

Summary
Autism spectrum disorder (ASD) is more prevalent in males; however, the etiology for this sex-bias is not well understood. Interestingly, many mutations on an X-linked cell adhesion molecule NLGN4X result in ASD or intellectual disability. NLGN4X is part of an X-Y pair with NLGN4Y sharing ~97% sequence homology. Using biochemistry, electrophysiology, and imaging, we show that NLGN4Y displays severe deficits in maturation, surface expression, and synaptogenesis regulated by one amino acid difference with NLGN4X. Furthermore, we identify a cluster of ASD-associated mutations surrounding the critical amino acid in NLGN4X, and these mutations phenocopy NLGN4Y. Importantly, we show that NLGN4Y cannot compensate for the functional deficits observed in ASD-associated NLGN4X mutations. Taken together, our data reveal a potential pathogenic mechanism for male bias in NLGN4X-associated ASD.
eTOC blurb
NLGN4X and NLGN4Y are thought to have similar function due to ~97% amino acid identity. Nguyen et al. demonstrate NLGN4Y has a deficit in trafficking due to one amino acid difference. The trafficking deficit phenotype is found in a cluster of autism-associated variants in NLGN4X near the critical amino acid.
Introduction
Neuroligins (NLGNs) are postsynaptic cell adhesion molecules that have crucial roles in synapse maturation and function (Südhof, 2008; Bemben, Shipman, et al., 2015; Jeong, Paskus and Roche, 2017). Although all NLGNs bind to presynaptic neurexins to drive synapse formation, there are multiple isoforms that display differential localization and functional effects. NLGN 1–3 are highly conserved in mammals and extensive research in mouse models shows that NLGN1 is localized at excitatory synapses, NLGN2 at inhibitory synapses, and NLGN3 is at both (Bemben, Shipman, et al., 2015; Jeong et al., 2017). Interestingly, there is divergence regarding NLGN4, with humans having a NLGN4X located on the X chromosome and a NLGN4Y on the Y chromosome forming an X-Y pair. In contrast, mouse NLGN4-like is more divergent and is located on the pseudoautosomal region of the X chromosome (Bolliger et al., 2001, 2008; Maxeiner et al., 2020). Although these are broadly referred to as NLGN4, NLGN4X and NLGN4-like, they are poorly conserved with only ~60% sequence identity.
Interestingly, many studies have linked NLGNs with neurodevelopmental disorders such as autism spectrum disorder (ASD) and intellectual disability (ID) (Singh and Eroglu, 2013). Before the explosion of next-generation sequencing, the X-linked NLGN3 and NLGN4X were heavily associated with ASD/ID (Jamain et al., 2003). In addition, a multi-generation study reported a NLGN4X variant that resulted in ASD/ID. Importantly, the variant consisted of a frameshift mutation that only affected male family members carrying the mutation, whereas female carriers displayed no symptoms of ASD/ID (Laumonnier et al., 2004). Subsequent studies from many ASD/ID cohorts revealed more mutations in NLGN4X, and a concomitant pattern of male bias (Yan et al., 2008; Pampanos et al., 2009; Zhang et al., 2009; Xu et al., 2014; Yuen et al., 2017) (Table S1).
To add further complexity, in females NLGN4X can escape X-inactivation, and in males NLGN4Y mRNA has been reliably detected in the brain (Jamain et al., 2003; Carrel and Willard, 2005; Trabzuni et al., 2013; Tukiainen et al., 2017). Because NLGN4X and NLGN4Y share 97% sequence identity (Figure 1A), they are presumed to have the same function and act as an X-Y gene pair, even though the genes are located outside of the pseudo-autosomal area on the sex chromosome. With genetic sequencing, the number of pathogenic mutations found in NLGN4X is increasing, whereas only one mutation has been identified in NLGN4Y (Yan et al., 2008). The male bias observed from mutations in NLGN4X is puzzling because NLGN4Y is thought to play a similar role to NLGN4X and thus should be able to compensate for NLGN4X ASD-associated mutations. This lack of compensation in males suggests that NLGN4Y may have an uncharacterized distinct function that needs to be explored.
Figure 1. NLGN4X and NLGN4Y are differentially expressed.
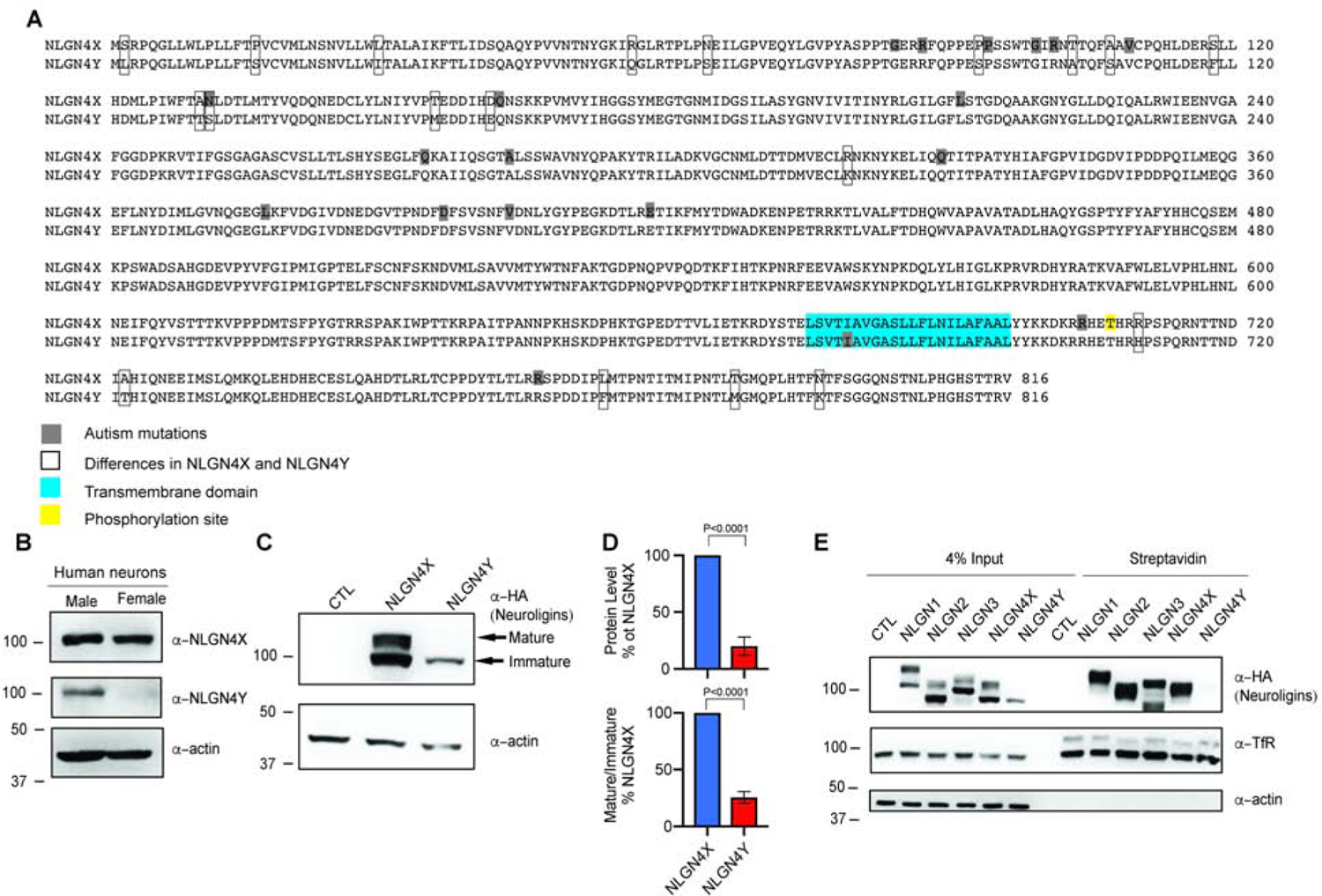
A. Alignment of NLGN4X and NLGL4Y. Differences between NLGN4X and NLGN4Y are marked with open boxes; ASD-associated mutations are boxed in gray; transmembrane domain is boxed in blue; phosphorylation site is boxed in yellow. B. Immunoblot of NLGN4X and NLGN4Y from male and female human differentiated neurons. C. Immunoblot of HA-tagged NLGN4X and NLGN4Y expressed in HEK293T cells. Arrows indicate mature and immature bands. D. Total protein level and ratio of mature/immature level (means ± S.E.M.) normalized to NLGN4X. P values were calculated using unpaired t test. (n=5). E. Surface expression of NLGN1, 2, 3, 4X, and 4Y analyzed by immunoblot of purified biotinylated proteins from transfected HEK293T cells.
In this study, we characterized the functional differences between NLGN4X and NLGN4Y. We show that both endogenous NLGN4X and NLGN4Y are expressed in human neurons. Despite having 97% sequence identity, we find that NLGN4Y has profound deficits in trafficking to the cell surface and related synaptic effects. This functional difference between NLGN4X and NLGN4Y is regulated by a single critical amino acid difference in the extracellular domain. Using publicly available human genetic data, as well as newly identified probands, we define a sensitive region in NLGN4X that contains a cluster of ASD-associated variants. This sensitive region surrounds the critical amino acid substitution in NLGN4Y. Furthermore, the disease-associated mutations phenocopy NLGN4Y. Thus, our study provides insight into the sex bias of NLGN4X-associated ASD/ID.
Results
NLGN4X and NLGN4Y are differentially processed
Is NLGN4Y expressed in humans? To test NLGN4Y protein expression, we first developed a NLGN4Y-specific antibody (Figure S1A). An immunoblot of lysates from Human Embryonic Kidney 293 (HEK293T) cells transfected with hemagglutinin (HA)-tagged NLGN1, 2, 3, 4X, and 4Y shows specific immunoreactivity recognizing NLGN4Y (Figure S1B). Next, we immunoblotted lysates from male and female differentiated neurons from human induced pluripotent stem cells (hiPSCs) and detected NLGN4Y only in male-derived neurons (Figure 1B). Similarly, we detected NLGN4Y in human brain lysate from males but not females (Figure S1C). Taken together, we showed here that NLGN4Y is expressed in humans.
Why is there a disproportionate number of ASD-associated variants in NLGN4X vs NLGN4Y? To answer this question, we directly compared NLGN4X and NLGN4Y in a variety of assays. First, we expressed HA-tagged NLGN4X or NLGN4Y in HEK293T cells and unexpectedly observed that NLGN4Y is differentially expressed from NLGN4X (Figure 1C and D). Interestingly, we only observed the lower molecular weight band, which is the presumptive immature form of the protein, whereas NLGN4X expressed both mature and immature forms (Fig. 1C and D). It is important to note here that both NLGN4X and NLGN4Y are inserted into the same mammalian expressing vector. To ensure that both plasmids express at a comparable level, we extracted mRNA from HEK293T cells expressing NLGN4X and NLGN4Y then used RT-PCR with primers that can recognize both NLGN4X and NLGN4Y. We saw a small, but significant, decrease in NLGN4Y expression compared to NLGN4X (Figure S1F and G). Next, we reasoned that the lack of mature NLGN4Y corresponded to a lack of NLGN4Y surface expression. To examine surface expression of NLGNs, we labeled live transfected HEK293T cells with sulfo-NHS-SS-biotin and isolated the surface NLGN pool with Streptavidin. As predicted, NLGN4Y was not detected on the surface compared to NLGN1–4X (Figure 1E). As a control, we used transferrin receptor (TfR) to ensure our assay adequately detected surface proteins. Because NLGN4Y was not at the surface, we hypothesized that NLGN4Y was retained in the endoplasmic reticulum (ER). We coexpressed NLGN4X or NLGN4Y with an ER marker to visualize their localization. The Mander’s coefficient for NLGN4Y overlapping with the ER marker was significantly higher than with NLGN4X revealing that NLGN4Y was more heavily localized to the ER compared to NLGN4X (Figure S2A and B). Because retention in the ER could lead to degradation, we tested if we could rescue NLGN4Y surface expression by blocking degradation using MG132 to inhibit proteasomal degradation or chloroquine to inhibit lysosomal degradation in HEK293T cells expressing NLGN4X or NLGN4Y. Treatment with either MG132 or chloroquine did not increase the mature band for NLGN4Y (Figure S1D). Although we observed an apparent increase in the NLGNY immature band following treatment with MG132, it did not reach statistical significance (Figure S1E). Taken together, we show that NLGN4X and NLGN4Y are differentially processed, which results in profound deficits in the maturation of NLGN4Y and trafficking to the cell surface.
The ECD of NLGN4X is important for surface expression
To determine whether the extracellular domain (ECD) or the intracellular domain (ICD) impairs NLGN4Y surface expression, we generated two chimeric constructs: NLGN4X ECD with NLGN4Y ICD (NLGN4X/Y) and NLGN4Y ECD with NLGN4X ICD (NLGN4Y/X) (Fig. 2A). Comparison of NLGN4X, 4Y, 4X/Y, or 4Y/X expressed in HEK293T cells revealed that the ECD of NLGN4X is important for protein maturation (Figure S1H and I). Next, we used a biotinylation assay to label surface-expressed proteins and found that NLGN4X/Y displayed significantly increased surface expression compared to NLGN4Y, whereas NLGN4Y/X decreased surface expression compared to NLGN4X (Figure 2B and 2C). With only 14 amino acid differences between the NLGN4X and NLGN4Y ECD, there must be a region or an amino acid difference that could account for the lack of NLGN4Y surface expression. We made six chimeric constructs to narrow down the critical region for NLGN4X mature band expression (Figure S1J). Swapping the signal peptide did not rescue expression of the NLGN4Y mature band; however, swapping the first 100 amino acids was sufficient to increase the NLGN4Y mature band (Figure S3K).
Figure 2. Differences in NLGN4X and NLGN4Y protein expression are primarily regulated by one amino acid difference.
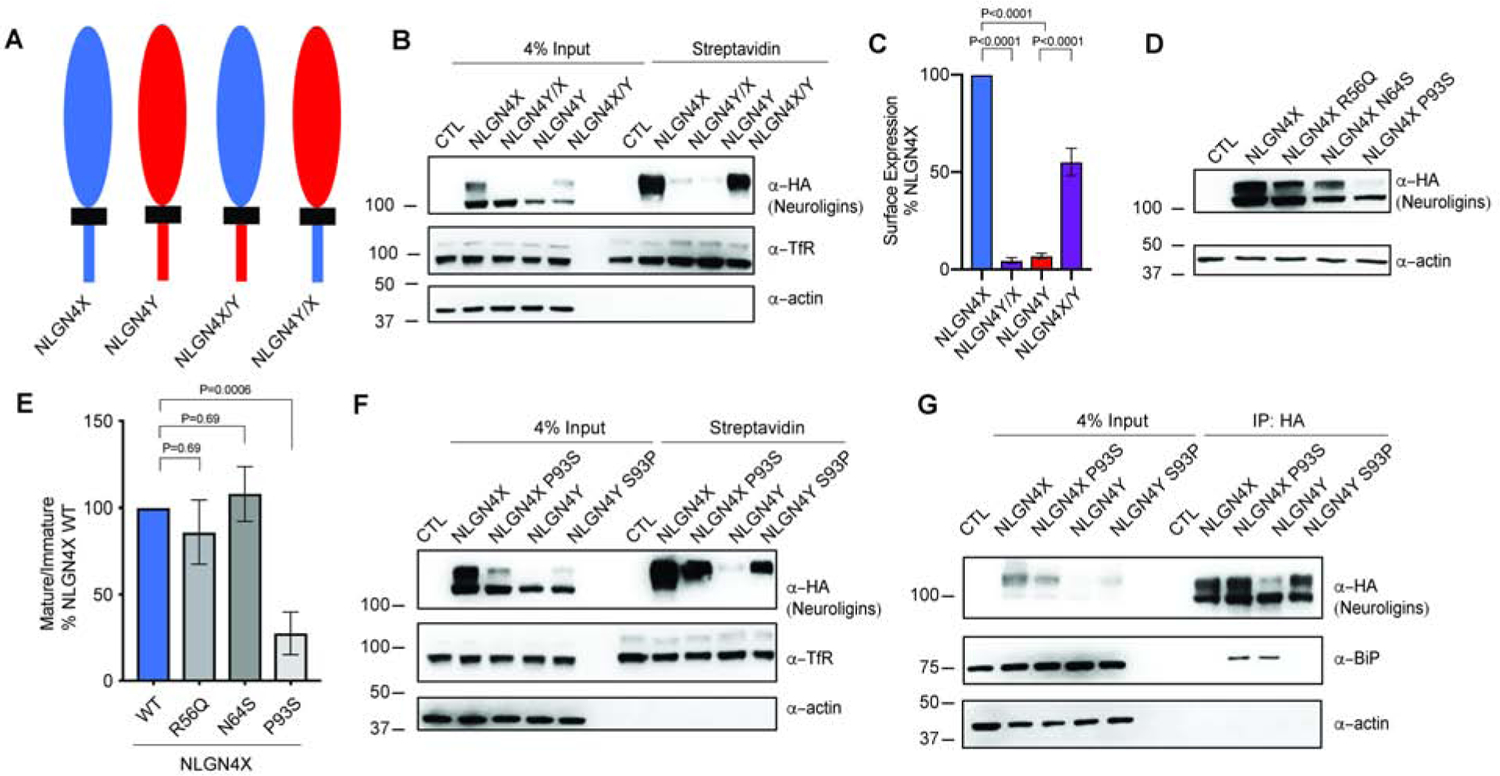
A. Schematic showing domain swap for NLGN4X, NLGN4Y, NLGN4X/Y, and NLGN4Y/X. NLGN4X/Y contains the extracellular domain (ECD) of NLGN4X and the intracellular domain (ICD) of NLGN4Y; NLGN4Y/X contains the ECD of NLGN4Y and the ICD of NLGN4X. B. Surface expression of NLGN4X, NLGN4Y, NLGN4X/Y, and NLGN4Y/X was analyzed by immunoblotting purified surface biotinylated proteins from transfected HEK293T cells. C. Surface intensity level (means ± S.E.M.) normalized to NLGN4X. P values were calculated by ANOVA with Bonferroni’s multiple comparison test. (n=5). D. Immunoblot of transfected NLGN4X (WT, R56Q, N64S, or P93S) and NLGN4Y from HEK293T cells. E. Mature/immature level (means ± S.E.M.) normalized to control. P values were calculated by one-way ANOVA with Bonferroni’s multiple comparison test. (n=7). F. Surface expression of NLGN4X (WT or P93S) and NLGN4Y (WT or S93P) were analyzed by immunoblotting isolated surface biotinylated proteins in transfected HEK293T cells. G. NLGN4X (WT and P93S) and NLGN4Y (WT and S93P) were transfected in HEK293T cells then immunoprecipitated with HA-antibody. Immunoblot was probed for BiP and HA from the immunoprecipitates.
Differential function of NLGN4X vs. NLGN4Y is regulated by one amino acid difference
Within the region from the end of the signal peptide to the 100th amino acid, there are three amino acid differences between NLGN4X and NLGN4Y. Next, we created three new constructs: NLGN4X (R56Q, N64S, or P93S), substituting the analogous NLGN4Y amino acids for NLGN4X residues. An immunoblot of transfected HEK293T cell lysates expressing NLGN4X (WT, R56Q, N64S, or P93S) revealed that NLGN4X P93S had a significant decrease in expression of the mature band (Figure 2D). Taking the ratio of mature/immature bands, we observed that NLGN4X P93S had ~75% decrease compared to WT, whereas R56Q and N64S showed no significant effect (Figure 2E). The proline residue is conserved in NLGN1 and NLGN3, whereas in NLGN2, it is an alanine. To confirm that this one amino acid difference can modulate differential surface expression of NLGN4X and NLGN4Y, we used a biotinylation assay to compare NLGN4X (WT, P93S) and NLGN4Y (WT, S93P). We found that NLGN4Y S93P rescued NLGN4Y surface expression (Figure 2F). In addition, we showed that NLGN4Y S93P did not strongly colocalize with ER markers, unlike NLGN4Y WT in Cos-7 cells. The Mander’s overlapping coefficient for NLGN4Y S93P was significantly lower than that of WT (Figure S2A and B). How is NLGN4Y retained in the ER? To better understand differential functions, we performed an unbiased proteomic screen for unique protein interactions. Untransfected HEK293T cells, or cells expressing NLGN4X or NLGN4Y were immunoprecipitated using HA antibody and we performed proteomic analyses using label-free liquid chromatography with tandem mass spectrometry (LC-MS/MS) to identify specific interactomes for NLGN4X and NLGN4Y (Figure S1L). We also performed quantitative analysis to compare the NLGN4X and NLGN4Y interactomes (Figure S1M). The ER chaperone protein BiP was enriched in the NLGN4Y interactome. Interestingly, BiP has been shown to robustly bind to the NLGN3 ASD-associated variant, R451C (De Jaco et al., 2010). Similar to NLGN4Y, NLGN3 R451C displays a reduction in maturation and surface expression, and it is retained in the ER. Using coimmunoprecipitation from HEK293T cells expressing NLGN4X (WT, P93S) and NLGN4Y (WT, S93P), we found that NLGN4Y and NLGN4X P93S bind strongly with BiP, whereas NLGN4X and NLGN4Y S93P do not (Figure 2G). Taken together, we demonstrated that the NLGN4Y ER retention phenotype is likely due to the interactions with BiP, and this interaction is regulated by the same residue affecting NLGN4Y maturation and trafficking.
To investigate NLGN4X and NLGN4Y expression in neurons, we used immunofluorescence confocal microscopy to visualize surface expression of NLGN4X, NLGN4Y, and NLGN4Y S93P expressed in primary rat hippocampal neurons. We exogenously expressed NLGN4X or NLGN4Y (WT and S93P) with NLGN microRNAs (NLmiRs), which we used to knockdown endogenous NLGN1, 2, and 3 to avoid potential heterodimerization. Consistent with our biochemical assays, NLGN4Y S93P showed an increase in surface expression compared to WT (Figure 3A and B). Next, we tested the functional roles of NLGN4X and NLGN4Y at synapses. We cocultured Cos-7 cells expressing NLGN4X (WT, P93S), NLGN4Y (WT, S93P), or CD4, a membrane protein used as a negative control, with rat hippocampal neurons. We labeled endogenous VGLUT1 and observed an increase in VGLUT1 in neurons in contact with NLGN4X, but not NLGN4Y (Figure S2C and D). We observed a reduction in VGLUT1 at NLGN4X P93S expressing Cos-7 cells compared to WT, and an increase in VGLUT1 at NLGN4Y S93P expressing cells compared to WT (Figure S2C and D). Next, we recorded miniature excitatory postsynaptic currents (mEPSCs) in hippocampal cultured neurons expressing NLmiRs alone or coexpressing NLmiRs with NLGN4X and NLGN4Y (WT and S93P) (Figure 3C). As expected, knockdown of NLGNs reduced mEPSC frequency and amplitude. NLGN4X, but not NLGN4Y, rescued the reduced mEPSC frequency and amplitude (Figure 3D–G). Consistent with our imaging and biochemical data, NLGN4Y S93P was also able to rescue mEPSC frequency and amplitude (Figure 3D–G). Our results indicate that WT NLGN4Y cannot traffic to the surface as efficiently as NLGN4X, and this trafficking deficit reduced the ability of NLGN4Y to increase synaptic transmission. Taken together, differences in function between NLGN4X and NLGN4Y are predominantly due to one amino acid substitution.
Figure 3. NLGN4Y S93 regulates differential surface expression and functional effects of NLGN4X vs. NLGN4Y.
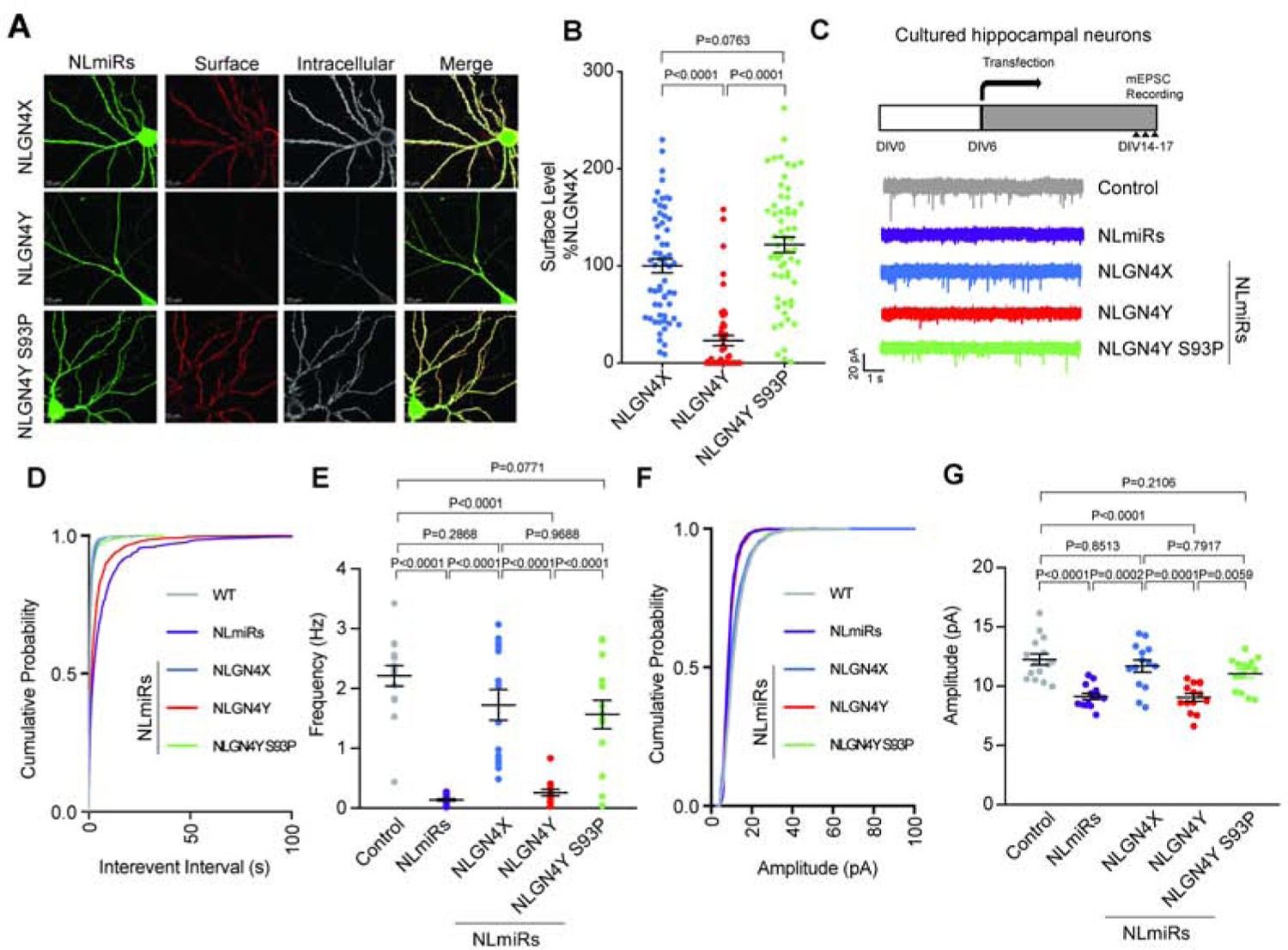
A. Representative images of expression of NLmiRs and NLGN4X, NLGN4Y (WT, S93P) in rat hippocampal neurons at DIV 13. Surface and intracellular NLGNs were labeled with anti-HA. B. Surface intensity level (means ± S.E.M.) normalized to NLGN4X. P values were calculated by one-way ANOVA Bonferroni’s multiple comparison test. (n=3). C. Experimental design for recording, and representative mEPSC traces recorded in cultured hippocampal neurons expressing NLmiRs or NLmiRs with NLGN4X, NLGN4Y (WT, S93P). D and E. mEPSC frequency cumulative probability and mean. P values were calculated by one-way ANOVA Bonferroni’s multiple comparison test. (n=3). F and G. mEPSC amplitude cumulative probability and mean. P values were calculated by one-way ANOVA with Bonferroni’s multiple comparison test. (n=3).
NLGN4Y heterodimerization with other NLGNs does not increase its maturation
Neuroligins exist as dimers, and neuroligin heterodimerization has been shown to be important for function (Poulopoulos et al., 2012; Shipman and Nicoll, 2012). To test if NLGN4Y can form heterodimers, we coexpressed HA-tagged NLGN1, 2, 3, or 4X with Myc-tagged NLGN4Y, and immunoprecipitated NLGN4Y with Myc antibody. We found that NLGN4Y could form heterodimers with all NLGNs (Figure S3A); however, heterodimer formation did not promote expression of the mature band of NLGN4Y, but coexpression with NLGN4Y dampened NLGN1, 2, 3, and 4X protein maturation (Figure S3B and C). Next, we tested NLGN4Y’s effect on synaptic strength by exogenously expressing GFP or GFP with NLGN4Y to preserve NLGN1–3 at synapses (Figure S3D). Expressing NLGN4Y in rat hippocampal neurons reduced both frequency and amplitude of mEPSCs (Figure 3SE–H). Together, our data show that NLGN4Y is distinct from NLGN4X in that it does not traffic to the surface and it can also act as a negative regulator of other NLGNs.
ASD-associated mutations in NLGN4X phenocopy NLGN4Y
The lack of NLGN4Y surface expression strikingly resembles a previously described ASD-associated mutation in NLGN4X R87W (Zhang et al., 2009). Due to the close proximity to the critical amino acid at NLGN4X P93, we wondered might there be other rare variants surrounding this residue. Using published research and databases for ASD patients, we found three additional ASD-associated mutations surrounding the critical amino acid (Figure 4A) (Yan et al., 2005; Zhang et al., 2009; Xu et al., 2014). Interestingly, two of the cases (NLGN4X G84R and NLGN4X R87W) were found in males; one case (NLGN4X G99S) was found in a female, but the proband also has an affected brother (Table S1). In addition, we now report two new independent cases of missense mutations in NLGN4X in ASD and ID patients (Sobreira et al., 2015). The probands are male and have a substitution at R101Q (c.302G>A) or V109L (c.325G>C). Interestingly, in both cases, the mothers are carriers with no known symptoms. Fisher exact test indicates that the region between amino acid 75 to 125 is enriched with ASD-associated variants compared to variants in the healthy population. Because these variants are in close proximity to NLGN4X P93, we hypothesized that these mutations would phenocopy NLGN4Y. Expressing these variants in HEK293T cells, we found that they all had a decrease in mature expression of NLGN4X (Fig. 4B and C). Interestingly, a mutation found in the healthy population within this region, NLGN4X E85D (Lek et al., 2016), displayed no changes in the mature band (Figure S4A) although the mature/immature ratio decreased modestly due to an increase in immature band expression (Figure 4B and 4C). Because multiple ASD-associated mutations displayed a similar protein maturation deficit, we chose to focus on two variants: NLGN4X P94L, the mutation adjacent to the critical residue in NLGN4X P93; and NLGN4X G99S, a well-documented case in the literature (Yan et al., 2005; Araç et al., 2007; Fabrichny et al., 2007). We conducted a biotinylation assay with HEK293T cells expressing NLGN4X (WT, E85D, P94L, or G99S) and showed that NLGN4X E85D did not have a decrease in surface expression compared to WT, whereas the ASD-associated variants NLGN4X P94L and NLGN4X G99S displayed significantly decreased surface expression (Figure S4B and S4C). In addition, we performed functional analyses by recording mEPSCs in hippocampal neurons expressing NLmiRs with NLGN4X (WT, E85D, P94L, or G99S). In agreement with our biochemical results, NLGN4X WT and NLGN4X E85D rescued the reduced mEPSCs frequency and amplitude, but NLGN4X P94L and NLGN4X G99S could not (Figure 4D–F, and S4D and S4E). Taken together, we found that the region surrounding NLGN4X P93 is highly intolerant, and most mutations within this region phenocopy NLGN4Y.
Figure 4. Cluster of ASD-associated mutations on NLGN4X surround the critical amino acid difference between NLGN4X and NLGN4Y.
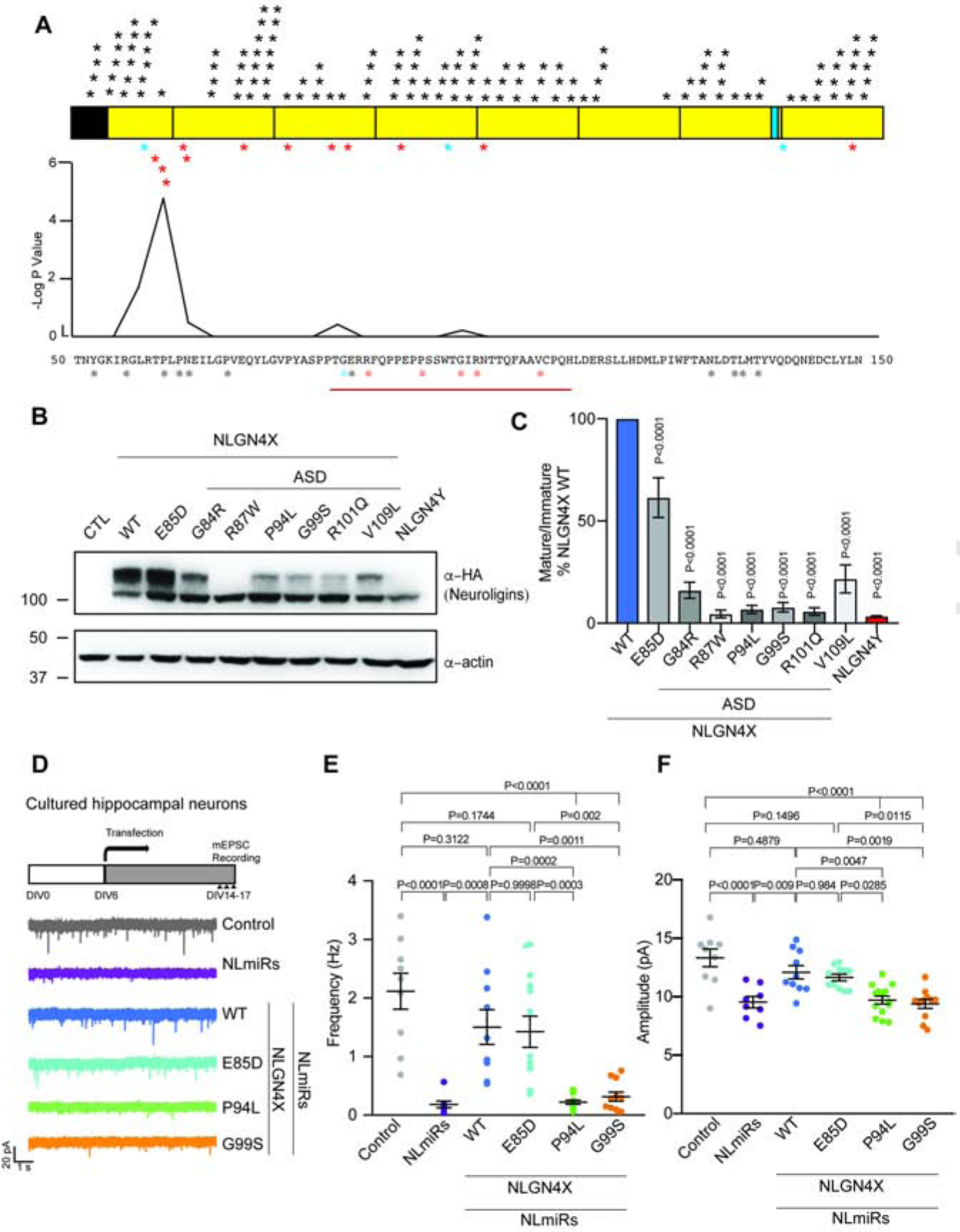
A. Schematic for NLGN4X showing mutations on NLGN4X. Black asterisks indicate variants in the normal population (gnomAD), blue asterisks indicate mutations in both normal population and ASD probands, and red asterisks indicate mutations in ASD and ID. Plot for -log p value from Fisher Exact Test to indicate the area surrounding NLGN4X P93 is significantly enriched in ASD-associated probands. B. Immunoblot of transfected NLGN4X WT, healthy population variant (E85D), and ASD-associated mutations (G84R, R87W, P94L, G99S, R101Q, and V109L). C. Ratio of mature/immature (means ± S.E.M.) normalized to NLGN4X WT. P values were calculated by one-way ANOVA with Bonferroni’s comparison test. (n=4). D. Experimental design for recording, and representative mEPSC traces recorded in cultured hippocampal neurons expressing NLmiRs or NLmiRs with NLGN4X (WT, E85D, P94L, G99S). E and F. Graph for the mean of mEPSC frequency and amplitude of cultured hippocampal neurons expressing NLmiRs or NLmiRs with NLGN4X (WT, E85D, P94L, or G99S). P values were calculated by one-way ANOVA Bonferroni’s multiple comparison test. (n=3)
Wild type NLGN4X, but not NLGN4Y, can increase ASD-associated mutations maturation
Most ASD/ID probands with mutations in NLGN4X are males, often with unaffected mothers who are carriers. We have shown here that NLGN4Y cannot efficiently traffic to the surface to induce synapses. We wondered if this lack of function can amplify the effect of ASD-associated mutations in NLGN4X in male probands, and if a WT NLGN4X can provide a protective effect in females carrying ASD-associated mutations. Coexpressing NLGN4X harboring ASD-associated mutations with WT NLGN4X or NLGN4Y showed that NLGN4X increased protein maturation for NLGN4X harboring ASD-associated mutations, whereas NLGN4Y did not (Figure S4F–H). Overall, our data demonstrate that the male-specific neuroligin NLGN4Y cannot compensate for variants that disrupt NLGN4X maturation and trafficking.
Discussion
The sex chromosomes have long been linked with neurodevelopmental disorders. While the X chromosome has been the focus of many studies, the Y chromosome receives far less attention. Recent investigation has revealed that the Y chromosome contains many important genes other than sex-determining genes (Bellott et al., 2014; Cortez et al., 2014). In particular, Y-linked genes that have an X-linked homolog are important because they are needed to maintain an ancestral gene dosage between sexes (Bellott et al., 2014). Thus, these studies reveal the importance of X-Y gene pairs. In our current study, we report a unique case for this X-Y gene pair hypothesis. Due to their sequence homology, NLGN4X and NLGN4Y are presumed to have the same function, thus forming an X-Y pair that can act as an autosomal gene. We showed here that NLGN4X and NLGN4Y are differentially processed. An amino acid difference between the two proteins led to a divergence in protein localization and function. Given that the chromosomal location of the gene is outside that of the pseudoautosomal region and thus has no ability to recombine, this has allowed for fixed genetic differences. Because NLGN4Y is not able to recombine, it is possible that differences in NLGN4Y are due to selection in males.
What is the role of NLGN4Y at synapses? We showed that NLGN4Y has a deficit in trafficking that hinders its ability to induce synapses. We narrowed down the NLGN4Y deficit to a single residue at S93. By mutating NLGN4Y S93P, we can rescue NLGN4Y surface trafficking. Furthermore, NLGN4Y S93P can also induce synapses to a comparable level to that of NLGN4X. It is important to note here that although NLGN4X P93S displayed a reduction in surface expression and a reduction in synapse formation in a coculture assay, NLGN4X P93S was still capable of modest levels of surface expression and clustering of VGLUT. Conversely, NLGN4Y S93P did not completely rescue NLGN4Y surface. It is possible that there is another amino acid difference, or region, between NLGN4X and NLGN4Y that can contribute to these observations. There might also be additional unidentified posttranslational modifications in NLGN4X or NLGN4Y that modulate maturation. NLGN4X can enhance synaptic strength when phosphorylated by PKC (Bemben et al. 2015). It is unclear if this same effect can be observed in NLGN4Y. Taken together, our data reveal a single residue that leads to a dramatic functional divergence between NLGN4X and NLGN4Y.
The structure of the extracellular domain of neuroligins has been solved, and the residues critical for neurexin binding are highly conserved (Araç et al., 2007; Fabrichny et al., 2007). Although NLGN4Y was not included in these studies, sequence comparison between NLGN4X and NLGN4Y reveals that the neurexin binding residues are completely conserved. With this information, our data suggest that the inability of NLGN4Y to induce synapse formation is not due to its inability to bind to its presynaptic partner, neurexin, but instead due to its inability to traffic to the surface. It is important to note that much of our study makes use of heterologous cells or rodent neurons. Importantly, a recent study showed that phenotypes from overexpressed NLGN4X observed in mouse and rat neurons were identical when the experiments were performed in human differentiated neurons (Marro et al., 2019). We chose to rely on the ectopic expression of NLGN4X and NLGN4Y because we can use a simple tag to directly compare NLGN4X and NLGN4Y.
ASD/ID are complex disorders with many associated genes. As such, NLGN3 and NLGN4X are only implicated in a small number of ASD/ID cases. Although there are many risk genes for ASD/ID, there is good evidence that some mutations in NLGN4X are pathogenic (Jamain et al., 2003). The best evidence showing the causality of NLGN4X with ASD/ID is that a variant of NLGN4X that encodes a frameshift mutation has complete penetrance in males in one pedigree through many generations. Interestingly, female carriers in this family did not show any ASD/ID phenotype (Laumonnier et al., 2004). We showed here that this male bias in NLGN4X is likely due to the lack of NLGN4Y function and its inability to compensate for any deficits in NLGN4X. We find each of a cluster of ASD-associated NLGN4X variants have severe deficits in protein maturation and that when coexpressed with WT NLGN4X there is a significant rescue of protein maturation. However, when the NLGN4X ASD-associated variants were coexpressed with WT NLGN4Y there was no rescue of the NLGN4X maturation.
There are several known ASD-associated NLGN4X variants. Notably, a study directly showed that NLGN4X R87W is retained in the ER (Zhang et al., 2009). We replicated the experiments showing a deficit in protein maturation with NLGN4X R87W, and then expanded to include other mutations found in the literature and in databases. Importantly, many NLGN4X-associated mutations cluster around the critical residue in NLGN4X at P93. One variant that was listed in ClinVar, was particularly interesting because it targeted a nearby proline. In this study, we also report two new probands with a variant in this region. Based on these data, we performed statistical analyses, which revealed this region is a sensitive region that is highly intolerant to variation. However, in a database of healthy individuals (gnomAD), there was a NLGN4X variant, NLGN4X E85D, in this region. Importantly, we did not observe a deficit in protein maturation, trafficking, nor deficits in mEPSCs with NLGN4X E85D. Thus, not every variant in this region results in a synaptic deficit or neurological disorders. The NLGN4X E85D mutation is quite conserved because the structures of glutamic acid and aspartic acid are similar, which likely allows for this mutation to be tolerated. Together, the identified sensitive region can be used in the future to screen for potential pathogenic mutations in NLGN4X.
While both NLGN3 and NLGN4X are X-linked and have been linked to ASD/ID, NLGN3 has been the focus of more studies to understand the etiology of ASD. NLGN3 research benefits from the high conservation of NLGN3 between mammals, facilitating the use of mouse models to study NLGN3 mutations in vivo (Tabuchi et al., 2007; Rothwell et al., 2014). However, NLGN4 is unusually divergent in mammals, with mouse and human NLGN4 only sharing ~60% sequence homology (Bolliger et al., 2008; Maxeiner et al., 2020). NLGN4-like knockout mice exhibit similar phenotypes to ASD, including social deficits (Jamain et al., 2008). However, mouse NLGN4-like is localized at glycinergic synapses and influences inhibitory synapses, whereas human NLGN4X is shown to localize exclusively at excitatory synapses (Hoon et al., 2011; Bemben, Nguyen, et al., 2015; Hammer et al., 2015; Chanda et al., 2016; Zhang et al., 2018; Marro et al., 2019). We now show that on a NLGN1–3 null background, NLGN4X expression in cultured rat hippocampal neurons increases excitatory synaptic transmission, whereas NLGN4Y does not. Although the stem cell technology to model human neurodevelopmental diseases is still in its infancy, it is exciting to speculate about the possibility of generating human derived neurons or brain organoid models to precisely depict human diseases. With the growing numbers of patients obtaining sequencing coupled with our identification of the sensitive region of NLGN4X, we expect that the numbers of reported affected individuals will increase and facilitate future human studies on this cohort of ASD/ID probands.
STAR Methods
Lead Contact and Materials Availability
Information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Katherine W. Roche (rochek@ninds.nih.gov).
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Dissociated neuronal culture
The use of animals in this study followed the guidelines of the NIH Animal Care and Use Committee (Protocol number: 1171). Primary cultured neurons were isolated from both male and female embryonic day 18 Sprague-Dawley rats. Hippocampal neurons were used for imaging and electrophysiology experiments. For imaging experiments, hippocampal neurons were plated on 12 well dishes at 150,000 cells/well; for electrophysiology experiments, hippocampal neurons were plated on 24 well dishes at 200,000 cells/well. For both set of experiments, the neurons were plated on glass coverslips coated with poly-D-lysine (Sigma, P7280) in Neurobasal media supplemented with B27 and L-glutamine. For transfection, 1 μg of NLmiRs and 1 μg of NLGNs were mixed with 2 μL of Lippofectamine 2000 (Invitrogen, 11668–019) in 100 μL of pre-warmed plain Neurobasal media. The mixture was incubated at RT for 15 minutes before added to individual wells. Hippocampal neurons were transfected at DIV13 and labeled at DIV17.
Human brain synaptosomes
Human brain samples were obtained from surgically resected regions of cortex in patients with epilepsy. For detection of NLGN4X and NLGN4Y in brain, 50–100 mg of human brain were homogenized in 20 mM HEPES (Sigma, H3375), 0.32 M sucrose (Sigma, S7903), 5 mM EDTA (Sigma, 681-92-6) with a protease inhibitor cocktail (Roche, 11 873 580 001) at pH 7.4. Homogenates were centrifuged at 1,000 x g for 10 minutes at 4°C, and the supernatant (S1) was saved. The synaptosome preps were prepared by centrifuge S1 at 10,000 x g for 20 minutes at 4°C, and the pellets (P2) were saved. The P2 pellets were solubilized with 1% SDS at 37°C for 30 minutes. Protein concentration was measured by BCA protein assay (Pierce, 23228). 20–100 μg of total homogenate and synaptosome preps were used for immunoblotting analysis.
Neuronal differentiation from iPSCs
Neural stem cells (NSCs) were differentiated from induced pluripotent stem cells (iPSCs) following our previous protocol (Wang et al., 2013). Briefly, iPSCs were seeded on a Matrigel-coated 6 well plate in E8 medium (Thermo Scientific, A1517001). After 24 hours, the E8 medium was replaced with neural induction media (Thermo Scientific, A1647801) for 7 days. The resulting NSCs were dissociated with accutase (Thermo Scientific, A1110501) treatment and replated for characterization by nestin immunostaining. The NSCs were further differentiated to neurons when incubated in neuronal differentiation medium (DMEM/F12 (Thermo Scientific, 11330057) containing 1xN2 supplement (Thermo Scientific, 17502048), 1xB27 supplement (Thermo Scientific, A3582801), 300 ng/ml cyclic adenosine monophosphate (Sigma, A6885) and 0.2 mM vitamin C (Sigma), 10 ng/ml brain derived neurotrophic factor (PeproTech, 450–02) and 10 ng/ml glial-derived neurotrophic factor (PeproTech, 450–10) for 14 days. The resulting neurons were confirmed by immunostaining for β-III-tubulin.
Method Details
Constructs and Antibody Generation
NLGN4Y was purchased from Origene (Origene, SC31009). NLGN4Y was then subcloned into the pCAG vector. The HA and Myc tag were inserted after the signal peptide at the exact same position as pCAG-NLGN4X. A standard two-step PCR cloning procedure was used to create chimeras and variants in NLGN4X and NLGN4Y.
For NLGN4Y antibody, rabbits were immunized with the synthetic peptide Ac-NTLMGMQPLHTFKTFS-amide which corresponds to amino acids 784–799. Sera were collected and purified. NLGN4Y antibody was generated by New England Peptide. Primary antibodies were used at 1 μg/mL unless otherwise stated.
Cell culture and transfection
HEK293T or Cos7 cells were grown and maintained in DMEM (Gibco, 11966025) supplemented with 10% fetal bovine serum (FBS) (Hyclone, SH30071.03) and 1% glutamine (Gibco, 25030081) in an incubator at 37°C and 5% CO2. For biochemistry, we used a standard transfection protocol. In brief, HEK293T cells were split into either 6-well plates with ~350,000 cells per well or in 6 cm dish with 1×106 cells. After letting the cells settle for 24 hours, 1 μg of DNA (6-well plate) or 2 μg of DNA (6 cm dish) were mixed with polyethylenimine (PEI) (Sigma, 408727) at a ratio of 1:3 in 100 μL of Opti-MEM (Gibco, 31985062). The mixture was incubated at room temperature (RT) for 15 minutes. The mixture was added to the 6-well plate or 6 cm dish. For immunofluorescence microscopy, glass cover slips in 12-well plates were plated with 50,000 cells per well. 24 hours after plating, 0.5 μg of DNA was mixed with PEI at a ratio of 1:3 in 50 μL of pre-warmed Opti-DMEM. The mixture was incubated at RT for 15 minutes, then added to an individual well of the 12-well plate.
Immunoblotting
Two days post transfection, heterologous cells were lysed using 150 μL of 4X SDS-PAGE sample buffer, sonicated, and centrifuged at 20,000 x g for 15 minutes to pellet cellular debris. Protein lysates (5 μL) were then denatured and were resolved by SDS-PAGE (7%). After separation, proteins were transferred to PVDF membranes (Bio-Rad, 1620177), blocked for 30 minutes with 5% milk in TBST, incubated with primary antibodies overnight at 4°C. The following day, membranes were washed three times with TBST, then incubated with HRP-conjugated secondary antibodies for 1 hour at RT. After secondary incubation, membranes were washed three times with TBST. Proteins were detected after one minute incubation with enhanced chemiluminescence (ECL) (Thermo Scientific, 34087) with an imager (KwanKwik). The digital images were then quantified using Adobe Photoshop and ImageJ.
Co-Immunoprecipitation
HA-NLGN1, 2, 3, 4X, 4Y were cotransfected with Myc-NLGN4Y into HEK293T cells plated on a 6 cm dish. Two days post transfection, cells were washed and collected in cold PBS. After centrifugation, the pelleted cells were lysed in TBS buffer with 1% Triton-X-100, protease inhibitors (Roche, 05056489001), phosphatase inhibitor cocktail 2 (Sigma, P5726), phosphatase inhibitor cocktail 3 (Sigma, P0044) for 1 hour at 4°C. Cells were centrifuged at 20,000 x g for 10 minutes to remove any insoluble debris. A fraction of each lysates was collected as input. 1 μg of Myc antibody was added to lysate and incubated overnight on a rocker at 4°C. The following day, lysates were added to protein G conjugated beads (GE Healthcare, 17-0618-01) and incubated at 4°C for 3 hours. The beads were then washed three times using cold IP buffer (TBS buffer with 0.01% Triton-X-100, 5 mM EDTA) with 1 minute of 2,000 x g centrifugation between each wash. The precipitated proteins were then eluted with 4X SDS loading buffer and boiled at 95°C for 5 minutes.
For co-immunoprecipitation with BiP, HA-NLGN4X (WT, P93S) and NLGN4Y (WT, S93P) were transfected into HEK293T cells plated on 6 cm dish. The above protocol was followed except 1 μg of HA antibody were added to lysate. Protein A conjugated beads (Sigma, P3391) were used to isolate immunoprecipitated proteins instead.
Biotinylation assay
NLGNs were transfected into HEK293T cells plated on 6 cm dishes. Two days post transfection, cells were washed with cold PBS buffer with 2 mM CaCl2 and 1 mM MgCl2 (this buffer was used for the rest of this experiment). Cells were then incubated with 1 μg/mL biotin (EZLink Sulfo-NHS-LC-Biotin) (Thermo Scientific, 21335) in PBS buffer for 30 minutes on ice, then incubated with 100 mM glycine in PBS buffer for 20 minutes on ice. Cells were washed with cold PBS buffer three times between incubation. After the last wash, cells were collected in cold PBS and centrifuge at 10,000 x g for 10 minutes. The pelleted cells were lysed using RIPA buffer with 150 mM NaCl, 50 mM Tris-HCL, pH7.4, 1 mM EDTA, 0.1% SDS, 0.1% DOC, and 1% Triton-X-100, protease inhibitors, and phosphatase inhibitor cocktails for 1 hour at 4°C. After incubation, lysates were briefly sonicated, and centrifuged at 20,000 x g for 15 minutes to remove insoluble debris. A portion of the lysates were collected for input, and the remaining lysates were incubated with Streptavidin agarose beads overnight at 4°C. The following morning, Streptavidin beads were washed three times with cold PBS with 1 minute of 2,000 x g centrifugation between each wash. Proteins were eluted by adding 4X SDS loading buffer and analyzed by immunoblotting.
Immunofluorescence microscopy
For hippocampal neurons, live cells were incubated with HA antibody (rat) for 15 minutes at RT to label surface proteins. Cells were then washed with PBS and fixed with 4% paraformaldehyde and 4% sucrose in PBS for 10 minutes, washed, blocked in 10% normal goat serum (NGS) (Vector, S-1000), washed, and incubated with Alexa 555-conjugated anti-rat secondary. After surface staining, cells were then permeabilized with 0.25% Triton-X-100 for 15 minutes, washed, blocked in 10% NGS, incubated with HA antibody and anti-GFP in 3% NGS, washed, then incubated with Alexa 647-conjugated anti-rabbit and Alexa 488-conjugated anti-chicken in 3% NGS. All blocking, primary, and secondary antibody incubations were performed using BioWave Pro system. After labeling, coverslips were mounted in Slowfade gold antifade reagent.
For heterologous cells, coverslips were washed with PBS and fixed with 4% paraformaldehyde and 4% sucrose in PBS for 10 minutes, washed, permeabilized with 0.25% Triton-X-100 for 15 minutes, and washed. After permeabilization, cells were blocked with 10% NGS, washed, and incubated with HA antibody (rabbit) in 3% NGS for 1 hour at RT. Cells were washed, and incubated with Alexa 488-conjugated (green) anti-rabbit secondary antibody and DAPI in 3% NGS for 1 hour at RT. Cells were washed, and coverslips were mounted in Slowfade gold antifade reagent (Invitrogen, P36934). Cells were imaged with a 63X objective on a Zeiss LSM800 confocal microscope.
Coculture assay
Cos-7 cells were transfected with HA-tagged NLGN4X (WT, P93S), NLGN4Y (WT, S93P), or CD4 using PEI as described above. Two days after transfection, Cos-7 cells were detached using trypsin. 10,000 cells were added to DIV 6 hippocampal neurons. 24 hours after adding Cos-7 cells to neurons, immunofluorescence labeling was performed as described above.
Gene expression
HEK293T cells were transfected with NLGN4X or NLGN4Y using PEI as described above. Two days after transfection, RNA was extracted from HEK293T using RNeasy per manufacturer instruction (Qiagen, 74106). 1 μg of RNA was used to generate cDNA using high-capacity cDNA reverse transcription kits. PCR amplification of HA-NLGN4X and NLGN4Y was performed using a primer set that can recognize both. The forward primer is 5’-CGACGTTCCGGACTACGCTCCAGTTG-3’, and the reverse primer is 5’-GTAAAGGCAGTCTTCATTTTGATCTTGAAC-3’. GAPDH was used as loading control. The forward primer is 5’-CCAGCCGAGCCACATCGCTC-3’, and the reverse primer is 5’ ATGAGCCCCAGCCTTCTCCAT-3’. PCR amplification was done using KOD (toyobo, F1696K) and standard PCR protocol.
Label-free Mass Spectrometry
Samples were prepared by immunoprecipitated HEK293T cells expressing HA-NLGN4X, HA-NLGN4Y, and using untransfected HEK293T as control (n=3). Samples were subjected to trypsin digestion after TECP reduction and NEM alkylation. Digests were extracted from the gel and desalted using Waters Oasis HLB μElution plate. LC-MS/MS data acquisition was performed in data-dependent acquisition (DDA) mode on an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) coupled with a 3000 Ultimate high pressure liquid chromatography instrument (Thermo Fisher Scientific). Peptides were separated over a 62-min gradient (5% – 35% MPB) on a ES802 column (Thermo Fisher Scientific) at a flow rate of 300 nl/min. The MS resolution is 120K at m/z 400, MS scan range is 300–1500 m/z, the automated gain control (AGC) target is 2 × 10e5. The quadrupole isolation window is 1.4 m/z. Precursors with charge states 2–6 and intensity higher than 1×10e4 within a 3 sec cycle between MS1 scans are selected for MS/MS acquisition in the linear ion trap, and activated with higher energy collisional dissociation (HCD) method.
Electrophysiology
Whole-cell voltage clamp recordings of mEPSCs were performed in DIV 14 to 17 cultured rat hippocampal neurons at 20–25°C using glass patch electrodes filled with an internal solution consisting of 135 mM CsMeSO4, 8 mM NaCl, 10 mM HEPES, 0.3 mM Na-GTP, 4 mM Mg-ATP, 0.3 mM EGTA, 5 mM QX-314, and 0.1 mM spermine. The external solutions contained 119 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM Na2PO4, 11 mM glucose, 2.5 mM CaCl2, and 1.3 mM MgCl2; 0.1 mM Picrotoxin and 0.5 μM TTX were added to the external solutions before recording. Transfected cells were visualized with fluorescence, and mEPSCs were measured at −70 mV. Series resistance was monitored and not compensated, and cells in which series resistance was more than 25 MΩ or varied by 25% during a recording session were discarded. Synaptic responses were collected with a Multiclamp 700B amplifier (Axon Instruments), filtered at 2 kHz, and digitized at 10 kHz. The analysis of the mEPSCs was done offline semiautomatically, using in-house software in Igor Pro (Wavemetrics) developed in Roger Nicoll’s laboratory at University of California, San Francisco (UCSF). Whole-cell voltage clamp recordings of mEPSCs were performed on cells expressing GFP.
Novel Mutation Identification
Clinical whole exome analysis was completed for a diagnosis of ASD or ID. The variants were reported as variants of uncertain significance by the clinical lab. Clinicians provided variant information through GENEMATCHER(Sobreira et al., 2015). Participants provided signed authorization to have genetic information included in the study.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and analysis of immunoblot and electrophysiology.
Immunoblots were analyzed by ImageJ by calculating the area under the curve. Intensity was normalized to actin and normalized to NLGN4X WT. Statistical analysis was done using student t-test or one-way ANOVA with Bonferroni’s multiple correction test when appropriate. For electrophysiology, one-way ANOVA with Bonferroni’s multiple correction test or student t-test were used to calculate statistical significance.
Quantification and analysis of immunocytochemistry
For surface expression in neurons, three regions from dendrites were randomly selected per neurons. The data were blindly obtained. Metamorph was used to measure surface and intracellular intensity level. Analysis was done using one-way ANOVA with Bonferroni’s multiple correction test. For coculture experiment, Metamorph was used to measure VGLUT and NLGN intensity level. Analysis was done using one-way ANOVA with Bonferroni’s multiple correction test. For colocalization experiment, Mander’s coefficients were calculated using JACoP plugin in Fiji. Analysis was done using one-way ANOVA with Bonferroni’s multiple correction
Quantification and analysis of label free mass spectrometry
The database search and label-free quantitation were performed with Proteome Discoverer 2.4. Parameters for analysis as followed: trypsin digestion with full specificity; 2 missed cleavages allowed; N-ethylmaleimide on cysteines as fixed modification; oxidation (M) as variable modification, the mass tolerance is 10 ppm for precursor ions and 0.6 Da for fragment ions. Using Percolator, the false discovery rate for peptide-spectrum matches (PSMs) is set to 0.05. Only unique peptides are considered for the quantification. The results are filtered by a FDR set at 1% at protein level. For protein ratio calculation, the summed intensity of the unique peptides matched to the protein is used. The maximum fold change allowed is set to 100. Normalization is applied based on total peptide abundance of each sample. The individual proteins ANOVA method is used for hypothesis test.
Statistical Analysis
Data analysis was carried out using ImageJ and GraphPad Prism. All experiments were performed at least three independent times. For human genetic analysis, we performed a sliding window analysis for frequency of variants in reported individuals with ASD compared to the gnomAD database. We compared 50 amino acid bins that overlapped by 25 amino acids. P values were calculated by Fisher’s exact Test.
Supplementary Material
Table S2. Related to Figure 2. Label-Free LC MS/MS.
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA (rabbit) | Abcam | Cat# ab9110 RRID:AB_307019Rabbit |
| HA (rat) | Roche | Cat# 11867423001, RRID:AB_390918 |
| NLGN4 | Abcam | Cat#: ab181251 |
| Myc | Cell Signaling Technology | Cat# 2276, RRID:AB_331783 |
| Actin | Abm | Cat# G043 |
| GFP | Thermo Fisher | Cat# A10262, RRID:AB_2534023 |
| pan-NLGN | Synaptic System | Cat# 129 011, RRID:AB_887745 |
| BiP | BD Biosciences | Cat# 610978, RRID:AB_398291 |
| Tau | Synaptic System | Cat# 314 011, RRID:AB_10805762 |
| Transferrin Receptor | Thermo Fisher | Cat# 13–6800, RRID:AB_2533029 |
| VGLUT1 | Millipore | Cat# AB5905, RRID:AB_2301751 |
| Mouse IgG HRP-linked | GE Healthcare | Cat#: NA931 RRID:AB_772210 |
| Rabbit IgG HRP-linked | GE Healthcare | Cat#: NA934 RRID:AB_772206 |
| Anti-chicken Alexa Fluor 488 | Thermo Fisher | Cat# A-11039, RRID:AB_2534096 |
| Anti-rabbit Alexa Fluor 488 | Thermo Fisher | Cat#:A11034 RRID:AB_2576217 |
| Anti-mouse Alexa Fluor 405 | Thermo Fisher | Cat# A-31553, RRID:AB_221604 |
| Anti-rabbit Alexa Fluor 555 | Thermo Fisher | Cat#: A21428 RRID:AB_141784 |
| Anti-rat Alexa Fluor 647 | Thermo Fisher | Cat# A-21247, RRID:AB_141778 |
| Anti-guinea pig Alexa Fluor 647 | Thermo Fisher | Cat# A-21450, RRID:AB_2735091 |
| NLGN4Y | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 | Thermo Fisher | Cat# 11668019 |
| Protease inhibitor mixture | Roche | Cat#: 11697498001 |
| Phosphatase inhibitor mixture II | Sigma-Aldrich | Cat#: P5726 |
| Phosphatase inhibitor mixture III | Sigma-Aldrich | Cat#: P0044 |
| Protein A-Sepharose | Sigma-Aldrich | Cat#: P3391 |
| Cell Lines | ||
| Human embryonic kydney (HEK) 293T | ATCC | Cat#: CRL11268 RRID:CVCL_1926 |
| COS 7 | ATCC | Cat# CRL-1651, RRID:CVCL_0224 |
| Organisms/Strains | ||
| Sprague Dawley outbred rat | Envigo | Cat#: 002 |
| Oligonucleotides | ||
| pCAG-NLGN4X-Myc FW primer GAACAAAAACTCATCTCAGAAGAGGATCTGCCAGTTGTCAACACAAATT | This study | N/A |
| pCAG-NLGN4X-Myc RW primer CAGATCCTCTTCTGAGATGAGTTTTTGTTCATACTGTGCTTGGCTGTCAATGA | This study | N/A |
| pCAG-NLGN4X-HA R56Q FW primer CAAATTATGGCAAAATCCAGGGCCTAAGAAC | This study | N/A |
| pCAG-NLGN4X-HA R56Q RW primer GGTGTTCTTAGGCCCTGGATTTTGCC | This study | N/A |
| pCAG-NLGN4X-HA N64S FW primer TAAGAACACCGTTACCCAGTGAGATCTTGGGTCCA | This study | N/A |
| pCAG-NLGN4X-HA N64S RW primer TGGACCCAAGATCTCACTGGGTAACGGTGTTCTTA | This study | N/A |
| pCAG-NLGN4X-HA P93S FW primer GCCCCCAGAAAGCCCGTCC | This study | N/A |
| pCAG-NLGN4X-HA P93S RW primer GAGGACGGGCTTTCTGGGGG | This study | N/A |
| pCAG-NLGN4X-HA E85D FW primer CACCCCCCACTGGAGATAGGCGGTTTC | This study | N/A |
| pCAG-NLGN4X-HA E85D RW primer GAAACCGCCTATCTCCAGTGGGGGGTG | This study | N/A |
| pCAG-NLGN4X-HA G84R FW primer CTCACCCCCCACTAGAGAGAGGCG | This study | N/A |
| pCAG-NLGN4X-HA G84R RW primer CGCCTCTCTCTAGTGGGGGGTGAG | This study | N/A |
| pCAG-NLGN4X-HA R87W FW primer CACTGGAGAGAGGTGGTTTCAGCCCC | This study | N/A |
| pCAG-NLGN4X-HA R87W RW primer GGGGCTGAAACCACCTCTCTCCAGTG | This study | N/A |
| pCAG-NLGN4X-HA P94L FW primer CCCAGAACCCCTGTCCTCCTGGAC | This study | N/A |
| pCAG-NLGN4X-HA P94L RW primer GTCCAGGAGGACAGGGGTTCTGGG | This study | N/A |
| pCAG-NLGN4X-HA G99S FW primer CCCGTCCTCCTGGACTAGCATCCGAAATAC | This study | N/A |
| pCAG-NLGN4X-HA G99S RW primer GTATTTCGGATGCTAGTCCAGGAGGACGGG | This study | N/A |
| pCAG-NLGN4X-HA/Myc R101Q FW primer CTCCTGGACTGGCATCCAAAATACTACTCAGTTTG | This study | N/A |
| pCAG-NLGN4X-HA/Myc R101Q RW primer CAAACTGAGTAGTATTTTGGATGCCAGTCCAGGAG | This study | N/A |
| pCAG-NLGN4X-HA/Myc V109L FW primer CTACTCAGTTTGCTGCTTTGTGCCCCCAG | This study | N/A |
| pCAG-NLGN4X-HA/Myc V109L RW primer CTGGGGGCACAAAGCAGCAAACTGAGTAG | This study | N/A |
| pCAG-NLGN4Y-HA FW primer TATCCATACGACGTTCCGGACTACGCTCCAGTTGTCAACACAAATTATGC | This study | N/A |
| pCAG-NLGN4Y-HA RW primer AGCGTAGTCCGGAACGTCGTATGGATAATACTGTGCTTGGCTGTCAATGAG | This study | N/A |
| pCAG-NLGN4Y-Myc FW primer GAACAAAAACTCATCTCAGAAGAGGATCTGCCAGTTGTCAACACAAATT | This study | N/A |
| pCAG-NLGN4Y-Myc RW primer CAGATCCTCTTCTGAGATGAGTTTTTGTTCATACTGTGCTTGGCTGTCAATGA | This study | N/A |
| pCAG-NLGN4Y-HA S93P FW primer GTTTCAGCCACCAGAACCCCCATCCTC | This study | N/A |
| pCAG-NLGN4Y-HA S93P RW primer GAGGATGGGGGTTCTGGTGGCTGAAAC | This study | N/A |
| pCAG-NLGN4X/Y or NLGN4Y/X-HA FW primer GCCTTTGCGGCGCTGTACTACAAAA | This study | N/A |
| pCAG-NLGN4X/Y or NLGN4Y/X-HA RW primer TTTTGTAGTACAGCGCCGCAAAGGC | This study | N/A |
| pCAG-NLGN4X-SP-Y or NLGN4Y-SP-X-HA FW primer CCTCATTGACAGCCAAGCACAG | This study | N/A |
| pCAG-NLGN4X-SP-Y or NLGN4Y-SP-X-HA RW primer CTGTGCTTGGCTGTCAATGAGG | This study | N/A |
| pCAG-NLGN4X-100-Y or NLGN4Y-100-X-HA FW primer CATGGGGGATCTTACATGGAGG | This study | N/A |
| pCAG-NLGN4X-100-Y or NLGN4Y-100-X-HA RW primer CCTCCATGTAAGATCCCCCATG | This study | N/A |
| pCAG-NLGN4X-180-Y or NLGN4Y-180-X-HA FW primer TCCTCCTGGACTGGCATCC | This study | N/A |
| pCAG-NLGN4X-180-Y or NLGN4Y-180-X-HA RW primer GGATGCCAGTCCAGGAGGA | This study | N/A |
| NLGN4X/Y RT-PCR primer FW CGACGTTCCGGACTACGCTCCAGTTG | This study | N/A |
| NLGN4X/Y RT-PCR primer RW GTAAAGGCAGTCTTCATTTTGATCTTGAAC | This study | N/A |
| GAPDH RT-PCR primer FW CCAGCCGAGCCACATCGCTC | This study | N/A |
| GAPDH RT-PCR primer RW ATGAGCCCCAGCCTTCTCCAT | This study | N/A |
| Recombinant DNA | ||
| pCAG-NLGN4X-HA | Bemben et al., 2014 | N/A |
| NLGN4Y | Origene | Cat#: SC31009 |
| pCAG-NLGN4Y-HA | Bemben et al., 2015 | N/A |
| pCAG-NLGN4Y-Myc | This study | N/A |
| pCAG-NLGN1-HA | Bemben et al., 2019 | N/A |
| pCAG-NLGN2-HA | Bemben et al., 2019 | N/A |
| pCAG-NLGN3-HA | Bemben et al., 2019 | N/A |
| pCAG-NLGN4X/4Y-HA | This study | N/A |
| pCAG-NLGN4Y/4X-HA | This study | N/A |
| pCAG-NLGN4X-180-Y-HA | This study | N/A |
| pCAG-NLGN4Y-180-X-HA | This study | N/A |
| pCAG-NLGN4X-100-Y-HA | This study | N/A |
| pCAG-NLGN4Y-100-X HA | This study | N/A |
| pCAG-NLGN4X-SP-Y-HA | This study | N/A |
| pCAG-NLGN4Y-SP-X-HA | This study | N/A |
| ER-Crimson | Addgene | Cat#: 104010 |
| pCAG-NLGN4X R56Q-HA | This study | N/A |
| pCAG-NLGN4X N64S-HA | This study | N/A |
| pCAG-NLGN4X P93S-HA | This study | N/A |
| pCAG-NLGN4X G84R-HA | This study | N/A |
| pCAG-NLGN4X R87W-HA | This study | N/A |
| pCAG-NLGN4X P94L-HA | This study | N/A |
| pCAG-NLGn4X G99S-HA | This study | N/A |
| pCAG-NLGN4X R101Q-HA | This study | N/A |
| pCAG-NLGN4X V109L-HA | This study | N/A |
| pCAG-NLGN4Y S93P-HA | This study | N/A |
| pCAG-NLGN4X-Myc | This study | N/A |
| pCAG-NLmiRs-GFP | Bemben et al., 2014 | N/A |
| pCAG-eGFP | Bemben et al., 2014 | N/A |
| CD4 | Li et al., 2017 | N/A |
| Software and Algorithms | ||
| Adobe Photoshop CC 2018 | Adobe | RRID:SCR_014199 |
| Adobe Illustrator CC 2018 | Adobe | RRID:SCR_010279 |
| Fiji | NIH | RRID:SCR_002285 |
| GraphPad Prism 8 | Graphpad | RRID:SCR_002798 |
| Proteome Discover 2.2 | Thermo Fisher | Cat# 30795 RRID:SCR_014477 |
| Metamorph | Molecular Devices | RRID: SCR_002368 |
Highlights:
Despite sharing ~97% amino acid identity, NLGN4X and −4Y are differentially regulated.
NLGN4Y cannot traffic to the surface due to one amino difference from NLGN4X.
A cluster of autism-associated variants in NLGN4X surround the critical amino acid.
NLGN4X autism-associated variants display a deficit in trafficking similar to NLGN4Y.
Acknowledgments
We are grateful to members of the Roche Lab for technical assistance and for discussions on the project and manuscript. We would like to thank the affected families that shared their clinical data with us. Funding: This research was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program (T.A.N., K.W., S.P., A.W.L., Y.L., M.A.B, J.D.B., T.W., W.L., K.W.R.) and the National Institute of Mental Health (A.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
References
- Araç D et al. (2007) ‘Structures of Neuroligin-1 and the Neuroligin-1/Neurexin-1β Complex Reveal Specific Protein-Protein and Protein-Ca2+ Interactions’, Neuron, 56(6), pp. 992–1003. 10.1016/j.neuron.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Bellott DW et al. (2014) ‘Mammalian y chromosomes retain widely expressed dosage-sensitive regulators’, Nature, 508(7497), pp. 494–499. 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben M. a., Nguyen Q-A, et al. (2015) ‘Autism-associated mutation inhibits protein kinase C-mediated neuroligin-4X enhancement of excitatory synapses’, Proceedings of the National Academy of Sciences, 707, p. 201500501 10.1073/pnas.1500501112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben M. a., Shipman SL, et al. (2015) ‘The cellular and molecular landscape of neuroligins’, Trends in neurosciences. Elsevier Ltd, 38(8), pp. 496–505. 10.1016/j.tins.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF et al. (2001) ‘Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression.’, The Biochemical journal, 356(Pt 2), pp. 581–588. 10.1042/0264-6021:3560581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF et al. (2008) ‘Unusually rapid evolution of Neuroligin-4 in mice.’, Proceedings of the National Academy of Sciences of the United States of America, 105(17), pp. 6421–6. 10.1073/pnas.0801383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel L and Willard HF (2005) ‘X-inactivation profile reveals extensive variability in X-linked gene expression in females’, Nature, 434(7031), pp. 400–404. 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Chanda S et al. (2016) ‘Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking’, Molecular Psychiatry, 21(2), pp. 169–177. 10.1038/mp.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D et al. (2014) ‘Origins and functional evolution of y chromosomes across mammals’, Nature, 508(7497), pp. 488–493. 10.1038/nature13151. [DOI] [PubMed] [Google Scholar]
- Fabrichny IP et al. (2007) ‘Structural Analysis of the Synaptic Protein Neuroligin and Its β-Neurexin Complex: Determinants for Folding and Cell Adhesion’, Neuron, 56(6), pp. 979–991. 10.1016/j.neuron.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M et al. (2015) ‘Perturbed Hippocampal Synaptic Inhibition and ??-Oscillations in a Neuroligin-4 Knockout Mouse Model of Autism’, Cell Reports. The Authors, 13(3), pp. 516–523. 10.1016/j.celrep.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon M et al. (2011) ‘Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina’, Proceedings of the National Academy of Sciences, 108(7), pp. 3053–3058. 10.1073/pnas.1006946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jaco A et al. (2010) ‘Neuroligin trafficking deficiencies arising from mutations in the α/β-hydrolase fold protein family’, Journal of Biological Chemistry, 285(37), pp. 28674–28682. 10.1074/jbc.M110.139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S et al. (2003) ‘Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism.’, Nature genetics, 34(1), pp. 27–29. 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S et al. (2008) ‘Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism’, Proceedings of the National Academy of Sciences, 105(5), pp. 1710–1715. 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Paskus JD and Roche KW (2017) ‘Posttranslational modifications of neuroligins regulate neuronal and glial signaling’, Current Opinion in Neurobiology. Elsevier Ltd, 45, pp. 130–138. 10.1016/j.conb.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F et al. (2004) ‘X-Linked Mental Retardation and Autism Are Associated with a Mutation in the NLGN4 Gene, a Member of the Neuroligin Family’, The American Journal of Human Genetics, 74(3), pp. 552–557. 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M et al. (2016) ‘Analysis of protein-coding genetic variation in 60,706 humans’, Nature. Nature Publishing Group, 536(7616), pp. 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marro SG et al. (2019) ‘Neuroligin-4 Regulates Excitatory Synaptic Transmission in Human Neurons’, Neuron, pp. 1–10. 10.1016/j.neuron.2019.05.043. [DOI] [PMC free article] [PubMed]
- Maxeiner S et al. (2020) ‘Evolution of the Autism-Associated Neuroligin-4 Gene Reveals Broad Erosion of Pseudoautosomal Regions in Rodents’, Molecular Biology and Evolution. 10.1093/molbev/msaa014. [DOI] [PMC free article] [PubMed]
- Pampanos A et al. (2009) ‘ A Substitution Involving the NLGN4 Gene Associated with Autistic Behavior in the Greek Population ‘, Genetic Testing and Molecular Biomarkers, 13(5), pp. 611–615. 10.1089/gtmb.2009.0005. [DOI] [PubMed] [Google Scholar]
- Poulopoulos A et al. (2012) ‘Homodimerization and isoform-specific heterodimerization of neuroligins.’, The Biochemical journal, 446(2), pp. 321–330. 10.1042/BJ20120808. [DOI] [PubMed] [Google Scholar]
- Rothwell PE et al. (2014) ‘Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors’, Cell, 158(1), pp. 198–212. 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL and Nicoll RA (2012) ‘Dimerization of postsynaptic neuroligin drives synaptic assembly via transsynaptic clustering of neurexin.’, Proceedings of the National Academy of Sciences of the United States of America, 109(47), pp. 19432–7. 10.1073/pnas.1217633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK and Eroglu C (2013) ‘Neuroligins provide molecular links between syndromic and nonsyndromic autism.’, Science signaling, 6(283), p. re4. 10.1126/scisignal.2004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N et al. (2015) ‘GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene’, Human Mutation, 36(10), pp. 928–930. 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2008) ‘Neuroligins and neurexins link synaptic function to cognitive disease’, Nature, 455(7215), pp. 903–911. 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K et al. (2007) ‘A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice’, Science, 318(5847), pp. 71–6. 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabzuni D et al. (2013) ‘Widespread sex differences in gene expression and splicing in the adult human brain.’, Nature communications. Nature Publishing Group, 4, p. 2771 10.1038/ncomms3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen T et al. (2017) ‘Landscape of X chromosome inactivation across human tissues’, Nature. Nature Publishing Group, 550(7675), pp. 244–248. 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T et al. (2013) ‘Derivation of neural stem cells from human adult peripheral CD34+ cells for an autologous model of neuroinflammation’, PLoS ONE, 8(11), pp. 1–18. 10.1371/journal.pone.0081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X et al. (2014) ‘Variations analysis of NLGN3 and NLGN4X gene in Chinese autism patients’, Molecular Biology Reports, 41(6), pp. 4133–4140. 10.1007/s11033-014-3284-5. [DOI] [PubMed] [Google Scholar]
- Yan J et al. (2005) ‘Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients.’, Molecular Psychiatry, 10(4), pp. 329–332. 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- Yan J et al. (2008) ‘Analysis of the neuroligin 4Y gene in patients with autism.’, Psychiatric Genetics, 18(4), pp. 204–207. 10.1097/YPG.0b013e3282fb7fe6. [DOI] [PubMed] [Google Scholar]
- Yuen RKC et al. (2017) ‘Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder’, Nature Neuroscience, 20(4), pp. 602–611. 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B et al. (2018) ‘Autism-associated neuroligin-4 mutation selectively impairs glycinergic synaptic transmission in mouse brainstem synapses’, The Journal of Experimental Medicine, 215(6), pp. 1543–1553. 10.1084/jem.20172162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C et al. (2009) ‘A Neuroligin-4 Missense Mutation Associated with Autism Impairs Neuroligin-4 Folding and Endoplasmic Reticulum Export’, Journal of Neuroscience, 29(35), pp. 10843–10854. 10.1523/JNEUROSCI.1248-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Related to Figure 2. Label-Free LC MS/MS.