenase mechanism and assembly, with an emphasis on the genetic manipulation of nitrogenase systems.
Markus W. Ribbe received a B.S. degree in Biology, a M.S. degree in Microbiology, and a Ph.D. degree in Microbiology from the University of Bayreuth, Germany. He was a postdoctoral fellow at the University of California, Irvine, and is now Chancellor’s Professor at the same institute. During the past 25 years, he has focused on the mechanistic investigation of nitrogenase catalysis and assembly by combined biochemical, spectroscopic, and structural approaches.
References
- (1).Dos Santos PC; Fang Z; Mason SW; Setubal JC; Dixon R Distribution of Nitrogen Fixation and Nitrogenase-Like Sequences Amongst Microbial Genomes. BMC Genomics 2012, 13, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mus F; Colman DR; Peters JW; Boyd ES Geobiological Feedbacks, Oxygen, and the Evolution of Nitrogenase. Free Radical Biol. Med 2019, 140, 250–259. [DOI] [PubMed] [Google Scholar]
- (3).McRose DL; Zhang X; Kraepiel AML; Morel FMM. Diversity and Activity of Alternative Nitrogenases in Sequenced Genomes and Coastal Environments. Front. Microbiol 2017, 8, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Affourtit J; Zehr JP; Paerl HW Distribution of Nitrogen-Fixing Microorganisms Along the Neuse River Estuary, North Carolina. Microb. Ecol 2001, 41, 114–123. [DOI] [PubMed] [Google Scholar]
- (5).Cleveland CC; Townsend AR; Schimel DS; Fisher H; Howarth RW; Hedin LO; Perakis SS; Latty EF; Von Fischer JC; Elseroad A et al. Global Patterns of Terrestrial Biological Nitrogen (N2) Fixation in Natural Ecosystems. Global Biogeochem. Cycles 1999, 13, 623–645. [Google Scholar]
- (6).Noda S; Ohkuma M; Usami R; Horikoshi K; Kudo T Culture-Independent Characterization of a Gene Responsible for Nitrogen Fixation in the Symbiotic Microbial Community in the Gut of the Termite Neotermes koshunensis. Appl. Environ. Microbiol 1999, 65, 4935–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Betancourt DA; Loveless TM; Brown JW; Bishop PE Characterization of Diazotrophs Containing Mo-Independent Nitrogenases, Isolated from Diverse Natural Environments. Appl. Environ. Microbiol 2008, 74, 3471–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bellenger JP; Xu Y; Zhang X; Morel FMM; Kraepiel AML Possible Contribution of Alternative Nitrogenases to Nitrogen Fixation by Asymbiotic N2-Fixing Bacteria in Soils. Soil Biol. Biochem 2014, 69, 413–420. [Google Scholar]
- (9).Joerger RD; Bishop PE; Evans HJ Bacterial Alternative Nitrogen Fixation Systems. Crit. Rev. Microbiol 1988, 16, 1–14. [DOI] [PubMed] [Google Scholar]
- (10).Raymond J; Siefert JL; Staples CR; Blankenship RE The Natural History of Nitrogen Fixation. Mol. Biol. Evol 2004, 21, 541–554. [DOI] [PubMed] [Google Scholar]
- (11).Boyd E; Peters J New Insights into the Evolutionary History of Biological Nitrogen Fixation. Front. Microbiol 2013, 4, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Soboh B; Boyd ES; Zhao D; Peters JW; Rubio LM Substrate Specificity and Evolutionary Implications of a NifDK Enzyme Carrying NifB-co at Its Active Site. FEBS Lett. 2010, 584, 1487–1492. [DOI] [PubMed] [Google Scholar]
- (13).Oda Y; Samanta SK; Rey FE; Wu L; Liu X; Yan T; Zhou J; Harwood CS Functional Genomic Analysis of Three Nitrogenase Isozymes in the Photosynthetic Bacterium Rhodopseudomonas palustris. J. Bacteriol 2005, 187, 7784–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schneider K; Müller A; Schramm U; Klipp W Demonstration of a Molybdenum- and Vanadium-Independent Nitrogenase in a nifHDK-Deletion Mutant of Rhodobacter capsulatus. Eur. J. Biochem 1991, 195, 653–661. [DOI] [PubMed] [Google Scholar]
- (15).Boison G; Steingen C; Stal LJ; Bothe H The Rice Field Cyanobacteria Anabaena azotica and Anabaena sp. Ch1 Express Vanadium-Dependent Nitrogenase. Arch. Microbiol 2006, 186, 367–376. [DOI] [PubMed] [Google Scholar]
- (16).Joerger RD; Jacobson MR; Premakumar R; Wolfinger ED; Bishop PE Nucleotide Sequence and Mutational Analysis of the Structural Genes (anfHDGK) for the Second Alternative Nitrogenase from Azotobacter vinelandii. J. Bacteriol 1989, 171, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mus F; Alleman AB; Pence N; Seefeldt LC; Peters JW Exploring the Alternatives of Biological Nitrogen Fixation. Metallomics 2018, 10, 523–538. [DOI] [PubMed] [Google Scholar]
- (18).Hamilton TL; Ludwig M; Dixon R; Boyd ES; Dos Santos PC; Setubal JC; Bryant DA; Dean DR; Peters JW Transcriptional Profiling of Nitrogen Fixation in Azotobacter vinelandii. J. Bacteriol 2011, 193, 4477–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Demtröder L; Pfänder Y; Schäkermann S; Bandow JE; Masepohl B NifA Is the Master Regulator of Both Nitrogenase Systems in Rhodobacter capsulatus. MicrobiologyOpen 2019, 8, e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Thiel T; Pratte SB Regulation of Three Nitrogenase Gene Clusters in the Cyanobacterium Anabaena variabilis ATCC 29413. Life 2014, 4, 944–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang X; McRose DL; Darnajoux R; Bellenger JP; Morel FMM; Kraepiel AML. Alternative Nitrogenase Activity in the Environment and Nitrogen Cycle Implications. Biogeochemistry 2016, 127, 189–198. [Google Scholar]
- (22).Darnajoux R; Zhang X; McRose DL; Miadlikowska J; Lutzoni F; Kraepiel AML; Bellenger J-P Biological Nitrogen Fixation by Alternative Nitrogenases in Boreal Cyanolichens: Importance of Molybdenum Availability and Implications for Current Biological Nitrogen Fixation Estimates. New Phytol. 2017, 213, 680–689. [DOI] [PubMed] [Google Scholar]
- (23).Rebelein JG; Lee CC; Newcomb M; Hu Y; Ribbe MW Characterization of an M-Cluster-Substituted Nitrogenase VFe Protein. mBio 2018, 9, e00310–00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Anbar AD; Knoll AH Proterozoic Ocean Chemistry and Evolution: A Bioinorganic Bridge? Science 2002, 297, 1137–1142. [DOI] [PubMed] [Google Scholar]
- (25).Boyd E; Hamilton T; Peters J An Alternative Path for the Evolution of Biological Nitrogen Fixation. Front. Microbiol 2011, 2, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Boyd ES; Anbar AD; Miller S; Hamilton TL; Lavin M; Peters JW A Late Methanogen Origin for Molybdenum-Dependent Nitrogenase. Geobiology 2011, 9, 221–232. [DOI] [PubMed] [Google Scholar]
- (27).Zhang X; Sigman DM; Morel FMM; Kraepiel AML Nitrogen Isotope Fractionation by Alternative Nitrogenases and Past Ocean Anoxia. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 4782–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Stüeken EE; Buick R; Guy BM; Koehler MC Isotopic Evidence for Biological Nitrogen Fixation by Molybdenum-Nitrogenase from 3.2 Gyr. Nature 2015, 520, 666–669. [DOI] [PubMed] [Google Scholar]
- (29).Zerkle AL; Junium CK; Canfield DE; House CH Production of 15N-Depleted Biomass During Cyanobacterial N2-Fixation at High Fe Concentrations. J. Geophys. Res.: Biogeosci 2008, 113, G03014. [Google Scholar]
- (30).Sickerman, Nathaniel S; Rettberg, Lee A; Lee, Chi C; Hu Y; Ribbe, Markus W. Cluster Assembly in Nitrogenase. Essays Biochem. 2017, 61, 271–279. [DOI] [PubMed] [Google Scholar]
- (31).Hu Y; Ribbe MW Biosynthesis of the Metalloclusters of Nitrogenases. Annu. Rev. Biochem 2016, 85, 455–483. [DOI] [PubMed] [Google Scholar]
- (32).Ribbe MW; Hu Y; Hodgson KO; Hedman B Biosynthesis of Nitrogenase Metalloclusters. Chem. Rev 2014, 114, 4063–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hu Y; Fay AW; Lee CC; Yoshizawa J; Ribbe MW Assembly of Nitrogenase MoFe Protein. Biochemistry 2008, 47, 3973–3981. [DOI] [PubMed] [Google Scholar]
- (34).Hu Y; Corbett MC; Fay AW; Webber JA; Hedman B; Hodgson KO; Ribbe MW Nitrogenase Reactivity with P-Cluster Variants. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 13825–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Blank MA; Lee CC; Hu Y; Hodgson KO; Hedman B; Ribbe MW Structural Models of the [Fe4S4] Clusters of Homologous Nitrogenase Fe Proteins. Inorg. Chem 2011, 50, 7123–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hu Y; Ribbe MW Biosynthesis of the Iron-Molybdenum Cofactor of Nitrogenase. J. Biol. Chem 2013, 288, 13173–13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu Y; Ribbe MW Maturation of Nitrogenase Cofactor—the Role of a Class E Radical SAM Methyltransferase NifB. Curr. Opin. Chem. Biol 2016, 31, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kennedy C; Dean D The nifU, nifS and nifV Gene Products Are Required for Activity of All Three Nitrogenases of Azotobacter vinelandii. Mol. Gen. Genet 1992, 231, 494–498. [DOI] [PubMed] [Google Scholar]
- (39).Zheng L; Dean DR Catalytic Formation of a Nitrogenase Iron-Sulfur Cluster. J. Biol. Chem 1994, 269, 18723–18726. [PubMed] [Google Scholar]
- (40).Shi H.-w.; Wang L.-y.; Li X.-x.; Liu X.-m.; Hao T.-y.; He X.-j.; Chen S.-f. Genome-Wide Transcriptome Profiling of Nitrogen Fixation in Paenibacillus sp. WLY78. BMC Microbiol. 2016, 16, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fay AW; Wiig JA; Lee CC; Hu Y Identification and Characterization of Functional Homologs of Nitrogenase Cofactor Biosynthesis Protein NifB from Methanogens. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 14829–14833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schwarz G; Mendel RR; Ribbe MW Molybdenum Cofactors, Enzymes and Pathways. Nature 2009, 460, 839–847. [DOI] [PubMed] [Google Scholar]
- (43).Wiig JA; Hu Y; Ribbe MW Nifen-B Complex of Azotobacter vinelandii Is Fully Functional in Nitrogenase FeMo Cofactor Assembly. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 8623–8627.21551100 [Google Scholar]
- (44).Corbett MC; Hu Y; Fay AW; Ribbe MW; Hedman B; Hodgson KO Structural Insights into a Protein-Bound Iron-Molybdenum Cofactor Precursor. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Fay AW; Blank MA; Lee CC; Hu Y; Hodgson KO; Hedman B; Ribbe MW Spectroscopic Characterization of the Isolated Iron–Molybdenum Cofactor (FeMoco) Precursor from the Protein NifEN. Angew. Chem. Int. Ed 2011, 50, 7787–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wiig JA; Hu Y; Lee CC; Ribbe MW Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337, 1672–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Boal AK; Rosenzweig AC A Radical Route for Nitrogenase Carbide Insertion. Science 2012, 337, 1617–1618. [DOI] [PubMed] [Google Scholar]
- (48).Boal AK; Grove TL; McLaughlin MI; Yennawar NH; Booker SJ; Rosenzweig AC Structural Basis for Methyl Transfer by a Radical SAM Enzyme. Science 2011, 332, 1089–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wiig JA; Hu Y; Ribbe MW Refining the Pathway of Carbide Insertion into the Nitrogenase M-Cluster. Nat. Commun 2015, 6, 8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Rettberg LA; Wilcoxen J; Lee CC; Stiebritz MT; Tanifuji K; Britt RD; Hu Y Probing the Coordination and Function of Fe4S4 Modules in Nitrogenase Assembly Protein NifB. Nat. Commun 2018, 9, 2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tanifuji K; Lee CC; Sickerman NS; Tatsumi K; Ohki Y; Hu Y; Ribbe MW Tracing the ‘Ninth Sulfur’ of the Nitrogenase Cofactor Via a Semi-Synthetic Approach. Nat. Chem 2018, 10, 568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jasniewski AJ; Wilcoxen J; Tanifuji K; Hedman B; Hodgson KO; Britt RD; Hu Y; Ribbe MW Spectroscopic Characterization of an Eight-Iron Nitrogenase Cofactor Precursor That Lacks the “9th Sulfur”. Angew. Chem. Int. Ed 2019, 58, 14703–14707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fugate CJ; Jarrett JT Biotin Synthase: Insights into Radical-Mediated Carbon–Sulfur Bond Formation. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 1213–1222. [DOI] [PubMed] [Google Scholar]
- (54).Zheng L; White RH; Cash VL; Dean DR Mechanism for the Desulfurization of L-Cysteine Catalyzed by the nifS Gene Product. Biochemistry 1994, 33, 4714–4720. [DOI] [PubMed] [Google Scholar]
- (55).Kertesz MA Riding the Sulfur Cycle – Metabolism of Sulfonates and Sulfate Esters in Gram-Negative Bacteria. FEMS Microbiol. Rev 2000, 24, 135–175. [DOI] [PubMed] [Google Scholar]
- (56).Hu Y; Corbett MC; Fay AW; Webber JA; Hodgson KO; Hedman B; Ribbe MW FeMo Cofactor Maturation on NifEN. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17119–17124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yoshizawa JM; Blank MA; Fay AW; Lee CC; Wiig JA; Hu Y; Hodgson KO; Hedman B; Ribbe MW Optimization of FeMoco Maturation on NifEN. J. Am. Chem. Soc 2009, 131, 9321–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hu Y; Corbett MC; Fay AW; Webber JA; Hodgson KO; Hedman B; Ribbe MW Nitrogenase Fe Protein: A Molybdate/Homocitrate Insertase. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17125–17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Fay AW; Blank MA; Rebelein JG; Lee CC; Ribbe MW; Hedman B; Hodgson KO; Hu Y Assembly Scaffold NifEN: A Structural and Functional Homolog of the Nitrogenase Catalytic Component. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 9504–9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Fay AW; Blank MA; Yoshizawa JM; Lee CC; Wiig JA; Hu Y; Hodgson KO; Hedman B; Ribbe MW Formation of a Homocitrate-Free Iron-Molybdenum Cluster on NifEN: Implications for the Role of Homocitrate in Nitrogenase Assembly. Dalton Trans. 2010, 39, 3124–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kaiser JT; Hu Y; Wiig JA; Rees DC; Ribbe MW Structure of Precursor-Bound NifEN: A Nitrogenase FeMo Cofactor Maturase/Insertase. Science 2011, 331, 91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Schmid B; Ribbe MW; Einsle O; Yoshida M; Thomas LM; Dean DR; Rees DC; Burgess BK Structure of a Cofactor-Deficient Nitrogenase MoFe Protein. Science 2002, 296, 352–356. [DOI] [PubMed] [Google Scholar]
- (63).Yoshizawa JM; Fay AW; Lee CC; Hu Y; Ribbe MW Insertion of Heterometals into the NifEN-Associated Iron–Molybdenum Cofactor Precursor. JBIC, J. Biol. Inorg. Chem 2010, 15, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wolfinger ED; Bishop PE Nucleotide Sequence and Mutational Analysis of the vnfENX Region of Azotobacter vinelandii. J. Bacteriol 1991, 173, 7565–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Howard JB; Rees DC Structural Basis of Biological Nitrogen Fixation. Chem. Rev 1996, 96, 2965–2982. [DOI] [PubMed] [Google Scholar]
- (66).Burgess BK; Lowe DJ Mechanism of Molybdenum Nitrogenase. Chem. Rev 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- (67).Kim J; Rees DC Crystallographic Structure and Functional Implications of the Nitrogenase Molybdenum–Iron Protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. [DOI] [PubMed] [Google Scholar]
- (68).Kim J; Rees DC Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 1992, 257, 1677–1682. [DOI] [PubMed] [Google Scholar]
- (69).Georgiadis MM; Komiya H; Chakrabarti P; Woo D; Kornuc JJ; Rees DC Crystallographic Structure of the Nitrogenase Iron Protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [DOI] [PubMed] [Google Scholar]
- (70).Schindelin H; Kisker C; Schlessman JL; Howard JB; Rees DC Structure of ADP·AlF4 – Stabilized Nitrogenase Complex and Its Implications for Signal Transduction. Nature 1997, 387, 370–376. [DOI] [PubMed] [Google Scholar]
- (71).Tezcan FA; Kaiser JT; Mustafi D; Walton MY; Howard JB; Rees DC Nitrogenase Complexes: Multiple Docking Sites for a Nucleotide Switch Protein. Science 2005, 309, 1377–1380. [DOI] [PubMed] [Google Scholar]
- (72).George GN; Coyle CL; Hales BJ; Cramer SP X-Ray Absorption of Azotobacter vinelandii Vanadium Nitrogenase. J. Am. Chem. Soc 1988, 110, 4057–4059. [Google Scholar]
- (73).Arber JM; Dobson BR; Eady RR; Stevens P; Hasnain SS; Garner CD; Smith BE Vanadium K-Edge X-Ray Absorption Spectrum of the VFe Protein of the Vanadium Nitrogenase of Azotobacter chroococcum. Nature 1987, 325, 372–374. [Google Scholar]
- (74).Sippel D; Einsle O The Structure of Vanadium Nitrogenase Reveals an Unusual Bridging Ligand. Nat. Chem. Biol 2017, 13, 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Rohde M; Trncik C; Sippel D; Gerhardt S; Einsle O Crystal Structure of VnfH, the Iron Protein Component of Vanadium Nitrogenase. JBIC, J. Biol. Inorg. Chem 2018, 23, 1049–1056. [DOI] [PubMed] [Google Scholar]
- (76).Krahn E; Weiss B; Kröckel M; Groppe J; Henkel G; Cramer S; Trautwein A; Schneider K; Müller A The Fe-Only Nitrogenase from Rhodobacter capsulatus: Identification of the Cofactor, an Unusual, High-Nuclearity Iron-Sulfur Cluster, by Fe K-Edge EXAFS and 57Fe Mössbauer Spectroscopy. JBIC, J. Biol. Inorg. Chem 2002, 7, 37–45. [DOI] [PubMed] [Google Scholar]
- (77).Jasniewski A; Sickerman N; Hu Y; Ribbe M The Fe Protein: An Unsung Hero of Nitrogenase. Inorganics 2018, 6, 25. [Google Scholar]
- (78).Lee CC; Stiebritz MT; Hu Y Reactivity of [Fe4S4] Clusters toward C1 Substrates: Mechanism, Implications, and Potential Applications. Acc. Chem. Res 2019, 52, 1168–1176. [DOI] [PubMed] [Google Scholar]
- (79).Broach RB; Rupnik K; Hu Y; Fay AW; Cotton M; Ribbe MW; Hales BJ Variable-Temperature, Variable-Field Magnetic Circular Dichroism Spectroscopic Study of the Metal Clusters in the ΔnifB and ΔnifH MoFe Proteins of Nitrogenase from Azotobacter vinelandii. Biochemistry 2006, 45, 15039–15048. [DOI] [PubMed] [Google Scholar]
- (80).Cotton MS; Rupnik K; Broach RB; Hu Y; Fay AW; Ribbe MW; Hales BJ Vtvh-Mcd Study of the ΔnifBΔnifZ MoFe Protein from Azotobacter vinelandii. J. Am. Chem. Soc 2009, 131, 4558–4559. [DOI] [PubMed] [Google Scholar]
- (81).Corbett MC; Hu Y; Naderi F; Ribbe MW; Hedman B; Hodgson KO Comparison of Iron-Molybdenum Cofactor-Deficient Nitrogenase MoFe Proteins by X-Ray Absorption Spectroscopy: Implications for P-Cluster Biosynthesis. J. Biol. Chem 2004, 279, 28276–28282. [DOI] [PubMed] [Google Scholar]
- (82).Hu Y; Fay AW; Dos Santos PC; Naderi F; Ribbe MW Characterization of Azotobacter vinelandii nifZ Deletion Strains: Indication of Stepwise MoFe Protein Assembly. J. Biol. Chem 2004, 279, 54963–54971. [DOI] [PubMed] [Google Scholar]
- (83).Seefeldt LC; Hoffman BM; Peters JW; Raugei S; Beratan DN; Antony E; Dean DR Energy Transduction in Nitrogenase. Acc. Chem. Res 2018, 51, 2179–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Walker JE; Saraste M; Runswick MJ; Gay NJ Distantly Related Sequences in the Alpha- and Beta-Subunits of ATP Synthase, Myosin, Kinases and Other ATP-Requiring Enzymes and a Common Nucleotide Binding Fold. EMBO J. 1982, 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Jang SB; Seefeldt LC; Peters JW Insights into Nucleotide Signal Transduction in Nitrogenase: Structure of an Iron Protein with MgADP Bound. Biochemistry 2000, 39, 14745–14752. [DOI] [PubMed] [Google Scholar]
- (86).Sen S; Krishnakumar A; McClead J; Johnson MK; Seefeldt LC; Szilagyi RK; Peters JW Insights into the Role of Nucleotide-Dependent Conformational Change in Nitrogenase Catalysis: Structural Characterization of the Nitrogenase Fe Protein Leu127 Deletion Variant with Bound MgATP. J. Inorg. Biochem 2006, 100, 1041–1052. [DOI] [PubMed] [Google Scholar]
- (87).Rebelein JG; Stiebritz MT; Lee CC; Hu Y Activation and Reduction of Carbon Dioxide by Nitrogenase Iron Proteins. Nat. Chem. Biol 2016, 13, 147–149. [DOI] [PubMed] [Google Scholar]
- (88).Walker GA; Mortenson LE Effect of Magnesium Adenosine 5’-Triphosphate on the Accessibility of the Iron of Clostridial Azoferredoxin, a Component of Nitrogenase. Biochemistry 1974, 13, 2382–2388. [DOI] [PubMed] [Google Scholar]
- (89).Ljones T; Burris RH Nitrogenase: The Reaction between Iron Protein and Bathophenanthrolinedisulfonate as a Probe for Interactions with MgATP. Biochemistry 1978, 17, 1866–1872. [DOI] [PubMed] [Google Scholar]
- (90).Deits TL; Howard JB Kinetics of Mgatp-Dependent Iron Chelation from the Fe-Protein of the Azotobacter vinelandii Nitrogenase Complex. Evidence for Two States. J. Biol. Chem 1989, 264, 6619–6628. [PubMed] [Google Scholar]
- (91).Chen L; Gavini N; Tsuruta H; Eliezer D; Burgess BK; Doniach S; Hodgson KO MgATP-Induced Conformational Changes in the Iron Protein from Azotobacter vinelandii, as Studied by Small-Angle X-Ray Scattering. J. Biol. Chem 1994, 269, 3290–3294. [PubMed] [Google Scholar]
- (92).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Yates MG Electron Transport to Nitrogenase in Azotobacter chroococcum: Azotobacter Flavodoxin Hydroquinone as an Electron Donor. FEBS Lett. 1972, 27, 63–67. [DOI] [PubMed] [Google Scholar]
- (94).Duyvis MG; Wassink H; Haaker H Nitrogenase of Azotobacter vinelandii: Kinetic Analysis of the Fe Protein Redox Cycle. Biochemistry 1998, 37, 17345–17354. [DOI] [PubMed] [Google Scholar]
- (95).Bennett LT; Jacobson MR; Dean DR Isolation, Sequencing, and Mutagenesis of the nifF Gene Encoding Flavodoxin from Azotobacter vinelandii. J. Biol. Chem 1988, 263, 1364–1369. [PubMed] [Google Scholar]
- (96).Thorneley RN; Deistung J Electron-Transfer Studies Involving Flavodoxin and a Natural Redox Partner, the Iron Protein of Nitrogenase. Conformational Constraints on Protein-Protein Interactions and the Kinetics of Electron Transfer within the Protein Complex. Biochem. J 1988, 253, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Martin AE; Burgess BK; Iismaa SE; Smartt CT; Jacobson MR; Dean DR Construction and Characterization of an Azotobacter vinelandii Strain with Mutations in the Genes Encoding Flavodoxin and Ferredoxin I. J. Bacteriol 1989, 171, 3162–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Watt GD; Reddy KRN Formation of an All Ferrous Fe4S4 Cluster in the Iron Protein Component of Azotobacter vinelandii Nitrogenase. J. Inorg. Biochem 1994, 53, 281–294. [Google Scholar]
- (99).Watt GD; Wang ZC; Knotts RR Redox Reactions of and Nucleotide Binding to the Iron Protein of Azotobacter vinelandii. Biochemistry 1986, 25, 8156–8162. [Google Scholar]
- (100).Lanzilotta WN; Ryle MJ; Seefeldt LC Nucleotide Hydrolysis and Protein Conformational Changes in Azotobacter vinelandii Nitrogenase Iron Protein: Defining the Function of Aspartate 129. Biochemistry 1995, 34, 10713–10723. [DOI] [PubMed] [Google Scholar]
- (101).Bergström J; Eady RR; Thorneley RNF The Vanadium- and Molybdenum-Containing Nitrogenases of Azotobacter chroococcum. Comparison of Mid-Point Potentials and Kinetics of Reduction by Sodium Dithionite of the Iron Proteins with Bound Magnesium Adenosine 5′-Diphosphate. Biochem. J 1988, 251, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Lindahl PA; Day EP; Kent TA; Orme-Johnson WH; Münck E Mössbauer, EPR, and Magnetization Studies of the Azotobacter vinelandii Fe Protein. Evidence for a [4Fe-4S]1+ Cluster with Spin S = 3/2. J. Biol. Chem 1985, 260, 11160–11173. [PubMed] [Google Scholar]
- (103).Orme-Johnson WH; Hamilton WD; Jones TL; Tso MYW; Burris RH; Shah VK; Brill WJ Electron Paramagnetic Resonance of Nitrogenase and Nitrogenase Components from Clostridium pasteurianum W5 and Azotobacter vinelandii Op. Proc. Natl. Acad. Sci. U. S. A 1972, 69, 3142–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Hiller CJ; Stiebritz MT; Lee CC; Liedtke J; Hu Y Tuning Electron Flux through Nitrogenase with Methanogen Iron Protein Homologues. Chem. Eur. J 2017, 23, 16152–16156. [DOI] [PubMed] [Google Scholar]
- (105).Lindahl PA; Gorelick NJ; Münck E; Orme-Johnson WH EPR and Mössbauer Studies of Nucleotide-Bound Nitrogenase Iron Protein from Azotobacter vinelandii. J. Biol. Chem 1987, 262, 14945–14953. [PubMed] [Google Scholar]
- (106).Lowery TJ; Wilson PE; Zhang B; Bunker J; Harrison RG; Nyborg AC; Thiriot D; Watt GD Flavodoxin Hydroquinone Reduces Azotobacter vinelandii Fe Protein to the All-Ferrous Redox State with a S = 0 Spin State. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17131–17136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Guo M; Sulc F; Ribbe MW; Farmer PJ; Burgess BK Direct Assessment of the Reduction Potential of the [4Fe–4S]1+/0 Couple of the Fe Protein from Azotobacter vinelandii. J. Am. Chem. Soc 2002, 124, 12100–12101. [DOI] [PubMed] [Google Scholar]
- (108).Angove HC; Yoo SJ; Burgess BK; Münck E Mössbauer and Epr Evidence for an All-Ferrous Fe4S4 Cluster with S = 4 in the Fe Protein of Nitrogenase. J. Am. Chem. Soc 1997, 119, 8730–8731. [Google Scholar]
- (109).Vincent KA; Tilley GJ; Quammie NC; Streeter I; Burgess BK; Cheesman MR; Armstrong FA Instantaneous, Stoichiometric Generation of Powerfully Reducing States of Protein Active Sites Using Eu(II) and Polyaminocarboxylate Ligands. Chem. Commun 2003, 2590–2591. [DOI] [PubMed] [Google Scholar]
- (110).Chakrabarti M; Deng L; Holm RH; Münck E; Bominaar EL Mössbauer, Electron Paramagnetic Resonance, and Theoretical Studies of a Carbene-Based All-Ferrous Fe4S4 Cluster: Electronic Origin and Structural Identification of the Unique Spectroscopic Site. Inorg. Chem 2009, 48, 2735–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Chakrabarti M; Münck E; Bominaar EL Density Functional Theory Study of an All Ferrous 4Fe-4S Cluster. Inorg. Chem 2011, 50, 4322–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Yoo SJ; Angove HC; Burgess BK; Hendrich MP; Münck E Mössbauer and Integer-Spin EPR Studies and Spin-Coupling Analysis of the [4Fe-4S]0 Cluster of the Fe Protein from Azotobacter vinelandii Nitrogenase. J. Am. Chem. Soc 1999, 121, 2534–2545. [Google Scholar]
- (113).Strop P; Takahara PM; Chiu H-J; Angove HC; Burgess BK; Rees DC Crystal Structure of the All-Ferrous [4Fe-4S]0 Form of the Nitrogenase Iron Protein from Azotobacter vinelandii. Biochemistry 2001, 40, 651–656. [DOI] [PubMed] [Google Scholar]
- (114).Musgrave KB; Angove HC; Burgess BK; Hedman B; Hodgson KO All-Ferrous Titanium(III) Citrate Reduced Fe Protein of Nitrogenase: An XAS Study of Electronic and Metrical Structure. J. Am. Chem. Soc 1998, 120, 5325–5326. [Google Scholar]
- (115).Schneider K; Gollan U; Dröttboom M; Selsemeier-Voigt S; Müller A Comparative Biochemical Characterization of the Iron-Only Nitrogenase and the Molybdenum Nitrogenase from Rhodobacter capsulatus. Eur. J. Biochem 1997, 244, 789–800. [DOI] [PubMed] [Google Scholar]
- (116).Spatzal T; Aksoyoglu M; Zhang L; Andrade SLA; Schleicher E; Weber S; Rees DC; Einsle O Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Einsle O; Tezcan FA; Andrade SLA; Schmid B; Yoshida M; Howard JB; Rees DC Nitrogenase MoFe-Protein at 1.16 Å Resolution: A Central Ligand in the FeMo-Cofactor. Science 2002, 297, 1696–1700. [DOI] [PubMed] [Google Scholar]
- (118).Chan MK; Kim J; Rees DC The Nitrogenase FeMo-Cofactor and P-Cluster Pair: 2.2 Å Resolution Structures. Science 1993, 260, 792–794. [DOI] [PubMed] [Google Scholar]
- (119).Lancaster KM; Roemelt M; Ettenhuber P; Hu Y; Ribbe MW; Neese F; Bergmann U; DeBeer S X-Ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Pierik AJ; Wassink H; Haaker H; Hagen WR Redox Properties and EPR Spectroscopy of the P Clusters of Azotobacter vinelandii MoFe Protein. Eur. J. Biochem 1993, 212, 51–61. [DOI] [PubMed] [Google Scholar]
- (121).Huynh BH; Henzl MT; Christner JA; Zimmermann R; Orme-Johnson WH; Münck E Nitrogenase XII. Mössbauer Studies of the MoFe Protein from Clostridium pasteurianum W5. Biochim. Biophys. Acta, Protein Struct 1980, 623, 124–138. [DOI] [PubMed] [Google Scholar]
- (122).Rupnik K; Hu Y; Lee CC; Wiig JA; Ribbe MW; Hales BJ P+ State of Nitrogenase P-Cluster Exhibits Electronic Structure of a [Fe4S4]+ Cluster. J. Am. Chem. Soc 2012, 134, 13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Mayer SM; Lawson DM; Gormal CA; Roe SM; Smith BE New Insights into Structure-Function Relationships in Nitrogenase: A 1.6 Å Resolution X-Ray Crystallographic Study of Klebsiella pneumoniae MoFe-Protein. J. Mol. Biol 1999, 292, 871–891. [DOI] [PubMed] [Google Scholar]
- (124).Morgan TV; Mortenson LE; McDonald JW; Watt GD Comparison of Redox and EPR Properties of the Molybdenum Iron Proteins of Clostridium pasteurianum and Azotobacter vinelandii Nitrogenases. J. Inorg. Biochem 1988, 33, 111–120. [DOI] [PubMed] [Google Scholar]
- (125).Surerus KK; Hendrich MP; Christie PD; Rottgardt D; Orme-Johnson WH; Munck E Möessbauer and Integer-Spin EPR of the Oxidized P-Clusters of Nitrogenase: Pox Is a Non-Kramers System with a Nearly Degenerate Ground Doublet. J. Am. Chem. Soc 1992, 114, 8579–8590. [Google Scholar]
- (126).Peters JW; Stowell MHB; Soltis SM; Finnegan MG; Johnson MK; Rees DC Redox-Dependent Structural Changes in the Nitrogenase P-Cluster. Biochemistry 1997, 36, 1181–1187. [DOI] [PubMed] [Google Scholar]
- (127).Danyal K; Dean DR; Hoffman BM; Seefeldt LC Electron Transfer within Nitrogenase: Evidence for a Deficit-Spending Mechanism. Biochemistry 2011, 50, 9255–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Chan JM; Christiansen J; Dean DR; Seefeldt LC Spectroscopic Evidence for Changes in the Redox State of the Nitrogenase P-Cluster During Turnover. Biochemistry 1999, 38, 5779–5785. [DOI] [PubMed] [Google Scholar]
- (129).Danyal K; Mayweather D; Dean DR; Seefeldt LC; Hoffman BM Conformational Gating of Electron Transfer from the Nitrogenase Fe Protein to MoFe Protein. J. Am. Chem. Soc 2010, 132, 6894–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Rawlings J; Shah VK; Chisnell JR; Brill WJ; Zimmermann R; Münck E; Orme-Johnson WH Novel Metal Cluster in the Iron-Molybdenum Cofactor of Nitrogenase. Spectroscopic Evidence. J. Biol. Chem 1978, 253, 1001–1004. [PubMed] [Google Scholar]
- (131).Burgess BK The Iron-Molybdenum Cofactor of Nitrogenase. Chem. Rev 1990, 90, 1377–1406. [Google Scholar]
- (132).Smith BE; Lowe DJ; Bray RC Studies by Electron Paramagnetic Resonance on the Catalytic Mechanism of Nitrogenase of Klebsiella pneumoniae. Biochem. J 1973, 135, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Siemann S; Schneider K; Dröttboom M; Müller A The Fe-Only Nitrogenase and the Mo Nitrogenase from Rhodobacter capsulatus. Eur. J. Biochem 2002, 269, 1650–1661. [DOI] [PubMed] [Google Scholar]
- (134).Watt GD; Burns A; Lough S; Tennent DL Redox and Spectroscopic Properties of Oxidized MoFe Protein from Azotobacter vinelandii. Biochemistry 1980, 19, 4926–4932. [DOI] [PubMed] [Google Scholar]
- (135).Watt GD; Burns A; Tennent DL Stoichiometry and Spectral Properties of the Molybdenum-Iron (MoFe) Cofactor and Noncofactor Redox Centers in the Molybdenum-Iron (MoFe) Protein of Nitrogenase from Azotobacter vinelandii. Biochemistry 1981, 20, 7272–7277. [DOI] [PubMed] [Google Scholar]
- (136).Lough S; Burns A; Watt GD Redox Reactions of the Nitrogenase Complex from Azotobacter vinelandii. Biochemistry 1983, 22, 4062–4066. [Google Scholar]
- (137).Schultz FA; Gheller SF; Burgess BK; Lough S; Newton WE Electrochemical Characterization of the Iron-Molybdenum Cofactor from Azotobacter vinelandii Nitrogenase. J. Am. Chem. Soc 1985, 107, 5364–5368. [Google Scholar]
- (138).Pickett CJ; Vincent KA; Ibrahim SK; Gormal CA; Smith BE; Fairhurst SA; Best SP Synergic Binding of Carbon Monoxide and Cyanide to the FeMo Cofactor of Nitrogenase: Relic Chemistry of an Ancient Enzyme? Chem. Eur. J 2004, 10, 4770–4776. [DOI] [PubMed] [Google Scholar]
- (139).Lydon BR; Lee CC; Tanifuji K; Sickerman NS; Newcomb MP; Hu Y; Ribbe MW; Yang JY Electrochemical Characterization of Isolated Nitrogenase Cofactors from Azotobacter vinelandii. ChemBioChem [Online Early Access]. DOI: 10.1002/cbic.201900425. Published Online: Aug 7, 2019. [DOI] [PubMed] [Google Scholar]
- (140).Hickey DP; Cai R; Yang Z-Y; Grunau K; Einsle O; Seefeldt LC; Minteer SD Establishing a Thermodynamic Landscape for the Active Site of Mo-Dependent Nitrogenase. J. Am. Chem. Soc 2019, 141, 17150–17157. [DOI] [PubMed] [Google Scholar]
- (141).Eady RR; Robson RL; Richardson TH; Miller RW; Hawkins M The Vanadium Nitrogenase of Azotobacter chroococcum. Purification and Properties of the VFe Protein. Biochem. J 1987, 244, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Hales BJ; Case EE; Morningstar JE; Dzeda MF; Mauterer LA Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25, 7251–7255. [DOI] [PubMed] [Google Scholar]
- (143).Eady RR Structure–Function Relationships of Alternative Nitrogenases. Chem. Rev 1996, 96, 3013–3030. [DOI] [PubMed] [Google Scholar]
- (144).Robson RL; Woodley PR; Pau RN; Eady RR Structural Genes for the Vanadium Nitrogenase from Azotobacter chroococcum. EMBO J. 1989, 8, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Fallik E; Robson RL Completed Sequence of the Region Encoding the Structural Genes for the Vanadium Nitrogenase of Azotobacter chroococcum. Nucleic Acids Res. 1990, 18, 4616–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Dyer DH; Rubio LM; Thoden JB; Holden HM; Ludden PW; Rayment I The Three-Dimensional Structure of the Core Domain of NafY from Azotobacter vinelandii Determined at 1.8-Å Resolution. J. Biol. Chem 2003, 278, 32150–32156. [DOI] [PubMed] [Google Scholar]
- (147).Homer MJ; Paustian TD; Shah VK; Roberts GP The nifY Product of Klebsiella pneumoniae Is Associated with Apodinitrogenase and Dissociates Upon Activation with the Iron-Molybdenum Cofactor. J. Bacteriol 1993, 175, 4907–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Blanchard CZ; Hales BJ Isolation of Two Forms of the Nitrogenase VFe Protein from Azotobacter vinelandii. Biochemistry 1996, 35, 472–478. [DOI] [PubMed] [Google Scholar]
- (149).Lee CC; Hu Y; Ribbe MW Unique Features of the Nitrogenase VFe Protein from Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 9209–9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A Bound Reaction Intermediate Sheds Light on the Mechanism of Nitrogenase. Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
- (151).Fisher K; Dilworth MJ; Kim C-H; Newton WE Azotobacter vinelandii Nitrogenases with Substitutions in the FeMo-Cofactor Environment of the MoFe Protein: Effects of Acetylene or Ethylene on Interactions with H+, HCN, and CN−. Biochemistry 2000, 39, 10855–10865. [DOI] [PubMed] [Google Scholar]
- (152).Fisher K; Dilworth MJ; Newton WE Differential Effects on N2 Binding and Reduction, HD Formation, and Azide Reduction with α-195His- and α-191Gln-Substituted MoFe Proteins of Azotobacter vinelandii Nitrogenase. Biochemistry 2000, 39, 15570–15577. [DOI] [PubMed] [Google Scholar]
- (153).Scott DJ; May HD; Newton WE; Brigle KE; Dean DR Role for the Nitrogenase MoFe Protein α-Subunit in FeMo-Cofactor Binding and Catalysis. Nature 1990, 343, 188–190. [DOI] [PubMed] [Google Scholar]
- (154).Kim C-H; Newton WE; Dean DR Role of the MoFe Protein A-Subunit Histidine-195 Residue in FeMo-Cofactor Binding and Nitrogenase Catalysis. Biochemistry 1995, 34, 2798–2808. [DOI] [PubMed] [Google Scholar]
- (155).Brown SD; Mehn MP; Peters JC Heterolytic H2 Activation Mediated by Low-Coordinate L 3Fe-(μ-N)-FeL3 Complexes to Generate Fe(μ-NH)(μ-H)Fe Species. J. Am. Chem. Soc 2005, 127, 13146–13147. [DOI] [PubMed] [Google Scholar]
- (156).Benediktsson B; Thorhallsson AT; Bjornsson R QM/MM Calculations Reveal a Bridging Hydroxo Group in a Vanadium Nitrogenase Crystal Structure. Chem. Commun 2018, 54, 7310–7313. [DOI] [PubMed] [Google Scholar]
- (157).Smith BE; Eady RR; Lowe DJ; Gormal C The Vanadium-Iron Protein of Vanadium Nitrogenase from Azotobacter chroococcum contains an Iron-Vanadium Cofactor. Biochem. J 1988, 250, 299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Pierik AJ In New Horizons in Nitrogen Fixation: Proceedings of the 9th International Congress on Nitrogen Fixation, Cancún, Mexico, December 6–12, 1992; Lowe DJ;Eldridge ME;Marritt S;Farrar JA;Thomson AJ;Eady RR, Eds.; Springer Netherlands: Dordrecht, 1993, 147 [Google Scholar]
- (159).Morningstar JE; Johnson MK; Case EE; Hales BJ Characterization of the Metal Clusters in the Nitrogenase Molybdenum-Iron and Vanadium-Iron Proteins of Azotobacter vinelandii Using Magnetic Circular Dichroism Spectroscopy. Biochemistry 1987, 26, 1795–1800. [DOI] [PubMed] [Google Scholar]
- (160).Morningstar JE; Hales BJ Electron Paramagnetic Resonance Study of the Vanadium-Iron Protein of Nitrogenase from Azotobacter vinelandii. J. Am. Chem. Soc 1987, 109, 6854–6855. [Google Scholar]
- (161).Ravi N; Moore V; Lloyd SG; Hales BJ; Huynh BH Mössbauer Characterization of the Metal Clusters in Azotobacter vinelandii Nitrogenase VFe Protein. J. Biol. Chem 1994, 269, 20920–20924. [PubMed] [Google Scholar]
- (162).Tittsworth RC; Hales BJ Oxidative Titration of the Nitrogenase VFe Protein from Azotobacter vinelandii: An Example of Redox-Gated Electron Flow. Biochemistry 1996, 35, 479–487. [DOI] [PubMed] [Google Scholar]
- (163).Moore VG; Tittsworth RC; Hales BJ Construction and Characterization of Hybrid Component 1 from V-Nitrogenase Containing FeMo Cofactor. J. Am. Chem. Soc 1994, 116, 12101–12102. [Google Scholar]
- (164).Tittsworth RC; Hales BJ Detection of EPR Signals Assigned to the 1-Equiv-Oxidized P-Clusters of the Nitrogenase MoFe-Protein from Azotobacter vinelandii. J. Am. Chem. Soc 1993, 115, 9763–9767. [Google Scholar]
- (165).Arber JM; Dobson BR; Eady RR; Hasnain SS; Garner CD; Matsushita T; Nomura M; Smith BE Vanadium K-Edge X-Ray-Absorption Spectroscopy of the Functioning and Thionine-Oxidized Forms of the VFe-Protein of the Vanadium Nitrogenase from Azotobacter chroococcum. Biochem. J 1989, 258, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Harvey I; Arber JM; Eady RR; Smith BE; Garner CD; Hasnain SS Iron K-Edge X-Ray-Absorption Spectroscopy of the Iron-Vanadium Cofactor of the Vanadium Nitrogenase from Azotobacter chroococcum. Biochem. J 1990, 266, 929–931. [PMC free article] [PubMed] [Google Scholar]
- (167).Chen J; Christiansen J; Tittsworth RC; Hales BJ; George SJ; Coucouvanis D; Cramer SP Iron EXAFS of Azotobacter vinelandii Nitrogenase Molybdenum-Iron and Vanadium-Iron Proteins. J. Am. Chem. Soc 1993, 115, 5509–5515. [Google Scholar]
- (168).Fay AW; Blank MA; Lee CC; Hu Y; Hodgson KO; Hedman B; Ribbe MW Characterization of Isolated Nitrogenase FeVco. J. Am. Chem. Soc 2010, 132, 12612–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Rees JA; Bjornsson R; Schlesier J; Sippel D; Einsle O; DeBeer S The Fe–V Cofactor of Vanadium Nitrogenase Contains an Interstitial Carbon Atom. Angew. Chem. Int. Ed 2015, 54, 13249–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Rees JA; Bjornsson R; Kowalska JK; Lima FA; Schlesier J; Sippel D; Weyhermüller T; Einsle O; Kovacs JA; DeBeer S Comparative Electronic Structures of Nitrogenase FeMoco and FeVco. Dalton Trans. 2017, 46, 2445–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Bjornsson R; Lima FA; Spatzal T; Weyhermuller T; Glatzel P; Bill E; Einsle O; Neese F; DeBeer S Identification of a Spin-Coupled Mo(III) in the Nitrogenase Iron-Molybdenum Cofactor. Chem. Sci 2014, 5, 3096–3103. [Google Scholar]
- (172).Bjornsson R; Neese F; Schrock RR; Einsle O; DeBeer S The Discovery of Mo(III) in FeMoco: Reuniting Enzyme and Model Chemistry. JBIC, J. Biol. Inorg. Chem 2015, 20, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Davis R; Lehman L; Petrovich R; Shah VK; Roberts GP; Ludden PW Purification and Characterization of the Alternative Nitrogenase from the Photosynthetic Bacterium Rhodospirillum rubrum. J. Bacteriol 1996, 178, 1445–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Müller A; Schneider K; Gollan U; Krahn E; Dröttboom M Characterization of the “Iron Only” Nitrogenase from Rhodobacter capsulatus. J. Inorg. Biochem 1995, 59, 551. [DOI] [PubMed] [Google Scholar]
- (175).Chisnell JR; Premakumar R; Bishop PE Purification of a Second Alternative Nitrogenase from a nifHDK Deletion Strain of Azotobacter vinelandii. J. Bacteriol 1988, 170, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Pau RN; Eldridge ME; Lowe DJ; Mitchenall LA; Eady RR Molybdenum-Independent Nitrogenases of Azotobacter vinelandii: A Functional Species of Alternative Nitrogenase-3 Isolated from a Molybdenum-Tolerant Strain Contains an Iron-Molybdenum Cofactor. Biochem. J 1993, 293, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Zheng Y; Harris DF; Yu Z; Fu Y; Poudel S; Ledbetter RN; Fixen KR; Yang Z-Y; Boyd ES; Lidstrom ME et al. A Pathway for Biological Methane Production Using Bacterial Iron-Only Nitrogenase. Nat. Microbiol 2018, 3, 281–286. [DOI] [PubMed] [Google Scholar]
- (178).Schneider K; Müller A In Catalysts for Nitrogen Fixation: Nitrogenases, Relevant Chemical Models and Commercial Processes; Smith BE;Richards RL;Newton WE, Eds.; Springer Netherlands: Dordrecht, 2004, 281–307 [Google Scholar]
- (179).Hales BJ In Catalysts for Nitrogen Fixation: Nitrogenases, Relevant Chemical Models and Commercial Processes; Smith BE;Richards RL;Newton WE, Eds.; Springer Netherlands: Dordrecht, 2004, 255–279 [Google Scholar]
- (180).Harris DF; Lukoyanov DA; Shaw S; Compton P; Tokmina-Lukaszewska M; Bothner B; Kelleher N; Dean DR; Hoffman BM; Seefeldt LC Mechanism of N2 Reduction Catalyzed by Fe-Nitrogenase Involves Reductive Elimination of H2. Biochemistry 2018, 57, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).McLean PA; Papaefthymiou V; Orme-Johnson WH; Münck E Isotopic Hybrids of Nitrogenase. Mössbauer Study of MoFe Protein with Selective 57Fe Enrichment of the P-Cluster. J. Biol. Chem 1987, 262, 12900–12903. [PubMed] [Google Scholar]
- (182).Yoo SJ; Angove HC; Papaefthymiou V; Burgess BK; Münck E Mössbauer Study of the MoFe Protein of Nitrogenase from Azotobacter vinelandii Using Selective 57Fe Enrichment of the M-Centers. J. Am. Chem. Soc 2000, 122, 4926–4936. [Google Scholar]
- (183).Harris TV; Szilagyi RK Comparative Assessment of the Composition and Charge State of Nitrogenase FeMo-Cofactor. Inorg. Chem 2011, 50, 4811–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Bjornsson R; Neese F; DeBeer S Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem 2017, 56, 1470–1477. [DOI] [PubMed] [Google Scholar]
- (185).Kowalska JK; Henthorn JT; Van Stappen C; Trncik C; Einsle O; Keavney D; DeBeer S X-Ray Magnetic Circular Dichroism Spectroscopy Applied to Nitrogenase and Related Models: Experimental Evidence for a Spin-Coupled Molybdenum(III) Center. Angew. Chem. Int. Ed 2019, 58, 9373–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Einsle O; Andrade SLA; Dobbek H; Meyer J; Rees DC Assignment of Individual Metal Redox States in a Metalloprotein by Crystallographic Refinement at Multiple X-Ray Wavelengths. J. Am. Chem. Soc 2007, 129, 2210–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Spatzal T; Schlesier J; Burger E-M; Sippel D; Zhang L; Andrade SLA; Rees DC; Einsle O Nitrogenase FeMoco Investigated by Spatially Resolved Anomalous Dispersion Refinement. Nat. Commun 2016, 7, 10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Thorneley RNF; Lowe DJ In Molybdenum Enzymes; Spiro TG, Ed.; Wiley: New York, 1985, 221–284 [Google Scholar]
- (189).Thorneley RNF; Ashby GA Oxidation of Nitrogenase Iron Protein by Dioxygen without Inactivation Could Contribute to High Respiration Rates of Azotobacter Species and Facilitate Nitrogen Fixation in Other Aerobic Environments. Biochem. J 1989, 261, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Barney BM; Yurth MG; Dos Santos PC; Dean DR; Seefeldt LC A Substrate Channel in the Nitrogenase MoFe Protein. JBIC, J. Biol. Inorg. Chem 2009, 14, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Pham DN; Burgess BK Nitrogenase Reactivity: Effects of pH on Substrate Reduction and Carbon Monoxide Inhibition. Biochemistry 1993, 32, 13725–13731. [DOI] [PubMed] [Google Scholar]
- (192).Imperial J; Hoover TR; Madden MS; Ludden PW; Shah VK Substrate Reduction Properties of Dinitrogenase Activated in vitro Are Dependent Upon the Presence of Homocitrate or Its Analogs During Iron-Molybdenum Cofactor Synthesis. Biochemistry 1989, 28, 7796–7799. [DOI] [PubMed] [Google Scholar]
- (193).Morrison CN; Hoy JA; Zhang L; Einsle O; Rees DC Substrate Pathways in the Nitrogenase MoFe Protein by Experimental Identification of Small Molecule Binding Sites. Biochemistry 2015, 54, 2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Dance I The Pathway for Serial Proton Supply to the Active Site of Nitrogenase: Enhanced Density Functional Modeling of the Grotthuss Mechanism. Dalton Trans. 2015, 44, 18167–18186. [DOI] [PubMed] [Google Scholar]
- (195).Siegbahn PEM A Major Structural Change of the Homocitrate Ligand of Probable Importance for the Nitrogenase Mechanism. Inorg. Chem 2018, 57, 1090–1095. [DOI] [PubMed] [Google Scholar]
- (196).Durrant MC Controlled Protonation of Iron–Molybdenum Cofactor by Nitrogenase: A Structural and Theoretical Analysis. Biochem. J 2001, 355, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Hoffman BM; Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Burgess BK In Molybdenum Enzymes, 1985, 161–219 [Google Scholar]
- (199).Barney BM; Yang T-C; Igarashi RY; Dos Santos PC; Laryukhin M; Lee H-I; Hoffman BM; Dean DR; Seefeldt LC Intermediates Trapped During Nitrogenase Reduction of N⋮N, CH3–NNH, and H2N–NH2. J. Am. Chem. Soc 2005, 127, 14960–14961. [DOI] [PubMed] [Google Scholar]
- (200).Seefeldt LC; Hoffman BM; Dean DR Mechanism of Mo-Dependent Nitrogenase. Annu. Rev. Biochem 2009, 78, 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Watt GD; Burns A Kinetics of Dithionite Ion Utilization and ATP Hydrolysis for Reactions Catalyzed by the Nitrogenase Complex from Azotobacter vinelandii. Biochemistry 1977, 16, 264–270. [DOI] [PubMed] [Google Scholar]
- (202).Burgess BK; Jacobs DB; Stiefel EI Large-Scale Purification of High Activity Azotobacter vinelandii Nitrogenase. Biochim. Biophys. Acta, Enzymol 1980, 614, 196–209. [DOI] [PubMed] [Google Scholar]
- (203).Hardy RWF; Holsten RD; Jackson EK; Burns RC The Acetylene-Ethylene Assay for N2 fixation: Laboratory and Field Evaluation. Plant Physiol. 1968, 43, 1185–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Dalton H; Whittenbury R The Acetylene Reduction Technique as an Assay for Nitrogenase Activity in the Methane Oxidizing Bacterium Methylococcus capsulatus Strain Bath. Arch. Microbiol 1976, 109, 147–151. [Google Scholar]
- (205).Miller RW; Eady RR Molybdenum and Vanadium Nitrogenases of Azotobacter chroococcum. Low Temperature Favours N2 Reduction by Vanadium Nitrogenase. Biochem. J 1988, 256, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Yates MG; Planqué , K. Nitrogenase from Azotobacter chroococcum. Eur. J. Biochem 1975, 60, 467–476. [DOI] [PubMed] [Google Scholar]
- (207).Dilworth MJ; Eady RR Hydrazine Is a Product of Dinitrogen Reduction by the Vanadium-Nitrogenase from Azotobacter chroococcum. Biochem. J 1991, 277, 465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Sippel D; Schlesier J; Rohde M; Trncik C; Decamps L; Djurdjevic I; Spatzal T; Andrade SLA; Einsle O Production and Isolation of Vanadium Nitrogenase from Azotobacter vinelandii by Molybdenum Depletion. JBIC, J. Biol. Inorg. Chem 2017, 22, 161–168. [DOI] [PubMed] [Google Scholar]
- (209).Bulen WA Proceedings of the First International Symposium on Nitrogen Fixation, Pullman, Washington, 1976; p 177–186. [Google Scholar]
- (210).Burgess BK; Wherland S; Newton WE; Stiefel EI Nitrogenase Reactivity: Insight into the Nitrogen-Fixing Process through Hydrogen-Inhibition and HD-Forming Reactions. Biochemistry 1981, 20, 5140–5146. [DOI] [PubMed] [Google Scholar]
- (211).Thorneley RNF; Eady RR; Lowe DJ Biological Nitrogen Fixation by Way of an Enzyme-Bound Dinitrogen-Hydride Intermediate. Nature 1978, 272, 557–558. [Google Scholar]
- (212).Hardy RWF In A Treatise on Dinitrogen Fixation; Bottomley F;Burns RC, Eds.; Wiley: New York, NY, 1979; Vol. Section 1–2, 515–568 [Google Scholar]
- (213).Davis LC; Henzl MT; Burris RH; Orme-Johnson WH Iron-Sulfur Clusters in the Molybdenum-Iron Protein Component of Nitrogenase. Electron Paramagnetic Resonance of the Carbon Monoxide Inhibited State. Biochemistry 1979, 18, 4860–4869. [DOI] [PubMed] [Google Scholar]
- (214).Lowe DJ; Fisher K; Thorneley RN Klebsiella pneumoniae Nitrogenase. Mechanism of Acetylene Reduction and Its Inhibition by Carbon Monoxide. Biochem. J 1990, 272, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Rivera-Ortiz JM; Burris RH Interactions among Substrates and Inhibitors of Nitrogenase. J. Bacteriol 1975, 123, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Simpson F; Burris R A Nitrogen Pressure of 50 Atmospheres Does Not Prevent Evolution of Hydrogen by Nitrogenase. Science 1984, 224, 1095–1097. [DOI] [PubMed] [Google Scholar]
- (217).Dilworth MJ; Eady RR; Eldridge ME The Vanadium Nitrogenase of Azotobacter chroococcum. Reduction of Acetylene and Ethylene to Ethane. Biochem. J 1988, 249, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Lehman LJ; Roberts GP Identification of an Alternative Nitrogenase System in Rhodospirillum rubrum. J. Bacteriol 1991, 173, 5705–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Dilworth MJ; Eady RR; Robson RL; Miller RW Ethane Formation from Acetylene as a Potential Test for Vanadium Nitrogenase in vivo. Nature 1987, 327, 167–168. [Google Scholar]
- (220).Harris DF; Lukoyanov DA; Kallas H; Trncik C; Yang Z-Y; Compton P; Kelleher N; Einsle O; Dean DR; Hoffman BM et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism to Achieve N2 Reduction. Biochemistry 2019, 58, 3293–3301. [DOI] [PubMed] [Google Scholar]
- (221).Cai R; Milton RD; Abdellaoui S; Park T; Patel J; Alkotaini B; Minteer SD Electroenzymatic C–C Bond Formation from CO2. J. Am. Chem. Soc 2018, 140, 5041–5044. [DOI] [PubMed] [Google Scholar]
- (222).Lee CC; Tanifuji K; Newcomb M; Liedtke J; Hu Y; Ribbe MW A Comparative Analysis of the CO-Reducing Activities of MoFe Proteins Containing Mo- and V-Nitrogenase Cofactors. ChemBioChem 2018, 19, 649–653. [DOI] [PubMed] [Google Scholar]
- (223).Siemann S; Schneider K; Oley M; Müller A Characterization of a Tungsten-Substituted Nitrogenase Isolated from Rhodobacter capsulatus. Biochemistry 2003, 42, 3846–3857. [DOI] [PubMed] [Google Scholar]
- (224).Hales BJ; Case EE Nitrogen Fixation by Azotobacter vinelandii in Tungsten-Containing Medium. J. Biol. Chem 1987, 262, 16205–16211. [PubMed] [Google Scholar]
- (225).Schöllhorn R; Burris RH Reduction of Azide by the N2-Fixing Enzyme System. Proc. Natl. Acad. Sci. U. S. A 1967, 57, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Dilworth MJ; Thorneley RNF Nitrogenase of Klebsiella pneumoniae. Hydrazine Is a Product of Azide Reduction. Biochem. J 1981, 193, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Rubinson JF; Burgess BK; Corbin JL; Dilworth MJ Nitrogenase Reactivity: Azide Reduction. Biochemistry 1985, 24, 273–283. [Google Scholar]
- (228).Guth JH; Burris RH Inhibition of Nitrogenase-Catalyzed Ammonia Formation by Hydrogen. Biochemistry 1983, 22, 5111–5122. [DOI] [PubMed] [Google Scholar]
- (229).Boughton JH; Keller RN Dissociation Constants of Hydropseudohalic Acids. J. Inorg. Nucl. Chem 1966, 28, 2851–2859. [Google Scholar]
- (230).Fisher K; Dilworth MJ; Newton WE Azotobacter vinelandii Vanadium Nitrogenase: Formaldehyde Is a Product of Catalyzed HCN Reduction, and Excess Ammonia Arises Directly from Catalyzed Azide Reduction. Biochemistry 2006, 45, 4190–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Hardy RWF; Knight E ATP-Dependent Reduction of Azide and HCN by N2-Fixing Enzymes of Azotobacter vinelandii and Clostridium pasteurianum. Biochim. Biophys. Acta, Enzymol 1967, 139, 69–90. [DOI] [PubMed] [Google Scholar]
- (232).Kelly M; Postgate JR; Richards RL Reduction of Cyanide and Isocyanide by Nitrogenase of Azotobacter chroococcum. Biochem. J 1967, 102, 1C–3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Li J; Burgess BK; Corbin JL Nitrogenase Reactivity: Cyanide as Substrate and Inhibitor. Biochemistry 1982, 21, 4393–4402. [DOI] [PubMed] [Google Scholar]
- (234).Izatt RM; Christensen JJ; Pack RT; Bench R Thermodynamics of Metal-Cyanide Coordination. I. pK, ΔH0, and ΔS0 Values as a Function of Temperature for Hydrocyanic Acid Dissociation in Aqueous Solution. Inorg. Chem 1962, 1, 828–831. [Google Scholar]
- (235).Rofer-DePoorter CK A Comprehensive Mechanism for the Fischer-Tropsch Synthesis. Chem. Rev 1981, 81, 447–474. [Google Scholar]
- (236).Cameron LM; Hales BJ Unusual Effect of CO on C2H2 Reduction by V Nitrogenase from Azotobacter vinelandii. J. Am. Chem. Soc 1996, 118, 279–280. [Google Scholar]
- (237).Hu Y; Lee CC; Ribbe MW Extending the Carbon Chain: Hydrocarbon Formation Catalyzed by Vanadium/Molybdenum Nitrogenases. Science 2011, 333, 753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Lee CC; Hu Y; Ribbe MW Vanadium Nitrogenase Reduces CO. Science 2010, 329, 642–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Rebelein JG; Lee CC; Hu Y; Ribbe MW The in Vivo Hydrocarbon Formation by Vanadium Nitrogenase Follows a Secondary Metabolic Pathway. Nat. Commun 2016, 7, 13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Seefeldt LC; Rasche ME; Ensign SA Carbonyl Sulfide and Carbon Dioxide as New Substrates, and Carbon Disulfide as a New Inhibitor, of Nitrogenase. Biochemistry 1995, 34, 5382–5389. [DOI] [PubMed] [Google Scholar]
- (241).Yang Z-Y; Moure VR; Dean DR; Seefeldt LC Carbon Dioxide Reduction to Methane and Coupling with Acetylene to Form Propylene Catalyzed by Remodeled Nitrogenase. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 19644–19648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Rebelein JG; Hu Y; Ribbe MW Differential Reduction of CO2 by Molybdenum and Vanadium Nitrogenases. Angew. Chem. Int. Ed 2014, 53, 11543–11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Rebelein JG; Hu Y; Ribbe MW Widening the Product Profile of Carbon Dioxide Reduction by Vanadium Nitrogenase. ChemBioChem 2015, 16, 1993–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Hu B; Harris DF; Dean DR; Liu TL; Yang Z-Y; Seefeldt LC Electrocatalytic CO2 Reduction Catalyzed by Nitrogenase MoFe and FeFe Proteins. Bioelectrochemistry 2018, 120, 104–109. [DOI] [PubMed] [Google Scholar]
- (245).Shah VK; Brill WJ Isolation of an Iron-Molybdenum Cofactor from Nitrogenase. Proc. Natl. Acad. Sci. U. S. A 1977, 74, 3249–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Fay AW; Lee C-C; Wiig JA; Hu Y; Ribbe MW In Nitrogen Fixation: Methods and Protocols; Ribbe MW, Ed.; Humana Press: Totowa, NJ, 2011, 239–248 [Google Scholar]
- (247).Lee CC; Hu Y; Ribbe MW ATP-Independent Formation of Hydrocarbons Catalyzed by Isolated Nitrogenase Cofactors. Angew. Chem. Int. Ed 2012, 51, 1947–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Lee CC; Hu Y; Ribbe MW Catalytic Reduction of CN−, CO, and CO2 by Nitrogenase Cofactors in Lanthanide-Driven Reactions. Angew. Chem. Int. Ed 2015, 54, 1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Lee CC; Hu Y; Ribbe MW Insights into Hydrocarbon Formation by Nitrogenase Cofactor Homologs. mBio 2015, 6, e00307–00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Sickerman NS; Tanifuji K; Lee CC; Ohki Y; Tatsumi K; Ribbe MW; Hu Y Reduction of C1 Substrates to Hydrocarbons by the Homometallic Precursor and Synthetic Mimic of the Nitrogenase Cofactor. J. Am. Chem. Soc 2017, 139, 603–606. [DOI] [PubMed] [Google Scholar]
- (251).Tanifuji K; Sickerman N; Lee CC; Nagasawa T; Miyazaki K; Ohki Y; Tatsumi K; Hu Y; Ribbe MW Structure and Reactivity of an Asymmetric Synthetic Mimic of Nitrogenase Cofactor. Angew. Chem 2016, 128, 15862–15865. [DOI] [PubMed] [Google Scholar]
- (252).Lee CC; Hu Y; Ribbe MW Reduction and Condensation of Aldehydes by the Isolated Cofactor of Nitrogenase. ACS Cent. Sci 2018, 4, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Fisher K; Lowe DJ; Petersen J Vanadium(V) Is Reduced by the ‘as Isolated’ Nitrogenase Fe-Protein at Neutral pH. Chem. Commun 2006, 2807–2809. [DOI] [PubMed] [Google Scholar]
- (254).Stiebritz MT; Hiller CJ; Sickerman NS; Lee CC; Tanifuji K; Ohki Y; Hu Y Ambient Conversion of CO2 to Hydrocarbons by Biogenic and Synthetic [Fe4S4] Clusters. Nat. Catal 2018, 1, 444–451. [Google Scholar]
- (255).Lowe DJ; Thorneley RN The Mechanism of Klebsiella pneumoniae Nitrogenase Action. Pre-Steady-State Kinetics of H2 Formation. Biochem. J 1984, 224, 877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Lowe DJ; Thorneley RNF The Mechanism of Klebsiella pneumoniae Nitrogenase Action. The Determination of Rate Constants Required for the Simulation of the Kinetics of N2 Reduction and H2 Evolution. Biochem. J 1984, 224, 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Thorneley RNF; Lowe DJ The Mechanism of Klebsiella pneumoniae Nitrogenase Action. Pre-Steady-State Kinetics of an Enzyme-Bound Intermediate in N2 Reduction and of NH3 Formation. Biochem. J 1984, 224, 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Thorneley RNF; Lowe DJ The Mechanism of Klebsiella pneumoniae Nitrogenase Action. Simulation of the Dependences of H2-Evolution Rate on Component-Protein Concentration and Ratio and Sodium Dithionite Concentration. Biochem. J 1984, 224, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Harris DF; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Kinetic Understanding of N2 Reduction Versus H2 Evolution at the E4(4H) Janus State in the Three Nitrogenases. Biochemistry 2018, 57, 5706–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Lukoyanov D; Khadka N; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Reductive Elimination of H2 Activates Nitrogenase to Reduce the N≡N Triple Bond: Characterization of the E 4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc 2016, 138, 10674–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Yang Z-Y; Ledbetter R; Shaw S; Pence N; Tokmina-Lukaszewska M; Eilers B; Guo Q; Pokhrel N; Cash VL; Dean DR et al. Evidence That the Pi Release Event Is the Rate-Limiting Step in the Nitrogenase Catalytic Cycle. Biochemistry 2016, 55, 3625–3635. [DOI] [PubMed] [Google Scholar]
- (262).Duval S; Danyal K; Shaw S; Lytle AK; Dean DR; Hoffman BM; Antony E; Seefeldt LC Electron Transfer Precedes ATP Hydrolysis During Nitrogenase Catalysis. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 16414–16419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Jensen BB; Burris RH Effect of High pN2 and High pD2 on Ammonia Production, Hydrogen Evolution, and Hydrogen Deuteride Formation by Nitrogenases. Biochemistry 1985, 24, 1141–1147. [DOI] [PubMed] [Google Scholar]
- (264).Burgess BK; Corbin JL; Rubinson JF; Li J. g.; Dilworth MJ; Newton WE. In Advances in Nitrogen Fixation Research: Proceedings of the 5th International Symposium on Nitrogen Fixation, Noordwijkerhout, the Netherlands, August 28 – September 3, 1983; Veeger C;Newton WE, Eds.; Springer Netherlands: Dordrecht, 1984, 146–146 [Google Scholar]
- (265).Yang Z-Y; Khadka N; Lukoyanov D; Hoffman BM; Dean DR; Seefeldt LC On Reversible H2 Loss Upon N2 Binding to FeMo-Cofactor of Nitrogenase. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 16327–16332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Raugei S; Seefeldt LC; Hoffman BM Critical Computational Analysis Illuminates the Reductive-Elimination Mechanism That Activates Nitrogenase for N2 reduction. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 10521–10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Thorhallsson AT; Benediktsson B; Bjornsson R A Model for Dinitrogen Binding in the E4 State of Nitrogenase. Chem. Sci 2019, 10, 11110–11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Igarashi RY; Laryukhin M; Dos Santos PC; Lee H-I; Dean DR; Seefeldt LC; Hoffman BM Trapping H− Bound to the Nitrogenase FeMo-Cofactor Active Site During H2 Evolution: Characterization by ENDOR Spectroscopy. J. Am. Chem. Soc 2005, 127, 6231–6241. [DOI] [PubMed] [Google Scholar]
- (269).Lukoyanov D; Barney BM; Dean DR; Seefeldt LC; Hoffman BM Connecting Nitrogenase Intermediates with the Kinetic Scheme for N2 Reduction by a Relaxation Protocol and Identification of the N2 Binding State. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Kinney RA; Saouma CT; Peters JC; Hoffman BM Modeling the Signatures of Hydrides in Metalloenzymes: ENDOR Analysis of a Di-Iron Fe(μ-NH)(μ-H)Fe Core. J. Am. Chem. Soc 2012, 134, 12637–12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC; Hoffman BM Is Mo Involved in Hydride Binding by the Four-Electron Reduced (E4) Intermediate of the Nitrogenase MoFe Protein? J. Am. Chem. Soc 2010, 132, 2526–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Doan PE; Telser J; Barney BM; Igarashi RY; Dean DR; Seefeldt LC; Hoffman BM 57Fe ENDOR Spectroscopy and ‘Electron Inventory’ Analysis of the Nitrogenase E4 Intermediate Suggest the Metal-Ion Core of FeMo-Cofactor Cycles through Only One Redox Couple. J. Am. Chem. Soc 2011, 133, 17329–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Hoeke V; Tociu L; Case DA; Seefeldt LC; Raugei S; Hoffman BM High-Resolution ENDOR Spectroscopy Combined with Quantum Chemical Calculations Reveals the Structure of Nitrogenase Janus Intermediate E4(4H). J. Am. Chem. Soc 2019, 141, 11984–11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem. Rev 2014, 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- (275).Wiedner ES Thermodynamic Hydricity of [FeFe]-Hydrogenases. J. Am. Chem. Soc 2019, 141, 7212–7222. [DOI] [PubMed] [Google Scholar]
- (276).Belkova NV; Epstein LM; Filippov OA; Shubina ES Hydrogen and Dihydrogen Bonds in the Reactions of Metal Hydrides. Chem. Rev 2016, 116, 8545–8587. [DOI] [PubMed] [Google Scholar]
- (277).Wiedner ES; Chambers MB; Pitman CL; Bullock RM; Miller AJM; Appel AM Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev 2016, 116, 8655–8692. [DOI] [PubMed] [Google Scholar]
- (278).Neese F The Yandulov/Schrock Cycle and the Nitrogenase Reaction: Pathways of Nitrogen Fixation Studied by Density Functional Theory. Angew. Chem. Int. Ed 2006, 45, 196–199. [DOI] [PubMed] [Google Scholar]
- (279).Chatt J; Pearman AJ; Richards RL Diazenido (Iminonitrosyl) (N2H), Hydrazido(2-) (N2H2, and Hydrazido(1-) (N2H3) Ligands as Intermediates in the Reduction of Ligating Dinitrogen to Ammonia. J. Organomet. Chem 1975, 101, C45–C47. [Google Scholar]
- (280).Chatt J; Pearman AJ; Richards RL The Reduction of Mono-Coordinated Molecular Nitrogen to Ammonia in a Protic Environment. Nature 1975, 253, 39–40. [Google Scholar]
- (281).Chatt J; Pearman AJ; Richards RL Relevance of Oxygen Ligands to Reduction of Ligating Dinitrogen. Nature 1976, 259, 204. [DOI] [PubMed] [Google Scholar]
- (282).Chatt J; Pearman AJ; Richards RL Conversion of Dinitrogen in Its Molybdenum and Tungsten Complexes into Ammonia and Possible Relevance to the Nitrogenase Reaction. J. Chem. Soc., Dalton Trans 1977, 1852–1860. [Google Scholar]
- (283).Yandulov DV; Schrock RR Reduction of Dinitrogen to Ammonia at a Well-Protected Reaction Site in a Molybdenum Triamidoamine Complex. J. Am. Chem. Soc 2002, 124, 6252–6253. [DOI] [PubMed] [Google Scholar]
- (284).Yandulov DV; Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- (285).Hinnemann B; Nørskov JK Catalysis by Enzymes: The Biological Ammonia Synthesis. Top. Catal 2006, 37, 55–70. [Google Scholar]
- (286).Tanabe Y; Nishibayashi Y Developing More Sustainable Processes for Ammonia Synthesis. Coord. Chem. Rev 2013, 257, 2551–2564. [Google Scholar]
- (287).Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Acc. Chem. Res 2005, 38, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).MacLeod KC; Holland PL Recent Developments in the Homogeneous Reduction of Dinitrogen by Molybdenum and Iron. Nat. Chem 2013, 5, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Rodriguez MM; Bill E; Brennessel WW; Holland PL N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Rittle J; Peters JC An Fe-N2 Complex That Generates Hydrazine and Ammonia Via Fe=NNH2: Demonstrating a Hybrid Distal-to-Alternating Pathway for N2 Reduction. J. Am. Chem. Soc 2016, 138, 4243–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Seefeldt LC; Dance IG; Dean DR Substrate Interactions with Nitrogenase: Fe Versus Mo. Biochemistry 2004, 43, 1401–1409. [DOI] [PubMed] [Google Scholar]
- (292).Davis LC Hydrazine as a Substrate and Inhibitor of Azotobacter vinelandii Nitrogenase. Arch. Biochem. Biophys 1980, 204, 270–276. [DOI] [PubMed] [Google Scholar]
- (293).Barney BM; McClead J; Lukoyanov D; Laryukhin M; Yang T-C; Dean DR; Hoffman BM; Seefeldt LC Diazene (HNNH) Is a Substrate for Nitrogenase: Insights into the Pathway of N2 Reduction. Biochemistry 2007, 46, 6784–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Barney BM; Lukoyanov D; Yang T-C; Dean DR; Hoffman BM; Seefeldt LC A Methyldiazene (HN=N-CH3)-Derived Species Bound to the Nitrogenase Active-Site FeMo Cofactor: Implications for Mechanism. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17113–17118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Dilworth MJ; Eldridge ME; Eady RR The Molybdenum and Vanadium Nitrogenases of Azotobacter chroococcum: Effect of Elevated Temperature on N2 reduction. Biochem. J 1993, 289, 395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Dilworth MJ; Eldridge ME; Eady RR Correction for Creatine Interference with the Direct Indophenol Measurement of NH3 in Steady-State Nitrogenase Assays. Anal. Biochem 1992, 207, 6–10. [DOI] [PubMed] [Google Scholar]
- (297).Robson RL Nitrogen Fixation in Strains of Azotobacter chroococcum Bearing Deletions of a Cluster of Genes Coding for Nitrogenase. Arch. Microbiol 1986, 146, 74–79. [Google Scholar]
- (298).Bishop PE; Hawkins ME; Eady RR Nitrogen Fixation in Molybdenum-Deficient Continuous Culture by a Strain of Azotobacter vinelandii carrying a Deletion of the Structural Genes for Nitrogenase (nifHDK). Biochem. J 1986, 238, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Spatzal T; Perez KA; Einsle O; Howard JB; Rees DC Ligand Binding to the FeMo-Cofactor: Structures of CO-Bound and Reactivated Nitrogenase. Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Spatzal T; Perez KA; Howard JB; Rees DC Catalysis-Dependent Selenium Incorporation and Migration in the Nitrogenase Active Site Iron-Molybdenum Cofactor. eLife 2015, 4, e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Dance I Mechanisms of the S/Co/Se Interchange Reactions at FeMo-Co, the Active Site Cluster of Nitrogenase. Dalton Trans. 2016, 45, 14285–14300. [DOI] [PubMed] [Google Scholar]
- (302).Dance I How Feasible Is the Reversible S-Dissociation Mechanism for the Activation of FeMo-CO, the Catalytic Site of Nitrogenase? Dalton Trans. 2019, 48, 1251–1262. [DOI] [PubMed] [Google Scholar]
- (303).Siegbahn PEM The Mechanism for Nitrogenase Including All Steps. Phys. Chem. Chem. Phys 2019, 21, 15747–15759. [DOI] [PubMed] [Google Scholar]
- (304).Pollock RC; Lee H-I; Cameron LM; DeRose VJ; Hales BJ; Orme-Johnson WH; Hoffman BM Investigation of CO Bound to Inhibited Forms of Nitrogenase MoFe Protein by 13C ENDOR. J. Am. Chem. Soc 1995, 117, 8686–8687. [Google Scholar]
- (305).Christie PD; Lee H-I; Cameron LM; Hales BJ; Orme-Johnson WH; Hoffman BM Identification of the CO-Binding Cluster in Nitrogenase MoFe Protein by ENDOR of 57Fe Isotopomers. J. Am. Chem. Soc 1996, 118, 8707–8709. [Google Scholar]
- (306).Lee H-I; Cameron LM; Hales BJ; Hoffman BM Co Binding to the FeMo Cofactor of CO-Inhibited Nitrogenase: 13CO and 1H Q-Band ENDOR Investigation. J. Am. Chem. Soc 1997, 119, 10121–10126. [Google Scholar]
- (307).George SJ; Ashby GA; Wharton CW; Thorneley RNF Time-Resolved Binding of Carbon Monoxide to Nitrogenase Monitored by Stopped-Flow Infrared Spectroscopy. J. Am. Chem. Soc 1997, 119, 6450–6451. [Google Scholar]
- (308).Yan L; Pelmenschikov V; Dapper CH; Scott AD; Newton WE; Cramer SP IR-Monitored Photolysis of CO-Inhibited Nitrogenase: A Major EPR-Silent Species with Coupled Terminal CO Ligands. Chem. Eur. J 2012, 18, 16349–16357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Yang Z-Y; Seefeldt LC; Dean DR; Cramer SP; George SJ Steric Control of the Hi-CO MoFe Nitrogenase Complex Revealed by Stopped-Flow Infrared Spectroscopy. Angew. Chem. Int. Ed 2011, 50, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Maskos Z; Hales BJ Photo-Lability of CO Bound to Mo-Nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem 2003, 93, 11–17. [DOI] [PubMed] [Google Scholar]
- (311).Yan L; Dapper CH; George SJ; Wang H; Mitra D; Dong W; Newton WE; Cramer SP Photolysis of Hi-CO Nitrogenase – Observation of a Plethora of Distinct CO Species Using Infrared Spectroscopy. Eur. J. Inorg. Chem 2011, 2011, 2064–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Barney BM; Igarashi RY; Dos Santos PC; Dean DR; Seefeldt LC Substrate Interaction at an Iron-Sulfur Face of the FeMo-Cofactor During Nitrogenase Catalysis. J. Biol. Chem 2004, 279, 53621–53624. [DOI] [PubMed] [Google Scholar]
- (313).Benton PMC; Laryukhin M; Mayer SM; Hoffman BM; Dean DR; Seefeldt LC Localization of a Substrate Binding Site on the FeMo-Cofactor in Nitrogenase: Trapping Propargyl Alcohol with an α-70-Substituted MoFe Protein. Biochemistry 2003, 42, 9102–9109. [DOI] [PubMed] [Google Scholar]
- (314).Yang Z-Y; Dean DR; Seefeldt LC Molybdenum Nitrogenase Catalyzes the Reduction and Coupling of CO to Form Hydrocarbons. J. Biol. Chem 2011, 286, 19417–19421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Lee CC; Fay AW; Weng T-C; Krest CM; Hedman B; Hodgson KO; Hu Y; Ribbe MW Uncoupling Binding of Substrate CO from Turnover by Vanadium Nitrogenase. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 13845–13849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Varley JB; Wang Y; Chan K; Studt F; Nørskov JK Mechanistic Insights into Nitrogen Fixation by Nitrogenase Enzymes. Phys. Chem. Chem. Phys 2015, 17, 29541–29547. [DOI] [PubMed] [Google Scholar]
- (317).Lee H-I; Sørlie M; Christiansen J; Yang T-C; Shao J; Dean DR; Hales BJ; Hoffman BM Electron Inventory, Kinetic Assignment (En), Structure, and Bonding of Nitrogenase Turnover Intermediates with C2H2 and CO. J. Am. Chem. Soc 2005, 127, 15880–15890. [DOI] [PubMed] [Google Scholar]
- (318).Dance I How Does Vanadium Nitrogenase Reduce CO to Hydrocarbons? Dalton Trans. 2011, 40, 5516–5527. [DOI] [PubMed] [Google Scholar]
- (319).Varley JB; Nørskov JK First-Principles Calculations of Fischer–Tropsch Processes Catalyzed by Nitrogenase Enzymes. ChemCatChem 2013, 5, 732–736. [Google Scholar]
- (320).Lee CC; Wilcoxen J; Hiller CJ; Britt RD; Hu Y Evaluation of the Catalytic Relevance of the CO-Bound States of V-Nitrogenase. Angew. Chem. Int. Ed 2018, 57, 3411–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Hiller CJ; Lee CC; Stiebritz MT; Rettberg LA; Hu Y Strategies Towards Capturing Nitrogenase Substrates and Intermediates Via Controlled Alteration of Electron Fluxes. Chem. Eur. J 2019, 25, 2389–2395. [DOI] [PubMed] [Google Scholar]