

Abstract
Regorafenib is a multi-targeting kinase inhibitor approved for the treatment of metastatic colorectal cancer patients in refractory to standard chemotherapy. Similarly to sorafenib, this agent was originally developed as a RAF1 inhibitor. However, the kinase inhibitory profile is distinct from sorafenib. A broad-spectrum of kinase inhibition induces wide-range drug sensitivity, irrespective of mutation status of major oncogenes. This agent’s main therapeutic effects are anti-angiogenesis and the remodeling of tumor microenvironment through several mechanisms of action. The dual blockade of VEGF receptors and TIE2 can lead to both additive anti-angiogenesis effects and the suggestive unique regulation of vessel stability. Additionally, it inhibits molecular escape pathways to VEGF inhibition (e.g., FGF, PIGF, and PDGF signaling), enabling its continuous antiangiogenic effect even in tumors resistant to VEGF inhibitors. Furthermore, regorafenib has the important effect of enhancing anti-tumor immunity via macrophage modulation. Based on this concept, clinical trials have been recently launched for the development of a combination strategy with immune checkpoint inhibitors. Contrary to regorafenib induced clinical benefits and advances in the novel strategy, currently no predictive biomarkers have been identified. In the present review, we revisit and summarize regorafenib’s unique mechanisms of action. The review could highlight molecular insights and provide some perspective for the search of predictive biomarkers used in metastatic colorectal cancer patients treated with regorafenib.
Keywords: Regorafenib, Colorectal cancer, Anti-angiogenesis, Tumor microenvironment, Immunotherapy, Biomarker
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related deaths worldwide [1]. Most metastatic CRC (mCRC) patients are incurable. The main therapeutic strategy for mCRC patients is palliative chemotherapy, while non-systematic therapy (such as surgery and, more optionally, radiation and ablative techniques) is selective for patients with resectable metastatic lesions in improving the survival time. Recent progress in the development of new chemotherapeutic drugs has prolonged the median survival time to around 30 months [2,3]. Of note, in treatment of mCRC, molecular targeting agents represent key players in providing a survival advantage.
Regorafenib is a novel targeting drug introduced into clinical setting of mCRC in the past few years. It was originally developed as a RAF1 inhibitor, similarly to sorafenib. A series of drug designs of RAF1 inhibitors identified regorafenib as the fifteenth compound, immediately following sorafenib [4]. Subsequently, preclinical experiments revealed regorafenib’s role as a multi-targeting kinase inhibitor with a broad range of therapeutic targets [5]. Based on its chemical structure, regorafenib is classified as a type II kinase inhibitor. It binds inactive DFG-out conformation of the kinase domain, which represents a hydrophobic space adjacent to the ATP binding pocket [6]. Regorafenib’s structure is similar to, but differs from sorafenib, by the addition of a fluorine atom in the proximal phenyl ring. This structural difference leads to very distinct properties among the two drugs [5]. Preclinically, regorafenib demonstrated a more robust anti-tumor activity than other specific angiogenic inhibitors, due to its broad-spectrum kinase inhibition [7].
Despite a promising anti-tumor effect shown in preclinical models, clinical benefit regorafenib provides to mCRC patients is modest. In two randomized phase III trials (CORRECT and CONCUR), regorafenib significantly increased overall survival in heavily-treated mCRC, with a reported survival benefit of 1.4 and 2.5 months over placebo [8,9]. Additionally, a large phase IIIb study (CONSIGN) confirmed its safety profile [10]. After the FDA approved regorafenib for the treatment of mCRC patients in refractory to standard chemotherapy, several post-marketing observational studies showed extensive real-world data of efficacy and toxicities (CORRELATE, Japanese post-marketing study, REBECCA, RECORA) [11–14]. These broad global clinical experiences showed that adverse events associated with regorafenib occur frequently in the early stages of treatment, especially within the first cycle [15]. Such findings have encouraged an investigation into how to optimize first cycle dosing to support tolerability and treatment continuation. To test this clinical question, a randomized phase II, ReDOS trial has been conducted. This trial evaluated the safety and efficacy of two regorafenib dosing schedules (flexible first cycle dose-optimization schedule; and standard-dose schedule). The primary endpoint was the proportion of evaluable patients initiating cycle 3, assumed to be superior in dose-optimization schedule. The primary endpoint was met, and dose-optimization schedule represented lower incidence of adverse events and a trend for higher efficacy, compared to standard-dose schedule [16]. Based on these findings, the National Comprehensive Cancer Network currently recommends the ReDOS dose-escalation schema as an option in its mCRC treatment guidelines [17].
The current challenge is patient selection. The identification of relevant predictive markers for clinical outcome associated with regorafenib treatment is critical. However, to date, no promising biomarkers have been identified. In the present review, the unique mechanisms of action of regorafenib are summarized. Additionally, the molecular background behind the pathway interacting with regorafenib is revisited based on preclinical and clinical findings. Specifically, it includes both well-known antiangiogenic actions and novel immunomodulation mechanisms. This could facilitate the understanding of the molecular insights for clinical use and aid biomarker discovery in regorafenib treatment for mCRC patients.
Broad-spectrum of kinase inhibition
Colorectal carcinogenesis is complex. It is based on multiple steps and signaling pathways that control cell division and survival [18]. Numerous altered cell surface ligand–receptor interactions induce the activation of intracellular signaling pathways involved in the several cancer growth hallmarks (e.g., angiogenesis, activating metastasis activation, cell proliferation maintenance, and immune suppression, Fig. 1). During the course of multistep tumorigenesis, tumors create a “tumor microenvironment (TME)”, which represents a tumor-host interactive structure consisting of many types of cells [e.g., cancer cells, cancer stem cells, endothelial cells, pericytes, immune inflammatory cells, cancer-associated fibroblast (CAF), and bone marrow-derived stromal cells] [19]. Regorafenib simultaneously targets several hallmarks of CRC development through broad kinase inhibition with anti-angiogenesis (by inhibiting VEGFR1, −2, −3, TIE2, PDGFR, and FGFR1 and −2), anti-proliferation (by inhibiting c-KIT, RAF1, BRAF, and RET), anti-metastasis (by inhibiting VEGFR2 and −3, and PDGFR), and anti-immunosuppression (by inhibiting CSF1R) effects [5]. These anti-tumor properties are closely associated with TME modulation, which leads to an improvement of therapeutic outcomes, even in highly aggressive CRC [7]. Additionally, in preclinical models, the broad-spectrum kinase inhibitory property is capable of achieving wide-ranging drug sensitivity, irrespective of RAS and BRAF mutation status [20]. Consistently, clinical trials data show that regorafenib provides survival benefits across all the patient subgroups, including those carrying major oncogenes mutations (e.g., RAS and BRAF) [21,22].
Fig. 1. Anti-tumor mechanisms of regorafenib.
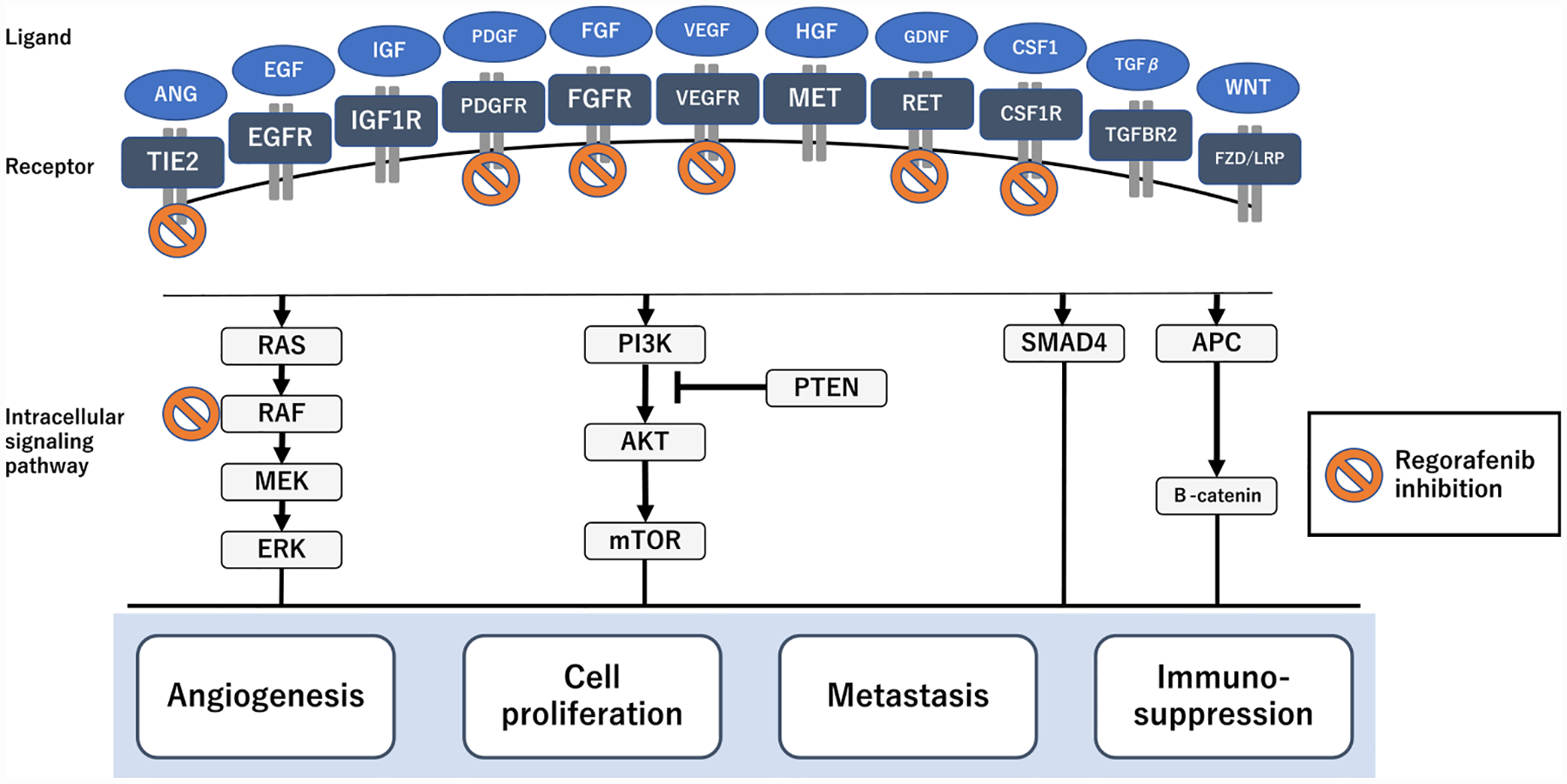
Regorafenib inhibits a broad range of activated pathways related to angiogenesis, metastasis, cell proliferation, and immunosuppression. This provides putative mechanisms for its anti-tumor efficacy in CRC.
Anti-angiogenetic effect by dual blocking of VEGFRs and TIE2
In CRC, angiogenesis is a highly complicated process essential for cell survival and tumor progression [23]. Tumor blood vessels grow abnormally, resulting in poorly functioning vasculature. The loose association between the endothelium and perivascular cells in the tumor vasculature leads to tumor cell intravasation through the vascular wall into the circulation. As a consequence, metastatic spreading to secondary sites is promoted. VEGF–VEGFR signaling predominantly promotes tumor angiogenesis, leading to vascular destabilization. Of note, VEGFR2 is a master receptor, capable of transmitting signals to the downstream pro-angiogenic pathway via stimuli of its ligands (VEGFA, C, and D) [24]. Antiangiogenic treatment is aimed at reducing the abundant tumor vessels required for the essential supply of nutrients and oxygen to proliferating cancer cells. Additionally, it normalizes tumor vasculature, resulting in reduced tissue hypoxia and the enhanced delivery of cytotoxic agents [25]. However, the limited efficacy of clinically approved anti-VEGF/VEGFR drugs has prompted searching for co-targetable angiogenesis regulators [26–28]. The angiopoietin (ANG) – TIE pathway has emerged as the second attractive target in anti-angiogenesis treatment [29]. This pathway uniquely controls vessel remodeling and stabilization by involving two receptors (i.e., TIE1 and TIE2) and two well-characterized ligands (i.e., ANG1 and ANG2). An understanding of the highly complicated system characterized by double-sided ligands and receptors working in concert with the VEGF pathway is necessary in order to identify more effective treatment strategies.
TIE2 is a well-studied tyrosine kinase receptor binding ANG1 and ANG2. ANG1 is released primarily from perivascular cells in a paracrine manner. On the contrary, ANG2 is released from endothelial cells in an autocrine manner [29]. ANG1 is a strong agonist of TIE2. It promotes angiogenesis via the downstream AKT signaling and stabilizes the vascular adherence junction component (VE-cadherin) via another downstream RhoA signaling. The latter interferes with the VEGF/VEGFR2/Src axis causing vascular destabilization through VE-cadherin internalization [30]. As opposed to ANG1, ANG2 works in a context-dependent manner serving as both TIE2 agonist and antagonist [31]. In a non-pathological condition or in the absence of ANG1, ANG2 functions as a weak TIE2 agonist [32]. While in inflammation or tumor settings, ANG2 acts as a TIE2 antagonist, promoting vessel destabilization and pericyte dropout [32]. ANG2 working as a TIE2 antagonist induces angiogenesis in the presence of VEGF, while leads to vessel regression in the absence of VEGF [33]. In tumor “angiogenesis switch”, cancer-secreted VEGFA induces the formation of motor endothelial cells, called tip cells, which invade the extracellular matrix and lead the new vascular sprouting. This process is supported by ANG2 [34]. ANG2 expression increases in response to VEGF and hypoxia [35]. In several cancer types, including CRC, there is a significant upregulation of ANG2 [36]. This leads to increased ANG2–TIE2 binding and consequently causes vascular dysfunction, hyper-permeability, poor perfusion, and tissue hypoxia [29]. In this context, tissue hypoxia promotes VEGF’s expression, which also results in an increased ANG2 spiral. Briefly, ANG1 – TIE2 signaling contributes to angiogenesis with vascular stabilization independently of VEGF. On the contrary, in tumors, ANG2 – TIE2 works as a vascular destabilizing pro-angiogenesis factor in concert with the VEGF pathway (Fig. 2).
Fig. 2. Different regulation of angiogenesis and vessel stability between VEGF and angiopoietin signaling, relating to regorafenib action.
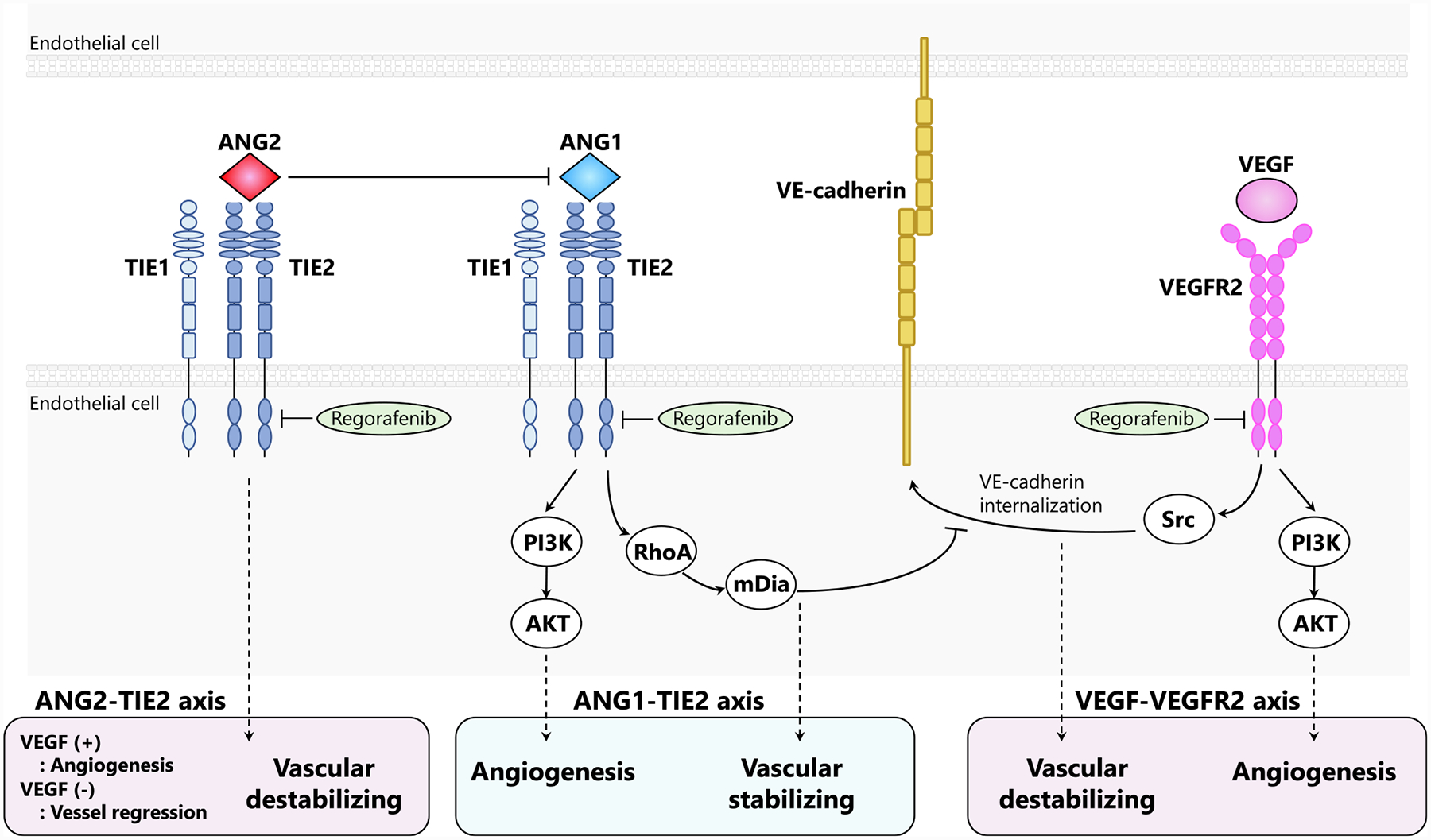
Both VEGF-VEGFR2 and ANG1-TIE2 pathways are pro-angiogenic. However, they have opposite effects on vascular stabilization. Specifically, VEGF-VEGFR2 signaling activates Src-dependent internalization of VE-cadherin. Therefore, it promotes the disruption of interendothelial adherens junctions. Meanwhile, ANG1-TIE2 signaling activates downstream RhoA and mDia limiting the access of Src to VEGFR2. As a consequence, it strengthens vasculature integrity. In tumors, ANG2 acts as an antagonist of ANG1-TIE2 pathway. Specifically, it prompts vessel destabilization and pericyte dropout. ANG2-TIE2 signaling induces angiogenesis in VEGF’s presence, while it leads to vessel regression in VEGF’s absence. TIE1 is an orphan receptor, interacting with TIE2. Regorafenib inhibits both VEGFR2 and TIE2.
While less well known than TIE2, TIE1 has been shown to play an important role during angiogenesis. TIE1 is an orphan receptor with no ligand to bind [37]. It appears to contribute to TIE2 signaling by heterodimerization, affecting angiopoietin binding to TIE2. Functionally, during angiogenesis, TIE1 can both negatively and positively regulate TIE2 signaling, depending on the cellular context. For example, TIE1 expression in angiogenic tip cells negatively regulates TIE2 surface presentation. On the contrary, it has been shown that TIE1 expression in remodeling stalk cells stabilizes and sustains TIE2 signaling [38]. In response to VEGF and TNF-α, TIE1 undergoes regulated ectodomain cleavage [39]. The cleaved product is associated with soluble TIE1 (sTIE1). In the context of acute inflammation, TIE2 ectodomain shedding prevents ANG2’s agonistic action, favoring its antagonistic action on TIE2. This, as a consequence, leads to vascular remodeling and destabilization [40]. Similar effects were observed in TIE1 deleted mice [41]. On the contrary, in the case of chronically growing tumors, TIE1 deletion induces TIE2 gain-of-function (upregulation of ANG1, ANG2, and TIE2, and shift of ANG1/ANG2 ratio toward ANG1) and VEGFR2 loss-of-function (strong downregulation of KDR, which is VEGFR2’s encoding gene). This leads to reduced angiogenesis and vascular normalization [42]. Such findings suggest that TIE1 works as a pro-angiogenetic factor in tumors, in contrast to acute inflammation.
Regorafenib is a distinctive agent, inhibiting both TIE2 and VEGFRs. ANG2 upregulation in the tumor vasculature has been suggested as a potential mechanism of resistance to VEGF pathway inhibition [43]. As a consequence, the inhibition of both ANG2–TIE2 and VEGF pathways may be a more effective strategy to overcome resistance compared to the exclusive inhibition of VEGFRs. Of note, it has been shown that the combination of ANG2- and VEGF-blocking increases anti-tumor effects, inducing an additional vascular stabilizing effect compared to VEGF blockade only [44]. However, the effect of ANG1 – TIE2 signaling inhibition is not comparable to ANG2 – TIE2 inhibition. Preclinical studies in CRC cell showed that selective ANG2 inhibition is effective for anti-angiogenesis and vascular normalization. On the contrary, selective ANG1 inhibition is much less effective [45]. In TIE2 and VEGFRs dual blocking, there is an obvious enhancement of the effect of tumor vessel reduction. However, it remains unclear whether the effect of vascular normalization becomes synergetic. Specifically, this is because while the ANG1 – TIE2 axis contributes to vessel stabilization, the ANG2–TIE2 and VEGF–VEGFR axis contribute to vessel destabilization. In CRC cells, the combined inhibition of ANG1 and ANG2 favors tumor vessel abnormalities, due to the absence of ANG1’s stabilizing effect [45]. Thus, it has been speculated that the balance between ANG1 – TIE2, ANG2 – TIE2, and VEGF – VEGFR plays an important role in vascular normalization after regorafenib. Importantly, this may affect anti-tumor effects since normalization of tumor vasculature is critically effective in anti-angiogenesis treatment. Based on this observation, the importance of TIE2 pathway inhibition as part of regorafenib’s therapeutic efficacy remains to be further investigated.
Inhibiting molecular escape pathways to VEGF inhibition
In combination with cytotoxic agents, VEGF axis inhibition is at the basis of the therapeutic strategy recommended across standard front-line treatments for mCRC patients, while anti-EGFR antibodies are also recommended and commonly used in standard treatment for RAS wild and left sided tumors [2,26–28]. Currently, three active VEGF targeting drugs (i.e., bevacizumab, ramucirumab, and zib-aflibercept) have been approved for use in clinical practice for mCRC patients [26–28]. In line with preclinical data suggesting that continuous VEGF suppression is key to achieving and maintaining tumor control [46], the continuous use of VEGF targeting drugs is an established treatment strategy providing a survival benefit [47]. On the contrary, long duration of VEGF inhibiting treatment can lead to the molecular escape of VEGF inhibition, resulting in acquired drug resistance [48]. One of the compensatory angiogenic factors is represented by the FGF family of ligands. Endothelial cells express high levels of FGFR1 and −2, both of which are potent pro-angiogenic factors [49]. It has been shown that an angiogenic rebound following VEGF inhibition in tumors is associated with an increased expression of FGF family. Of note, the blockage of the FGF signaling minimizes the acquired resistance to VEGF targeting therapy [50]. PIGF is a member of the VEGF family, selectively binding to VEGFR1. It contributes to the molecular escape to VEGFA inhibition. It has been shown that PIGF has increased plasma levels following VEGF signaling blockade [51]. PDGF-BB is another redundant factor modulating angiogenesis by activating VEGF and FGF and by stimulating the endothelial cells [52]. In CRC, PDGF-BB is related to increasing pericytes within the tumor, resulting from the interaction with PDGFR on pericytes [53]. There is a close association between these angiogenic factors and TME. For example, it has been shown that colon CAFs secrete FGF1 and PDGF [54], and PIGF plays an important role in the recruitment of macrophages, promoting the angiogenic response [55]. By targeting VEGFR1 (receptor for PIGF), PDGFR (receptor for PDGF-BB), and FGFR (receptor for FGF), regorafenib can overcome these molecular escape pathways to VEGF inhibition [5]. Preclinical and clinical studies have supported the evidence of regorafenib’s continuous antiangiogenic effect in tumors that have become resistant to other VEGF inhibitors, as well as its role in TME remodeling [8,56].
Promoting anti-tumor immunity
Increasing data indicate a close link between angiogenesis and the suppression of anti-tumor immune system [57]. This close relationship has a connection with TME, in a two-way process. Angiogenic factors impair the vessel’s normal function relating to trafficking and extra-vasation of effector T cells and change the TME into an immunosuppressive state. Of note, several immune-components of TME [e.g., dendric cells (DC), regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), and tumor-associated macrophages (TAM] produce angiogenetic factors and can promote angiogenesis [57,58]. VEGF plays a key role in attenuating tumor immune response by negatively affecting the antigen-presenting cells (APC, such as DC) and effector T cells. Meanwhile, it also positively affects immune suppressive cells (e.g., Treg, MDSC, and TAM) [57]. VEGF increases T cell exhaustion by enhancing the expression of inhibitory checkpoints (e.g., PD-L1, CTLA-4, TIM3) on T cell [59]. Furthermore, since immune cells (e.g., dendric cell, effector T cell, Treg, and TAM) also express VEGFRs, VEGF signaling additionally functions as a chemokine that recruits the subsets of immune cells to TME [57]. ANG2 is another important component capable of strongly driving immune resistance [57]. The mechanism of ANG2-induced immunosuppression takes place through the recruitment of myelocytes. TIE2-expressing monocytes/macrophages (TEM) are a subpopulation of myeloid cells, characterized by both the expression of monocyte/macrophage markers and the TIE2 [60]. By interacting with TIE2, ANG2 increases TEMs recruitment to the endothelium. Additionally, it stimulates IL-10 secretion by TEMs, promoting Treg expansion and suppressing T cell proliferation (Fig. 3) [61].
Fig. 3. TME modulation and promotion of anti-tumor immunity by regorafenib.
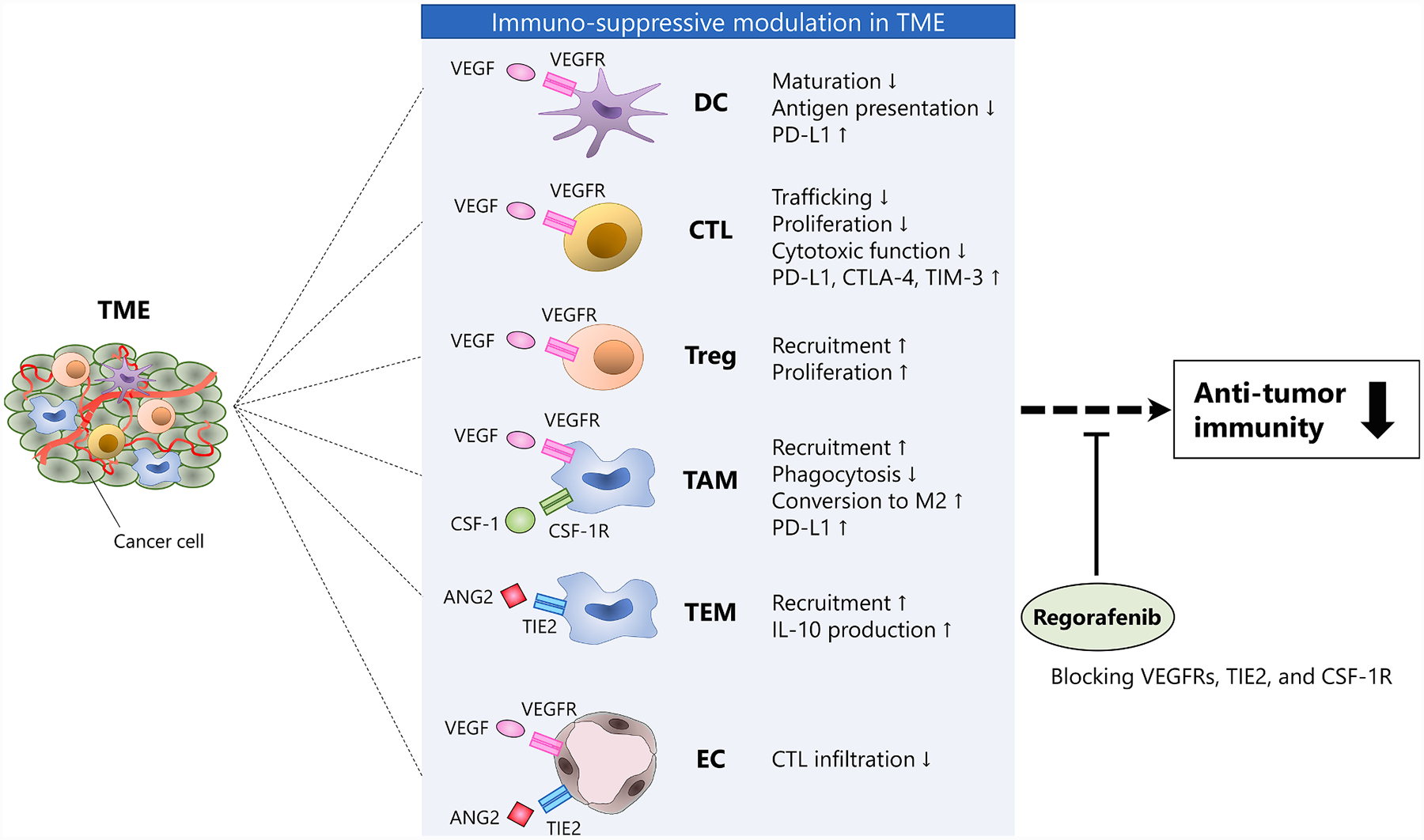
Several TME’s components promote an immune suppressive environment. Regorafenib modulates immuno-suppressive TME by blocking VEGFRs, TIE2, and CSF-1R, enhancing anti-tumor immunity. Abbreviations: CTL, cytotoxic T lymphocyte; DC, dendric cell; EC, endothelial cell; TAM, tumor-associated macrophage; TEM, TIE2-expressing monocyte/macrophage; TME, tumor microenvironment; Treg, regulatory T cell.
Antiangiogenic agents have an immune-modulating effect that restores anti-tumor activity by paving the TME and reprogramming the vascular function in favor of T cell trafficking improvement [58]. It has been shown that the dual inhibition of ANG2 and VEGFA promotes anti-tumor immunity, sensitizing to PD-1 checkpoint blockade better than single VEGFA inhibition [62]. This immune enhancement may be due to the anti-ANG2 effect, leading to vascular normalization and suppression of TEMs recruitment. An earlier study on a CRC murine model has found that regorafenib had a more pronounced reduction in TAMs and TEMs compared to selective VEGFR2 inhibitor [7]. Additionally, recent evidence in CRC has shown that regorafenib reduces the levels of TAM. Specifically, this occurs through the inhibition of the CSF-1 – CSF-1R axis which promotes monocytes differentiation into TAM [63]. Such preclinical data suggest an immunomodulatory effect of regorafenib, by both vascular normalization and macrophage modulation (Fig. 3).
Recently, an additional mechanism of immune-modulation by regorafenib was reported in a melanoma model [64]. Specifically, the authors found that regorafenib has most potent effect, among a number of kinase inhibitors, in promoting anti-tumor immunity by blocking the IFN-γ induced expression of immune suppressive factors. Mechanistically, regorafenib suppressed IFN-γ induced PD-L1 and IDO1 expression by inhibiting the RET – Src axis. [64]. Of note, it has been shown that immune-enhancing treatment by dual ANG2 and VEGFA inhibition also has negative feedback potential which leads to immunosuppressive PD-L1 upregulation in response to T cell-derived IFN-γ [62]. As a consequence, regorafenib may reset such negative feedback through this newly-reported function of suppressing IFN-γ induced PD-L1 expression through RET – Src inhibition, probably rendering its distinct immune-promoting function.
Following such promising preclinical findings showing regorafenib’s anti-immunosuppressive property, regorafenib in conjunction with immune checkpoint inhibitors is being used in novel combination strategies. A Japanese phase Ib trial (REGONIVO trial; NCT03406871) evaluated the simultaneous combination therapy of regorafenib and anti-PD-1 monoclonal antibody nivolumab in 50 patients (50% of the patients with mCRC, and 50% with advanced gastric cancer) refractory to standard chemotherapy. Importantly, enrolled individuals had microsatellite stability (MSS) tumors, which do not seem to benefit from nivolumab monotherapy, except for one patient with MSI high CRC. The results of this trial showed that this combination therapy had an excellent efficacy. Specifically, authors observed a 36% objective response rate in mCRC patients and 44% of that in advanced gastric cancer patients [65]. This promising efficacy is encouraging further studies on such combination strategy. Of note, currently there are additional ongoing clinical trials, as follows: French phase I/II trial (REGOMUNE trial; NCT03475953) evaluating regorafenib plus anti-PD-L1 monoclonal antibody avelumab in advanced digestive solid tumors, and American phase I/II trial (NCT03657641) evaluating regorafenib plus anti-PD-1 monoclonal antibody pembrolizumab in mCRC.
Inhibiting c-KIT and RET
C-KIT and RET are proto-oncogenic receptor tyrosine kinases (RTKs) potently inhibited by regorafenib. But, the content of inhibiting these RTKs remains incompletely understood in CRC, because their roles in colonic tumorigenesis have not yet fully elucidated. In this section, we discuss about this content based on limited evidences.
C-KIT expresses in specialized goblet cells that contribute to the stem cell niche in the murine colon crypt base [66]. In human CRC, c-KIT expression has been controversial [67–69]. While activating oncogenic mutations in c-KIT are well-documented in gastrointestinal stromal tumor and melanoma, it is not significantly mutated in CRC. Recently, Chen et al presented important evidence that c-KIT plays a functional role in CRC driven by autocrine and/or paracrine manner of its ligand (named as KITLG) rather than activating mutations. They found c-KIT-expressing CRC cells, which were highly tumorigenic especially in the CD44+ fraction. Their findings could propose the hypothesis that a subset of CRC patients expressing c-KIT may benefit from c-KIT inhibiting drugs such as regorafenib and imatinib, but clinical evidence is lacked so far [69,70].
In contrast to other RTKs, RET does not bind directly its ligands (GDNF family). Instead, the ligands bind to co-receptors, subsequently resulting in RET homodimerization and activation of downstream signaling. Constitutive oncogenic activation of RET can occur by two primary mechanisms; chromosomal rearrangements and mutations [71].
RET rearrangements are identified in 1–2% of non-small-cell lung cancers NSCLCs) and 5–10% of papillary thyroid carcinomas [72–75]. In mCRC, RET rearrangements are very rare (0.2%) and some types of fusion are identified (NCOA4-RET, CCDC6-RET, TRIM24-RET, TNIP1-RET and SNRNP70-RET) [76,77]. RET rearrangements independently predict a poorer prognosis of mCRC patients, likely to BRAF mutation, leading to a new precision medicine targeting this actionable gene alteration [77]. Multi-kinase inhibitors with activity against RET, such as cabozantinib, vandetanib and lenvatinib, have demonstrated clinical efficacy in a subtype of thyroid cancers and NSCLCs having RET rearrangement [75,78]. Of note, regorafenib provided a therapeutic response to a mCRC patient carrying CCDC6-RET fusion even though with a quarter-to-half dose of the recommended dose [76,79]. To date, no substantial differences in the activity of multi-kinase inhibitors against RET rearrangements with different upstream partners or breakpoints have been identified in preclinical studies [71]. Further investigation is needed to confirm the regorafenib’s activity in RET fusion positive mCRC patients.
Somatic RET mutations are also identified in a small subset of CRC patients [80]. However, not all RET mutations are activating and amenable to RET targeted therapy. For instance, RET G533C variant is clearly oncogenic, whereas P1047S variant is not, in CRC [80]. In addition, not all active RET mutations can be inhibiting by multi-kinase inhibitors having activity against RET. For instance, V804M/L gate-keeper mutations decrease access of agents such as cabozantinib and vandetanib to the hydrophobic ATP-binding pocket of the RET kinase [71]. To our knowledge, regorafenib have just reported to have activity against C634W mutant in preclinical assay [5]. Further accumulation of knowledge on this content is warranted.
Combination with cytotoxic agents
The results from clinical trials have shown the less successful activity of regorafenib in combination with standard oxaliplatin- or irinotecan-based chemotherapy for mCRC [81,82]. A single-arm phase II trial (CORDIAL trial: NCT01289821) evaluated efficacy and safety of regorafenib plus mFOLFOX6 as a first-line treatment. However, this treatment did not improve the objective response rate over historical controls [81]. Another placebo-controlled phase II trial (NCT01298570) assessed the efficacy of regorafenib plus FOLFIRI as a second-line treatment. As a result, despite the primary endpoint for progression-free survival (PFS) was met, the corresponding improvement in median PFS was only 0.8 months, and the benefit of overall survival (OS) was lacked [82]. In another setting, a phase I trial (RECAP trial: NCT02910843) is ongoing to evaluate efficacy and safety of neoadjuvant treatment with regorafenib and capecitabine combined with radiotherapy in locally advanced rectal cancer.
Biomarker discovery to predict clinical outcome
Numerous exploratory studies aimed at identifying predictive markers of regorafenib efficacy have been focusing on the following factors: oncogene mutation, gene expression, plasma protein, circulating tumor DNA level, MSI status, and primary tumor sidedness (Table 1) [21,83–94]. Currently, validated biomarkers for regorafenib are not available. However, findings from biomarker studies of CORRECT and CONCUR trials are informative because both phase III trials include a controlling placebo arm. This allows for the evaluation of each bio-marker’s predictive value for regorafenib benefit compared to placebo. Of note, a novel approach using non-invasive liquid biopsy was conducted on samples from the CORRECT trial [21]. Tumor genotyping of circulating DNA offers distinct advantages compared to DNA analysis from archival tumor tissue. Specifically, its value is based on the fact that it is not limited by intratumor heterogeneity. Additionally, non-invasive sampling allows for real-time evaluation, while archival tissue analysis is unable to show genotypic changes occurring through the treatment period [95]. In the CORRECT trial, KRAS and PIK3CA mutation status identified from circulating tumor DNA did not have a predictive value. Regorafenib’s benefit was shown across all subgroups based on mutation status. BRAF mutation was not evaluated in the correlative analysis because of its low mutational frequency [21]. Among the 15 plasma proteins evaluated in this trial, sTIE1 had a predictive potential for OS. Of note, its high concentration was associated with greater regorafenib benefit. However, in the multivariate analysis, this association was not significant [21]. Such observation is supported by another trial in renal cell carcinoma, showing that regorafenib induced greater tumor shrinkage in patients with high sTIE1 concentration compared to those with low concentration [96]. As previously mentioned, TIE1 works as a pro-angiogenic factor in tumors. However, the clinical meaning of this protein’ soluble conformation remains unclear. In preliminary analyses of single-nucleotide polymorphisms (SNPs) conducted in the CORRECT trial, VEGFA SNPs were not associated with regorafenib benefit. On the contrary, a SNP of TIE2 (rs7024233) seemed to have some predictive value [83]. Another exploratory analysis in this trial suggested that a molecular subtype based on comprehensive gene expression data might derive a differential benefit from regorafenib. However, it was not possible to rule out the potential bias linked to the small number of patients in some subtypes [84]. Additionally, although there seemed to be an inferior clinical benefit in patients with MSI high tumor, a significant association was not observed between MSI status and regorafenib benefit [85]. In the CONCUR trial, the authors reported the analysis of plasma protein biomarkers, which were almost the same as those evaluated in the CORRECT trial. In this biomarker study, ANG2 was shown to be just a prognostic marker. Its elevated levels were associated with poor OS. Of note, none of the plasma proteins analyzed were predictive of regorafenib benefit [86]. Taken together, based on these findings, the outcome of regorafenib treatment currently cannot be predicted by validated biomarkers.
Table 1.
Previous finding in the exploration of regorafenib’s predictive biomarkers in mCRC.
| Author (Year) | Trial name (Design) | N | Sample detail | Biomarker | Findings |
|---|---|---|---|---|---|
| Tabernero J (2015) [21] | CORRECT (Phase III) | 503 | DNA (blood) | Mutation status cfDNA level | KRAS and PIK3CA mutation status and cfDNA levels did not show a predictive value for regorafenib benefit on OS and PFS. |
| Tabernero J (2015) [21] | CORRECT (Phase III) | 729 | DNA (tumor) | Mutation status | KRAS mutation status did not show a predictive value for regorafenib benefit on OS and PFS. |
| Tabernero J (2015) [21] | CORRECT (Phase III) | 611 | Blood | Protein level | In the univariate analyses, high sTIE1 levels showed greater regorafenib benefits on OS, and a low VWF levels showed greater regorafenib benefits on PFS. However, these associations were not significant in the multivariate analyses. |
| Lambrechts D (2016) [83] | CORRECT (Phase III) | 528 | DNA (blood) | SNPs | No VEGFA SNPs were predictive for PFS, whereas TIE2 rs7024233 was correlated with regorafenib benefits on OS. |
| Teufel M (2015) [84] | CORRECT (Phase III) | 281 | RNA (tumor) | Molecular subtypes (Marisa, CMS) | High-risk subgroup (Marisa’s C4 + C6) showed greater regorafenib benefits on PFS vs. the low-risk subgroup (C1 + C2 + C3 + C5). CMS3 subgroup showed greater regorafenib benefits on PFS. |
| Köchert K (2017) [85] | CORRECT (Phase III) | 229 | DNA (tumor) | MSI status | The MSI-H subgroup appeared to have a smaller regorafenib benefit (OS and PFS) vs. the non-MSI-H subgroup. However, no significant interaction was shown between MSI status and regorafenib benefit (OS and PFS). |
| Teufel M (2016) [86] | CONCUR (Phase III) | 121 | Blood | Protein level | No plasma proteins were predictive for PFS and OS, while ANG2 showed prognostic value for OS and VWF for PFS (after correction for multiple testing). |
| Suenaga M (2018) [87] | No name (Retrospective) | 229 | DNA (blood/tumor) | SNPs | CCL4 rs1634517 and CCL3 rs1130371 were associated with PFS in the evaluation cohort, and PFS and OS in the validation cohort. |
| Suenaga M (2016) [88] | No name (Retrospective) | 54 | Blood | Protein level | A high baseline CCL5 level was associated with worse tumor shrinkage, PFS, and OS. Decreased VEGFA levels on day 21 were associated with better PFS. |
| Suenaga M (2018) [89] | No name (Retrospective) | 228 | DNA (blood/tumor) | SNPs |
MMP2 res1477017 was correlated with OS in the univariate analysis. MMP9 rs2274755 was correlated with PFS and OS in the multivariate analysis |
| Ducreux M (2019) [90] | CORRELATE (Prospective) | 967 | – | Primary tumor location | Primary tumor location was not predictive for PFS and OS. |
| Giampieri R (2016) [91] | ALICE-3 (Retrospective) | 138 | DNA (blood/tumor) | SNPs | VEGFA rs2010963 was correlated with PFS and OS. |
| Wong AL (2015) [92] | No name (Phase II) | 35 | DNA (blood)Tumor (FNA biopsy*) *pre- and post (day21) - treatment |
Mutation statuscfDNA level IHC analysis Proteomic analysis |
KRAS mutation were associated with shorter PFS. High baseline cfDNA levels were associated with shorter PFS. IHC downregulation: pVEGFR2, pAKT/upregulation: pMEK, pJUN, pSRC Downregulation of PI3K (proteomic analysis) was associated with longer PFS. |
| Vandeputte C (2018) [93] | RegARd-C (Phase II) | 20 | DNA (blood) | cfDNA level ctDNA change |
High baseline cfDNA levels were associated with shorter OS. An early increase in ctDNA copy number was associated with worse PFS and OS. |
| Khan K (2018) [94] | PROSPECT-R (Phase II) | 27 | DNA (blood) | ctDNA change | RAS mutant clones drop in ctDNA after 8 weeks was associated with better PFS and OS. |
Abbreviation: cfDNA, cell free DNA; CMS, consensus molecular subtype; ctDNA, circulating tumor DNA; OS, overall survival; PFS, progression-free survival.
Concluding remarks
Regorafenib is the first tyrosine kinase inhibitor with proven survival benefits for the treatment of mCRC patients. Its distinct mechanisms of action provide continuous anti-angiogenetic effects, and address a broad range of drug sensitivity. The potent effect of TME modulation leads to enhancing anti-tumor immunity. A number of novel concepts combining regorafenib with immune checkpoint inhibitors are being tested in ongoing clinical trials. This combination strategy has great potential for future development. There is a high clinical significance in this drug’s use as a personalized therapy. Therefore, biomarker strategies are critical to guide patient selection in mCRC.
Acknowledgements
H.-J.L. was supported by the National Cancer Institute (grant P30CA014089), Gloria Borges WunderGlo FoundationeThe Wunder Project, Dhont Family Foundation, San Pedro Peninsula Cancer Guild, Daniel Butler Research Fund, and Call to Cure Fund.
Footnotes
Declaration of Competing Interest
Heinz-Josef Lenz reports receiving speakers bureau honoraria from and is a consultant/advisory board member for Merck Serono, Bayer, and Genentech. The other authors have declared no conflicts of interest.
References
- [1].GLOBOCAN. All cancer fact sheet 2018, http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. 2018.
- [2].Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med 2014;371(17):1609–18. 10.1056/NEJMoa1403108. [DOI] [PubMed] [Google Scholar]
- [3].Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15(10):1065–75. 10.1016/s1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- [4].Miura K, Satoh M, Kinouchi M, Yamamoto K, Hasegawa Y, Philchenkov A, et al. The preclinical development of regorafenib for the treatment of colorectal cancer. Expert Opin Drug Discov 2014;9(9):1087–101. 10.1517/17460441.2014.924923. [DOI] [PubMed] [Google Scholar]
- [5].Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 2011;129(1):245–55. 10.1002/ijc.25864. [DOI] [PubMed] [Google Scholar]
- [6].Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2(7):358–64. 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- [7].Abou-Elkacem L, Arns S, Brix G, Gremse F, Zopf D, Kiessling F, et al. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther 2013;12(7):1322–31. 10.1158/1535-7163.MCT-12-1162. [DOI] [PubMed] [Google Scholar]
- [8].Grothey A, Cutsem EV, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. The Lancet 2013;381(9863):303–12. 10.1016/s0140-6736(12)61900-x. [DOI] [PubMed] [Google Scholar]
- [9].Li J, Qin S, Xu R, Yau TCC, Ma B, Pan H, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2015;16(6):619–29. 10.1016/s1470-2045(15)70156-7. [DOI] [PubMed] [Google Scholar]
- [10].Van Cutsem E, Martinelli E, Cascinu S, Sobrero A, Banzi M, Seitz JF, et al. Regorafenib for patients with metastatic colorectal cancer who progressed after standard therapy: results of the large, single-arm, open-label phase IIIb CONSIGN study. Oncologist 2019;24(2):185–92. 10.1634/theoncologist.2018-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].O’Connor Juan M, Öhler L, Scheithauer W, Metges J-P, Dourthe L-M, de Groot Jan W, et al. PD-025Real-world dosing of regorafenib in metastatic colorectal cancer (mCRC): interim analysis from the prospective, observational CORRELATE study. Ann Oncol 2017;28(suppl_3). 10.1093/annonc/mdx263.024. [DOI] [Google Scholar]
- [12].Komatsu Y, Muro K, Yamaguchi K, Satoh T, Uetake H, Yoshino T, et al. Safety and efficacy of regorafenib post-marketing surveillance (PMS) in Japanese patients with metastatic colorectal cancer (mCRC). J Clin Oncol 2017;35(4_suppl):721 10.1200/JCO.2017.35.4_suppl.721. [DOI] [Google Scholar]
- [13].Adenis A, de la Fouchardiere C, Paule B, Burtin P, Tougeron D, Wallet J, et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer 2016;16:412 10.1186/s12885-016-2440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schulz H, Janssen J, Strauss UP, Langen M, Dworschak KU, Fiala-Buskies S, et al. Clinical efficacy and safety of regorafenib (REG) in the treatment of metastatic colorectal cancer (mCRC) in daily practice in Germany: final results of the prospective multicentre non-interventional RECORA study. J Clin Oncol 2018;36(4_suppl):748 10.1200/JCO.2018.36.4_suppl.748. [DOI] [Google Scholar]
- [15].Grothey A, George S, van Cutsem E, Blay JY, Sobrero A, Demetri GD. Optimizing treatment outcomes with regorafenib: personalized dosing and other strategies to support patient care. Oncologist 2014;19(6):669–80. 10.1634/theoncologist.2013-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bekaii-Saab TS, Ou F-S, Ahn DH, Boland PM, Ciombor KK, Heying EN, et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open-label, phase 2 study. Lancet Oncol 2019;20(8):1070–82. 10.1016/s1470-2045(19)30272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].NCCN. Clinical Practice Guidelines in Oncology (Colon Cancer Version 2. 2019); 2019.
- [18].Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 2009;361(25):2449–60. 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [20].Lange F, Franz B, Maletzki C, Linnebacher M, Huhns M, Jaster R. Biological and molecular effects of small molecule kinase inhibitors on low-passage human colorectal cancer cell lines. Biomed Res Int 2014;2014:568693 10.1155/2014/568693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tabernero J, Lenz H-J, Siena S, Sobrero A, Falcone A, Ychou M, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol 2015;16(8):937–48. 10.1016/s1470-2045(15)00138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garcia-Alfonso P, Benavides M, Falco E, Munoz A, Gomez A, Sastre J, et al. Single-agent regorafenib in metastatic colorectal cancer patients with any RAS or BRAF mutation previously treated with FOLFOXIRI plus Bevacizumab (PREVIUM Trial). Oncologist 2018;23(11):1271–e128. 10.1634/theoncologist.2018-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clarke JM, Hurwitz HI, Rangwala F. Understanding the mechanisms of action of antiangiogenic agents in metastatic colorectal cancer: a clinician’s perspective. Cancer Treat Rev 2014;40(9):1065–72. 10.1016/j.ctrv.2014.07.001. [DOI] [PubMed] [Google Scholar]
- [24].Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis 2017;20(4):409–26. 10.1007/s10456-017-9562-9. [DOI] [PubMed] [Google Scholar]
- [25].Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol 2013;3:211 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol: Official J Am Soc Clin Oncol 2008;26(12):2013–9. 10.1200/JCO.2007.14.9930. [DOI] [PubMed] [Google Scholar]
- [27].Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol: Off J Am Soc Clin Oncol 2012;30(28):3499–506. 10.1200/JCO.2012.42.8201. [DOI] [PubMed] [Google Scholar]
- [28].Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol 2015;16(5):499–508. 10.1016/s1470-2045(15)70127-0. [DOI] [PubMed] [Google Scholar]
- [29].Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 2017;16(9):635–61. 10.1038/nrd.2016.278. [DOI] [PubMed] [Google Scholar]
- [30].Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 2008;14(1):25–36. 10.1016/j.devcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- [31].Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019;8(5). 10.3390/cells8050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thurston G, Daly C. The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb Perspect Med 2012;2(9):a006550 10.1101/cshperspect.a006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci USA 2002;99(17):11205–10. 10.1073/pnas.172161899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer 2003;3(6):401–10. 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- [35].Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y. Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine micro-vascular endothelial cells. J Biol Chem 1999;274(22):15732–9. 10.1074/jbc.274.22.15732. [DOI] [PubMed] [Google Scholar]
- [36].Goede V, Coutelle O, Neuneier J, Reinacher-Schick A, Schnell R, Koslowsky TC, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer 2010;103(9):1407–14. 10.1038/sj.bjc.6605925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mueller SB, Kontos CD. Tie1: an orphan receptor provides context for angiopoietin-2/Tie2 signaling. J Clin Invest 2016;126(9):3188–91. 10.1172/JCI89963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Savant S, La Porta S, Budnik A, Busch K, Hu J, Tisch N, et al. The orphan receptor Tie1 controls angiogenesis and vascular remodeling by differentially regulating Tie2 in Tip and stalk cells. Cell Rep 2015;12(11):1761–73. 10.1016/j.celrep.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Singh H, Hansen TM, Patel N, Brindle NP. The molecular balance between receptor tyrosine kinases Tie1 and Tie2 is dynamically controlled by VEGF and TNFalpha and regulates angiopoietin signalling. PLoS ONE 2012;7(1):e29319 10.1371/journal.pone.0029319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest 2016;126(9):3511–25. 10.1172/JCI84871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest 2016;126(9):3495–510. 10.1172/JCI84923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].La Porta S, Roth L, Singhal M, Mogler C, Spegg C, Schieb B, et al. Endothelial Tie1-mediated angiogenesis and vascular abnormalization promote tumor progression and metastasis. J Clin Invest 2018;128(2):834–45. 10.1172/JCI94674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow Ching C, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to vegf signaling blockade. Cell Reports 2014;8(3):696–706. 10.1016/j.celrep.2014.06.059. [DOI] [PubMed] [Google Scholar]
- [44].Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci USA 2016;113(16):4476–81. 10.1073/pnas.1525360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Falcon BL, Hashizume H, Koumoutsakos P, Chou J, Bready JV, Coxon A, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol 2009;175(5):2159–70. 10.2353/ajpath.2009.090391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bagri A, Berry L, Gunter B, Singh M, Kasman I, Damico LA, et al. Effects of anti-VEGF treatment duration on tumor growth, tumor regrowth, and treatment efficacy. Clin Cancer Res 2010;16(15):3887–900. 10.1158/1078-0432.CCR-09-3100. [DOI] [PubMed] [Google Scholar]
- [47].Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 2013;14(1):29–37. 10.1016/s1470-2045(12)70477-1. [DOI] [PubMed] [Google Scholar]
- [48].Ellis LM, Hicklin DJ. Pathways mediating resistance to vascular endothelial growth factor-targeted therapy. Clin Cancer Res 2008;14(20):6371–5. 10.1158/1078-0432.CCR-07-5287. [DOI] [PubMed] [Google Scholar]
- [49].Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10(2):116–29. 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- [50].Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8(4):299–309. 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- [51].Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA 2007;104(43):17069–74. 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Manzat Saplacan RM, Balacescu L, Gherman C, Chira RI, Craiu A, Mircea PA, et al. The role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflamm 2017;2017:4708076 10.1155/2017/4708076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest 2007;117(8):2114–22. 10.1172/JCI31334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tommelein J, Verset L, Boterberg T, Demetter P, Bracke M, De Wever O. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front Oncol 2015;5:63 10.3389/fonc.2015.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007;131(3):463–75. 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- [56].Zhao P, Wang Y, Kang X, Wu A, Yin W, Tang Y, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci 2018;9(10):2674–89. 10.1039/c7sc04853j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res 2019. 10.1158/1078-0432.CCR-18-1543. [DOI] [PubMed] [Google Scholar]
- [58].Schmittnaegel M, De Palma M. Reprogramming tumor blood vessels for enhancing immunotherapy. Trends Cancer 2017;3(12):809–12. 10.1016/j.trecan.2017.10.002. [DOI] [PubMed] [Google Scholar]
- [59].Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015;212(2):139–48. 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lewis CE, Ferrara N. Multiple effects of angiopoietin-2 blockade on tumors. Cancer Cell 2011;19(4):431–3. 10.1016/j.ccr.2011.03.016. [DOI] [PubMed] [Google Scholar]
- [61].Coffelt SB, Chen YY, Muthana M, Welford AF, Tal AO, Scholz A, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol 2011;186(7):4183–90. 10.4049/jimmunol.1002802. [DOI] [PubMed] [Google Scholar]
- [62].Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 2017;9(385). 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- [63].Hoff S, Grünewald S, Röse L, Zopf D. 1198PImmunomodulation by regorafenib alone and in combination with anti PD1 antibody on murine models of colorectal cancer. Ann Oncol 2017;28(suppl_5). 10.1093/annonc/mdx376.060. [DOI] [Google Scholar]
- [64].Wu RY, Kong PF, Xia LP, Huang Y, Li ZL, Tang YY, et al. Regorafenib promotes antitumor immunity via inhibiting PD-L1 and IDO1 expression in melanoma. Clin Cancer Res 2019;25(14):4530–41. 10.1158/1078-0432.CCR-18-2840. [DOI] [PubMed] [Google Scholar]
- [65].Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric (GC) or colorectal cancer (CRC): an open-label, dose-finding, and dose-expansion phase 1b trial (REGONIVO, EPOC1603). J Clin Oncol 2019;37(15_suppl):2522 10.1200/JCO.2019.37.15_suppl.2522. [DOI] [PubMed] [Google Scholar]
- [66].Rothenberg ME, Nusse Y, Kalisky T, Lee JJ, Dalerba P, Scheeren F, et al. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology 2012;142(5):1195–205. 10.1053/j.gastro.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Reed J, Ouban A, Schickor FK, Muraca P, Yeatman T, Coppola D. Immunohistochemical staining for c-Kit (CD117) is a rare event in human colorectal carcinoma. Clin Colorectal Cancer 2002;2(2):119–22. 10.3816/CCC.2002.n.018. [DOI] [PubMed] [Google Scholar]
- [68].Yorke R, Chirala M, Younes M. c-kit proto-oncogene product is rarely detected in colorectal adenocarcinoma. J Clinical Oncol: Off J Am Soc Clin Oncol 2003;21(20):3885–6. 10.1200/JCO.2003.99.213. [DOI] [PubMed] [Google Scholar]
- [69].Chen EC, Karl TA, Kalisky T, Gupta SK, O’Brien CA, Longacre TA, et al. KIT signaling promotes growth of colon xenograft tumors in mice and is up-regulated in a subset of human colon cancers. Gastroenterology 2015;149(3):705–17 e2 10.1053/j.gastro.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Shah YM, van den Brink GR. c-Kit as a novel potential therapeutic target in colorectal cancer. Gastroenterology 2015;149(3):534–7. 10.1053/j.gastro.2015.07.027. [DOI] [PubMed] [Google Scholar]
- [71].Drilon A, Hu ZI, Lai GGY, Tan DSW. Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol 2018;15(3):151–67. 10.1038/nrclinonc.2017.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50. 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159(3):676–90. 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bronte G, Ulivi P, Verlicchi A, Cravero P, Delmonte A, Crino L. Targeting RET-rearranged non-small-cell lung cancer: future prospects. Lung Cancer 2019;10:27–36. 10.2147/LCTT.S192830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, et al. Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 2015;6(30):28929–37. 10.18632/oncotarget.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, Fuca G, et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol: Off J Eur Soc Med Oncol. 2018;29(6):1394–401. 10.1093/annonc/mdy090. [DOI] [PubMed] [Google Scholar]
- [78].Wells SA Jr., Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol: Off J Am Soc Clin Oncol 2012;30(2):134–41. 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sunakawa Y, Furuse J, Okusaka T, Ikeda M, Nagashima F, Ueno H, et al. Regorafenib in Japanese patients with solid tumors: phase I study of safety, efficacy, and pharmacokinetics. Invest New Drugs 2014;32(1):104–12. 10.1007/s10637-013-9953-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mendes Oliveira D, Grillone K, Mignogna C, De Falco V, Laudanna C, Biamonte F, et al. Next-generation sequencing analysis of receptor-type tyrosine kinase genes in surgically resected colon cancer: identification of gain-of-function mutations in the RET proto-oncogene. J Exp Clin Cancer Res: CR 2018;37(1):84 10.1186/s13046-018-0746-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Argiles G, Saunders MP, Rivera F, Sobrero A, Benson A 3rd, Guillen Ponce C, et al. Regorafenib plus modified FOLFOX6 as first-line treatment of metastatic colorectal cancer: a phase II trial. Eur J Cancer 2015;51(8):942–9. 10.1016/j.ejca.2015.02.013. [DOI] [PubMed] [Google Scholar]
- [82].Sanoff HK, Goldberg RM, Ivanova A, O’Reilly S, Kasbari SS, Kim RD, et al. Multicenter, randomized, double-blind phase 2 trial of FOLFIRI with regorafenib or placebo as second-line therapy for metastatic colorectal cancer. Cancer 2018;124(15):3118–26. 10.1002/cncr.31552. [DOI] [PubMed] [Google Scholar]
- [83].Lambrechts D, Koechert K, Schulz A, Vonk R, Rutstein M, Kobina S, et al. PD-003Analysis of single-nucleotide polymorphisms (SNPs) in the phase 3 CORRECT trial of regorafenib vs placebo in patients with metastatic colorectal cancer (mCRC). Ann Oncol 2016;27(suppl_2):ii102–ii. 10.1093/annonc/mdw200.03. [DOI] [Google Scholar]
- [84].Teufel M, Schwenke S, Seidel H, Beckmann G, Reischl J, Vonk R, et al. Molecular subtypes and outcomes in regorafenib-treated patients with metastatic colorectal cancer (mCRC) enrolled in the CORRECT trial. J Clin Oncol 2015;33(15_suppl):3558 10.1200/jco.2015.33.15_suppl.3558. [DOI] [Google Scholar]
- [85].Köchert K, Beckmann G, Teufel M. 534PExploratory analysis of baseline microsatellite instability (MSI) status in patients with metastatic colorectal cancer (mCRC) treated with regorafenib (REG) or placebo in the phase 3 CORRECT trial. Ann Oncol 2017;28(suppl_5). 10.1093/annonc/mdx393.060. [DOI] [Google Scholar]
- [86].Teufel M, Kalmus J, Rutstein MD, Koechert K, Seidel H, Reischl J, et al. Analysis of plasma protein biomarkers from the phase 3 CONCUR study of regorafenib in Asian patients with metastatic colorectal cancer (mCRC). J Clin Oncol 2016;34(4_suppl):672 10.1200/jco.2016.34.4_suppl.672. [DOI] [Google Scholar]
- [87].Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Ning Y, et al. Gene polymorphisms in the CCL5/CCR5 pathway as a genetic biomarker for outcome and hand-foot skin reaction in metastatic colorectal cancer patients treated with regorafenib. Clin Colorectal Cancer 2018;17(2):e395–414. 10.1016/j.clcc.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Suenaga M, Mashima T, Kawata N, Wakatsuki T, Horiike Y, Matsusaka S, et al. Serum VEGF-A and CCL5 levels as candidate biomarkers for efficacy and toxicity of regorafenib in patients with metastatic colorectal cancer. Oncotarget 2016;7(23):34811–23. 10.18632/oncotarget.9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Ning Y, et al. Matrix metalloproteinase-related gene polymorphisms to predict efficacy of regorafenib in patients with metastatic colorectal cancer. J Clin Oncol 2018;36(4_suppl):692 10.1200/JCO.2018.36.4_suppl.692. [DOI] [Google Scholar]
- [90].Ducreux M, Petersen LN, Öhler L, Bergamo F, Metges J-P, Groot JWd, et al. Outcomes by tumor location in patients with metastatic colorectal cancer (mCRC) treated with regorafenib (REG): final analysis from the prospective, observational CORRELATE study. J Clin Oncol 2019;37(4_suppl):539 10.1200/JCO.2019.37.4_suppl.539. [DOI] [Google Scholar]
- [91].Giampieri R, Salvatore L, Del Prete M, Prochilo T, D’Anzeo M, Loretelli C, et al. Angiogenesis genotyping and clinical outcome during regorafenib treatment in metastatic colorectal cancer patients. Sci Rep 2016;6:25195 10.1038/srep25195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wong AL, Lim JS, Sinha A, Gopinathan A, Lim R, Tan CS, et al. Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J Transl Med 2015;13:57 10.1186/s12967-015-0405-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Vandeputte C, Kehagias P, El Housni H, Ameye L, Laes JF, Desmedt C, et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi-kinase inhibitor. Oncotarget 2018;9(25):17756–69. 10.18632/oncotarget.24879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Khan K, Rata M, Cunningham D, Koh DM, Tunariu N, Hahne JC, et al. Functional imaging and circulating biomarkers of response to regorafenib in treatment-refractory metastatic colorectal cancer patients in a prospective phase II study. Gut 2018;67(8):1484–92. 10.1136/gutjnl-2017-314178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Diaz LA Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol: Off J Am Soc Clin Oncol. 2014;32(6):579–86. 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Eisen T, Joensuu H, Nathan PD, Harper PG, Wojtukiewicz MZ, Nicholson S, et al. Regorafenib for patients with previously untreated metastatic or unresectable renal-cell carcinoma: a single-group phase 2 trial. Lancet Oncol 2012;13(10):1055–62. 10.1016/s1470-2045(12)70364-9. [DOI] [PubMed] [Google Scholar]