

Abstract
Radial glial maintenance is essential for the proper development of the cortex. It is known that the evolutionarily conserved Notch signaling pathway is required for maintaining the pool of radial glial stem cells although the mechanisms involved are not entirely understood. Here, we study the Notch ligand, Jagged1, in the mouse ventricular zone at a late stage of embryonic development. We use a conditional loss of function allele to show that Jagged1 is required for maintaining radial glial cells and when absent, leads to defects in the cortical proliferation zone and expression of intermediate progenitor cells. Using in-vitro approaches, we found that depletion of Jagged1 reduced the size of primary neurospheres and their capacity to self-renewal. Finally, Jagged1 mutants also showed precocious neuronal differentiation and cortical defects. Together, these data support a role for Jagged1 in radial glia maintenance in the neocortex.
Keywords: Notch, Jagged1, corticogenesis, neocortex, Pax6, Tbr2, Tbr1, glia
Introduction
At the onset of neocortical neurogenesis, neuroepithelial cells become radial glial cells (RGCs) (Kriegstein et al., 2006). RGCs likely act as stem cells that divide in an asymmetric manner to self-renew and generate restricted intermediate progenitor cells (IPCs) and cortical neurons (Englund et al., 2006; Kriegstein et al., 2006; Merkle and Alvarez-Buylla, 2006; Miyata et al., 2001; Tamamaki et al., 2001). Although the ability of RGC to generate neurons while self-maintaining is one of their essential functions during corticogenesis, the basic mechanisms that regulate this process are not fully understood.
Notch signaling has been implicated in neurodevelopmental, neurodegenerative, and psychiatric disorders (Dieset et al., 2012; Masek and Andersson, 2017; Oda et al., 1997; Song et al., 1999). For example, Notch signaling can alter neurogenic potential of RGCs and promote neuronal differentiation, cell division, and neuronal migration (Dang et al., 2006; Gaiano and Fishell, 2002; Justice and Jan, 2002). Loss of Notch signaling leads to the premature differentiation of neural stem cells into neurons in most regions of the CNS (Hatakeyama et al., 2004). However, the role of Notch signaling in the regulation of RGCs during development has not been completely clarified.
In mammals, there are four Notch receptors (Notch1-4) that are activated by five ligands (Delta-like1, 3, and 4, and Jagged1 and 2) (Artavanis-Tsakonas et al., 1995; Kopan and Ilagan, 2009). Ligand binding to a Notch receptor leads to a series of cleavage events that results in the release of the Notch intracellular domain (NICD). Translocation of the NICD to the nucleus leads to the formation of a transcriptional complex that drives the expression of Notch target genes. Mutation of Foxp1 was previously suggested to promote neuronal differentiation in neurosphere cultures by suppressing Jag1 transcriptionally (Braccioli et al., 2017), however, a role for Jag1 in neurogenesis in vivo has yet to be established. Moreover, Jag1 is essential for maintaining progenitor division in the postnatal and adult subventricular zone (Nyfeler et al., 2005). However, whether Jag1 plays a similar role during embryonic development, and whether or not Jag1 is involved in the radial glia maintenance is unknown.
We utilize the Foxg1Cre driver (Hebert and McConnell, 2000) and a conditional loss-of-function allele of Jag1 (Kiernan et al., 2006) to drive a decrease in the expression of Jag1. We show that decrease Jag1 is associated with reduced numbers of RGCs and proliferation in the Ventricular zone (VZ). We further show the loss of Jag1 causes precocious intermediate progenitor cells, which in turn leads to defects in cortical migration during late embryonic development. Our results demonstrate that Jag1 is required for maintaining RGCs and for the proper development of the neocortex.
Materials and methods
2.1. Mice
All animal procedures were performed in accordance with the guidelines outlined in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (eighth Edition, https://guide-for-the-care-and-use-of-laboratory-animals.pdf) and were approved by Cornell University’s Animal Care and Use Committee (IACUC). Jagged1flox (Jag1flox) (Kiernan et al., 2006b) and Foxg1Cre (Hebert and McConnell, 2000b) mice were maintained on a mixed 129Sv/C57BL/6 background.
2.2. RNA in situ hybridization
Embryonic day 17.5 (E17.5; E0.5 was defined as the first detection of the vaginal plug) embryos were dissected and decapitated. Heads were embedded in OCT (Tissue Tek) and fresh-frozen in liquid nitrogen cooled isopentane. Briefly, RNA in situ hybridization was done as previously described (Rodriguez et al., 2008). Probes for Notch1-3 and Jag1 were identical to those previously reported (Rodriguez et al., 2008; Williams et al.,2011). Other probes (Dll1, Jag2, Vimentin, Tbr1) were derived from the BMAP (Brain Molecular Anatomy Project) or NIA (National Institute of Aging) 15k or 7.4k clone sets.
2.3. Histology and immunohistochemistry
Embryos (E17.5) were fresh-frozen in Optimal Cutting Temperature OCT compound (Tissue-Tek) using liquid nitrogen cooled isopentane and sectioned (20 micron). Sections were dried for two hours and fixed in 4% phosphate buffered formaldehyde (pH 7.4). After three rinses with distilled water and treatment with 0.2% acetate buffer (pH 4.0) (2 min), slides were stained using 0.1% cresyl violet (5-10 min) before rinsing with water and mounting using 70% glycerol.
Next, antigen retrieval was performed by incubation with citrate buffer (10 mM sodium citrate, pH 6.0) and microwaved (2 min, 70% power followed by 8 min, 20% power). Slides were cooled to room temperature for 3-5 minutes, washed with 1x PBS, and blocked in 10% heat-inactivated normal goat serum in PBS. Primary antibody was applied overnight at 4 °C, washed with PBS, and hybridized with secondary antibody in 5% goat serum/PBS for two hours. After washing, samples were mounted in 70% glycerol/PBS with 10 μg/ml DAPI. Slides were imaged on a Zeiss Axioskop2 Plus fitted with appropriate filter cubes. Primary antibodies used were: Ki67 (1:100, NovoCastra, NCL-Ki67p); rabbit anti-phospho-HistoneH3 (1:200, Millipore, 06570); rabbit TBR2 (1:1:000, Millipore, 42-6600); rabbit Anti-PCNA (1:500, Abcam, ab18197); and mouse anti-Pax6 (1:250, Abcam; ab78545). Secondary antibodies used were rabbit and mouse Alexa Fluor 568 conjugated (Thermo-Fisher; A-11036), goat anti-rabbit-HRP (1:500; Santa Cruz; sc-2054). TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining was performed following the manufacturer’s protocol (Roche).
2.4. Neurosphere and Differentiation Assays
Dorsal proliferative zone tissue from E17.5 mice were dissected and mechanically dissociated. 10,000 cells were placed in 48 well plates. Cells were grown for 5-7 days in vitro (DIV) with medium containing DMEM (ThermoFisher Scientific; 11320033), B27 (ThermoFisher Scientific; 875713), and 10 ng/ml EGF (ThermoFisher Scientific; PHG0311). To generate secondary neurospheres, primary neurospheres were passaged on sixth DIV and incubated in 0.25% Trypsin/EDTA (ThermoFisher; 25200-0056) for 5-10mins. They were then incubated in an equal volume of Trypsin inhibitor (Sigma-Aldrich; T6522) and triturated with gauge needles (Becton Dickinson 305190), and plated at 10,000 cells per well (48 well plate) and DMEM/F12 (ThermoFisher Scientific; 11320-033) containing 10% FBS (ThermoFisher Scientific; 26400044) was added every three DIV. Neurospheres were induced to differentiate by plating on poly-L-lysine-coated coverslips in DMEM/F12 without B27 or EGF.
2.5. Immunofluorescence
Neurospheres were fixed with 4% paraformaldehyde, and blocked in goat serum containing 0.5% Triton. Immunofluorescent analysis of protein expression was performed using mouse anti-PSA-NCAM (Millipore; MAB5324; 1:100). Secondary antibodies used were biotinylated goat anti-mouse (Abcam; ab64255; 1:1000), and streptavidin texas 568 conjugate (ThermoFisher Scientific; S11226; 1:500).
2.6. Data acquisition and statistics
Nissl, Vimentin, Pax6, Tbr1, and Tbr2 quantitation were carried out using imageJ64 (version 1.48a; Bethesda, MD) and Adobe (version CS3; San Jose, CA) software. Images were imported; a standardized region of interest of the same size was selected for all images. Using the ruler tool function the area were calculated by measuring the length of the positive signaling and the width of the expression per picture view. Ki67, p-HH3, and PCNA quantitation were carried manually by counting the number of positive cells in the VZ. The VZ was defined as 4-5 cells in length and a minimal of 3mm in width from the first cell layer of the dorsal VZ and dividing by the number of DAPI-positive cells to generate a percentage (Ki67 and p-HH3). Neurosphere and differentiation assays quantitation were carried out using Adobe CS3. Size measurements or neuron counts were taken by measuring the diameter using the ruler tool or the count function. A minimal of 3 random wells were selected and pictured using a Cannon EOS Digital Rebel XTi digital camera. Data were analyzed using PRISM 8 (GraphPad Software; San Diego, CA). All data are based on a minimum of three independent experiments and expressed as means ± SEM and regarded statistically significant if p<0.05 by performing student’s t-test.t-test.
Results
3.1. Jag1 expression is maintained in the ventricular zone at E17.5
Notch signaling is critical for maintaining stem and progenitor cells in the VZ during corticogenesis (Dave et al., 2011). While virtually no radial glial stem cells are present at E10-E11 their numbers begin to peak at later stages of corticogenesis. Radial glia generates intermediate progenitor cells (IPC) expressing Tbr2 (Englund et al., 2005). Tbr2+ are first expressed from E10.0/12.5 and most abundant from E13.5-16.5, during the peak of neurogenesis and their expression is essential for the sequential progression into migrating cortical neuron (Englund et al., 2005; Nadarajah and Parnavelas, 2002; Turrero Garcia and Harwell, 2017). We examined the expression of Notch receptors and ligands at E17.5, after IPC have peak. We found that Notch1, Notch2, and Notch3 receptors were all expressed (Fig. 1A-C), as were Jagged1 (Jag1), Jagged2 (Jag2), Delta-like1 (Dll1) (Fig. 1D-F). In addition, Jag1 expression is maintained in the VZ (Fig 1D; arrow) and RNAseq studies have revealed higher Jag1 than Delta1 mRNA expression in neurons (Zhang et al., 2014). We thus investigated the potential roles of Jag1 during late embryonic development.
Figure 1. Notch receptor and ligand expression patterns in the ventricular zone.
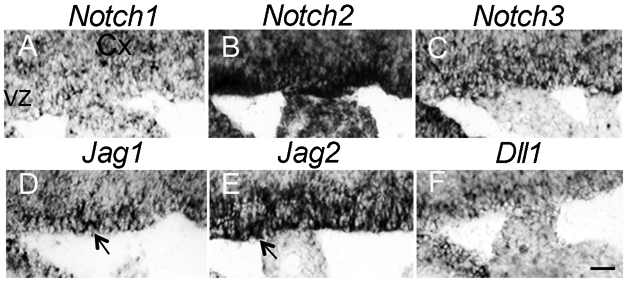
(A-F) RNA in situ hybridization of sagittal brain sections at embryonic day 17.5. Arrows in D and E indicates positive expression of Jag1 and Jag2 in the VZ. Scale bar = 30μm. Cx, cortex; VZ, ventricular zone.
3.2. Deletion of Jag1 reduces proliferative zone size
Null mutations of Jag1 are embryonic lethal at E10 (Xue et al., 1999). Therefore, we crossed a conditional Jag1 allele (Jag1f/f) (Kiernan et al., 2006) with the Foxg1Cre driver (Hebert and McConnell, 2000) to ablate Jag1 from telencephalic cells around embryonic day 8.5/10 (Fig. 2A). We used a lacZ reporter strain to confirm that Foxg1Cre was active in tissue that will give rise to the VZ (data not shown). Using this previously established conditional loss-of-function model of Jag1 (Brooker et al., 2006; Pan et al., 2010) and the chi square test (26 Jag1 mutants out of 120 total embryos; p < 0.05), we found that the Mendelian ratios of homozygous (Foxg1Cre/+; Jag1fl/fl) embryos were obtained at E17.5. However, homozygous Jag1 mutant mice die shortly after birth for undefined reasons.
Figure 2. Loss of Jag1 leads to thinner cortical proliferative zone.
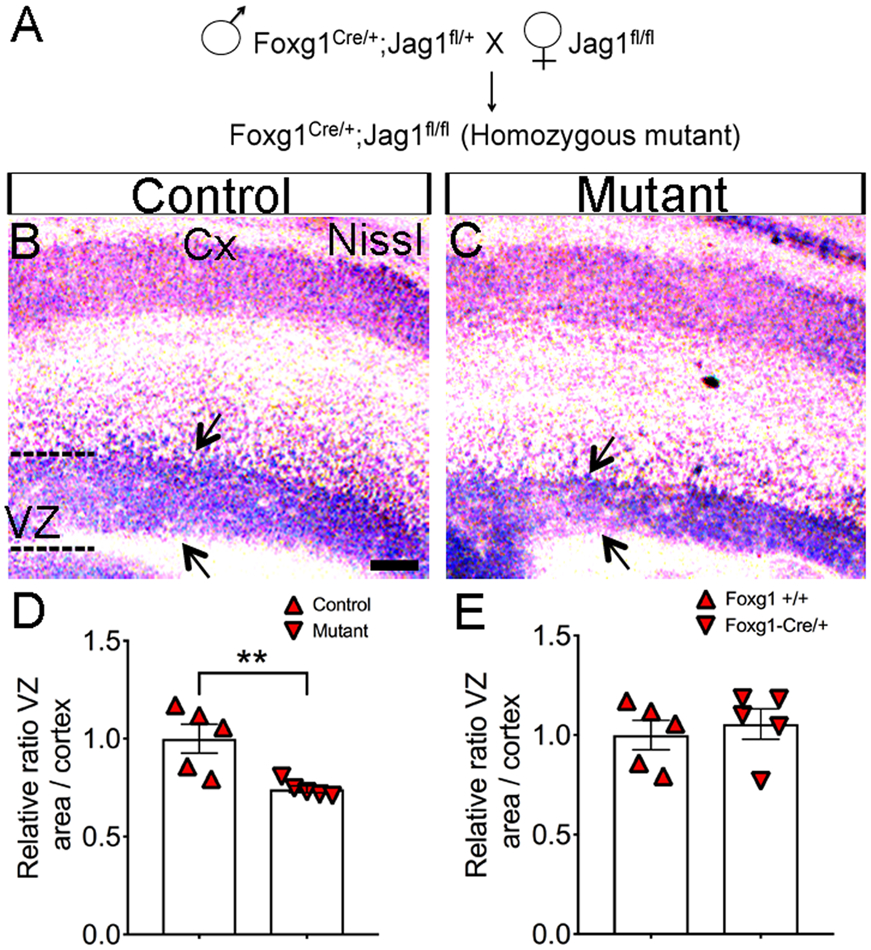
(A) Schematic cross for the generation of Jag1 homozygous mutants. (B-C, E) Reduction of VZ size as demonstrated by Nissl staining performed on sagittal brain sections at E17.5 (Arrows). (D) Quantification of the VZ area compared to control. Scale bar = 100μm. (E) Nissl Quantification of the VZ size in Foxg1 and Foxg1Cre/+ mice. All graphs show means ± SEM. **, p<0.01; Cx, cortex;
We first used Nissl staining to examine the overall structure of the neocortex at E17.5. We measured the area of the cortex between controls (234.8mm2 ± 2; n=5) and mutants (228.8mm2 ± 2; n=5) and found no significant differences (p=0.0545) (data not shown). In contrast, we observed a significant decrease in the cortical proliferative zone size in the Jag1 mutants (9.74mm2 ± 1; n=5) relative to the controls (13.14mm2 ± 0.5; n=5) (defined as littermates without the Foxg1Cre driver bearing one or two copies of the Jagged1 floxed allele) (Fig. 2B-D). Examination of the brains of mutants revealed that the mutant has a thinner proliferative zone (~ 25.8% decrease compared with control; (p=0.009). However, no significant (p=0.615) changes were observed in proliferative zone size of Foxg-cre/+ in comparison to wild-type mice (Fig. 2E).
3.3. Loss of Jag1 leads to reduced numbers of radial glial stem cells and proliferation in the VZ
The VZ gives rise to RGCs, which have been shown to be essential for the generation of cortical neurons in the neocortex (Gotz et al., 1998). To quantify and confirm these findings, we examined whether depletion of Jag1 would have an effect on Vimentin and Pax6 expression of RGCs. Using a probe for Vimentin we found a ~52% reduction (p=0.0005) in mutants (26.3mm2 ± 4; n=5) compared to control (55.6mm2 ± 3; n=5) (Fig. 3A-B, I). We also observed a ~58% reduction (p=0.0007) in the area of Pax6 expression in mutants (54.29mm2 ± 11; n=3) compared to controls (130.5mm2 ± 7; n=5) (Fig. 3C-D, J).
Figure 3. Reduce expression of Vimentin, Pax6, and proliferation suggest a loss of radial glial stem cell in Jag1 mutants.

(A-B) RNA In situ hybridization using a Vimentin probe and (C-D) Immunohistochemistry using anti-Pax6 antibody on sagittal sections show thinner expression in Jag1 mutant. Quantification of Vimentin (I) and Pax6+ (J) labeling shows decrease expression in mutants compared to control. Antibodies against p-HH3 (E-F, K) and Ki67 (G-H, L) show decrease expression in the VZ. Scale bar = 200μm. All graphs show means ± SEM. *, p<0.05; **, p<0.01; ***, p<0.001. Cx, cortex; dLGE, dorsal lateral ganglionic eminence.
Because RGCs undergo division, we examined whether cell proliferation was affected in the Jag1 mutant. We found that expression of phosphohistone H3 (p-HH3), a mitotic marker (Hendzel et al., 1997) (Fig. 3E-F, K) showed reduction(−67%; p=0.0007) in mutants (27% ± 2 cells/Dapi; n=4) compared to controls (9% ± 1 cells/Dapi; n=4). Similarly, using Ki67, a proliferation marker (Gerlach et al., 1997) (Fig. 3G-H, L), we observed a substantial reduction (−23%; p=0.0456) in mutants (155 ± 7 cells/Dapi; n=4) compared to controls (119 ± 2 cells/Dapi; n=4). Moreover, using an anti-PCNA antibody to label dividing cells (Bravo, 1986), we found significantly (~59%; p <0.0001) reduced proliferation in the mutants (185 ± 20 cells / μm2; n=4) compared to controls (454 ± 16 cells / μm2; n=4) in the VZ (Figs. 6A-B and 3M). Next, we asked whether or not programmed cell death could contribute to the reduced numbers of RGCs in the VZ. To this end, no significant differences (p= 0.9778) were found in the number of apoptotic cells between mutants (4 ± 1 cells; n=6) and controls (4 ± 1 cells; n=6) cells in the VZ at E17.5 (Fig. 3M).
Figure 6. Tbr2+ progenitor and Tbr1+ neuron show a defects in the neocortex.

(A-B, E) Immunohistochemistry on using anti-Tbr2 antibody shows increase expression in the VZ (yellow box). Scale bar = 200μm. (C-D, H) In situ hybridization using a probe for Tbr1 shows decreased expression in the cortical plate (arrow head) and increased expression in IZ (arrows) compared to control. Scale bar = 200μm. (E-F, I) Immunolabeling with anti-PCNA antibody shows decease proliferation in IZ (yellow arrow head). Scale bar = 100μm. All graphs show means ± SEM. *, p<0.05; **, p<0.01; ***, p<0.001. All sections are sagittal. VZ, ventricular zone Cx, cortex; MZ, marginal zone; dLGE, dorsal lateral ganglionic eminence, CP; cortical plate, IZ; intermediate zone.
3.4. Jag1 deficiency leads to reduced large neurospheres
Because we observed a reduction in proliferation and in RGCs in the VZ, we next examined the capacity of stem/progenitor cells to grow into spheres in vitro. To achieve this goal, we isolated tissue from the dorsal proliferative zone and cultured the cells, then monitored spheres growth using the neurospheres assay (Louis and Reynolds, 2005) for 12 days in vitro. We measured the sizes of the neurospheres because large-size neurospheres are presumably derived from stem cells due to the unlimited ability to divide (Golmohammadi et al., 2008; Louis et al., 2008) in vitro, we found that spheres derived from control mice showed substantially increased (~72% and ~49%, respectively) number of neurospheres that had diameters that were greater than 300μm (control, 5 ± 1 spheres, n=9; and mutant, 1 ± 0 spheres, n=9; p<0.0001) (Fig. 4A-B, E) and diameters that were between 200-300μm (control, 4 ± 0 spheres, n=9; and mutant, 2 ± 0 spheres, n=9; p <0.0006) (Fig. 4A-B, D) in size compared to the diameters of mutant mice. In contrast, the control mice showed a significantly decreased number of neurospheres that had diameters that were less than 200μm (control, 2 ± 0 spheres, n=9; and mutant, 5 ± 0 spheres, n=9; p<0.0001) (Fig. 4-B, C).
Figure 4. Jag1 mutants show fewer large size neurospheres after 12 days in vitro.

(A) Control and (B) Jag1 mutant primary neurospheres (NS) after 12 days in vitro. (C-E) Quantification of the NS that were less than 200μm, between 200-300μm, and greater than 300μm. Scale bar = 200μm. All graphs show means ± SEM; ***, p<0.001.
3.5. Jagged1 mutants showed decreased formation of secondary neurospheres
RGCs are known to undergo self-renewal. We therefore dissociated primary neurospheres and tested whether the neurospheres have ability to self-renew and generate secondary neurospheres. After 5-7 days in vitro, we found no significances (p = 0.2480) between the number of primary (23 ± 1 spheres; n=10) and secondary (21± 1 spheres; n=10) control neurospheres (Fig. 5A-C, E). In contrast, we observed a substantial decrease (−56%; p=0.0042) between the numbers of primary (9 ± 1 spheres; n=9) and secondary (4± 1 spheres; n=9) mutant neurospheres (Fig. 5B-D, F).
Figure 5. Loss of Jag1 affects self-renewal of neurospheres.
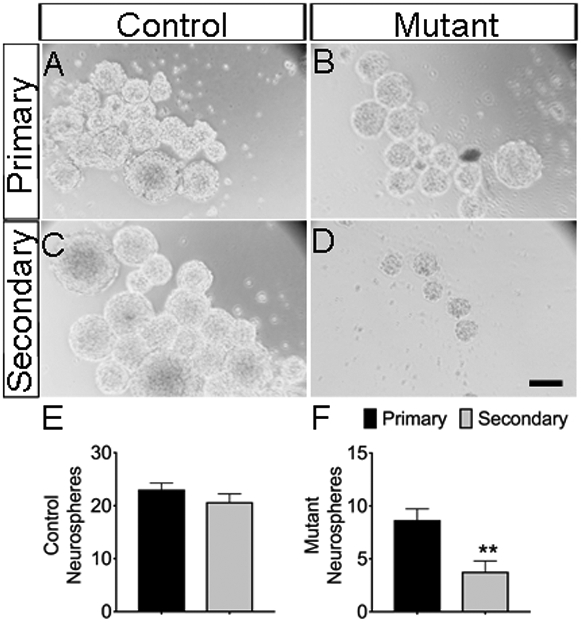
The growth of primary and secondary neurospheres after 5-7 days in vitro derived from control (A, C) and Jag1 mutant (B, D). Quantification of the primary and secondary neurospheres in control (E) and Jag1 mutant (F). Scale bar = 100μm. All graphs show means ± SEM; **, p<0.01.
3.6. Jag1 mutant shows defects in IPCs and Tbr1+ neurons
Given the earlier decrease in the numbers of RGCs (Fig. 3), we tested whether its downregulated expression may affect neurons in the cortical intermediate zone (IZ) layer. Therefore, we tested whether the loss of Jag1 leads to decreases in proliferating cells in the IZ. Using an anti-PCNA antibody, we found a −61.8% reduction (p = 0.0048) in mutants (91 ± 3 cells / μm2; n=4) compared to controls (239 ± 34 cells / μm2; n=4) (Fig. 6A-B, G; yellow arrow head). Next, we examined if the decreased proliferation in the IZ affected the production of proliferating IPC expressing Tbr2+. We chose to focus Tbr2+ because they are derived from RGCs expressing Pax6+ (Englund et al., 2005). Interestingly, the Jag1 mutants showed disruptions in the expression of Tbr2 (Fig. 6D; yellow arrow head) in the IZ layer. Surprisingly, we found a dramatically increased (p < 0.0001) Tbr2 expression in mutant mice (943 ± 32 μm2; n=4) relative to control mice (2 ± 1 μm2; n=3) in the VZ (Fig. 6C-D, H; yellow box). Tbr2+ neurons progress into Tbr1+ cortical neurons (Englund et al., 2005). Although Tbr1+ neurons are generated at ~E12.5/13.5 and by E17.5 their migration to their final destination are vastly complete. We therefore, investigated whether or not Tbr1+ cortical neurons had any apparent cortical defects by labeling its expression at E17.5. In the IZ, we observed significant increase (~64%; p= 0.003) in expression of Tbr1 in Jag1 mutants (132.88 ± 4 mm2; n=4) (6F; arrows) compared to controls (80 ± 6 mm2; n=4) (Fig. 6E-F, I). In contrast, in the cortical plate, we found a substantial decrease (~24%; p= 0.001) in expression of Tbr1 in mutants (145.72 ± 7 mm2; n=4) compared to controls (193.56 ± 8 mm2; n=4) (6D; arrowheads). Using the same Tbr1 probe, we also confirmed that the Foxg-cre/+ mice showed no changes (p=0.9988) in Tbr1 expression in the cortical plate (CP) in comparison to wild-type mice (data not shown).
3.7. Jagged1 mutant neurospheres show premature neuronal differentiation
To test in vitro whether the loss of Jag1 from neurospheres have an effect on neuronal differentiation. We cultivated neurospheres for 5-7 DIV under normal conditions. Afterwards, neurospheres were cultivated without supplement and growth factors to induce differentiation for 2 DIV and the numbers of differentiated neurons were quantified. We found substantial increase (~67%; p < 0.0001) in differentiated neurons in the mutants (87 ± 10 neurons; n=17) compared to controls (28 ± 4 neurons; n=17) (Fig. 7A-B; black arrow heads and 7E). We used NCAM to visualize the differentiated neurons (Fig. 7C-D; white arrow head).
Figure 7. Loss of Jag1 leads to neuronal differentiation in vitro.
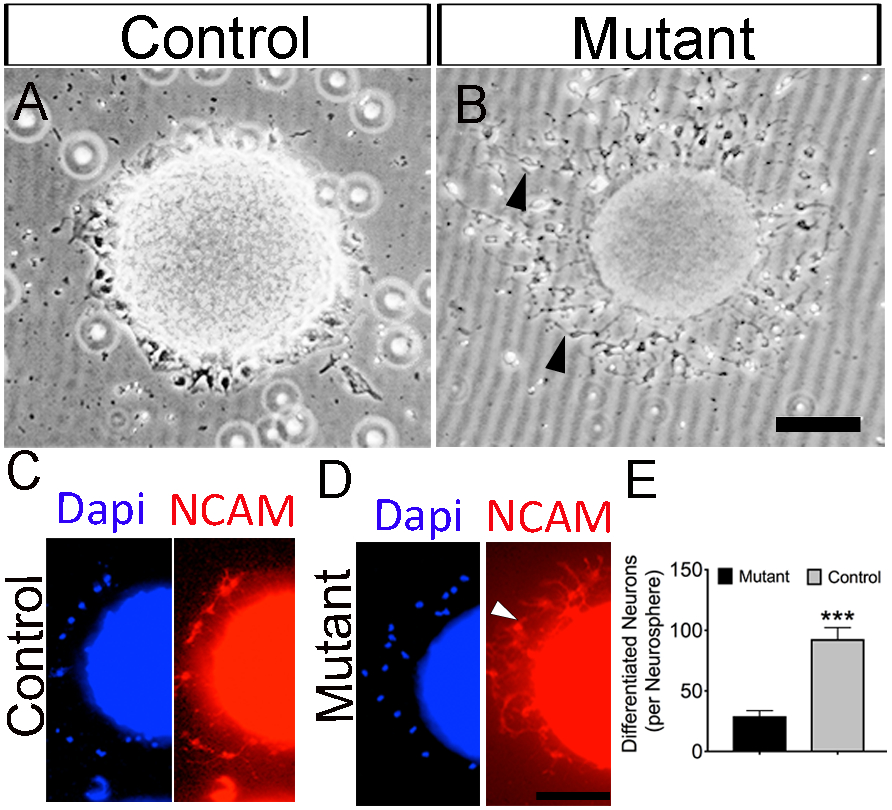
Visual representative images of neuronal differentiation of (A) control and (B) mutant neurospheres after 12 days in vitro. Visual representation of immunocytochemistry staining of (C) control and (D) mutant neurospheres using anti-NCAM co-stained with Dapi. (E) Quantification shows increase neuronal differentiation in mutants compared to control. Graphs shows means ± SEM. ***, p<0.001. Scale bar = 100μm.
Discussion
This study provides new insights into Notch signaling by examining the loss of Jag1 in corticogenesis. The depletion of Jag1 led to a thinner proliferative zone and decreased numbers of radial glial stem cells. Moreover, we showed that Jag1 promotes growth and self-renewal of neurospheres. Lastly, we observed that Jag1 deficiency is associated with precocious generation of Tbr2+ intermediate progenitors and cortical defects. These data detail a key role of Jag1 in the neocortex.
Our data that the loss of Jag1 leads to reduced numbers of RGCs (Fig. 3). Interestingly, RGCs are known to divide asymmetrically, giving rise to another RGC and either an intermediate progenitor cell or a neuron (Englund et al.2005; Gotz and Huttner, 2005; Noctor et al., 2004; Shitamukai et al., 2011). A possible explanation for the reduction of RGCs is that Jag1 promotes self-renewal. Our findings that the loss of Jag1 reduces the formation of secondary neurospheres support the role of Jag1 in promoting self-renewal (Fig. 5). This notion is consistent with previous reports demonstrating that Jag1 is essential for self-renewal of neural stem cells both in the postnatal subventricular zone (Nyfeler et al., 2005) and in the adult dentate gyrus (Lavado and Oliver, 2014). This idea is further supported by studies that demonstrate that inhibition of Notch can reduce the self-renewal of neural stem cells (Imayoshi et al., 2010) and diminish radial glial identify (Gaiano et al., 2000).
Studies have shown that continuous growing (large) neurospheres are derived from neural stem cells (Louis et al., 2008). In their report, the authors demonstrated that large neurospheres still maintained their unlimited growth potential after several passaged in vitro and can reform into a large neurosphere. Our results demonstrating that Jag1 mutants showed reduced numbers of large neurospheres (200-300μm or <300μm) after 12 days in vitro (DIV) suggests the possibility that there may be less neural stem cells in the proliferative zone (Fig. 4). This is in line with our observation that the loss of Jag1 leads to reduce RGCs (Fig. 3). Taken together, this suggests that Jag1 is essential to prevent the exhaustion of radial glial stem cells pool presumably by promoting self-renewal.
Our findings of precocious intermediate progenitor cells (Tbr2+) in the VZ of Jag1 mutants suggest that Jag1 may be, in part, required to prevent premature differentiation of RGCs. This is consistent with our observation that Jag1 mutants show early neuronal differentiation in vitro (Fig. 7). This is further supported by studies that Jag1 has been shown to rescues neuronal differentiation in other animal models (Braccioli et al., 2017) and reports that have shown that Notch activation promotes the expression of transcription factors that inhibit pro-neural differentiation, thereby acting to maintain the stem cell population (Kageyama et al., 2009). Thus, Jag1 may also play a role in preventing premature differentiation.
Our data implicate Jag1 as an important agent for migration of cortical neurons. Tbr1+ neurons that are normally expressed in the cortical plate were also found misplaced in the intermediate zone of the neocortex in Jag1 mutants even though Jag1 loss does not completely impact all Tbr1+ neurons (Fig. 6C). This discussion suggests that there may be additional Jag1-independent signals that may also participate in the migration of these neurons. Interestingly, our in situ hybridization studies show that Jag2 is also expressed in the VZ (Fig. 1E; arrow). Although not tested, it is possible that Jag2 or other Notch ligands may, in part, compensate for the loss of Jag1 in the migration of cortical neurons.
Given the known compensatory role of Notch ligands in Notch signaling, it is of interest to note that Jag1 mutants do not exhibit identical phenotype to delta1 mutants during development of the forebrain. Specifically, conditional deletion of Jag1 led to decreased Pax6 expression (Fig. 3) and premature neuronal differentiation (Fig. 7). In contrast, conditional deletion of Delta1 led to increased Pax6 expression and suppression of neuronal differentiation (Kawaguchi et al, 2008). Taken together, these observations suggest that Jag1 and Delta1 may play distinct roles during corticogenesis. Jag1 expression has been observed at earlier stages of development (E12.5) (Braccioli et al., 2017) resulting in the prediction that Jag1 may be essential throughout corticogenesis.
In humans, mutations in Jag1 cause Alagille Syndrome (ALGS) (Alagille et al., 1975; Krantz et al., 1997). Complications associated with ALGS patients that suffer from neurological defects including intellectual disability (Alagille et al., 1975; Krantz et al., 1997; Oda et al., 1997). Interestingly, Jag1 null mice provide a partial phenocopy of ALGS syndrome in human (Xue et al., 1999). Unfortunately, in the null mice identifying the role for Jag1 in the central nervous system has been challenging because the complete loss of Jag1 is embryonically lethal (Xue et al., 1999). Our approach using a conditional loss-of-function of Jag1 may begin to shed some light onto the possible mechanisms associated with ALGS. Finally, a clearer understanding of the factors involved in the maintenance of radial glial stem cells might inform delivery approaches that target neurons in brain regions that show neurodegeneration.
Figure 8. Schematic illustration: the possible roles of Jagged1 in radial glia maintenance in corticogenesis.

The possible roles of Notch ligand, Jag1 during late cortical development. Jag1 promotes self-renewal of radial glial stem cells, prevents premature neuronal differentiation, and may potentially play a role in cortical migration.
Highlights.
Conditional loss-of-function of Jagged1 shows reduced cortical proliferative zone
Loss of Jagged1 decreases the number of radial glial stem cells
Jagged1 deficiency leads to defects in self-renewal and growth of neurospheres
Jagged1 mutants display cortical defects
Acknowledgements
This work was supported by NIH Grants (1F31DC011709-01A1, K12GM102779) to C.A.B. C.A.B. was also supported by fellowships from the Ford Foundation, Cornell University’s Provost Office and IRACDA. The author would like to thank Thomas Gridley for providing the Jagfl/fl mouse and David M. Lin for his assistance in providing tissue samples. The author graciously thanks David Lin, Jean Hebert, Sayan Nandi, and Jean Lud Cadet for providing space, financial support, editorial support, reagents, and equipment.
Footnotes
Competing Interests: The author declares no competing interests.
References
- Alagille D, Odievre M, Gautier M, and Dommergues JP (1975). Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr 86, 63–71. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Matsuno K, and Fortini ME (1995). Notch signaling. Science 268, 225–232. [DOI] [PubMed] [Google Scholar]
- Braccioli L, Vervoort SJ, Adolfs Y, Heijnen CJ, Basak O, Pasterkamp RJ, Nijboer CH, and Coffer PJ (2017). FOXP1 Promotes Embryonic Neural Stem Cell Differentiation by Repressing Jagged1 Expression. Stem Cell Reports 9, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R (1986). Synthesis of the nuclear protein cyclin (PCNA) and its relationship with DNA replication. Exp Cell Res 163, 287–293. [DOI] [PubMed] [Google Scholar]
- Brooker R, Hozumi K, and Lewis J (2006). Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development 133, 1277–1286. [DOI] [PubMed] [Google Scholar]
- Dang L, Yoon K, Wang M, and Gaiano N (2006). Notch3 signaling promotes radial glial/progenitor character in the mammalian telencephalon. Dev Neurosci 28, 58–69. [DOI] [PubMed] [Google Scholar]
- Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, Bartlett PF, Cooper HM, Reynolds BA, and Wainwright BJ (2011). Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS One 6, e14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieset I, Djurovic S, Tesli M, Hope S, Mattingsdal M, Michelsen A, Joa I, Larsen TK, Agartz I, Melle I, et al. (2012). Up-regulation of NOTCH4 gene expression in bipolar disorder. Am J Psychiatry 169, 1292–1300. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, and Hevner RF (2005). Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, and Fishell G (2002). The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci 25, 471–490. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, and Fishell G (2000). Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26, 395–404. [DOI] [PubMed] [Google Scholar]
- Gerlach C, Golding M, Larue L, Alison MR, and Gerdes J (1997). Ki-67 immunoexpression is a robust marker of proliferative cells in the rat. Lab Invest 77, 697–698. [PubMed] [Google Scholar]
- Golmohammadi MG, Blackmore DG, Large B, Azari H, Esfandiary E, Paxinos G, Franklin KB, Reynolds BA, and Rietze RL (2008). Comparative analysis of the frequency and distribution of stem and progenitor cells in the adult mouse brain. Stem Cells 26, 979–987. [DOI] [PubMed] [Google Scholar]
- Gotz M, and Huttner WB (2005). The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6, 777–788. [DOI] [PubMed] [Google Scholar]
- Gotz M, Stoykova A, and Gruss P (1998). Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 21, 1031–1044. [DOI] [PubMed] [Google Scholar]
- Hatakeyama J, Bessho Y, Katoh K, Ookawara S, Fujioka M, Guillemot F, and Kageyama R (2004). Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539–5550. [DOI] [PubMed] [Google Scholar]
- Hebert JM, and McConnell SK (2000a). Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol 222, 296–306. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, and Allis CD (1997). Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106, 348–360. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, and Kageyama R (2010). Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci 30, 3489–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice NJ, and Jan YN (2002). Variations on the Notch pathway in neural development. Curr Opin Neurobiol 12, 64–70. [DOI] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, and Imayoshi I (2009). Dynamic regulation of Notch signaling in neural progenitor cells. Curr Opin Cell Biol 21, 733–740. [DOI] [PubMed] [Google Scholar]
- Kawaguchi D, Yoshimatsu T, Hozumi K, & Gotoh Y (2008). Selection of differentiating cells by different levels of delta-like 1 among neural precursor cells in the developing mouse telencephalon. Development, 135(23), 3849–3858. doi: 10.1242/dev.024570 [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Xu J, and Gridley T (2006b). The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet 2, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, and Ilagan MX (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz ID, Piccoli DA, and Spinner NB (1997). Alagille syndrome. J Med Genet 34, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, and Martinez-Cerdeno V (2006). Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat Rev Neurosci 7, 883–890. [DOI] [PubMed] [Google Scholar]
- Lavado A, and Oliver G (2014). Jagged1 is necessary for postnatal and adult neurogenesis in the dentate gyrus. Dev Biol 388, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis SA, and Reynolds BA (2005). Generation and differentiation of neurospheres from murine embryonic day 14 central nervous system tissue. Methods Mol Biol 290, 265–280. [DOI] [PubMed] [Google Scholar]
- Louis SA, Rietze RL, Deleyrolle L, Wagey RE, Thomas TE, Eaves AC, and Reynolds BA (2008). Enumeration of neural stem and progenitor cells in the neural colony-forming cell assay. Stem Cells 26, 988–996. [DOI] [PubMed] [Google Scholar]
- Masek J, and Andersson ER (2017). The developmental biology of genetic Notch disorders. Development 144, 1743–1763. [DOI] [PubMed] [Google Scholar]
- Merkle FT, and Alvarez-Buylla A (2006). Neural stem cells in mammalian development. Curr Opin Cell Biol 18, 704–709. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Okano H, and Ogawa M (2001). Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron 31, 727–741. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, and Parnavelas JG (2002). Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci 3, 423–432. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, and Kriegstein AR (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7, 136–144. [DOI] [PubMed] [Google Scholar]
- Nyfeler Y, Kirch RD, Mantei N, Leone DP, Radtke F, Suter U, and Taylor V (2005). Jagged1 signals in the postnatal subventricular zone are required for neural stem cell self-renewal. EMBO J 24, 3504–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, et al. (1997). Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16, 235–242. [DOI] [PubMed] [Google Scholar]
- Pan W, Jin Y, Stanger B, and Kiernan AE (2010). Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc Natl Acad Sci U S A 107, 15798–15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Sickles HM, Deleonardis C, Alcaraz A, Gridley T, and Lin DM (2008). Notch2 is required for maintaining sustentacular cell function in the adult mouse main olfactory epithelium. Dev Biol 314, 40–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A, Konno D, and Matsuzaki F (2011). Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci 31, 3683–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Nadeau P, Yuan M, Yang X, Shen J, and Yankner BA (1999). Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A 96, 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Nakamura K, Okamoto K, and Kaneko T (2001). Radial glia is a progenitor of neocortical neurons in the developing cerebral cortex. Neurosci Res 41, 51–60. [DOI] [PubMed] [Google Scholar]
- Turrero Garcia M, and Harwell CC (2017). Radial glia in the ventral telencephalon. FEBS Lett 591, 3942–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EO, Sickles HM, Dooley AL, Palumbos S, Bisogni AJ, and Lin DM (2011). Delta Protocadherin 10 is Regulated by Activity in the Mouse Main Olfactory System. Front Neural Circuits 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, and Gridley T (1999). Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8, 723–730. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]