

Abstract
DNA double-strand break repair allows cells to survive both exogenous and endogenous insults to the genome. In yeast, the recombinases Rad51 and Rad52 are central to multiple forms of homology-dependent repair. Classically, Rad51 and Rad52 are thought to act cooperatively, with formation of the functional Rad51 nucleofilament facilitated by the mediator function of Rad52. Several studies have now identified functions for the interaction between Rad51 and Rad52 that are independent of the mediator function of Rad52 and affect a seemingly diverse array of functions in de novo telomere addition, global chromosome mobility following DNA damage, Rad51 nucleofilament stability, checkpoint adaptation, and microhomology-mediated chromosome rearrangements. Here, we review these functions with an emphasis on our recent discovery that the Rad51-Rad52 interaction influences the probability of de novo telomere addition at sites preferentially targeted by telomerase following a double-strand break (DSB). We present data addressing the prevalence of sites within the yeast genome that are capable of stimulating de novo telomere addition following a DSB and speculate about the potential role such sites may play in genome stability.
Keywords: DNA repair, homologous recombination, Rad51, Rad52, telomere, telomerase
Double-strand break (DSB) repair is crucial to a cell’s ability to survive exogenous DNA damage, spontaneous chromosome breaks resulting from normal cellular processes such as DNA replication, and programmed breaks such as those occurring during meiosis, mating type switching, or class-switch recombination. A number of factors including the nature of the chromosome ends, cell cycle stage, and availability of homology impact the ultimate outcome of repair. Broadly, DSB repair occurs through homology-dependent mechanisms, the majority of which demonstrate high fidelity, or through mechanisms that require limited or no homology and are therefore more subject to error [non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ)] (Aylon and Kupiec 2004; Mehta and Haber 2014).
In some situations, repair of DSBs generates large deletions, inversions, or translocations (so-called gross chromosomal rearrangements or GCRs). For example, break-induced replication occurs when homology is present on only one side of a DNA break, precluding second end capture. The invading 3’ end serves as a primer for replicative copying of the invaded sequence, resulting in a non-reciprocal translocation (Kramara et al. 2018). Single-strand annealing can generate large deletions through the association of directly repeated sequences flanking a DSB, even when those sequences are many kilobases from the original break (Bhargava et al. 2016). Terminal deletions are generated following a DSB by the action of telomerase to create a new, or de novo, telomere (Pennaneach et al. 2006). Such events are highly mutagenic, but depending on the nature of the sequences that are lost, duplicated, or rearranged, may nonetheless allow a cell to survive a level or type of DNA damage that would otherwise be lethal.
In this mini-review, we focus on studies in the budding yeast, Saccharomyces cerevisiae. Isolation of mutants with increased sensitivity to ionizing radiation identified multiple genes required for homology-dependent repair (the RAD52 epistasis group including RAD52, RAD50, RAD51, RAD54, RAD55, RAD57, RAD59, RDH54/TID1, MRE11, and XRS2). The contributions of many years of work have led to the following general model for the early steps of HDR [for review, see Aylon and Kupiec 2004; Krogh and Symington 2004; Jasin and Rothstein 2013; Piazza and Heyer 2019; Waterman et al. 2020]. Following a DSB, the 5’ terminating strands are progressively degraded by a combination of nucleases to generate 3’ overhangs. In the absence of repair, resection is estimated to occur at a rate of ~4 kb per hour, with the potential to expose long regions of single-stranded DNA (ssDNA) (Fishman-Lobell et al. 1992). Exposed ssDNA is rapidly bound by the three-protein complex Replication Protein A (RPA) that serves both to protect the DNA and, when the amount of ssDNA is sufficient, to initiate checkpoint signaling. RPA is subsequently replaced by Rad51 to form a nucleoprotein filament that initiates the search for homologous sequences and effects strand invasion. Although Rad51 is capable of binding ssDNA, its association is inhibited by RPA such that replacement of RPA with Rad51 to form the nucleofilament requires the “mediator” activity of Rad52. Additional complexities underlying the interactions between RPA, Rad51, and Rad52 and the influence of those interactions on the repair process are becoming increasingly apparent.
RAD51 and RAD52 are classically defined as belonging to the same epistasis group, a result attributed to the requirement for Rad52’s mediator role in formation of the Rad51 nucleofilament. Several recent studies (and one older observation) reveal that Rad51 and Rad52 act antagonistically under some circumstances. As discussed here, these studies highlight novel roles for the interaction between yeast Rad51 and Rad52 in modulating cellular responses that occur after the initiation of resection. For example, our recent studies show that direct protein interaction between Rad51 and Rad52 modulates the probability with which telomerase induces new telomere formation following a DSB. We conclude this review by discussing the prevalence of sites within the yeast genome that are capable of stimulating de novo telomere addition following a DSB and speculate about the potential role such sites may play in genome stability.
SiRTAs: Hotspots of de novo telomere addition
Classic assays monitoring the frequencies and types of spontaneous GCR events identified de novo telomere addition as a common event resulting in loss of the terminal chromosome arm (Myung et al. 2001; Putnam et al. 2004). Because these assays take advantage of the counter-selectable marker CAN1 on the left arm of chromosome 5, scrutiny was directed toward events occurring in the non-essential sequences proximal to CAN1. Several studies revealed a “hotspot” of de novo telomere addition associated with a short region of TG-rich sequence (Stellwagen et al. 2003; Zhang and Durocher 2010). It was later shown that both this sequence on chromosome 5L and another TG-rich region on chromosome 9L undergo de novo telomere addition at rates ~ 200-fold higher than neighboring sequences (Obodo et al. 2016). These sites, which we have named SiRTAs (Sites of Repair-associated Telomere Addition), are not prone to breakage, but are capable of stimulating telomere addition even when the initiating chromosome break is located several kilobases distal to the eventual site at which telomerase acts.
As described in more detail in the last section of this review, we monitor de novo telomere addition in haploid cells by inducing a persistent DSB ~2 kb distal to the SiRTA. Cells that survive cleavage (i.e. have repaired the break in a manner that destroys the recognition site for the nuclease) are screened for those that have lost the non-essential sequences distal to the break. GCR events are constrained to occur between the cleavage site and the most distal essential gene on the chromosome arm. While the SiRTA itself typically comprises less than 1% of this region, it can account for 20–40% of the observed GCR events and nearly all GCR events at the SiRTA are the result of telomere addition (Obodo et al. 2016).
The SiRTAs on chromosomes 5 and 9 bear some similarity to the TG1–3 repeat found at budding yeast telomeres and are oriented such that resection of the 5’ terminating strand centromere-proximal to the break will place the TG-rich sequence on the 3’ overhang. Both SiRTAs are bipartite, with two separate TG-rich tracts. One tract (the core) is more distal on the chromosome arm and is the site at which telomerase typically acts to add a new telomere. However, the core alone is insufficient; a proximal “stim” sequence enhances telomere addition within the core. As discussed in more detail below, the stim sequence is recognized by the single-stranded telomeric DNA binding protein Cdc13 and can be functionally replaced by artificial recruitment of Cdc13 (Obodo et al. 2016). The bipartite structure of SiRTAs is reminiscent of observations by the Haber lab that insertion of an artificial TG-rich tract (originally intended to serve as a “seed” sequence for telomere addition) stimulates de novo telomere addition at endogenous TG sequences located distal to the seed (Kramer and Haber 1993). Mangahas et al. likewise observed telomere addition near, but not within, an endogenous telomere-like sequence (Mangahas et al. 2001). It seems likely, therefore, that this bipartite structure is a common attribute of SiRTAs. Mutational analysis of the chromosome 5 SiRTA suggests that the distance between the core and stim negatively correlates with the frequency of de novo telomere addition (Obodo et al. 2016).
The Rad51-Rad52 interaction affects the frequency of de novo telomere addition
Telomerase action requires a 3’ single-stranded overhang, implying that resection must proceed through the SiRTA before telomerase can act and suggesting that proteins associated with the ssDNA after resection might influence the probability of telomere addition. Indeed, previous work suggested that deletion of RAD51 reduces the frequency of de novo telomere addition (Oza et al. 2009), a result that we reproduced at SiRTAs on chromosome 5 and 9 (Epum et al. 2020). Surprisingly, deletion of RAD52 did not reduce, and in some cases modestly increased, de novo telomere addition at SiRTAs (Obodo et al. 2016; Epum et al. 2020). Given that RAD51 and RAD52 generally function in the same genetic pathway, this result was puzzling and suggested that the defect in the rad51Δ strain could not be a simple consequence of failure to form the Rad51 nucleofilament (which also cannot form in the absence of Rad52). We considered the possibility that Rad51 directly stimulates de novo telomere addition in a manner independent of its function in recombinational repair. However, the telomere addition defect of the rad51 deletion strain is completely suppressed by additional deletion of RAD52. Mutations in other genes of the RAD52 epistasis group that act in parallel or downstream of Rad51 (RAD55/57 and RAD54) have no effect on de novo telomere addition either alone or in combination with deletion of RAD51 (Epum et al. 2020), suggesting that the effect is highly specific to Rad52. The lack of involvement of Rad55/57 in particular supports the idea that Rad51 loading is not required for de novo telomere addition, since these genes affect the efficiency of nucleofilament formation (Hays et al. 1995; Sugawara et al. 2003). RAD51 and RAD52 affect the intranuclear trafficking of telomerase in response to DSBs (Ouenzar et al. 2017), but the epistatic relationship between the genes differs for the trafficking and SiRTA phenotypes, suggesting that the phenomenon observed at SiRTAs is not directly related to the influence of Rad51 and Rad52 on telomerase localization. Taken together, our genetic results are consistent with a model in which Rad52 actively suppresses de novo telomere addition in a manner that is counteracted or prevented by Rad51.
In an attempt to understand this unusual relationship between Rad51 and Rad52, a series of rad51 alleles was characterized. Mutations Y388H and G393D, both previously demonstrated to abrogate interaction between Rad51 and Rad52 (Krejci et al. 2001; Seong et al. 2009), decreased de novo telomere addition, suggesting that physical contact of Rad52 with Rad51 is required to prevent Rad52 from inhibiting de novo telomere addition. To assay the importance of nucleofilament formation, we examined the effect of mutating RAD51 lysine 191 to either alanine or arginine. These mutations lie in the Walker A motif of Rad51 and abolish ATP hydrolysis (Sung and Stratton 1996). The K191A variant is unable to bind ssDNA, does not form visible foci upon the induction of DNA damage, and eliminates the detection of Rad51 at a break by chromatin immunoprecipitation (ChIP) (Sung and Stratton 1996; Lee et al. 2003; Fung et al. 2006). Cells expressing this variant display damage sensitivity equivalent to the null allele (Sung and Stratton 1996). Given these phenotypes, it is remarkable that the rad51-K191A allele (and rad51-K191R) supports normal levels of de novo telomere addition (indistinguishable from WT RAD51), even when integrated at the endogenous locus (Epum et al. 2020). Both mutant proteins are expressed and retain interaction with Rad52, albeit at reduced levels compared to WT (Fung et al. 2006; Epum et al. 2020). Since these data suggest that Rad51 must interact directly with Rad52 to prevent Rad52 from inhibiting de novo telomere addition, we infer that either this functionally important interaction can occur off DNA or that association of the Rad51-K191A variant with DNA-associated Rad52 is too transient or diffuse to detect at breaks.
The results described above predict that mutation of Rad52 to disrupt interaction with Rad51 should phenocopy the loss of Rad51 by releasing Rad52 from negative regulation. We tested the effect of rad52-Δ409-412 (lacking amino acids YEKF; see Box 1 for a discussion of nomenclature), shown by the Sung laboratory to be severely compromised for interaction with Rad51, but to retain the capacity to oligomerize, bind DNA, and facilitate strand annealing (a property implying normal interaction with RPA)(Krejci et al. 2002). Expression of rad52-Δ409-412 in a RAD51 background decreases de novo telomere addition at SiRTAs and there is no additional decrease observed upon deletion of RAD51 (Epum et al. 2020). This epistatic relationship suggests that Rad52-Δ409-412 retains the ability to suppress de novo telomere addition, but can no longer be inhibited by Rad51.
BOX1: A note on Rad52 nomenclature.
RAD52 contains a total of five start codons (ATG) within the first 34 amino acids of the open reading frame. The rad52Δ409-412 allele (first described in Krejci et al. 2002) is named based on the assumption that the first ATG is utilized for translation. However, the major transcript starts 10 nucleotides downstream of the second ATG (Adzuma et al. 1984). In 2006, the Rothstein lab demonstrated that Rad52 protein is translated from the last three start sites (Antúnez de Mayolo et al. 2006). The Saccharomyces genome database now shows the protein sequence of Rad52 starting from the third ATG to generate a polypeptide of 471 amino acids. By this new numbering, the rad52Δ409-412 allele deletes amino acids 376–379. Inconsistency in the literature regarding the use of the “old” versus “new” numbering system has created confusion. Here, we refer to the deletion mutation by its original name (Δ409-412). Mutation of the first residue of this motif is referred to here as rad52-Y409A, but in Ma et al. as rad52-Y376A (Ma et al. 2018). Conversion to specific allele names rather than amino acid numbers would be beneficial where multiple isoforms exist.
Several groups have recently provided additional insight into the effects of deleting or mutating the Rad52 YEKF motif. Coïc and colleagues find that while the interaction between Rad51 and Rad52-Y409A is profoundly reduced both in vivo and in vitro, mediator activity of Rad52-Y409A is relatively normal as assessed both through in vivo assays of recombination proficiency and assembly of Rad51 nucleofilaments on RPA-coated DNA in vitro (Ma et al. 2018). Like WT Rad52, the Rad52-Y409A variant retains interaction with Rad51 nucleofilaments in vitro and is detected at breaks by ChIP (Ma et al. 2018). Smith et al. observe Rad51 foci in a homozygous rad52-Δ409-412 diploid after DNA damage, consistent with retention of mediator activity, although foci are less frequent and intense than in WT cells. Analyses of the total number of foci and the colocalization of Rad51 and Rad52-Δ409-412 led these authors to propose that the interaction between Rad51 and Rad52 contributes to the dissociation of Rad52 from the Rad51 nucleofilament (Smith et al. 2018). Together, these results are consistent with our observation that Rad52-Δ409-412 retains the ability to block de novo telomere addition, a function likely to require continued association of Rad52 with the nucleofilament.
Other reported roles for the Rad51-Rad52 interaction
Over the past few years, several additional functions have been attributed to the Rad51-Rad52 interaction. We describe those studies here, considering in each case whether there is a direct functional link between these roles and the observed effects on de novo telomere addition.
Protection of the Rad51 nucleofilament from disassembly by Srs2:
Cells lacking the Srs2 translocase are defective for repair by single-strand annealing (SSA), but that sensitivity is suppressed by deletion of RAD51 (Vaze et al. 2002). The failure of srs2 cells to complete repair by SSA or BIR reflects a Rad51-mediated inability to successfully resynthesize long regions of single-stranded DNA generated by resection (Vasianovich et al. 2017; Elango et al. 2017). Stability of the Rad51 nucleofilament against disruption by Srs2 is affected by the Rad51-Rad52 interaction. Ma et al. report that mutations in the Rad52 YEKF motif suppress the extreme sensitivity of a strain lacking SRS2 to DNA damage and that Rad51 filaments formed in the presence of Rad52-Y409A in vitro are more sensitive to disruption by Srs2 than those formed with WT Rad52 (Ma et al. 2018). A modest reduction in Rad51 association with a break in the rad52-Y409A strain is completely suppressed by deletion of Srs2, consistent with a model in which the Rad51-Rad52 interaction increases nucleofilament stability, but is not required for mediator activity per se (Ma et al. 2018). This alteration of nucleofilament properties is unlikely to directly account for decreased de novo telomere addition in the rad52-Δ409-412 strain since deletion of RAD52 also suppresses damage sensitivity of the srs2Δ strain (Ma et al. 2018), while having no effect on de novo telomere addition (Epum et al. 2020).
Regulation of global mobility following DNA damage:
Smith et al. found that the Rad51-Rad52 interaction regulates global mobility after DNA damage (Smith et al. 2018). Extensive or prolonged DNA damage causes increased mobility of DNA in the nucleus in a manner dependent on checkpoint activation. Although the effect is most dramatic for sequences immediately surrounding the break, mobility of all sequences (global mobility) is increased (Bordelet and Dubrana 2019; Zimmer and Fabre 2019). These changes are thought to facilitate the homology search during repair (Miné-Hattab and Rothstein 2013). Deletion of RAD51 or expression of the rad52-Δ409-412 allele eliminates global mobility, a result superficially consistent with the idea that Rad52 blocks mobility unless inhibited by direct interaction with Rad51. However, deletion of RAD52 alone prevents global mobility, implying that the Rad51-Rad52 interaction itself is needed to facilitate this response (Smith et al. 2018). Cells lacking both RAD51 and RAD52 display constitutive checkpoint activation and increased global mobility, suggesting that this regulatory mechanism can be bypassed under high levels of checkpoint activation (Smith et al. 2018). In particular, the opposite effects on global mobility in the RAD51 rad52 and rad51 rad52 strains argue that this phenomenon is mechanistically distinct from that affecting de novo telomere addition (where these strains behave identically), even though both phenotypes are regulated by interaction between Rad51 and Rad52.
Suppression of microhomology-mediated rearrangements:
During the course of our studies of de novo telomere addition, we fortuitously observed a dramatic increase in GCR events occurring centromere-proximal to the SiRTA on chromosome 9 in the absence of RAD51 (Epum et al. 2020). These events require RAD52 and are suppressed by the rad51-K191A variant, again suggesting that interaction between Rad52 and Rad51 is critical to limit GCR formation. These events require RAD59 and POL4 and consist of either non-reciprocal translocations or extremely large deletions involving 15–18 bp of microhomology. Here again, Rad51 appears to restrain an activity of Rad52 that promotes BIR and/or single-strand annealing type events. In that light, it is interesting to note that Rad51 inhibits strand annealing by Rad52 in vitro, even under conditions in which Rad51 is not directly associated with the single-stranded DNA (Wu et al. 2008; Sugiyama and Kantake 2009).
Regulation of checkpoint adaptation:
The genetic interactions described here are mirrored by effects observed by the Haber lab during the process of checkpoint adaptation (Lee et al. 2003). Cells that incur an irreparable DNA break accumulate high levels of ssDNA and arrest in G2/M. Despite failing to repair the break, cells eventually (after ~8 hours) release from the checkpoint and proceed through mitosis (Sandell and Zakian 1993; Toczyski et al. 1997; Lee et al. 1998; Coutelier and Xu 2019). The Haber lab reported that cells lacking RAD51 display a moderate checkpoint adaptation defect (cells remain arrested even after 24 hours) that is completely suppressed by loss of RAD52 (Lee et al. 2003). In perfect agreement with the effects on de novo telomere addition, the adaptation defect is observed for variants of Rad51 that fail to interact with Rad52, but adaptation is unaffected by the Rad51-K191A protein that retains Rad52 interaction but fails to form a Rad51 nucleofilament. These results suggest that engagement of Rad52 with the checkpoint is normally “turned off” through interaction with Rad51 (Lee et al. 2003). Whether the mechanism of this effect is directly related to that affecting de novo telomere addition is unclear. Haber and colleagues find that the rad51Δ adaptation phenotype is partially suppressed by rfa1-t11 (rfa1-K45E) (Umezu et al. 1998). The combination of rad52Δ with the rfa1-t11 allele results in an adaptation defect that is not seen with either single mutant, a result that is difficult to reconcile with a simple model in which sustained checkpoint signaling results solely from a Rad52-driven effect suppressed by interaction with Rad51. As suggested by Haber and colleagues, the phenotype of the rad52Δ rfa1-t11 strain may indicate that at least two signals converge to sustain the checkpoint after DNA damage, both of which have some dependence on RPA (Lee et al. 2003).
The striking similarity in the effects of Rad51 and Rad52 on adaptation and de novo telomere addition raise the possibility that failure to adapt to the checkpoint arrest is directly responsible for the reduced recovery of cells in which survival occurs through telomere addition at the SiRTA. Although deletion of RAD51 prevents cells with an unrepaired break from exiting the checkpoint, cells that repair the break are able to proceed through the cell cycle normally, so defective checkpoint recovery does not explain the lack of repair by telomere addition (Vaze et al. 2002; Lee et al. 2003). Importantly, de novo telomere addition frequencies at SiRTAs are unchanged from WT in strains lacking YKU80 or TID1, genes that are required for adaptation through different mechanisms from RAD51 (i.e. deletion of RAD52 does not suppress the adaptation defect in either strain) (Lee et al. 1998, 2001). Therefore, although the genetic requirements are very similar and interaction with Rad51 is required in each case to relieve an action by Rad52 (sustained checkpoint signaling or suppression of telomere addition), these two outcomes are independent.
Proposed mechanism for the regulation of de novo telomere addition by Rad51/Rad52
What then might account for the inhibitory effect of Rad52 on de novo telomere addition? One intriguing possibility is suggested by elegant single-molecule experiments from the Sung and Greene laboratories in which Rad52 is shown to alter the dissociation properties of RPA (Gibb et al. 2014b). Although RPA can persist on ssDNA for long periods of time in the absence of competing protein, additional RPA (or even bacterial single-strand binding protein; SSB) is sufficient to displace the bound protein (Gibb et al. 2014a). Since RPA binding stability results from multiple relatively weak DNA binding sites, transient partial (or micro-) dissociation provides an opportunity for other proteins to compete with RPA before reassociation occurs (Gibb et al. 2014a). Strikingly, addition of Rad52 to RPA-coated DNA substantially blocks displacement, suggesting that the micro-dissociative properties of the RPA complex are altered (Gibb et al. 2014b). The location of stabilized RPA along the ssDNA correlates with where Rad52 is physically bound, suggesting that this effect requires interaction between the two proteins. Remarkably, even in this context, addition of Rad51 results in efficient conversion to a Rad51 nucleofilament punctuated by “islands” of RPA/Rad52 (Gibb et al. 2014b).
Our prior work on the mechanism of telomere addition at SiRTAs indicated that binding of Cdc13 to the ssDNA generated by resection is crucial to stimulate de novo telomere addition (Obodo et al. 2016). Cdc13 is best understood as a telomere binding protein that recognizes the TG-rich telomeric repeat (Nugent et al. 1996). Cdc13, complexed with its binding partners Stn1 and Ten1, protects the 5’ terminating strand of the telomere from extensive resection and facilitates the recruitment of telomerase via interactions with the Est1 protein (Evans and Lundblad 1999; Rice and Skordalakes 2016). By ChIP, Cdc13 associates with single-stranded DNA generated on yeast chromosome 3 following a DSB induced at the MAT locus and association is reduced upon deletion of RAD51 (Oza et al. 2009). We speculated that binding of Cdc13 at a SiRTA would correlate with the level of de novo telomere addition and would be modulated by the RAD51/RAD52 genetic context. Indeed, while the association of Cdc13 is reduced upon RAD51 deletion, further removal of RAD52 completely suppresses that effect. Furthermore, recruiting Cdc13 to the SiRTA as a fusion protein with the Gal4 DNA binding domain (after replacing critical SiRTA sequences with the cis-acting DNA binding sequence for Gal4) eliminates the effect of RAD51 deletion (Epum et al. 2020), suggesting that the reason for reduced telomere addition in the absence of RAD51 is reduced recruitment of Cdc13 to the SiRTA.
Bringing these ideas together, we suggest that, in the absence of RAD51, the association of RPA with the 3’ ssDNA tail is altered through interaction with Rad52 and that the ability of Cdc13 to associate directly with SiRTA sequences is thereby compromised. In some manner that is not yet understood, the interaction of Rad51 with Rad52 (even in cases where Rad51 itself is incapable of binding the single-stranded DNA), is sufficient to relieve this inhibition, a prediction that could be tested using single-molecule approaches. Gibb et al. speculate that direct interaction between Rad52 and RPA is required to effect this change in RPA behavior since the stabilization of RPA against turnover is only observed on regions of the ssDNA simultaneously occupied by Rad52 (Gibb et al. 2014b). To address this idea, we examined the effect of the rfa1-44 allele (rfa1-G77D) (Firmenich et al. 1995; Hays et al. 1998) on de novo telomere addition. This mutation in the largest subunit of yeast RPA displays phenotypes indicative of defects in recombinational repair, but is strongly suppressed by Rad52 overexpression, suggesting that defects arise through failure to correctly interact with Rad52 (Firmenich et al. 1995). A strain expressing rfa1-44 displays normal levels of de novo telomere addition at SiRTA, but remarkably, combination of this allele with deletion of RAD51 shows complete suppression of the de novo telomere addition defect (Epum et al. 2020). This result is consistent with the hypothesis that de novo telomere addition is repressed in the absence of Rad51 through the action of Rad52 on RPA. In cells in which the Rfa1-Rad52 interaction is impaired, Rad52 no longer exerts this effect.
Are hotspots of de novo telomere addition common in the yeast genome?
Results described above show that the Rad51-Rad52 interaction regulates the probability of de novo telomere addition at two specific sites previously identified as hotspots for these events. The SiRTA located 35 kilobases from the left arm of chromosome 5 (SiRTA 5L-35) is a preferential target of de novo telomere addition in spontaneous GCR assays (Myung et al. 2001; Stellwagen et al. 2003; Zhang and Durocher 2010), indicating that repair at SiRTAs is relevant in wild-type cells under normal growth conditions. However, it is unclear how common such sequences are in the genome and therefore what impact such sequences might have on genome stability.
We have not done an exhaustive search for sequences that function as SiRTAs, but data we present here suggest that sequences with the capacity to stimulate de novo telomere addition at far above baseline levels are quite common in yeast. To identify additional SiRTAs, we have targeted sequences that lie in the non-essential terminal region of the chromosome arm and contain a high ratio of TG dinucleotides on the correct strand (i.e. these TG-sequences will be exposed by resection; Table 1). Since we had previously shown that function of a SiRTA is maintained when it is moved to new genomic location (Obodo et al. 2016), we have developed a “test site” for SiRTA activity on the left arm of chromosome 7 in a region lacking an endogenous hotspot of de novo telomere addition. Sequences of interest are integrated at the test site using CRISPR/Cas9 (see Methods in supplementary material). Distal to this location, the chromosome arm contains a recognition site for the HO endonuclease and the URA3 gene (Fig. 1A). Since this strain expresses the HO endonuclease under control of a galactose-inducible promoter, plating cells on media containing galactose results in a cycle of persistent cleavage that cells escape only by mutating or eliminating the HO cleavage site. Surviving cells are screened for loss of URA3, indicating truncation of the chromosome arm (a GCR event), and PCR is used to determine what fraction of the total GCR events have occurred within the test sequence. The efficiency of the SiRTA is defined as the percent of GCR events occurring within the integrated sequence. RAD52 is deleted in all strains to prevent the recovery of BIR-mediated non-reciprocal translocation events between the sequence inserted on chromosome 7 and its endogenous site.
Table 1.
Sequences tested for ability to stimulate de novo telomere addition.
| SiRTAa | sequence |
|---|---|
| 14L-35 | GTGTGGGCGGCAAGTGTTGTTGGTGCTCGTGGTGGTGCAGCGTGTTGTTGCTGCTATGTACGATTGGTGATT |
| 4L-13/10R-13b | GTGGTTTTGGTATATATGGTTGGTGCAATTATTCAGATCAGTTCGAATCACAAATGGTACCAATACTTTGTCGGTAAGATCATCTACGGTCTTGGTGCTGGTGGCTGTTCGGTGTTGTGT |
| 3R-07 | AAAATCGGTGACCGTGTTGGTGTTGGTGCCCAAGCCTTGGCGTGTTTTGAGTGTGAACGTTGCAAAAGTGACAACG |
| 9L-44 | TTCATCGACAAGGACTCGGGTAAAGGCGGTGGCGGTGGTGGTGGTGGAACGGCGCCACAAG |
| 5L-35 | GGATGAGTTGGTGTGGTGTTACTACTAGGATTTGGCGTGGATGAAGGACCTGCAGTGGAGGGTGTTGTTGTGGAGTTTT |
| 8L-50 | TGTGTCTCACAGTCAACGTCAGGTCCGTACAGAACGGTGTCTCGAGCCTTATCGCTGCACGCCATATACTGTGGATATAATCCGTAACGTCGCAGTAGCAACCGTTCGATGTTGTTGTGCTGTTGTTGTGCTGTTGTTGTTGTTGTTGTTGTTGTTGTTGCCGTTG |
| 14L-13/6L-11b | TAGTTTTATATATGTGTGTGTGTGTGTGTGAAATTCATTCCACA |
| 15L-20 | CTTTGTAGTCGCTGGGTCGCTTCTTGGTGTCATCGGAATGGGTCTTTTGTACAAATACAGGGTGGTATATGACGGGATTTCGGGTCTTATTGGCGCTGAGATTGTGGTCGGTATAGCTGGTGGT |
| 6L-22 | GTCATTTTTGGTGTTGGTGGTAATCTTCAAGCAACTGTAACAAAAGGTAGTGGTGGTTCCTATGAATCCCTATCATTTGCATGGGGGTTCGGTTGTATGCTTGGTGT |
Potential SiRTA sequences are shown. Sequences are named according to the chromosome arm (e.g. 14L) and distance in kilobases from the nearest telomere (e.g. 35). Sequences are oriented so that the nearest telomere is 3’ of the indicated sequence (to the right) to emphasize the TG-rich nature.
The identical sequence is found in two locations in the S. cerevisiae genome.
Figure 1. Naturally occurring sequences stimulate de novo telomere addition in response to a DSB.
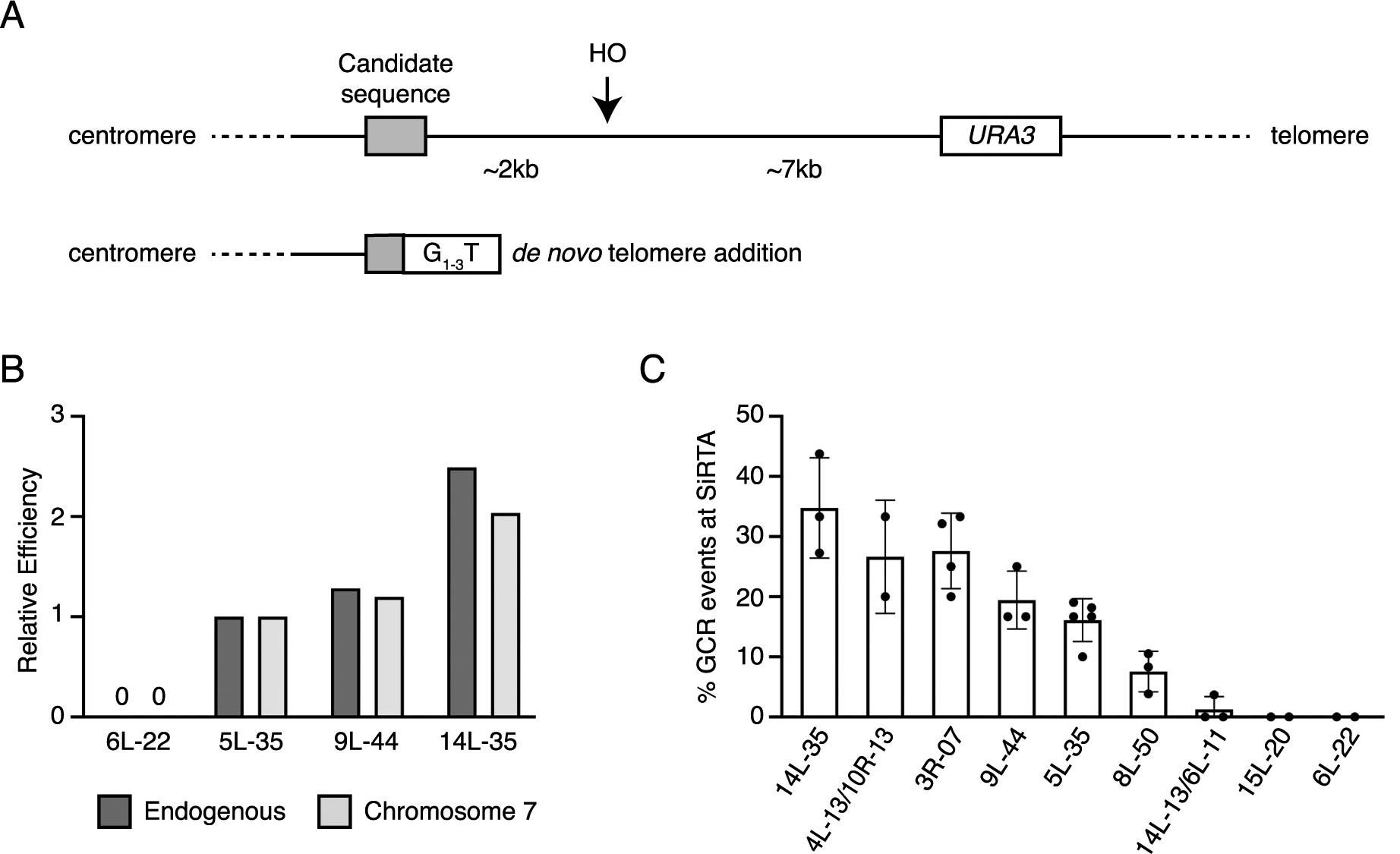
A) Schematic of HO cleavage assay. A DSB is generated at the HO cut site via the galactose-inducible HO endonuclease. Surviving colonies lacking the URA3 marker are selected on media containing 5-fluoroorotic acid (5-FOA) and repair events are mapped to the candidate SiRTA by PCR. Candidate SiRTAs can be tested in their endogenous location by integration of the HO recognition sequence and URA3 marker or can be integrated into a strain that already contains the HO cleavage site and URA3 marker on chromosome 7 using the CRISPR/Cas9 system (see Methods in supplementary materials). B) Relative efficiencies of the indicated SiRTAs tested on chromosome 7 and in their endogenous locations. Efficiencies are calculated as the percentage of GCR events that occur within the SiRTA. Events are normalized to the efficiency of SiRTA 5L-35 at each location. Data for SiRTAs 5L-35 and 9L-44 in their endogenous locations are from (Obodo et al. 2016). C) Percent of GCR events in each of the indicated candidate SiRTAs as measured on chromosome 7. Sequences analyzed are in Table 1. Data corresponding to this figure are in supplementary Table S1.
To verify this strategy, we measured telomere addition within four sequences at both their endogenous locations and after integration at the test site on chromosome 7 [published SiRTAs 5L-35 and 9L-44 (Obodo et al. 2016; Epum et al. 2020), unpublished SiRTA 14L-35, and a sequence found to be devoid of SiRTA activity]. For each active SiRTA, the efficiency measured on chromosome 7 is lower than in the endogenous location (for SiRTA 9L-44 this corresponds to efficiencies of 20% and 33%, respectively). However, when efficiencies are normalized to those of 5L-35, the relative efficiencies observed in the endogenous and chromosome 7 locations are similar (Fig. 1B). By Southern blot, 13 of 13 events observed at 5L-35 on chromosome 7 involve telomere addition (Ngo and Friedman, unpublished data), suggesting that most, if not all, GCR events identified in this assay involve telomere addition. A TG-rich sequence from chromosome 6 (6L-22) does not stimulate GCR formation in either its endogenous location or on chromosome 7 and serves as a negative control for the assay (Fig. 1B, C). Together, these results support use of the chromosome 7 test site to identify SiRTAs, although the extent to which these sites are utilized in their endogenous locations may be underestimated. In total, we have tested seven additional sequences for the capacity to function as SiRTAs. Of these, four are active and three demonstrate little or no ability to stimulate de novo telomere addition (including the negative control sequence described above). Three of the four new SiRTAs are substantially more active than SiRTAs 5L-35 and 9-44 that we have previously characterized (Fig. 1C). Because these newly identified SiRTAs are TG-rich, it is likely that they bind Cdc13 following resection and are subject to the same regulation by Rad51-Rad52 interaction described for SiRTAs 5L-35 and 9L-44 (Epum et al. 2020).
Final thoughts
As discussed in this review, the interaction between Rad51 and Rad52 serves to regulate multiple aspects of DNA repair including de novo telomere addition, micro-homology mediated recombination, resistance of the Rad51 nucleofilament to Srs2, global mobility, and checkpoint adaptation. Although it is tempting to attribute these phenomena to a common mechanism, differences in the genetic requirements in each case make it difficult to identify a single underlying theme. It is likely that Rad51 and Rad52 each affect the other upon their mutual interaction, perhaps accounting for the multiple outcomes regulated by this single protein-protein contact. The commonality between those situations in which the Rad51-Rad52 interaction is functionally relevant is the presence of extensive resection and activation of the checkpoint due to a single unrepaired break or to multiple breaks. Therefore, the cell may utilize interactions between these key repair proteins to modulate alternative outcomes, such as de novo telomere addition, when damage is persistent.
Multiple mechanisms inhibit de novo telomere addition (Churikov and Géli 2017), so it is surprising that interactions between Rad51 and Rad52 instead appear to stimulate these events. Importantly, the effects that we report are observed at specific DNA sequences that are telomere-like in sequence and support much higher rates of de novo telomere addition than surrounding sequences. It is an important unresolved question whether the interaction between Rad51 and Rad52 also affects telomere addition immediately adjacent to the HO cleavage site or at rare sites of telomere addition that are not associated with a hotspot. Answering this question will require detailed analysis of GCR events occurring outside of the SiRTA.
Why would a mechanism exist to increase de novo telomere addition despite the inevitable loss of terminal sequences? If SiRTAs were rare, we might speculate that the consequences of telomere addition at these sites would be small and that the observed effects of the Rad51-Rad52 interaction might instead be a secondary consequence of a different phenomenon. In contrast, results presented here suggest that endogenous sequences with the potential to stimulate de novo telomere addition are easily identified. Future work must focus on determining the genome-wide distribution of such sequences. One intriguing possibility is that SiRTAs might allow the cell to “cap” a chromosome break that would otherwise result in loss of essential sequences. If true, we would predict that sequences with the potential to act as SiRTAs might be preferentially found in the non-essential terminal regions of most chromosome arms. Although the SiRTAs shown in Figure 1 do follow this pattern, this is a consequence of our current bias to search for SiRTAs in these regions and highlights the need to develop methods to predict SiRTA function. If SiRTAs indeed play a role in mediating tolerance to DNA damage by allowing cells to survive an otherwise unrepairable break, the Rad51-Rad52 interaction may facilitate these events when significant resection has occurred.
Supplementary Material
Acknowledgements
We thank Sara Conwell and Blake Conwell for technical assistance. Many thanks to Dr. James Haber for gifts of strains and plasmids and for insightful comments on the manuscript. This work was supported by National Institutes of Health award R01GM123292 to KLF.
Footnotes
Conflicts of interest/Competing interests: The authors have declared that no competing interests exist
Availability of data and material (data transparency): Data summarized in Figure 1 are provided in supplementary table S1.
Ethics approval: N/A
Consent to participate: N/A
Consent for publication: N/A
Code availability: N/A
References
- Adzuma K, Ogawa T, Ogawa H (1984) Primary structure of the RAD52 gene in Saccharomyces cerevisiae. Mol Cell Biol 4:2735–2744. 10.1128/mcb.4.12.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antúnez de Mayolo A, Lisby M, Erdeniz N, et al. (2006) Multiple start codons and phosphorylation result in discrete Rad52 protein species. Nucleic Acids Res 34:2587–2597. 10.1093/nar/gkl280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Kupiec M (2004) DSB repair: the yeast paradigm. DNA Repair (Amst) 3:797–815. 10.1016/J.DNAREP.2004.04.013 [DOI] [PubMed] [Google Scholar]
- Bhargava R, Onyango DO, Stark JM (2016) Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32:566–575. 10.1016/j.tig.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordelet H, Dubrana K (2019) Keep moving and stay in a good shape to find your homologous recombination partner. Curr. Genet 65:29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churikov D, Géli V (2017) De novo telomere addition at chromosome breaks: Dangerous liaisons. J Cell Biol 216:2243–2245. 10.1083/jcb.201705156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutelier H, Xu Z (2019) Adaptation in replicative senescence: a risky business. Curr. Genet 65:711–716 [DOI] [PubMed] [Google Scholar]
- Elango R, Sheng Z, Jackson J, et al. (2017) Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8:1–13. 10.1038/s41467-017-01987-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epum EA, Mohan MJ, Ruppe NP, Friedman KL (2020) Interaction of yeast Rad51 and Rad52 relieves Rad52-mediated inhibition of de novo telomere addition. PLoS Genet 16:e1008608 10.1371/journal.pgen.1008608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SK, Lundblad V (1999) Est1 and Cdc13 as comediators of telomerase access. Science 286:117–120. 10.1126/science.286.5437.117 [DOI] [PubMed] [Google Scholar]
- Firmenich AA, Elias-Arnanz M, Berg P (1995) A novel allele of Saccharomyces cerevisiae RFA1 that is deficient in recombination and repair and suppressible by RAD52. Mol Cell Biol 15:1620–1631. 10.1128/mcb.15.3.1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Rudin N, Haber JE (1992) Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12:1292–1303. 10.1128/mcb.12.3.1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung CW, Fortin GS, Peterson SE, Symington LS (2006) The rad51-K191R ATPase-defective mutant Is impaired for presynaptic filament formation. Mol Cell Biol 26:9544–9554. 10.1128/mcb.00599-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B, Ye LF, Gergoudis SC, et al. (2014a) Concentration-dependent exchange of replication protein A on single-stranded DNA revealed by single-molecule imaging. PLoS One 9:e87922 10.1371/journal.pone.0087922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B, Ye LF, Kwon Y, et al. (2014b) Protein dynamics during presynaptic complex assembly on individual ssDNA molecules. Nat Struct Mol Biol 21:893 10.1038/NSMB.2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SL, Firmenich AA, Berg P (1995) Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55, and Rad57 proteins. Proc Natl Acad Sci U S A 92:6925–6929. 10.1073/pnas.92.15.6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SL, Firmenich AA, Massey P, et al. (1998) Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol Cell Biol 18:4400–4406. 10.1128/mcb.18.7.4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5:a012740 10.1101/cshperspect.a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, Malkova A (2018) Break-Induced Replication: The where, the why, and the how. Trends Genet 34:518–531. 10.1016/j.tig.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer KM, Haber JE (1993) New telomeres in yeast are initiated with a highly selected subset of TG1–3 repeats. Genes Dev 7:2345–2356. 10.1101/gad.7.12a.2345 [DOI] [PubMed] [Google Scholar]
- Krejci L, Damborsky J, Thomsen B, et al. (2001) Molecular dissection of interactions between Rad51 and members of the recombination-repair group. Mol Cell Biol 21:966–976. 10.1128/MCB.21.3.966-976.2001 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Krejci L, Song B, Bussen W, et al. (2002) Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J Biol Chem 277:40132–40141. 10.1074/jbc.M206511200 [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38:233–271. 10.1146/annurev.genet.38.072902.091500 [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, et al. (1998) Saccharomyces Ku70, Mre11/Rad50, and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94:399–409. 10.1016/S0092-8674(00)81482-8 [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Malkova A, et al. (2001) The Saccharomyces recombination protein Tid1p is required for adaptation from G2/M arrest induced by a double-strand break. Curr Biol 11:1053–1057. 10.1016/S0960-9822(01)00296-2 [DOI] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Vaze MB, et al. (2003) Yeast Rad52 and Rad51 recombination proteins define a second pathway of DNA damage assessment in response to a single double-strand break. Mol Cell Biol 23:8913–8923. 10.1128/mcb.23.23.8913-8923.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Dupaigne P, Maloisel L, et al. (2018) Rad52-Rad51 association is essential to protect Rad51 filaments against Srs2, but facultative for filament formation. Elife 7:. 10.7554/eLife.32744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangahas JL, Alexander MK, Sandell LL, Zakian VA (2001) Repair of chromosome ends after telomere loss in Saccharomyces. Mol Biol Cell 12:4078–4089. 10.1091/mbc.12.12.4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Haber JE (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6:a016428 10.1101/cshperspect.a016428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné-Hattab J, Rothstein R (2013) DNA in motion during double-strand break repair. Trends Cell Biol. 23:529–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Chen C, Kolodner RD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411:1073–1076. 10.1038/35082608 [DOI] [PubMed] [Google Scholar]
- Nugent CI, Hughes TR, Lue NF, Lundblad V (1996) Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 274:249–252. 10.1126/science.274.5285.249 [DOI] [PubMed] [Google Scholar]
- Obodo UC, Epum EA, Platts MH, et al. (2016) Endogenous hot spots of de novo telomere addition in the yeast genome contain proximal enhancers that bind Cdc13. Mol Cell Biol 36:1750–1763. 10.1128/mcb.00095-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouenzar F, Lalonde M, Laprade H, et al. (2017) Cell cycle-dependent spatial segregation of telomerase from sites of DNA damage. J Cell Biol 216:2355–2371. 10.1083/jcb.201610071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, et al. (2009) Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23:912–927. 10.1101/gad.1782209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennaneach V, Putnam CD, Kolodner RD (2006) Chromosome healing by de novo telomere addition in Saccharomyces cerevisiae. Mol Microbiol 59:1357–1368. 10.1111/j.1365-2958.2006.05026.x [DOI] [PubMed] [Google Scholar]
- Piazza A, Heyer WD (2019) Moving forward one step back at a time: reversibility during homologous recombination. Curr. Genet 65:1333–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Pennaneach V, Kolodner RD (2004) Chromosome healing through terminal deletions generated by de novo telomere additions in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 101:13262–13267. 10.1073/pnas.0405443101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice C, Skordalakes E (2016) Structure and function of the telomeric CST complex. Comput. Struct. Biotechnol. J 14:161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandell LL, Zakian VA (1993) Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell 75:729–739 [DOI] [PubMed] [Google Scholar]
- Seong C, Colavito S, Kwon Y, et al. (2009) Regulation of Rad51 recombinase presynaptic filament assembly via interactions with the Rad52 mediator and the Srs2 anti-recombinase. J Biol Chem 284:24363–24371. 10.1074/jbc.M109.032953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Bryant EE, Rothstein R (2018) Increased chromosomal mobility after DNA damage is controlled by interactions between the recombination machinery and the checkpoint. Genes Dev 32:1242–1251. 10.1101/gad.317966.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE (2003) Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 17:2384–2395. 10.1101/gad.1125903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Wang X, Haber JE (2003) In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell 12:209–219 [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kantake N (2009) Dynamic regulatory interactions of Rad51, Rad52, and Replication Protein-A in recombination intermediates. J Mol Biol 390:45–55. 10.1016/J.JMB.2009.05.009 [DOI] [PubMed] [Google Scholar]
- Sung P, Stratton SA (1996) Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J Biol Chem 271:27983–27986. 10.1074/jbc.271.45.27983 [DOI] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, Hartwell LH (1997) CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90:1097–1106. 10.1016/S0092-8674(00)80375-X [DOI] [PubMed] [Google Scholar]
- Umezu K, Sugawara N, Chen C, et al. (1998) Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics 148:989–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasianovich Y, Altmannova V, Kotenko O, et al. (2017) Unloading of homologous recombination factors is required for restoring double‐stranded DNA at damage repair loci. EMBO J 36:213–231. 10.15252/embj.201694628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, et al. (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10:373–385. 10.1016/S1097-2765(02)00593-2 [DOI] [PubMed] [Google Scholar]
- Waterman DP, Haber JE, Smolka MB (2020) Checkpoint responses to DNA double-strand breaks. Annu Rev Biochem 89:. 10.1146/annurev-biochem-011520-104722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC (2008) Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem 283:14883–14892. 10.1074/jbc.M801097200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Durocher D (2010) De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes Dev 24:502–515. 10.1101/gad.1869110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C, Fabre E (2019) Chromatin mobility upon DNA damage: state of the art and remaining questions. Curr. Genet 65 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.