

Abstract
Background.
Cystic fibrosis (CF) patients develop severe lung disease including chronic airway infections, neutrophilic inflammation, and progressive fibrotic remodeling in airways. However, cellular and molecular processes that regulate excessive collagen deposition in airways in these patients remain unclear. Fibrocytes are bone marrow (BM)-derived mesenchymal cells that express the hematopoietic cell marker CD45, and mesenchymal cell markers and implicated in collagen deposition in several fibrotic diseases. It is unknown whether fibrocytes accumulate in the lungs of CF patients, so the current study evaluates the presence of fibrocytes in the fibrotic lesions of airways in explanted CF lungs compared to non-CF unused donor lungs (control).
Methods.
We used immunofluorescence staining to determine if fibrocytes accumulate in explanted CF lungs compared to healthy donor lungs. Simultaneously, we evaluated cells collected by bronchoalveolar lavage (BAL) in CF patients using multi-color flow cytometry. Finally, we analyzed transcripts differentially expressed in fibrocytes isolated from the explanted CF lungs compared to control to assess fibrocyte-specific pro-fibrotic gene networks.
Results.
Our findings demonstrate fibrocyte accumulation in CF lungs compared to non-CF lungs. Additionally, fibrocytes were detected in the BAL of all CF children. Transcriptomic analysis of fibrocytes identified dysregulated genes associated with fibrotic remodeling in CF lungs.
Conclusions.
With significantly increased fibrocytes that show increased expression of pro-fibrotic gene transcripts compared to control, our findings suggest an intervention for fibrotic remodeling as a potential therapeutic target in CF.
Keywords: Cystic fibrosis, pulmonary fibrosis, fibrocytes, lung, CFTR
1. Introduction
Cystic fibrosis (CF) is an inheritable multi-system disease due to deficient chloride ion transport at the cell membrane of epithelial cells, especially in the lower airways, that leads to significant lung disease. CF is caused by mutations in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR) protein (1, 2). CF-associated mutations result in defective CFTR protein synthesis, processing or function, and often lead to fibrosis in multiple organs including the liver, pancreas and lung (1, 2). In addition to defective CFTR-dependent chloride transport, bicarbonate transport may be impaired and sodium absorption may be enhanced in the airways. Thick mucus gland secretions, airway mucus obstruction and susceptibility to chronic infection then ensue (2-4). The pathophysiologic mechanisms underlying CF lung disease that lead to bronchiectasis, myofibroblast accumulation and progressive collagen deposition, also referred to as bronchial wall thickening, remain undefined (3, 5). Previous fetal and animal work in CF suggests CF pro-fibrotic abnormalities manifest during the early stages of CF lung diseases before the establishment of chronic infection of the lower airways (6, 7). However, strong evidence indicates that the CF host inflammatory response is excessive and chronic inflammation and fibrosis directly cause bronchiectasis and respiratory failure as the disease progresses (6).
The mechanisms governing fibrotic lung remodeling due to the CFTR mutations in CF are not clearly defined. Currently, there is uncertainty whether early infection and inflammation initiate this pro-fibrotic phenotype. Also, the lack of an animal model that mimics clinical features of CF lung disease has reduced the progress on identifying mechanisms underlying fibrosis and other clinical abnormalities in CF. However, recent development of CF pigs that lack CFTR or express mutant CFTR (F508del) has created a phenotype similar to human disease including intestinal, pancreatic and airway disease. Moreover, newborn piglets with CF have developed airflow obstruction and air trapping in part due to tracheal structural abnormalities, in the absence of inflammation and mucus obstruction (8). Several studies provide evidence that pro-fibrotic growth factors, such as TGFβ, function as modifier genes in CF and may contribute to the severity of lung disease by activating pro-fibrotic pathways (3, 5). Morphometric analysis of TGFβ signaling, myofibroblast differentiation, and collagen deposition in the lungs of CF, IPF and non-CF patients demonstrate that pulmonary fibrosis is prominent in CF and is comparable to IPF (3). Additionally, unresolved neutrophilic inflammation, and altered IL-17A- and Th2 cytokine-driven signaling may also contribute to fibrosis and severe lung disease (3, 5, 9). Other studies have suggested that EGFR ligands, such as TGFα and cytokines including TNFα, IL-13, and IL-17, are abundant in CF compared to non-CF lungs (5, 10). Experimental models of chemical-induced lung injury or overexpression of TGFα have demonstrated that mesenchymal cells accumulate and excessive production of ECM leads to fibrotic remodeling (11, 12). Further understanding of the cellular and molecular mechanisms underlying fibrosis in CF should be a priority as the development of therapies that can inhibit or delay the onset of fibrosis and subsequent organ failure in CF could be essential in extending the life and/or delaying the need for lung transplantation.
Several fibrotic lung diseases are characterized by the progressive accumulation of lung mesenchymal cells, the subsequent expansion of fibrotic lesions and the ultimate loss of lung function (13). In CF, the cellular sources of the lung fibroblasts in fibrotic lesions remain unknown. Fibrocytes are bone marrow (BM)-derived stromal cells that express a variety of cell-surface markers related to leukocytes, hematopoietic progenitor cells, and fibroblasts (14). In tissues, fibrocytes have been shown to become alpha-smooth muscle actin (αSMA)-positive myofibroblasts, but this occurs in limited numbers (15). In patients with interstitial lung disease, the number of circulating fibrocytes has been shown to correlate with lung remodeling (16). Modulating the mechanisms that drive fibrocyte recruitment to the lung, and targeting chemokines involved in fibrocyte migration, have been shown to reduce fibrotic burden in mice (17). Studies from our laboratory and others have identified a specific, endogenous mechanism of pronounced and progressive pulmonary fibrosis driven by fibrocytes in the lung (15, 18). Specifically, we have found that fibrocytes are tissue invasive and sufficient to augment the proliferative expansion of lung-resident fibroblasts of the fibrotic lesions in the lung (15).
Currently, it is not clear whether fibrocytes accumulate in the pathogenesis of fibrotic lung disease in humans with defective CFTR. In this study, we tested whether fibrocytes accumulate in explanted CF lungs with the F508del mutation from patients who underwent lung transplantation. We performed systematic validation of fibrocyte presence in the lungs and identified fibrocyte-specific gene networks that may involve in fibrotic lung remodeling.
2. Materials and methods
2.1. Human tissue samples
Cells from BAL fluid were collected according to the Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Review Board (IRB) approved protocol (IRB # 2014-3309). In addition, de-identified explanted lung tissue specimens collected from the distal areas of lung were acquired using research protocols approved by the University of North Carolina and CCHMC (IRB # 2017-6850). CF was diagnosed according to clinical standards with an elevated sweat chloride test and two copies of a CFTR-deficient mutation. Lung tissue samples were obtained from CF patients with F508del CFTR homozygous mutation who had undergone lung transplantation. In addition, lung tissue was acquired from donors with no history of prior chronic lung disease (control group) (age range 16-39 years, attempting to match the CF cohort).
2.2. Lung mesenchymal cell culture
Human lung stromal cell culture was performed as described previously (19). We performed a detailed macroscopic evaluation of the explanted lung that was cut sagittally resulting in two gross sections to identify normal or remodeled areas in the distal lung. From the normal donor lungs, we selected distal lung tissue with a soft consistency to finger palpation, appeared red in color due to normal tissue perfusion and had no mucus plugging or cyst formation. We used the CF lung tissues with moderate to severe damage that had a firm consistency to finger palpation, appeared pale in color (due to scar) and that had airways filled with mucus. Thus, the distal lung parenchyma selected with small airways was cut into 2X2 cm pieces. Each piece was finely minced and digested in DMEM media containing collagenase (2mg/ml) by incubating at 37°C for 1 hr. Digested tissue was passed through a 100μm filter, washed twice by centrifugation at 100 X g for 5 min. After the digestion, ten million cells were cultured onto 100-mm tissue-culture plates in 10 ml DMEM supplemented with 10% bovine calf serum, 2 mM L-glutamine and a cocktail of antibiotics including Ceftazidime (100μg/ml), Tobramycin (80μg/ml), Vancomycin (100μg/ml), Ciprofloxacin (20μg/ml), Nystatin (100U/ml) and Doxycycline (4μg/ml). Mesenchymal cells adhered to plates and migrate away from the larger remaining tissue pieces. The lung mesenchymal cells were maintained in culture until they reached 80% confluency, and non-adherent floating cells and dead cells were washed away with regular media changes. Fibrocytes (CD45+ Col1+) from control and CF lung cultures were isolated on day 9 using anti-CD45 magnetic beads and the cell purity was greater than 94%, as assessed by flow cytometry (15).
2.3. RNA-sequencing, bioinformatics analysis and RT-PCR
Total RNA was prepared from fibrocytes purified from distal lung mesenchymal cell cultures of explanted end-stage CF lungs and healthy donor lungs. Total cellular RNA was isolated using RNeasy midi kit (Qiagen, Germantown, MD) and the purity of the total RNA was assessed using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE). We confirmed the integrity of RNA using an Agilent 2100 Bioanalyzer and performed RNA-seq using an Illumina HiSeq-1000 sequencer (Illumina, San Diego, CA) as described previously (20). Using TopHat aligner (21), reads were aligned to the reference genome based on the current gene definitions, and those aligned with low confidence were filtered out using SAM tools (22). These processes resulted in approximately 25 million reads on an average per sample. The number of reads (read counts) aligning to the gene’s coding region was summarized using Short Read (23) and various R- Bioconductor (24) packages (IRanges, GenomicRanges, Biostrings, Rsamtools). The differential gene expression analysis between groups was performed using the negative binomial statistical model of reading counts as implemented in the edgeR Bioconductor package (25). The cluster analysis of all genes differentially expressed in individual comparisons is performed using the Bayesian infinite mixture models (26). Gene transcripts were selected based on log2 fold change of ±1.5 (up or down) with a P-value cut-off of 0.05, resulting in 2818 genes in total (N=3/group) and a heat map was generated. The ToppFun application of ToppGene Suite (27) was utilized to identify the top enriched biological processes regulated by differentially expressed genes in CF fibrocytes compared to normal fibrocytes. Real-time PCR was performed using the CFX384 Touch Real Time PCR detection system and SYBR select master mix (Bio-Rad, Hercules, CA) as described previously (19). We have included human beta actin gene as an invariant endogenous control to correct sample to sample variations and have avoided errors in the normalization by using good quality RNA in equal amounts and also performed melt curve analysis to exclude primer sets producing non-specific PCR products. Real-Time PCR primer details are provided in table S1.
2.4. Flow Cytometry
The total BAL fluid cells or primary fibroblasts from lung cultures (0.5-1×106) were stained using anti-CD45 (clone HI30 & 30-F11), and collagen I-FITC (clone 5D8-G9) antibodies as described in our previous studies (20). Data were acquired using LSRII flow cytometer (BD Biosciences). BAL cells stained with isotype IgG and single stain control antibodies were used for compensation and gating. Data were analyzed using FlowJo software (TreeStar, Ashland,OR).
2.5. Immunofluorescence and confocal imaging
Human lung tissue sections were embedded in OCT medium and frozen lung sections were prepared as described previously (20). Lung section were co-immunostained using antibodies against CD45 (ab10558, Abcam) and vimentin (sc-7557, Santa Cruz Biotechnology) followed by secondary antibodies conjugated with Alexa Fluor 488 and Alexa Fluor 568. Lung sections were stained with DAPI for nucleus visualization and confocal Z-stack images were obtained using a Nikon AIR-A1 laser scanning microscope. 3D rendering from confocal Z-stacks was performed using the “animation” tool in Imaris (version 9.2, Bitplane) software. To quantify the vimentin or CD45 and vimentin-positive cells, six random high magnification images were obtained from each lung section and quantified using MetaMorph imaging software (Molecular Devices, CA, USA) as described (19). Also, immunostainings were performed on serial lung sections with antibodies against vimentin (anti-human vimentin, ab137321; Abcam), and CD45 (rabbit anti-CD45; Ventana, Tucson, AZ, USA) as described previously (15). Images were obtained using a Leica DM2700 M bright-field microscope (Leica Microsystems, Buffalo Grove, IL, USA).
2.6. Statistics
All data were analyzed using Prism (Version 5; GraphPad, La Jolla, CA, USA). Student’s t-test was used to compare two experimental groups. Data were considered statistically significant for P values less than 0.05.
3. Results
3.1. Fibrocytes accumulate in fibrotic lung lesions of CF patients
In order to identify fibrocytes, we co-immunostained the lung sections from F508del CFTR homozygous (n=6) and non-CF (n=4) donors with antibodies against CD45 and vimentin. We observed an increased number of mesenchymal cells that are dual positive for vimentin and CD45 in CF lungs compared to non-CF controls (Figure 1A). Also, we observed a significant increase in the total number of vimentin-positive mesenchymal cells in CF lungs compared to non-CF controls (Figure 1B). Further, we quantified the number of fibrocytes and observed a significantly greater total number of fibrocytes in CF lungs compared to non-CF controls (Figure 1C). 3D rendering of confocal images reveals the presence of fibrocytes that are positive for both CD45 and vimentin in CF lungs (Supplementary Video 1). In order to further identify an increase in mesenchymal cells, we performed immunostaining on serial sections of CF and non-CF lungs using antibodies against vimentin and CD45. In the serial sections, we observed an increase in cells positive for both vimentin and CD45 in CF lungs compared to non-CF disease controls (Figure S1).
Figure 1. Presence of fibrocytes in the lungs of cystic fibrosis patients.

(A) Frozen lung sections from non-CF (n=4) and CF (n=6) individuals were co immunostained using CD45 and vimentin. Images are representative of each group. Arrows indicating CD45 & vimentin double-positive cells. (60X, Scale bar: 50μm) (B) Percent of vimentin-positive cells were quantified from each group using metamorph imaging software. (C) Percent fibrocytes (CD45+ and Vimentin+) were calculated using metamorph imaging software. DAPI stained cells were quantified for total cell number. *P < 0.05, **P < 0.005. Unpaired t-test.
3.2. Fibrocytes identified in BAL specimens of pediatric CF patients
To determine whether fibrocytes accumulate in the lower airways of patients, we obtained cryopreserved BAL from CF patients. CF patients were recruited at the CCHMC CF Center, with all patients between 2-16 years of age. The clinical characteristics of the CF patients are summarized in Table 1. To identify fibrocytes in the BAL fluid of CF patients, cells were stained for CD45, and Collagen1 and analyzed using flow cytometry. FACS analysis identified that about 10.3% of the population in the total BAL cells were positive for fibrocyte markers, CD45, and Col1 (Figure 2 & Table 1).
Table 1:
Clinical parameters of cystic fibrosis patients
| Clinical Parameters | CF (N=11) |
|---|---|
| Age (years) median [range] | 11 [2.8-16.7] |
| Sex (male/female) | 5/6 |
| FEV1 % Predicted | 92 ± 6.5 |
| FEV1 (L) | 1.8 ± 0.15 |
| FVC (L) | 2.3 ± 0.23 |
| Macrophages (%) | 19 ± 0.05 |
| PMNs (%) | 79 ± 0.05 |
| Lymphocytes (%) | 3 ± 0.01 |
| Fibrocytes (%) | 9.5 ± 1.5 |
Figure 2. Identification of fibrocytes in BAL cells of cystic fibrosis patients.

Representative flow cytometry plots showing the gating strategy used to identify fibrocytes that are double-positive for CD45 and collagen 1 observed using FACS analysis of total BAL cells isolated from cystic fibrosis patients. For FACS analysis, the gates were set based on isotype antibody stained controls.
3.3. Differential expression of fibrosis-associated transcripts by fibrocytes in CF patients.
To identify fibrocyte-specific transcripts involved in CF lung disease, we isolated fibrocytes from lung stromal cell cultures of non-CF and F508del CFTR homozygous donor lung tissue and compared transcripts that were differentially expressed more than 1.5-fold (Table S2). As shown in figure 3, there was a larger group of genes subject to change in the fibrocytes of CF lungs compared to non-CF lungs. The F508del CFTR mutation resulted in up- and down-regulation of 879 and 1939 transcripts, respectively. Gene ontology analysis revealed the upregulation of genes involved in positive regulation of cell migration, negative regulation of wound healing, muscle tissue contraction and development, and downregulation of genes involved in CFTR dependent ion channels, F508del interactome, Nrf2 interactome, and epithelial-stromal cell crosstalk. In addition, we performed the validation of differentially expressed genes using RT-PCR. Transcripts of genes involved in CFTR dependent regulation of ion channels such as CFTR, KCNN4, and SPINT1 are downregulated in CF lung derived fibrocytes (Figure 4A). Notably, gene transcripts associated with the F508del interactome (CDH3 and CTSD), and Nrf2 interactome (SERPINA1 and ELN) were significantly downregulated in CF lung fibrocytes compared to control fibrocytes (Figure 4B and 4C). Conversely, the transcripts of profibrotic genes such as SELP, SERPINB2, and FGF1 are upregulated in CF fibrocytes compared to normal fibrocytes. These genes have been implicated in increased migration, delayed wound healing, and muscle tissue development and contraction (Figure 4D).
Figure 3. Differential gene expression analysis of CF Fibrocytes shows fibrosis-associated transcriptional changes in CF lungs.

Heat map of fibrocyte-specific transcripts isolated from fibrotic CF lungs compared to normal lungs was analyzed using RNA-sequencing (N=3). Gene ontology classification of highly regulated processes and selected example genes are listed on the right.
Figure 4. CF fibrocytes show dysregulated transcriptome.
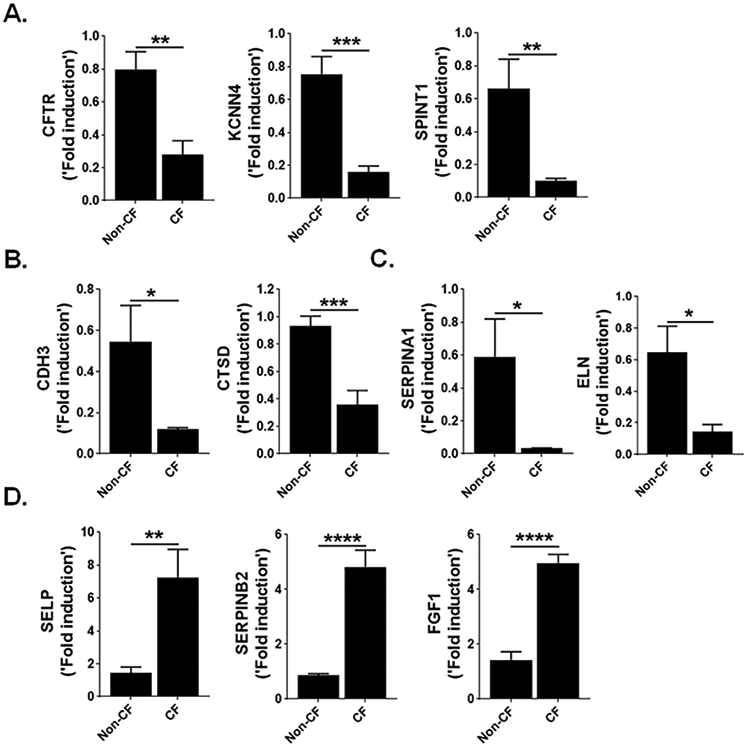
(A) Quantification of genes CFTR, KCNN4 and SPINT1 in fibrocytes isolated from non-CF and CF patient lungs using RT-PCR. (B) Quantification of genes CDH3 and CTSD in fibrocytes isolated from non-CF and CF patient lungs using RT-PCR. (C) Quantification of genes SERPINA1 and ELN in fibrocytes isolated from non-CF and CF patient lungs using RT-PCR. (D) Quantification of genes SELP, SERPINB2 and FGF1 in fibrocytes isolated from non-CF and CF patient lungs using RT-PCR. The relative expression level is expressed as fold induction relative to that in corresponding non-CF fibrocytes following normalization to the expression of human β-actin. All data are presented as mean ± SEM (n=6). *P < 0.05, **P < 0.005, ***P < 0.0005, and ****P < 0.00005. Unpaired t-test.
4. Discussion
Fibrocytes are a subset of macrophages that originate from bone marrow and migrate to injured areas to perform pro-fibrotic functions. Fibrocytes have been shown to increase in circulation and progressively accumulate in injured lungs in multiple chronic lung diseases including asthma, acute respiratory distress syndrome (ARDS), systemic sclerosis, and IPF(28-31). We identified that a significant number of fibrocytes accumulate in explanted CF lungs that are extensively remodeled.
Unremitting inflammation and fibrosis of the lower airways is a major cause of morbidity and mortality in CF patients. For the first time, we identified that fibrocytes marked with CD45+ and Col1 accumulate in the distal areas of the CF lungs. Importantly, we also identified that fibrocytes are present in the BAL fluid of younger CF patients. Although we cannot define that fibrocytes are associated with disease due to our study design, our findings suggest that their presence could be contributing to CF lung disease as their levels are typically undetectable in BAL fluid of healthy controls (32). Therefore, our findings support the need for additional studies with a larger cohort of CF patients to further investigate if fibrocytes in BAL fluid or circulation of CF accumulate and can contribute to lung disease. Multiple studies using chemical-induced lung injury models demonstrate that circulating fibrocytes migrate to the lung in response to the local release of cytokine and chemoattractants such as the C-C motif chemokine ligand CCL2, CCL5 and CXCL12/CXCR4 axis (17, 33, 34). Notably, disruption of these pathways that drive recruitment of circulating fibrocytes to the lung, and targeting chemokines involved in fibrocyte migration, reduce fibrotic burden in mouse models of pulmonary fibrosis (15, 17, 35). Studies from our laboratory and others show that multiple factors secreted by fibrocytes act in a paracrine manner to activate resident fibroblasts, stimulating their proliferation and differentiation during the pathogenesis of pulmonary fibrosis (20, 36). In particular, the adoptive transfer of fibrocytes was sufficient to augment the fibrotic burden in injured lungs but not in uninjured lungs during bleomycin-induced pulmonary fibrosis (20, 37). However, it is plausible that lung resident macrophages or fibrocytes expanded locally to contribute to increased fibrocyte numbers in CF lungs or other fibrotic lung diseases. The lack of CFTR expression has been shown to cause impaired resolution of inflammatory cells and increased damage in CF airway epithelia and lung fibrosis with LPS challenge or naphthalene injury in CF mice compared to wildtype mice (38, 39). Therefore, it is important to understand the mechanisms underlying fibrocyte recruitment or activation, and their contribution to the fibrotic burden in the CF lung.
Here, starting from the identification of fibrocytes in CF airways, we identified gene networks that were selectively dysregulated in CF fibrocytes compared to control fibrocytes using next-generation RNA-seq analysis. The loss of CFTR expression in CF fibrocytes suggests that CFTR exerts its regulatory role in fibrocyte-driven fibrotic lung disease. Specifically, gene ontology analysis revealed the downregulation of CFTR-dependent ion channels, delta-508 interactome, Nrf2 interactome, and genes involved in stromal-epithelial crosstalk. The expression of CFTR by fibrocytes is consistent with previous reports on CFTR expression by non-epithelial cells including macrophages, smooth muscle cells, and endothelial cells (40, 41). Zhang et al showed that macrophages derived from peripheral blood monocytes of CF patients have low levels of CFTR expression compared to non-CF macrophages (42). Moreover, previous studies show that during oxidative stress decreased CFTR expression is due to the transcriptional repression but not due to decreased stability (43, 44). However, future studies are warranted to understand the transcriptional regulation and functions of CFTR in fibrocytes. Detailed transcriptome analysis of CF fibrocytes allowed us to hypothesize that the loss of CFTR was associated with the activation of a pro-fibrotic gene program in CF fibrocytes. Also, the observed decrease in the expression of genes associated with delta-508 interactome (CDH3, CTSD), nrf2 interactome (SERPINA1, ELN), and stromal epithelial cross-talk (MMP9, LCN2) suggest that CFTR in fibrocytes might play a role in fibrotic lung remodeling. Therefore, future studies are warranted to identify the potential role of CFTR in fibrocyte activity and fibrotic remodeling in the CF lung. Additionally, various genes involved in positive regulation of cell migration (SELP, SMOC2, and PTGER3), negative regulation of wound healing (SERPINB2, AJAP1, GP5), and muscle tissue development and contraction (FGF1, MYH13, GATA4) were upregulated in CF fibrocytes. CF fibrocytes display an increase in the transcripts of FGF1, a pro-fibrotic growth factor implicated in muscle tissue development and regeneration. Published studies have shown FGF1 to cause growth and migration of both endothelial and smooth muscle cells. Moreover, vascular abnormalities and smooth muscle remodeling are significant factors that contribute to lung pathology in patients with CF (45-47).
CF lung disease in humans shares several features of fibrotic remodeling seen in IPF, including myofibroblast accumulation and excessive collagen deposition (48). Fibrocytes have been implicated in the pathogenesis of several chronic inflammatory diseases such as necrotizing enterocolitis (NEC), asthma, Graves’ disease, and rheumatoid arthritis (49-52). Consistent with previous work, we detected a significant number of fibrocytes in the BAL fluid of CF patients. In particular, fibrocyte induction in Grave’s disease and NEC has been shown to aggravate an inflammatory response. Nonetheless, defining the presence and role of fibrocytes in chronic fibrotic lung regions and BAL fluid warrants future investigations into the contribution of fibrocytes in CF lung fibrosis and inflammation.
In summary, we found fibrocyte accumulation in the lungs of patients with endstage CF lung disease. Our data show fibrocyte presence in the BAL cells of children with CF. Importantly, fibrocytes isolated from CF lungs have dysregulated transcriptome and pathways associated with CFTR dysfunction and lung pathology. Further studies are needed to investigate the mechanistic contribution of fibrocytes to fibrotic lung remodeling in CF.
Supplementary Material
Highlights.
Fibrocytes accumulate in the lungs of end-stage CF patients compared to control lungs.
Fibrocytes presence is observed in total BAL cells of CF patients.
CF fibrocyte gene expression profiles demonstrate marked changes in fibrosis-associated gene networks.
Acknowledgments
This research was supported by funds from Cystic Fibrosis Foundation (CFF) CLANCY15R0 (JPC), CFF BOUCHE15R0 (SHR), DoD W81XWH-17-1-0666 (SKM), NIH 1R01 HL134801 (SKM), NIH AI137309 (SKM) and NIH P30DK065988 (SHR). We would like to thank Flow Cytometry Core at Cincinnati Children’s Hospital Medical Center for assistance with flow cytometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no financial and personal relationships with other people or organizations that could influence our work.
REFERENCES
- 1.Zhang W, Fujii N, Naren AP. Recent advances and new perspectives in targeting CFTR for therapy of cystic fibrosis and enterotoxin-induced secretory diarrheas. Future medicinal chemistry. 2012;4(3):329–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowe SM, Clancy JP. Advances in cystic fibrosis therapies. Current opinion in pediatrics. 2006;18(6):604–13. [DOI] [PubMed] [Google Scholar]
- 3.Harris WT, Kelly DR, Zhou Y, Wang D, MacEwen M, Hagood JS, et al. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PloS one. 2013;8(8):e70196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicologic pathology. 2007;35(1):116–29. [DOI] [PubMed] [Google Scholar]
- 5.Hardie WD, Bejarano PA, Miller MA, Yankaskas JR, Ritter JH, Whitsett JA, et al. Immunolocalization of transforming growth factor alpha and epidermal growth factor receptor in lungs of patients with cystic fibrosis. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 1999;2(5):415–23. [DOI] [PubMed] [Google Scholar]
- 6.Hubeau C, Puchelle E, Gaillard D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. The Journal of allergy and clinical immunology. 2001;108(4):524–9. [DOI] [PubMed] [Google Scholar]
- 7.Awadalla M, Miyawaki S, Abou Alaiwa MH, Adam RJ, Bouzek DC, Michalski AS, et al. Early airway structural changes in cystic fibrosis pigs as a determinant of particle distribution and deposition. Annals of biomedical engineering. 2014;42(4):915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adam RJ, Michalski AS, Bauer C, Abou Alaiwa MH, Gross TJ, Awadalla MS, et al. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med. 2013;188(12):1434–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184(2):252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2013;187(6):621–9. [DOI] [PubMed] [Google Scholar]
- 11.Singh B, Kasam RK, Sontake V, Wynn TA, Madala SK. Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L796–L806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L741–9. [DOI] [PubMed] [Google Scholar]
- 13.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136–51. [DOI] [PubMed] [Google Scholar]
- 14.Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PloS one. 2009;4(10):e7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madala SK, Edukulla R, Schmidt S, Davidson C, Ikegami M, Hardie WD. Bone marrow-derived stromal cells are invasive and hyperproliferative and alter transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;50(4):777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(7):588–94. [DOI] [PubMed] [Google Scholar]
- 17.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, et al. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166(3):675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keeley EC, Mehrad B, Strieter RM. Fibrocytes: bringing new insights into mechanisms of inflammation and fibrosis. Int J Biochem Cell Biol. 2010;42(4):535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasam RK, Reddy GB, Jegga AG, Madala SK. Dysregulation of Mesenchymal Cell Survival Pathways in Severe Fibrotic Lung Disease: The Effect of Nintedanib Therapy. Front Pharmacol. 2019;10:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sontake V, Shanmukhappa SK, DiPasquale BA, Reddy GB, Medvedovic M, Hardie WD, et al. Fibrocytes Regulate Wilms Tumor 1-Positive Cell Accumulation in Severe Fibrotic Lung Disease. J Immunol. 2015;195(8):3978–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan M, Anders S, Lawrence M, Aboyoun P, Pages H, Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25(19):2607–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ihaka Ra, G R. R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics 1996;5:299–344. [Google Scholar]
- 25.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8(9):1765–86. [DOI] [PubMed] [Google Scholar]
- 26.Freudenberg JM, Sivaganesan S, Wagner M, Medvedovic M. A semi-parametric Bayesian model for unsupervised differential co-expression analysis. BMC Bioinformatics. 2010;11:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37(Web Server issue):W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore BB, Kolb M. Fibrocytes and progression of fibrotic lung disease. Ready for showtime? Am J Respir Crit Care Med. 2014; 190(12):1338–9. [DOI] [PubMed] [Google Scholar]
- 29.LaPar DJ, Burdick MD, Emaminia A, Harris DA, Strieter BA, Liu L, et al. Circulating fibrocytes correlate with bronchiolitis obliterans syndrome development after lung transplantation: a novel clinical biomarker. Ann Thorac Surg. 2011;92(2):470–7; discussion 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest. 2010;90(6):812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353(1):104–8. [DOI] [PubMed] [Google Scholar]
- 32.Borie R, Quesnel C, Phin S, Debray MP, Marchal-Somme J, Tiev K, et al. Detection of alveolar fibrocytes in idiopathic pulmonary fibrosis and systemic sclerosis. PLoS One. 2013;8(1):e53736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, et al. New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114(3):438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inomata M, Kamio K, Azuma A, Matsuda K, Kokuho N, Miura Y, et al. Pirfenidone inhibits fibrocyte accumulation in the lungs in bleomycin-induced murine pulmonary fibrosis. Respiratory research. 2014;15:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sontake V, Gajjala PR, Kasam RK, Madala SK. New therapeutics based on emerging concepts in pulmonary fibrosis. Expert Opin Ther Targets. 2019;23(1):69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleaveland KR, Velikoff M, Yang J, Agarwal M, Rippe RA, Moore BB, et al. Fibrocytes are not an essential source of type I collagen during lung fibrosis. J Immunol. 2014;193(10):5229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruscia EM, Zhang PX, Barone C, Scholte BJ, Homer R, Krause DS, et al. Increased susceptibility of Cftr−/− mice to LPS-induced lung remodeling. Am J Physiol Lung Cell Mol Physiol. 2016;310(8):L711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carvalho-Oliveira IM, Charro N, Aarbiou J, Buijs-Offerman RM, Wilke M, Schettgen T, et al. Proteomic analysis of naphthalene-induced airway epithelial injury and repair in a cystic fibrosis mouse model. J Proteome Res. 2009;8(7):3606–16. [DOI] [PubMed] [Google Scholar]
- 40.Tousson A, Van Tine BA, Naren AP, Shaw GM, Schwiebert LM. Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol. 1998;275(6):C1555–64. [DOI] [PubMed] [Google Scholar]
- 41.Vandebrouck C, Melin P, Norez C, Robert R, Guibert C, Mettey Y, et al. Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir Res. 2006;7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, Shrestha CL, Kopp BT. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci Rep. 2018;8(1):17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rab A, Bartoszewski R, Jurkuvenaite A, Wakefield J, Collawn JF, Bebok Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am J Physiol Cell Physiol. 2007;292(2):C756–66. [DOI] [PubMed] [Google Scholar]
- 44.Bartoszewski R, Rab A, Twitty G, Stevenson L, Fortenberry J, Piotrowski A, et al. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem. 2008;283(18):12154–65. [DOI] [PubMed] [Google Scholar]
- 45.Hannon K, Kudla AJ, McAvoy MJ, Clase KL, Olwin BB. Differentially expressed fibroblast growth factors regulate skeletal muscle development through autocrine and paracrine mechanisms. J Cell Biol. 1996;132(6):1151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook DP, Rector MV, Bouzek DC, Michalski AS, Gansemer ND, Reznikov LR, et al. Cystic Fibrosis Transmembrane Conductance Regulator in Sarcoplasmic Reticulum of Airway Smooth Muscle. Implications for Airway Contractility. Am J Respir Crit Care Med. 2016;193(4):417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hays SR, Ferrando RE, Carter R, Wong HH, Woodruff PG. Structural changes to airway smooth muscle in cystic fibrosis. Thorax. 2005;60(3):226–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris WT, Boyd JT, McPhail GL, Brody AS, Szczesniak RD, Korbee LL, et al. Constrictive Bronchiolitis in Cystic Fibrosis Adolescents with Refractory Pulmonary Decline. Ann Am Thorac Soc. 2016;13(12):2174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Qingjuan S, Gao Z, Deng C, Wang Y, Guo C. Circulating fibrocytes are involved in inflammation and leukocyte trafficking in neonates with necrotizing enterocolitis. Medicine (Baltimore). 2017;96(26):e7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Douglas RS, Afifiyan NF, Hwang CJ, Chong K, Haider U, Richards P, et al. Increased generation of fibrocytes in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2010;95(1):430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shipe R, Burdick MD, Strieter BA, Liu L, Shim YM, Sung SS, et al. Number, activation, and differentiation of circulating fibrocytes correlate with asthma severity. J Allergy Clin Immunol. 2016;137(3):750–7 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galligan CL, Siminovitch KA, Keystone EC, Bykerk V, Perez OD, Fish EN. Fibrocyte activation in rheumatoid arthritis. Rheumatology (Oxford). 2010;49(4):640–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.