

Abstract
Background:
There is growing evidence that the anticonvulsant topiramate is efficacious in reducing alcohol consumption. Further, an intronic single nucleotide polymorphism (rs2832407, C ➔ A) in the GRIK1 gene, which encodes the GluK1 subunit of the excitatory kainate receptor, predicted topiramate’s effectiveness in reducing heavy drinking in a clinical trial. The molecular correlates of GRIK1 genotype that may relate to topiramate’s ability to reduce drinking remain unknown.
Methods:
We differentiated induced pluripotent stem cells (iPSCs) characterized by GRIK1 rs2832407 genotype from 8 A/A and 8 C/C donors into forebrain-lineage neural cultures. Our differentiation protocol yielded mixed neural cultures enriched for glutamatergic neurons. Basal mRNA expression of the GRIK1 locus was examined via qPCR. The effects of acute topiramate exposure on excitatory spontaneous synaptic activity was examined via whole cell patch-clamp electrophysiology. Results were compared and contrasted between iPSC donor genotypes.
Results:
Although characterization of the GRIK1 locus revealed no effect of rs2832407 genotype on GRIK1 isoform mRNA expression, a significant difference was observed on GRIK1 antisense-2 expression, which was greater in C/C neural cultures. Differential effects of acute exposure to 5 μM topiramate were observed on spontaneous synaptic activity in A/A vs. C/C neurons, with a smaller reduction in excitatory event frequency observed in C/C donor neurons.
Conclusions:
This work highlights the use of iPSC technologies to study pharmacogenetic treatment effects in psychiatric disorders and furthers our understanding of the molecular effects of topiramate exposure in human neural cells.
Keywords: electrophysiology, pharmacogenetics, induced pluripotent stem cells, GluK1, alcohol use disorder, gene expression
Introduction
Alcohol use disorder (AUD) is a complex, debilitating, and highly prevalent disorder, affecting up to ~14% of the U.S. population in a one-year period (Grant et al., 2015, Grant et al., 2017). The factors contributing to the risk of developing AUD vary among individuals and include both environmental and genetic influences, as well as gene-gene and gene-environment interactions (Gelernter and Kranzler, 2009, Dick and Agrawal, 2008). Despite the detrimental effects of AUD, the majority of affected individuals never seek treatment (Cohen et al., 2007). Furthermore, the heterogeneity of the disorder among individuals may, in part, contribute to the variable efficacy of currently approved pharmacologic treatments. A better understanding of molecular mechanisms contributing to the heterogeneity of AUD could lead to improved therapeutics and personalized treatments (Heilig et al., 2011).
Topiramate, an anticonvulsant (Lyseng-Williamson and Yang, 2007), a prophylactic treatment for migraine (Parikh and Silberstein, 2019), and in combination with phentermine, a weight-loss medication (Hurt et al., 2014), exerts effects via a broad array of molecular actions that modulate synaptic transmission and neuronal excitability (Shank et al., 2000). The medication has also been efficacious in reducing drinking in individuals with an AUD (Johnson et al., 2003, Johnson et al., 2007, Baltieri et al., 2008, Kranzler et al., 2014a). In one study (Kranzler et al., 2014a), the ability of topiramate to reduce drinking in treatment-seeking individuals was moderated by a single nucleotide polymorphism (SNP) in the GRIK1 gene (rs2832407, C ➔ A) which had been previously associated with AUD (Kranzler et al., 2009). GRIK1 encodes the GluK1 subunit of the excitatory kainate receptor (Contractor et al., 2011, Collingridge et al., 2009), one of the molecular targets of topiramate (Gryder and Rogawski, 2003, Braga et al., 2009). Kranzler et al. found that topiramate reduced the frequency of heavy drinking in individuals homozygous for the rs2832407 C allele, while there was no significant difference from placebo among A-allele carriers (Kranzler et al., 2014a), an effect that persisted for 6 months following completion of the initial 12-week study (Kranzler et al., 2014b). The molecular basis for these long-lasting effects of topiramate in GRIK1 C-allele homozygotes remains to be elucidated. Because topiramate reduces excitability via its antagonism of GluK1-containing kainate receptors (Gryder and Rogawski, 2003, Braga et al., 2009), the ability of topiramate to reduce drinking in humans may relate to differential effects on synaptic transmission associated with the rs2832407 genotype.
Pluripotent stem cells derived from human fibroblasts (Takahashi et al., 2007) offer the potential to generate multiple cell types of the central nervous system (Mertens et al., 2016) that manifest the donor subject’s genetic background. This method can be used to explore the molecular actions of pharmacogenetic treatments for complex psychiatric disorders in a human cell model system, which may reveal underlying mechanisms leading to differential behavioral effects. Although the application of iPSC technology to study the molecular aspects of personalized medicine for psychiatric conditions is limited, prior work has shown that iPSC-derived neurons can recapitulate differential responses to lithium in bipolar disorder patients, allowing for the exploration of molecular differences underlying the heterogeneity of the treatment response (Tobe et al., 2017, Mertens et al., 2015). With respect to alcohol use disorder, cultures of induced GABAergic inhibitory neurons generated via overexpression of transcription factors in human iPS and isogenic hES and iPS cells co-cultured on a layer of mouse astrocytes were used to demonstrate functional pre-synaptic effects of the N40D mu-opioid receptor variant (Halikere et al., 2019), which has been associated in some samples with treatment response for AUD to naltrexone (Chamorro et al., 2012). Our group has applied growth factor-driven human iPSC differentiation to study the effects of alcohol exposure at the electrophysiologic and transcriptomic levels in a mixed cell type neural culture system (Lieberman et al., 2012, Lieberman et al., 2015, Lieberman et al., 2017, Jensen et al., 2019). Others have applied iPSC technology to the study of opioid (Sheng et al., 2016) and nicotine (Oni et al., 2016) addiction.
The objective of the current study was to explore the effects of topiramate in human iPSC-derived neural cultures generated from donors homozygous for the GRIK1 rs2832407 A or C allele. Neural cultures derived from 8 A/A and 8 C/C donors were utilized to explore genotype-associated differences in RNA expression within the GRIK1 locus and examine the effects of acute topiramate exposure on excitatory synaptic transmission. Results from this study highlight the potential utility of iPSCs for furthering our understanding of pharmacogenetic treatments for complex psychiatric disorders including AUD.
Materials and Methods
Generation of iPSCs from GRIK1 rs2832407 A/A and C/C donors
iPSCs were generated from fibroblasts obtained via skin punch biopsies of the inner, upper arm from participants in clinical studies at UCONN Health (UC, Farmington, CT), either non-alcoholic participants enrolled in a study examining the subjective effects of acute alcohol intoxication (Milivojevic et al., 2014, Covault et al., 2014) or individuals with a DSM-IV diagnosis of alcohol dependence (ADs) enrolled in a pharmacological treatment study examining topiramate’s ability to reduce drinking (Kranzler et al., 2014a). Biopsy samples were minced and cultured in Dulbecco’s modified eagles medium (DMEM, Thermo Fisher Scientific) supplemented with 20% fetal bovine serum (FBS, Thermo Fisher Scientific), 1x non-essential amino acids (Thermo Fisher Scientific) and 1x penicillin/streptomycin (Thermo Fisher Scientific). Fibroblast cultures were expanded and passaged using trypsin (Thermo Fisher Scientific) prior to being frozen or sent for reprogramming. Fibroblast DNA was genotyped at rs2832407 using a commercial TaqMan genotyping assay (c_2962029_10, Thermo Fisher Scientific).
The UC Stem Cell Core reprogrammed fibroblasts to pluripotency using retrovirus to express five factors (OCT4, SOX2, KLF4, c-MYC, and LIN28) or sendai virus to express four factors (OCT4, SOX2, KLF4, and c-MYC). Two to four weeks after viral transduction, multiple pluripotent stem cell colonies for each subject were selected and expanded as individual iPSC lines. Expression of pluripotency markers by iPSCs was verified by immunocytochemistry for SSEA-3/4 and NANOG by the UC Stem Cell Core. iPSCs were cultured on a feeder layer of irradiated mouse embryo fibroblasts using human embryonic stem cell media containing DMEM with F12 (DMEM/F12, 1:1 ratio, Thermo Fisher Scientific) supplemented with 20% Knockout Serum Replacer (Thermo Fisher Scientific), 1x non-essential amino acids, 1 mM L-glutamine (Thermo Fisher Scientific), 0.1 mM β-mercaptoethanol (MP Biomedicals), and 4 ng/mL of basic fibroblast growth factor (bFGF, Millipore). Media was fully replaced daily and cells were cultured to confluency before being passaged using 1 mg/mL Dispase (Thermo Fisher Scientific) in DMEM/F12. iPSC lines from 8 A/A and 8 C/C subjects were used for our study. Two distinct iPSC clones were included from 2 A/A and 4 C/C donors, for a total of 10 A/A and 12 C/C lines used as part of the current experiments.
iPSC neural differentiation
iPSCs were differentiated into neural cell cultures as previously described (Lieberman et al., 2012) using a protocol developed by the WiCell Institute for the differentiation of human embryonic stem cells into neural cells of a forebrain lineage (#SOP-CH-207, REV A, www.wicell.org, Madison, WI). Gene expression eQTLs in these neural cultures are more closely aligned with eQTLs identified in human brain cortical vs. subcortical brain regions (Jensen et al., 2019). This method utilized an embryoid body-based protocol wherein iPSC colonies are removed from the feeder layer substrate and cultured in suspension prior to neural induction. Neuroepithelial cells were generated by culturing for 3 weeks in neural induction media containing 1x N2 supplement (Life Technologies) and 2 μg/mL heparin (Sigma Aldrich), following which cells were dissociated and cultured at low density in 24-well plates on poly-L-ornithine and Matrigel (BD Biosciences, Bedford, MA) coated glass coverslips in neural differentiation media containing neural growth factors 1x B27 supplement (Life Technologies), 1 μg/mL laminin (Sigma-Aldrich), and 10 ng/mL each of brain-derived neurotrophic factor (BDNF, Peprotech, Rocky Hill, NJ), glial-derived neurotrophic factor (GDNF, Peprotech), and insulin-like growth factor 1 (IGF-1, Peprotech). This differentiation protocol yields cultures in which approximately 70% of neurons have immunostaining for glutamatergic neuron transcription factor TBR1, 25% for GABAergic marker GAD65/67, and 12% of total cells stain for the astrocyte marker S100b (Fink et al., 2017). All cells were incubated at 37° in 5% CO2. Neural cell differentiation times are referenced to the time post-plating of neural progenitor cells at low density.
Immunocytochemistry
Twelve weeks after being plated onto glass coverslips, neural cultures derived from 3 A/A and 5 C/C donors were fixed using 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized using 0.2% triton X-100 (Sigma-Aldrich) in PBS for 10 min. Following a 1-hr block using 5% donkey serum (Jackson ImmunoResearch, West Grove, PA), cultures were incubated for 24-48 hours at 4° with the following primary antibodies diluted in 5% donkey serum in PBS: mouse anti-beta III-tubulin (1:500, Covance, Dedham, MA), mouse anti-GFAP (1:500, Millipore, Brillerica, CA), rabbit anti-MAP2 (1:500, Millipore), rabbit anti-TBR1 [a transcription factor, T-box brain protein 1, that is a marker for deep cortical glutamatergic neurons (Hevner et al., 2001)]; 1:1000, ProteinTech Group, Chicago, IL, incubation included 0.1% triton X-100), and rabbit-anti GluK1 (1:100, Thermo Fisher). Cells were then washed and incubated at room temperature for 2 h in donkey anti-mouse alexa fluor 594 (1:1000, Life Technologies) and donkey anti-rabbit alexa fluor 488 (1:1000, Life Technologies) secondary antibodies diluted in 3% donkey serum in PBS, and mounted in DAPI-containing media for visualization.
Electrophysiology
Whole cell patch-clamp electrophysiology was performed on neurons differentiated from rs2832407 A/A and C/C iPSCs using previously described techniques (Lemtiri-Chlieh and Levine, 2010, Fink et al., 2017). Neurons were selected for recording based on morphology, including pyramidal-shaped soma and the presence of neurites. Artificial cerebrospinal fluid (aCSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2-6H2O, 25 mM NaHCO3, 2 mM CaCl2, and 25 mM dextrose was perfused through the recording chamber at 1 ml/min at room temperature. An internal recording solution containing 4 mM KCl, 125 mM K-gluconate, 10 mM HEPES, 10 mM phosphocreatine, 1 mM EGTA, 0.2 mM CaCl2, 4 mM Na2-ATP, and 0.3 mM Na-GTP (pH 7.3) was used for all recordings. Characterization of basal neuronal properties was performed on 15 A/A and 27 C/C neurons derived from 1 A/A and 2 C/C donors and cultured for 17-19 weeks after plating onto glass coverslips. Upon break-in, neurons were noted for their resting membrane potential by injection with 0 current and corrected post hoc for liquid junction potential. Action potentials were evoked in current clamp mode at ~−70 mV by applying 500-ms duration current steps from −20 pA to +40 pA in 5 pA intervals.
The acute effect of topiramate on spontaneous synaptic activity in pyramidal-like neurons was examined in voltage clamp mode. We explored the effects of 5 μM topiramate based on in vitro studies showing 50% antagonism of GluK1-containing kainate receptors by 0.5 μM topiramate, consideration of trough serum topiramate concentrations (7-24 μM) in a sample of 344 patients given topiramate 300 mg/d for the treatment of epilepsy (May et al., 2002), and consideration of the experimental design demonstrating moderation by rs2832407 of topiramate’s efficacy to reduce heavy drinking, which utilized a 12-week escalating dose starting at 25 mg/day and increasing weekly to a maximal daily dose of 200 mg (Kranzler et al., 2014a). In total, 19 A/A and 20 C/C neurons derived from 3 A/A and 4 C/C donors were used for analysis. All neurons were cultured for a minimum of 12 weeks prior to recording based on prior results examining the maturation profile of this neural differentiation protocol (Fink et al., 2017). Neurons were validated by the presence of voltage-gated inward and outward currents, and ability to fire an action potential. To observe excitatory synaptic activity, neurons were held at −70 mV. A 10-min baseline recording was performed (a 5-min equilibration period and a 5-min experimental period), followed by a 30-min perfusion of aCSF supplemented with 5 μM topiramate. Only one neuron was examined per coverslip following perfusion with topiramate. The last 5 min of the baseline recording period was used as the time zero pre-treatment baseline measurement and the number of synaptic events recorded during topiramate exposure was binned into 10-min intervals for statistical analysis. Percent change from baseline is presented for graphical presentation. Cells were excluded from the analysis if they were unable to fire an action potential or had 30 or fewer spontaneous events during the baseline recording (0.1 Hz). All electrophysiological recordings were performed using a HEKA EPC9 amplifier and PatchMaster software (version 2×67). Analysis and quantification were performed using Axon Clampfit software (version 10.3.1.4).
RNA extraction and quantitative polymerase chain reaction (qPCR)
Twelve-week old neural cultures were used to examine GRIK1 RNA expression. Neural cell lines derived from 15 donor subjects (8 A/A donors and 7 C/C donors) were available for qPCR gene expression analysis, with RNA from 6 coverslips processed separately per subject. RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. RNA was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Pittsburgh, PA) and cDNA was synthesized from 2 μg RNA using a High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific).
cDNA was analyzed by quantitative real-time polymerase chain reaction using an Applied Biosystems 7500 instrument (Thermo Fisher Scientific) and TaqMan Assays-on-Demand (Thermo Fisher Scientific) FAM-labeled probe and primer sets for GRIK1 isoform 1 (exons 9-10, Hs01081331_m1), GRIK1 isoform 2 (exons 8-10, Hs01081334_m1 and exons 16-18, Hs01081332_m1), total GRIK1 (exons 1-2, Hs00168165_m1) and GRIK1 antisense-2 (Hs00370612_m1). Expression was quantified relative to a VIC-labeled TaqMan probe for the reference gene GUSB (4326320E). cDNA synthesized from RNA extracted from each culture well was assayed in triplicate 20-μL reactions containing GRIK1 FAM-labeled and GUSB VIC-labeled assays using Gene Expression Master Mix (Thermo Fisher Scientific) per the manufacturer’s protocol.
PCR cycles were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec. A standard curve consisting of a 5-level serial dilution of 200%, 100%, 50%, 25%, and 12.5% of a cDNA pool from untreated 12-week old neural cultures differentiated from four subjects was included on each plate to determine the relative mRNA expression across different qPCR plates. Data are displayed as mRNA abundance relative to this cDNA pool and to the housekeeping gene GUSB (where a unit of 1 is equivalent to the abundance of the target gene relative to GUSB in the reference RNA sample). For each cell line, the expression of specific GRIK1 isoforms and antisense-2 was normalized to the expression of total GRIK1. Data generated from clones derived from the same donor subject was averaged and considered as a single point for analysis and visual representation.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (V5.0f for Mac, GraphPad Software, www.graphpad.com) or SPSS (v21, IBM, Armonk, NY). Student’s t-tests were used to compare TBR1+ staining, basal neuronal properties EPSC frequency, and mRNA expression between A/A and C/C neural cell cultures. Chi-square analysis was used to compare the action potential phenotype between A/A and C/C neurons. Generalized linear mixed models containing GRIK1 rs2832407 genotype (A/A vs. C/C), topiramate treatment time, and their interaction as factors and with baseline EPSC counts as a co-variate for each cell were utilized to examine the effect of acute topiramate exposure on the number of spontaneous excitatory synaptic events. Post-hoc analysis of genotype comparing EPSC frequency at baseline to the last 10-minute topiramate treatment bin was conducted via paired t-tests. Statistical significance was defined as p < 0.05.
Results
iPSCs from rs2832407 A/A and C/C donors differentiate into functional neurons
Using immunofluorescence, GluK1 could be visualized on the soma and on neurites of beta III-tubulin-positive neurons (Figure 1A–B) generated from GRIK1 rs2832407 A/A and C/C donors. The differentiation protocol produces mixed neural cultures containing both MAP2-positive neurons and GFAP-positive astrocytes (Figure 1C). There was no significant difference in the efficacy of GRIK1 A/A or C/C iPSCs to differentiate into neuronal cultures enriched for TBR1-positive deep layer cortical-like neurons (p = 0.54) (Figure 1D–E), with ~50% of the total cells staining positive for TBR1.
Figure 1. GRIK1 rs2832407 A/A and C/C iPSCs differentiate into functional neurons.
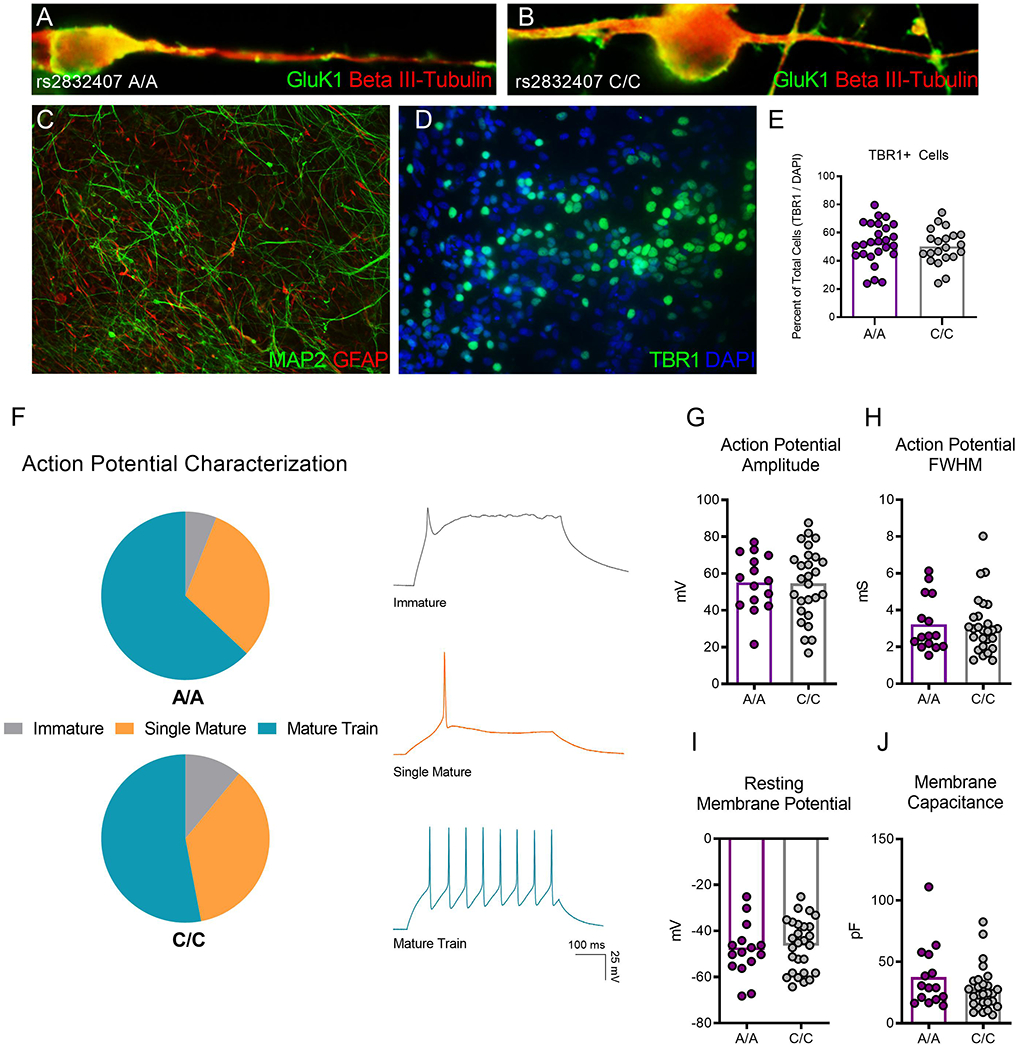
(A-B) A/A and C/C neurons 12 weeks after plating express GluK1 (green), which can be observed localizing to the soma and along beta-III tubulin positive neurites. (C) Neural differentiation produces mixed cultures containing MAP2-positive neurons and GFAP-positive astrocytes. (D-E) Cultures are enriched for TBR1-positive glutamate neurons. The percentage of TBR1-positive cells relative to the total cell population does not differ in neural cultures derived from 3 A/A and 5 C/C donors. 7,163 total cells total were counted for analysis. (F) iPSC-derived neurons (17-19 weeks post plating) generate mature action potential trains in response to a depolarizing current injection. The percentage of neurons that generate immature, single mature, or mature action potentials did not differ between 1 A/A and 2 C/C donors. Example traces for each action potential category are shown. (G-J) No difference between GRIK1 genotypes was observed in neuronal properties including (G) action potential amplitude, (H) action potential full width at half-maximum (FWHM), (I) resting membrane potential, and (J) membrane capacitance. Each dot on the graphs depicting distribution represents an individual neuron.
Neurons from A/A and C/C donors were characterized using whole-cell patch clamp electrophysiology. Upon the injection of a depolarizing current, iPSC-derived neurons generated an action potential, with the majority of neurons from both donor groups exhibiting mature action potentials. The percentage of neurons generating an immature, single mature spike, or a mature train did not differ between neurons from the A/A and C/C donors examined (A/A; mature train: 63%, single mature: 32%, immature: 6%, C/C; mature train: 53%, single mature: 36%, immature: 11%, x2 = 0.35, df = 2, p = 0.8) (Figure 1F), and there were no significant differences in either action potential amplitude (t = 0.1, df = 40, p = 0.93) (Figure 1G), or action potential full width at half-maximum (t = 0.1, df = 40, p = 0.93) (Figure 1H). We also observed no difference between genotypes in resting membrane potential (t = 0.55, df = 40, p = 0.59) (Figure 1I) or membrane capacitance (t = 1.42, df = 40, p = 0.16) (Figure 1J). Taken together, the immunostaining and electrophysiology results showed that our iPSC differentiation protocol generated functional neural cultures from both A/A and C/C donors.
Gene expression characterization of the GRIK1 locus
Alternative splicing of the GRIK1 transcript gives rise to GluK1-containing kainate receptors with different C-terminal domains and conductance properties (Jaskolski et al., 2005, Hirbec et al., 2003). We used TaqMan probe and primer sets spanning specific exon/intron boundaries to investigate the total GRIK1 and isoform-specific expression in 12-week old neural cells differentiated from 8 A/A and 7 C/C donors. We also examined the expression of an antisense RNA (GRIK1-AS2) that resides ~800 bp from rs2832407 (Figure 2A). Rs2832407 genotype did not associate with differences in mRNA expression of GRIK1 isoform 1 (exons 9-10 probe: t = 0.62, df = 13, p = 0.54), isoform 2 (exons 8-10 probe: t = 0.32, df = 13, p = 0.75; exons 16-18 probe: t = 0.83, df = 13, p = 0.42), or total GRIK1 (exons 1-2 probe: t = 0.22, df = 13, p = 0.83) (Figure 2B–E). There was a significant genotype difference in expression of GRIK1 antisense-2 (t = 3.27, df = 13, p = 0.006), with higher expression observed in neural cultures generated from C/C lines (Figure 2F).
Figure 2. GRIK1 gene expression in A/A and C/C iPSC-derived neural cultures.
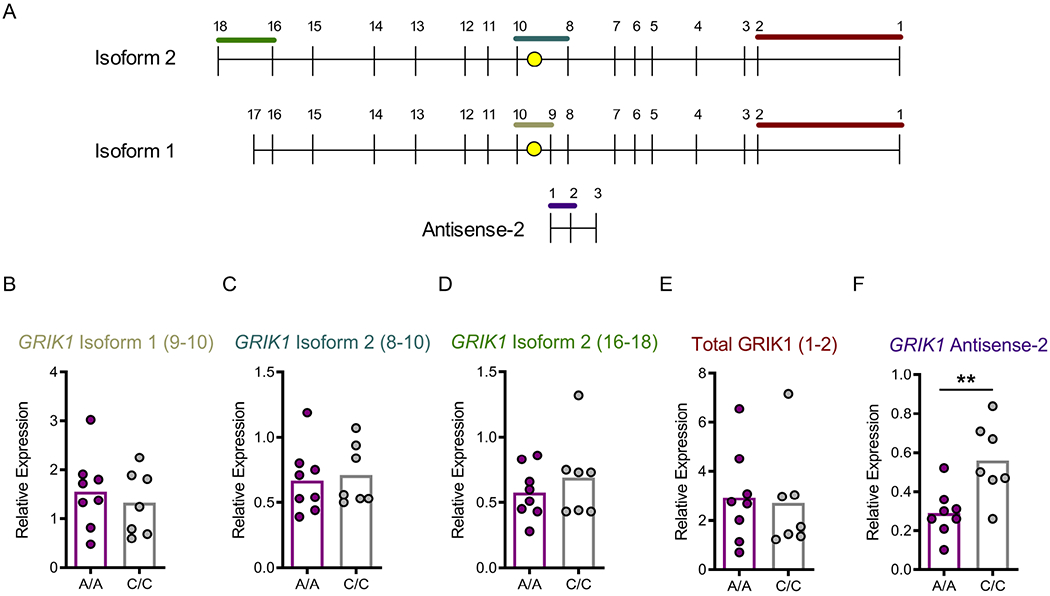
(A) Schematic representing the two major GRIK1 isoforms, which differ by the exclusion of exons 9 and 17 in isoform 2 (accession ID U16125), and exclusion of exon 18 in isoform 1 (accession ID NM-000830) as well as the antisense-2 RNA (Ref Seq accession ID NR_033368.1; which is also annotated as BACH1-207 in the Ensemble transcript database – ENST00000462262.1). Approximate location of rs2832407 is indicated by the yellow dot, which resides in the intronic region between exons 9-10 in isoform 1 and 8-10 in isoform 2. (B-F) iPSC-derived neural cultures were examined 12 weeks post plating. No significant effect of rs2832407 genotype was observed on the expression of GRIK1 isoform 1 (B), isoform 2 (C, D), or total GRIK1 (E). A significant effect of genotype was observed on the expression of GRIK1 antisense-2 (F). Each dot represents the average RNA expression collected from a donor subject.
Effects of acute topiramate exposure on frequency of spontaneous excitatory synaptic events
GluK1-containing receptors have been reported to enhance the release of neurotransmitter from excitatory presynaptic terminals (Aroniadou-Anderjaska et al., 2012). To examine genotype effects on presynaptic release in the cultures, we examined the frequency of spontaneous excitatory events following bath application of topiramate. Whole cell patch-clamp recordings of spontaneous excitatory postsynaptic currents (EPSCs) were obtained from neurons differentiated from 3 A/A donors (19 neurons) and 4 C/C donors (20 neurons) at a holding potential of −70 mV. EPSCs were blocked by the application of the AMPA/kainate receptor antagonist DNQX (Figure 3A). There was no significant difference between A/A and C/C neurons in the frequency of EPSCs at baseline (t = 1.4, df = 37, p = 0.17). However, a significant interaction of genotype with time of 5 μM topiramate exposure was observed for EPSC frequency (F = 3.4, df = 2, 110; p = 0.038), with C/C neurons showing a smaller reduction than A/A neurons (~5% vs. ~30%, respectively, at the end of treatment) (Figure 3C). Post-hoc analysis using paired t-tests and EPSC frequency data comparing baseline and the 20-30 minute treatment bin revealed no effect of topiramate in C/C neurons (t = 1.5, df = 19, p = 0.14), but a significant reduction of EPSC frequency in A/A neurons (t = 3.1, df = 18, p = 0.006).
Figure 3. Topiramate exposure attenuates the frequency of spontaneous excitatory synaptic activity.
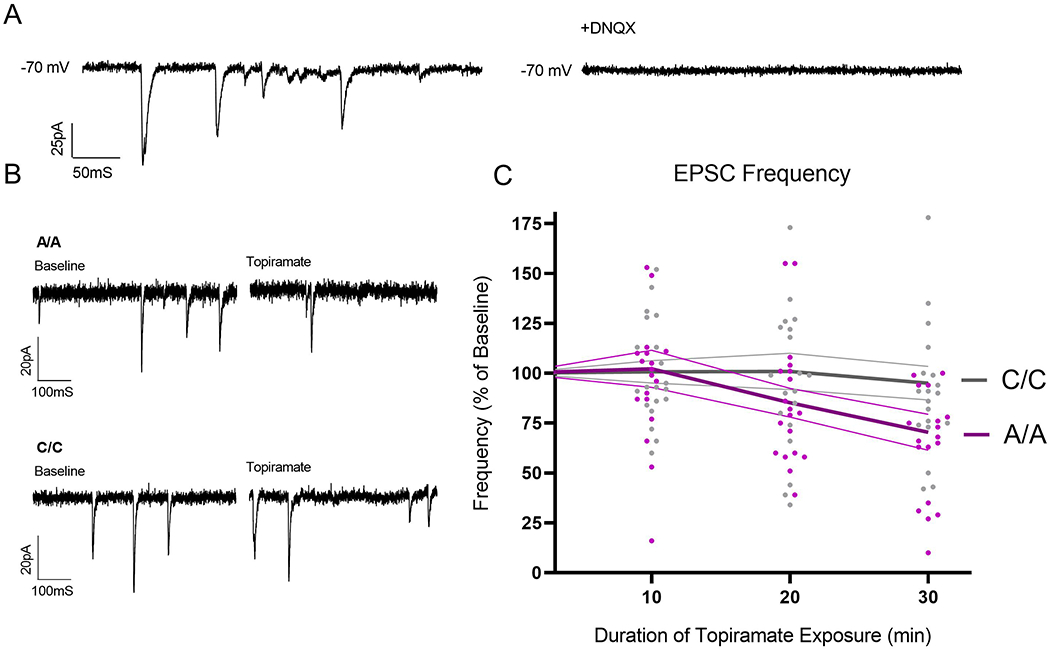
(A) Example trace representing the spontaneous excitatory postsynaptic currents (EPSCs) that were quantified, which were validated as excitatory by observing their attenuation in the presence of the AMPA/kainate receptor antagonist DNQX (10 μM). (B) Representative traces of EPSCs recorded from an A/A neuron and C/C neuron in the absence and presence of 5 μM topiramate for 20 min. (C) 12+ week old neurons generated from 3 A/A and 4 C/C donors were exposed to 5 μM topiramate for 30 min, and EPSCs were recorded and frequency plotted in 10 min bins. There was a significant interactive effect of topiramate exposure time and GRIK1 genotype on the frequency of EPSCs. Neurons derived from A/A donors showed a larger reduction in the frequency of EPSCs than neurons derived from C/C donors. Each dot represents an individual neuron. Thick lines represent the mean frequency per genotype. Thin lines depict ± SEM.
Discussion
Kainate receptors are a class of excitatory glutamate receptors comprised of tetrameric combinations of five subunit types (GluK1-5), with complex biological roles related to temporal changes in regional expression and involvement in both postsynaptic and presynaptic neurotransmission (Contractor et al., 2011). Topiramate, initially approved by the FDA as an anticonvulsant, reduces alcohol consumption in treatment-seeking individuals (Johnson et al., 2003, Johnson et al., 2007, Kranzler et al., 2014a) and non-treatment-seeking volunteers (Miranda et al., 2008). The pharmacologic actions of topiramate are diverse, with in vitro studies highlighting inhibitory effects on voltage-gated sodium channels (Zona et al., 1997), L-type calcium channels (Zhang et al., 2000), and non-NMDA glutamate (AMPA/kainate) receptors (Gibbs et al., 2000, Braga et al., 2009), and potentiating effects on GABAA receptors (White et al., 2000, Braga et al., 2009). The inhibitory effects of topiramate on non-NMDA glutamate receptors appear to be mediated primarily by kainate receptors containing the GluK1 subunit (Gryder and Rogawski, 2003, Braga et al., 2009), encoded by the GRIK1 gene. A single nucleotide polymorphism (rs2832407, C ➔ A) located in intron 9 of GRIK1 was associated with the risk of alcohol dependence in European Americans (Kranzler et al., 2009) and moderated the efficacy of topiramate in reducing heavy drinking days in treatment-seeking European Americans with AUD (Kranzler et al., 2014a, Kranzler et al., 2014b). However, the potential biological mechanisms underlying the association of rs2832407 to the risk of psychopathology and topiramate’s efficacy in reducing alcohol consumption are unknown.
We utilized iPSCs to identify the molecular correlates of the AUD-associated GRIK1 SNP rs2832407 in human neural cell cultures. The GRIK1 gene (21q22.1) is comprised of 18 exons, generating multiple mRNA isoforms and two antisense RNAs. The two most common isoforms in humans differ by the exclusion of exons 9 and 17 in isoform 2 (accession ID U16125), while isoform 1 (accession ID NM-000830) contains exons 1-17 but lacks exon 18 (Barbon and Barlati, 2000). Investigation in rodents revealed that alternative splicing of exon 17 and 18 results in amino acid sequence differences in the C-terminal domain, influencing membrane trafficking of the receptor (Jaskolski et al., 2005). Furthermore, the inclusion of exon 18, but not 17, results in PDZ-binding and PKC-phosphorylation sites that can alter kainate receptor localization and function at the synapse (Hirbec et al., 2003). Because of the molecular consequences of alternative splicing, we investigated whether rs2832407 genotype was associated with differential GRIK1 isoform expression but found no difference in mRNA expression of isoform 1, isoform 2, or total GRIK1 between A/A and C/C neural cultures. These results suggest that putative genetic effects linked to the rs2832407 SNP do not act via differences in the C-terminal the amino acid sequence of the GluK1 protein as a result of differential splicing of exon 17 vs 18.
We observed a significant association of GRIK1 genotype and antisense-2 RNA expression, with higher expression in neural cultures derived from C/C donors. Antisense transcripts are a subset of long non-coding RNAs which have been reported to impact cellular functions via multiple mechanisms including complementary binding to target RNAs, resulting in RNA degradation and/or the inhibition of translation, transcriptional modulation, effects on splicing, and translational effects at localized dendritic and synaptic sites. The antisense transcriptome has roles in neural development, response to the environment and in psychiatric and neurological disease (Mills et al., 2016; Rusconi, et al., 2020). The 1073 nt GRIK1-AS2 RNA has three exons, the first of which overlaps the GRIK1 transcript exon 9. Because antisense-2 RNA was elevated in neural cultures derived from C/C donors, an intriguing possibility is that rs2832407, or functional SNPs in linkage disequilibrium (LD) with it, alter the availability of GluK1-containing kainate receptors in C/C subjects by affecting antisense RNA transcription/splicing or stability. The effects of GRIK1 genotype on antisense-2 expression specifically and GluK1 subunit availability was previously proposed by Kranzler et al. (2014a) as a potential mechanism underlying the pharmacogenetic effects of topiramate on the reduction of alcohol consumption given the proximity of the rs2832407 haplotype block and GRIK1 antisense-2 exon 1, including being in near complete linkage with rs363431, which is located within exon 2 of the antisense transcript. The LD (r2) between rs2832407 and rs363431 in European Americans is 0.99. The imputed genotype at rs363431 in the antisense transcript is C/C for rs2832407 A/A lines and T/T for C/C lines. While antisense-2 is encoded within the intron 9 to 7 region of the GRIK1 gene, a recent Ensemble annotation (Ensemble release version 100) links the transcript to the BACH1 gene whose primary promoter is ~ 200 kilobases away. The BACH1 (BTB domain and CND homology 1) gene is a transcription factor expressed throughout the body that can act as either a transcriptional repressor or enhancer depending on context and whose target genes are involved in cellular oxidative stress responses (Warnatz et al., 2011; Zhang et al., 2018). The putative BACH1 associated transcript, BACH1-207 (ENST00000462262.1), does not encode an open reading frame and has limited supporting evidence. The most recent NCBI Ref Seq release 109.2 continues to annotate this transcript (NR_033368.1) as GRIK1-AS2. Direct studies to characterize this transcript, including mapping transcription start and end sites and demonstrating its potential effects on GRIK1 and/or neighboring genes is needed to better characterize this spliced transcript.
Acute exposure to 5 μM topiramate significantly reduced the frequency of EPSCs. The time course of topiramate effects in reducing EPSC frequency is consistent with the reported 10-20-minute delay following bath application of topiramate for the full inhibition of kainate receptors (Braga et al., 2009, Gibbs et al., 2000). This slow onset of inhibition is thought to relate to topiramate’s molecular mechanism of action via binding to kainate receptor phosphorylation site(s) when in the dephosphorylated state (Gibbs et al., 2000, Angehagen et al., 2004). We note that responses to topiramate exposure were variable and speculate that this may relate to heterogeneity of presynaptic inputs in this culture model. We observed a significant interaction between topiramate treatment time and GRIK1 genotype on the frequency of EPSCs following topiramate application, with neurons derived from C/C donors showing a smaller reduction in EPSC frequency than A/A donors. The effect of topiramate in reducing EPSCs is consistent with reports that GluK1-containing receptors on excitatory presynaptic terminals enhance glutamate release, and their inhibition decreases principal neuron EPSC frequency (Aroniadou-Anderjaska et al., 2012). Based on this prior study, our finding that spontaneous EPSC frequencies were significantly reduced by topiramate for A/A but not C/C neurons suggests that in the basal state, GluK1-facilitated glutamate release from excitatory presynaptic nerve terminals is lower in C/C than A/A neurons. A limitation of the current study is that we examined a single concentration of topiramate. Therefore, it is unclear whether neurons generated from C/C donors would respond if exposed to higher concentrations of the compound. The generation of dose-response curves in neurons from C/C and A/A donors would address whether topiramate has different pharmacodynamic properties in neurons of contrasting rs2832407 genotypes.
In view of our EPSC frequency results with respect to our gene expression findings, it may be that GRIK1 rs2832407 genotype effects on antisense-2 expression mediate GluK1 availability locally at presynaptic excitatory synapses, and that the higher antisense-2 expression in C/C neurons generates terminals with a lower potential to be moderated by topiramate. Although our results do not provide a clear model for how rs2832407 genotype might modulate topiramate treatment response for alcohol use, we speculate that rs2832407 genotype effects related to the greater reduction in heavy alcohol consumption in C/C subjects by topiramate (Kranzler et al., 2014a, Kranzler et al., 2014b) could be due, in part, to differential presynaptic effects of topiramate as a function of rs2832407 genotype. Alternatively, GRIK1 genotype and consequent biological effects may have developmental origins related to effects on the relative strength of interactions between brain regions that contribute to both an association of C/C genotype with alcohol dependence (Kranzler et al., 2009) and the reduction in drinking by topiramate (Kranzler et al., 2014a).
The findings reported here must be viewed in the context of several limitations. First, we examined the effect of acute topiramate exposure on non-pharmacologically isolated spontaneous post-synaptic events in pyramidal-like excitatory neurons that contained a mixture of action potential-dependent and non-action potential-dependent events. Therefore, it may be that the synaptic effects of topiramate observed in our iPSC-derived neurons were due in part to topiramate’s effect on receptors or channels other than GluK1-containing kainate receptors, e.g., voltage-gated sodium and calcium channels residing on the non-voltage clamped presynaptic neurons, although such components would not be expected to show GRIK1 genotype effects. Methods developed in in vitro rodent models to isolate kainate receptors pharmacologically (Gryder and Rogawski, 2003, Braga et al., 2009) may be more useful for examining the specific effects of topiramate in iPSC-derived neurons. Additionally, our acute experiments do not model the potential neuroadaptive effects of sustained topiramate exposure in the clinical treatment setting. Second, because iPSC neural cultures more closely resemble early brain development than adult neural tissue, with transcription profiles from iPSC neural cultures most closely resembling those of first-trimester brain tissue (Brennand et al., 2015), the effects of topiramate and GRIK1 genotype identified in this culture system may not reflect their effects in mature neural tissue. The neural differentiation protocol we utilized generates cultures enriched for forebrain-type glutamate neurons. Because the expression of kainate receptors varies by brain region (Contractor et al., 2011) and the effects of topiramate differ between excitatory and inhibitory neurons (Braga et al., 2009, Aroniadou-Anderjaska et al., 2012), future work should consider use of additional neural differentiation protocols to generate cultures enriched for inhibitory, excitatory, or dopaminergic neurons, among others (Mertens et al., 2016), to explore the effects of topiramate treatment on other neuronal cell types. Third, our results are correlational and do not address potential functional relationships between the observed higher antisense-2 RNA and reduced topiramate effects on the frequency of spontaneous EPSCs in C/C genotype neurons. Further our results do not speak to the issue of whether the rs2832407 sentinel variant (vs. others linked to this variant) have direct functional effects. Gene editing to generate isogenic lines could be used to investigate directly the effect of GRIK1 genetic variation on the two cellular phenotypes reported here. Finally, it remains to be determined how the effects of topiramate observed in our culture system in A/A vs. C/C donor cells relate to the differential efficacy of topiramate by genotype in reducing heavy drinking.
In summary, we have shown that iPSC-derived neural cells are a model system to explore the molecular actions of topiramate in relevant cell types in vitro. In particular, the technology allows one to probe mechanisms of pharmacogenetic treatment effects, even though the functional genetic element is not yet known, by utilizing cells that express the unmodified donor genome. Future studies can use novel protocols to generate specific populations of neural derivatives to identify between-cell pharmacologic sensitivity and the influence of genetic variation on neural activity. While there are clear challenges, iPSC technologies may provide a valuable tool for examining the molecular mechanisms underlying the pharmacogenetic effects of other pharmacological treatments for AUD (Jones et al., 2015).
Acknowledgments
We would like to thank Leann Crandall at the UC Stem Cell Core for her valued assistance in generating iPSC lines.
Funding and conflicts of interest: Supported by NIH grants R21 AA023212 (JC), R01 AA23192 (HK), R01 AA015606 (JC), P60 AA03510, and M01 RR06192.
Dr. Kranzler is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which over the past three years was supported by AbbVie, Alkermes, Amygdala Neurosciences, Arbor Pharmaceuticals, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, and Pfizer. Dr. Kranzler is named as an inventor on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. Since participating in this research, Dr. Jensen has become an employee of Celgene Corporation and declares no conflict of interest. The remaining authors have no conflicts of interest to declare.
References
- ANGEHAGEN M, BEN-MENACHEM E, SHANK R, RONNBACK L & HANSSON E 2004. Topiramate modulation of kainate-induced calcium currents is inversely related to channel phosphorylation level. J Neurochem, 88, 320–5. [DOI] [PubMed] [Google Scholar]
- ARONIADOU-ANDERJASKA V, PIDOPLICHKO VI, FIGUEIREDO TH, ALMEIDA-SUHETT CP, PRAGER EM & BRAGA MF 2012. Presynaptic facilitation of glutamate release in the basolateral amygdala: a mechanism for the anxiogenic and seizurogenic function of GluK1 receptors. Neuroscience, 221, 157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALTIERI DA, DARO FR, RIBEIRO PL & DE ANDRADE AG 2008. Comparing topiramate with naltrexone in the treatment of alcohol dependence. Addiction, 103, 2035–44. [DOI] [PubMed] [Google Scholar]
- BARBON A & BARLATI S 2000. Genomic organization, proposed alternative splicing mechanisms, and RNA editing structure of GRIK1. Cytogenet Cell Genet, 88, 236–9. [DOI] [PubMed] [Google Scholar]
- BRAGA MF, ARONIADOU-ANDERJASKA V, LI H & ROGAWSKI MA 2009. Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABAA receptors on principal neurons. J Pharmacol Exp Ther, 330, 558–66. [DOI] [PubMed] [Google Scholar]
- BRENNAND K, SAVAS JN, KIM Y, TRAN N, SIMONE A, HASHIMOTO-TORII K, BEAUMONT KG, KIM HJ, TOPOL A, LADRAN I, ABDELRAHIM M, MATIKAINEN-ANKNEY B, CHAO SH, MRKSICH M, RAKIC P, FANG G, ZHANG B, YATES JR 3RD & GAGE FH 2015. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry, 20, 361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMORRO AJ, MARCOS M, MIRON-CANELO JA, PASTOR I, GONZALEZ-SARMIENTO R & LASO FJ 2012. Association of micro-opioid receptor (OPRM1) gene polymorphism with response to naltrexone in alcohol dependence: a systematic review and meta-analysis. Addict Biol, 17, 505–12. [DOI] [PubMed] [Google Scholar]
- COHEN E, FEINN R, ARIAS A & KRANZLER HR 2007. Alcohol treatment utilization: findings from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend, 86, 214–21. [DOI] [PubMed] [Google Scholar]
- COLLINGRIDGE GL, OLSEN RW, PETERS J & SPEDDING M 2009. A nomenclature for ligand-gated ion channels. Neuropharmacology, 56, 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONTRACTOR A, MULLE C & SWANSON GT 2011. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci, 34, 154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVAULT J, POND T, FEINN R, ARIAS AJ, ONCKEN C & KRANZLER HR 2014. Dutasteride reduces alcohol’s sedative effects in men in a human laboratory setting and reduces drinking in the natural environment. Psychopharmacology (Berl), 231, 3609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICK DM & AGRAWAL A 2008. The genetics of alcohol and other drug dependence. Alcohol Res Health, 31, 111–8. [PMC free article] [PubMed] [Google Scholar]
- FINK JJ, ROBINSON TM, GERMAIN ND, SIROIS CL, BOLDUC KA, WARD AJ, RIGO F, CHAMBERLAIN SJ & LEVINE ES 2017. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat Commun, 8, 15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GELERNTER J & KRANZLER HR 2009. Genetics of alcohol dependence. Hum Genet, 126, 91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIBBS JW 3RD, SOMBATI S, DELORENZO RJ & COULTER DA 2000. Cellular actions of topiramate: blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia, 41 Suppl 1, S10–6. [DOI] [PubMed] [Google Scholar]
- GRANT BF, CHOU SP, SAHA TD, PICKERING RP, KERRIDGE BT, RUAN WJ, HUANG B, JUNG J, ZHANG H, FAN A & HASIN DS 2017. Prevalence of 12-Month Alcohol Use, High-Risk Drinking, and DSM-IV Alcohol Use Disorder in the United States, 2001–2002 to 2012–2013: Results From the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRANT BF, GOLDSTEIN RB, SAHA TD, CHOU SP, JUNG J, ZHANG H, PICKERING RP, RUAN WJ, SMITH SM, HUANG B & HASIN DS 2015. Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry, 72, 757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRYDER DS & ROGAWSKI MA 2003. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci, 23, 7069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALIKERE A, POPOVA D, SCARNATI MS, HAMOD A, SWERDEL MR, MOORE JC, TISCHFIELD JA, HART RP & PANG ZP 2019. Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEILIG M, GOLDMAN D, BERRETTINI W & O’BRIEN CP 2011. Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci, 12, 670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEVNER RF, SHI L, JUSTICE N, HSUEH Y, SHENG M, SMIGA S, BULFONE A, GOFFINET AM, CAMPAGNONI AT & RUBENSTEIN JL 2001. Tbr1 regulates differentiation of the preplate and layer 6. Neuron, 29, 353–66. [DOI] [PubMed] [Google Scholar]
- HIRBEC H, FRANCIS JC, LAURI SE, BRAITHWAITE SP, COUSSEN F, MULLE C, DEV KK, COUTINHO V, MEYER G, ISAAC JT, COLLINGRIDGE GL & HENLEY JM 2003. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron, 37, 625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HURT RT, EDAKKANAMBETH VARAYIL J & EBBERT JO 2014. New pharmacological treatments for the management of obesity. Curr Gastroenterol Rep, 16, 394. [DOI] [PubMed] [Google Scholar]
- JASKOLSKI F, COUSSEN F & MULLE C 2005. Subcellular localization and trafficking of kainate receptors. Trends Pharmacol Sci, 26, 20–6. [DOI] [PubMed] [Google Scholar]
- JENSEN KP, LIEBERMAN R, KRANZLER HR, GELERNTER J, CLINTON K & COVAULT J 2019. Alcohol-responsive genes identified in human iPSC-derived neural cultures. Transl Psychiatry, 9, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON BA, AIT-DAOUD N, BOWDEN CL, DICLEMENTE CC, ROACHE JD, LAWSON K, JAVORS MA & MA JZ 2003. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet, 361, 1677–85. [DOI] [PubMed] [Google Scholar]
- JOHNSON BA, ROSENTHAL N, CAPECE JA, WIEGAND F, MAO L, BEYERS K, MCKAY A, AIT-DAOUD N, ANTON RF, CIRAULO DA, KRANZLER HR, MANN K, O’MALLEY SS, SWIFT RM, TOPIRAMATE FOR ALCOHOLISM ADVISORY, B. & TOPIRAMATE FOR ALCOHOLISM STUDY, G. 2007. Topiramate for treating alcohol dependence: a randomized controlled trial. JAMA, 298, 1641–51. [DOI] [PubMed] [Google Scholar]
- JONES JD, COMER SD & KRANZLER HR 2015. The pharmacogenetics of alcohol use disorder. Alcohol Clin Exp Res, 39, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRANZLER HR, COVAULT J, FEINN R, ARMELI S, TENNEN H, ARIAS AJ, GELERNTER J, POND T, ONCKEN C & KAMPMAN KM 2014a. Topiramate treatment for heavy drinkers: moderation by a GRIK1 polymorphism. Am J Psychiatry, 171, 445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRANZLER HR, GELERNTER J, ANTON RF, ARIAS AJ, HERMAN A, ZHAO H, BURIAN L & COVAULT J 2009. Association of markers in the 3’ region of the GluR5 kainate receptor subunit gene to alcohol dependence. Alcohol Clin Exp Res, 33, 925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRANZLER HR, WETHERILL R, FEINN R, POND T, GELERNTER J & COVAULT J 2014b. Posttreatment effects of topiramate treatment for heavy drinking. Alcohol Clin Exp Res, 38, 3017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEMTIRI-CHLIEH F & LEVINE ES 2010. BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. J Neurophysiol, 104, 1923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBERMAN R, KRANZLER HR, JOSHI P, SHIN DG & COVAULT J 2015. GABRA2 Alcohol Dependence Risk Allele is Associated with Reduced Expression of Chromosome 4p12 GABAA Subunit Genes in Human Neural Cultures. Alcohol Clin Exp Res, 39, 1654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBERMAN R, KRANZLER HR, LEVINE ES & COVAULT J 2017. Examining the effects of alcohol on GABAA receptor mRNA expression and function in neural cultures generated from control and alcohol dependent donor induced pluripotent stem cells. Alcohol, 66, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBERMAN R, LEVINE ES, KRANZLER HR, ABREU C & COVAULT J 2012. Pilot Study of iPS-Derived Neural Cells to Examine Biologic Effects of Alcohol on Human Neurons In Vitro. Alcohol Clin Exp Res, 36, 1678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYSENG-WILLIAMSON KA & YANG LP 2007. Topiramate: a review of its use in the treatment of epilepsy. Drugs, 67, 2231–56. [DOI] [PubMed] [Google Scholar]
- MAY TW, RAMBECK B & JURGENS U 2002. Serum concentrations of topiramate in patients with epilepsy: influence of dose, age, and comedication. Ther Drug Monit, 24, 366–74. [DOI] [PubMed] [Google Scholar]
- MERTENS J, MARCHETTO MC, BARDY C & GAGE FH 2016. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci, 17, 424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERTENS J, WANG QW, KIM Y, YU DX, PHAM S, YANG B, ZHENG Y, DIFFENDERFER KE, ZHANG J, SOLTANI S, EAMES T, SCHAFER ST, BOYER L, MARCHETTO MC, NURNBERGER JI, CALABRESE JR, ODEGAARD KJ, MCCARTHY MJ, ZANDI PP, ALDA M, NIEVERGELT CM, PHARMACOGENOMICS OF BIPOLAR DISORDER, S., MI S, BRENNAND KJ, KELSOE JR, GAGE FH & YAO J 2015. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature, 527, 95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILIVOJEVIC V, FEINN R, KRANZLER HR & COVAULT J 2014. Variation in AKR1C3, which encodes the neuroactive steroid synthetic enzyme 3alpha-HSD type 2 (17beta-HSD type 5), moderates the subjective effects of alcohol. Psychopharmacology (Berl), 231, 3597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLS JD, CHEN BJ, UEBERHAM U, ARENDT A & JANITZ M 2016. The antisense transcriptome and the human brain. J Mol Neurosci, 58, 1–15 [DOI] [PubMed] [Google Scholar]
- MIRANDA R JR., MACKILLOP J, MONTI PM, ROHSENOW DJ, TIDEY J, GWALTNEY C, SWIFT R, RAY L & MCGEARY J 2008. Effects of topiramate on urge to drink and the subjective effects of alcohol: a preliminary laboratory study. Alcohol Clin Exp Res, 32, 489–97. [DOI] [PubMed] [Google Scholar]
- ONI EN, HALIKERE A, LI G, TORO-RAMOS AJ, SWERDEL MR, VERPEUT JL, MOORE JC, BELLO NT, BIERUT LJ, GOATE A, TISCHFIELD JA, PANG ZP & HART RP 2016. Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele. Sci Rep, 6, 34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARIKH SK & SILBERSTEIN SD 2019. Current Status of Antiepileptic Drugs as Preventive Migraine Therapy. Curr Treat Options Neurol, 21, 16. [DOI] [PubMed] [Google Scholar]
- RUSCONI F, BATTAGLIOLI E & VENTURIN M 2020. Psychiatrtic disorders and lncRNAs: A synaptic match. Int J Mol Sci, 21(9), 3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHANK RP, GARDOCKI JF, STREETER AJ & MARYANOFF BE 2000. An overview of the preclinical aspects of topiramate: pharmacology, pharmacokinetics, and mechanism of action. Epilepsia, 41 Suppl 1, S3–9. [PubMed] [Google Scholar]
- SHENG Y, FILICHIA E, SHICK E, PRESTON KL, PHILLIPS KA, COOPERMAN L, LIN Z, TESAR P, HOFFER B & LUO Y 2016. Using iPSC-derived human DA neurons from opioid-dependent subjects to study dopamine dynamics. Brain Behav, 6, e00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKAHASHI K, TANABE K, OHNUKI M, NARITA M, ICHISAKA T, TOMODA K & YAMANAKA S 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–72. [DOI] [PubMed] [Google Scholar]
- TOBE BTD, CRAIN AM, WINQUIST AM, CALABRESE B, MAKIHARA H, ZHAO WN, LALONDE J, NAKAMURA H, KONOPASKE G, SIDOR M, PERNIA CD, YAMASHITA N, WADA M, INOUE Y, NAKAMURA F, SHERIDAN SD, LOGAN RW, BRANDEL M, WU D, HUNSBERGER J, DORSETT L, DUERR C, BASA RCB, MCCARTHY MJ, UDESHI ND, MERTINS P, CARR SA, ROULEAU GA, MASTRANGELO L, LI J, GUTIERREZ GJ, BRILL LM, VENIZELOS N, CHEN G, NYE JS, MANJI H, PRICE JH, MCCLUNG CA, AKISKAL HS, ALDA M, CHUANG DM, COYLE JT, LIU Y, TENG YD, OHSHIMA T, MIKOSHIBA K, SIDMAN RL, HALPAIN S, HAGGARTY SJ, GOSHIMA Y & SNYDER EY 2017. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci U S A, 114, E4462–E4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNATZ HJ, SCHMIDT D, MANKE T, PICCINI I, SULTAN M, BORODINA T, BALZEREIT D, WRUCK W, SOLDATOV A, VINGRON M, LEHRACH H, YASPO ML 2011. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. 286(26):23521–23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE HS, BROWN SD, WOODHEAD JH, SKEEN GA & WOLF HH 2000. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia, 41 Suppl 1, S17–20. [PubMed] [Google Scholar]
- ZHANG X, GUO J, WEI X, NIU C, JIA M, LI Q, & MENG D 2018. Bach1: Function, regulation and involvement in disease. Oxid Med Cell Longevity, 2018, Article ID 1347969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, VELUMIAN AA, JONES OT & CARLEN PL 2000. Modulation of high-voltage-activated calcium channels in dentate granule cells by topiramate. Epilepsia, 41 Suppl 1, S52–60. [DOI] [PubMed] [Google Scholar]
- ZONA C, CIOTTI MT & AVOLI M 1997. Topiramate attenuates voltage-gated sodium currents in rat cerebellar granule cells. Neurosci Lett, 231, 123–6. [DOI] [PubMed] [Google Scholar]